

Abstract
Cumulative progress in nanoparticle development has opened a new era of targeted delivery of therapeutics to cancer cells and tissue. However, developing proper detection methods has lagged behind resulting in the lack of precise evaluation and monitoring of the systemically administered nanoparticles. RNA nanoparticles derived from the bacteriophage phi29 DNA packaging motor pRNA have emerged as a new generation of drugs for cancer therapy. Multifunctional RNA nanoparticles can be fabricated by bottom-up self-assembly of engineered RNA fragments harboring targeting (RNA aptamer or chemical ligand), therapeutic (siRNA, miRNA, ribozymes, and small molecule drugs), and imaging (fluorophore, radiolabels) modules. We have recently demonstrated that RNA nanoparticles can reach and target intracranial brain tumors in mice upon systemic injection with little or no accumulation in adjacent healthy brain tissues or in major healthy internal organs. Herein, we describe various functional imaging methods (fluorescence confocal microscopy, flow cytometry, fluorescence whole body imaging, and magnetic resonance imaging) to evaluate and monitor RNA nanoparticle targeting to intracranial brain tumors in mice. Such imaging techniques will allow in-depth evaluation of specifically delivered RNA therapeutics to brain tumors.
Keywords: RNA nanoparticle, pRNA, MRI, Fluorescence imaging, Bioluminescence imaging, Confocal microscopy, Glioma, Xenograft
1 Introduction
For high-grade gliomas, conventional treatments involving surgical resection, radiation, and concurrent chemotherapy often result in poor survival and high recurrence [1, 2]. A number of nanoparticle-based strategies mainly based on lipids, polymers, inorganic, and organic materials are being developed for targeting glioma [3–9]. For effective glioma therapy, nanoparticles must be delivered systemically and specifically target intracranial tumors with minimal toxicity and side effects. Recently, RNA nanotechnology has emerged as a new generation platform for biological and medical applications [10–12]. The packaging RNA (pRNA) of the bacteriophage phi29 DNA packaging motor is a highly versatile molecule [13] and can be used to construct varieties of RNA nanoparticles with precise control of shape, size, and stoichiometry [10, 14–21]. Various functional groups, such as imaging agents; targeting ligands; and therapeutic modules including siRNA, miRNA, or ribozymes can be easily integrated into the pRNA scaffold while retaining the three-dimensional folding and authentic functionalities of the incorporated functional modules [15–18]. More practically, nontoxic and nonimmunogenic properties [21] coupled with favorable biodistribution and pharmacokinetic profiles [15–18, 21, 22] make the RNA nanoparticles an attractive candidate for the development of cancer therapeutic agents. Recently, we demonstrated that RNA nanoparticles fabricated using the ultra-stable pRNA-3WJ motif as a scaffold [15] can target intracranial glioma tumors in mice with little or no collateral damage to healthy tissues [22]. Herein, various functional imaging techniques are described to detect and monitor the targeted delivery of RNA nanoparticles harboring imaging agents and therapeutic siRNAs to glioma-bearing mice. These functional detection methods will be beneficial to researchers for developing multifunctional RNA nanoparticles for disrupting pathways involved in glioma pathogenesis.
2 Materials
All reagents should be analytical grade and RNase- and DNase-free to prevent RNA degradation. All the solutions should be prepared using Milli-Q water (18.2 MΩ•cm at 25 °C resistivity) treated with diethylprocarbonate (DEPC) and then autoclaved. All tubes and glassware used in preparation of buffers and reagents should be autoclaved. Gloves and lab coats should be worn at all times.
2.1 Materials
V1 grade MICA substrate, 25 mm × 75 mm (Ted Pella).
3-Aminopropyltriethoxysilane (APTES) (Life Technologies).
N,N-di-isopropylethylamine (DIPEA) (Life Technologies).
Argon, ultrapure.
2 L vacuum desiccator.
Tris base.
Magnesium chloride.
Potassium chloride.
Sodium chloride.
PBS (Phosphate Buffer Saline) tablets.
5 min epoxy (Ted Pella).
Magnetic AFM support disks (Ted Pella).
HPLC grade water.
AFM cantilevers (Bruker).
Milli-Q water (18.2 MΩ•cm at 25 °C resistivity).
Urea, for molecular biology, DNase-free, RNase-free, and protease-free.
DMEM (Dulbecco's Modified Eagle Medium).
Ethanol.
EDTA (Ethylenediaminetetraacetic acid).
SDS (Sodium Dodecyl Sulfate).
Ammonium acetate.
Sodium acetate.
APS (Ammonium Persulfate).
TEMED (Tetramethylethylenediamine).
Boric acid.
Bis-acrylamide.
Acrylamide, 99 + %, electrophoresis grade.
RPMI-1640 medium.
Folate-free RPMI 1640 medium.
Fetal bovine serum.
0.25 % Trypsin–EDTA.
Athymic female nu/nu mice (Jackson Laboratory).
Isoflurane.
Buprenorphine.
ProLong® Gold Antifade Mountant with DAPI (Life Technologies).
Glass cover slides.
Lab-TekII 8-well chamber.
Luciferin.
PFA (Paraformaldehyde).
PermaFluor Mountant (Thermo Scientific).
DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) (Sigma-Aldrich).
Ketamine (Phoenix Pharmaceuticals).
Magnevist (Bayer Health Care Pharmaceuticals).
2.2 Equipment
Typhoon FLA 7000 gel imaging system (GE Healthcare).
Spectrophotometer.
pH meter.
Freezer (−80 °C and −20 °C) and 4 °C refrigerator.
Milli-Q water purification system.
Microwave.
PCR thermocycler.
Centrifuges.
PAGE system.
Handheld UV lamp.
Hamilton syringes.
Biosafety cabinet.
Flow cytometer.
Cell culture fl asks, plates, and other supplies.
Cell viability analyzer (Vi-Cell XR, Beckman Coulter).
Cell culture incubator.
Olympus 4-filter-based FluoView FV1000-Filter Confocal Microscope System (Olympus).
IVIS Spectrum in vivo Imaging System.
Magnetic Resonance Imaging system (BioSpec 94/30).
Multimode SPM with Nanoscope IV controller (Bruker).
Stereotactic apparatus.
Mouse restrainer.
2.3 Reagent Preparation
Phosphate-buffered saline (PBS, pH 7.4, consisting of 0.01 M phosphate buffer, 0.0027 M potassium chloride, and 0.137 M sodium chloride): Dissolve PBS tablets (Sigma-Aldrich) in Milli-Q water and filter with 0.22 μm filter for sterilization (see Note 1).
TMS buffer: 50 mM Tris, pH 8.0, 100 mM NaCl, and 10 mM MgCl 2 (see Note 2).
Synthesize RNA fragments by enzymatic transcription or chemical synthesis using RNA synthesizer (see Note 3).
Urea-denaturing PAGE gel, 10–15 % (w/v): Combine 10–15 % (w/v; 37.5:1) acrylamide, 8 M urea, 10 % (w/v) APS and TEMED.
Native PAGE gel (TBM or TBE): 10–15 % (w/v): Combine 10–15 % (w/v; 37.5:1) acrylamide, 1× Tris–borate buffer (pH 7.8), 10 mM MgCl2 (or 2 mM EDTA), 10 % (w/v) APS and TEMED.
Paraformaldehyde (PFA) solution 4 % (w/v): Prepare by diluting 16 % (w/v) PFA solution (EM grade, Electron Microscopy Sciences) in PBS (see Note 4).
Tris–borate EDTA (TBE) buffer: 89 mM Tris, 200 mM boric acid, 2 mM EDTA.
Tris–borate magnesium (TBM) buffer: 89 mM Tris, 200 mM boric acid, 5 mM MgCl2.
Elution buffer, 1×: Mix 0.5 M ammonium acetate, 10 mM EDTA, and 0.1 % (w/v) SDS in 0.05 % (v/v) DEPC-treated water.
3 Methods
3.1 In vitro RNA Synthesis and Construction of RNA Nanoparticles
Synthesize pRNA-3WJ building block modules as desired by in vitro transcription or chemical synthesis (IDT or Trilink), as previously described [15–18]. For end-labeled Folate (targeting ligand) or Alexa647 (imaging module), chemically synthesize the RNA fragments (IDT or Trilink).
Check the synthesized RNA fragments using 8–12 % polyacrylamide gel electrophoresis (PAGE) under denaturing (8 M Urea) conditions.
Locate and excise the corresponding bands from the gel under UV shadow (see Note 5).
Incubate the gel pieces in the elution buffer over 4 h at 37 °C (see Note 6).
Ethanol precipitate overnight at −20 °C by adding 2.5 volume of 100 % ethanol and 1/10 volume of 3 M NaOAc (see Note 7).
Pellet the precipitated RNA by centrifugation at 16,500 × g for 30 min (see Note 8).
Wash the RNA pellet with 70 % ethanol, and collect the RNA pellet by centrifugation at 16,500 × g for 30 min.
Dry the RNA pellet by using speed vacuum (see Note 9).
Resuspend the dried RNA pellet in 0.05 % DEPC-treated water (or TMS or PBS buffer) and store at −20 °C until further use (see Note 3).
Check RNA nanoparticle assembly from individual RNA fragments using 8–12 % native PAGE gels [15–18].
3.2 Atomic Force Microscopy Imaging to Characterize the Structure of RNA Nanoparticles
3.2.1 APTES Functionalization of MICA Substrates
Assemble a mount that can hold the MICA sheets inside the desiccator (see Note 10).
Cut two plastic caps from Eppendorf tubes and place at the bottom of the desiccator.
Place the desiccator under vacuum and fill with argon (see Note 11).
Freshly cleave a sheet of MICA to 0.05–0.1 mm thickness and mount inside the desiccator (see Note 12).
Fill the plastic caps with 30 μl APTES and 10 μL DIPEA, respectively (see Note 13).
Close the lid and leave for ∼2 h to functionalize.
Remove the reagents and purge the desiccator with Argon gas for 5 min.
Leave the mica sheets overnight to cure and store under Argon atmosphere.
3.2.2 AFM Imaging in Tapping Mode in Air (Fig. 1b)
Fig. 1.
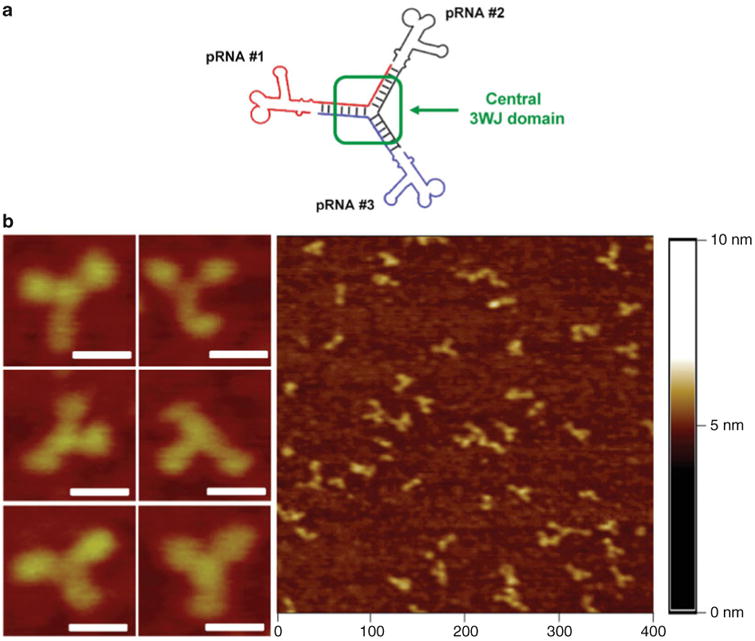
Construction of thermodynamically stable trivalent pRNA 3-way junction (3WJ) nanoparticles. (a) Three pRNA molecules bound at the pRNA-3WJ core sequence (black, red, and blue), and (b) its accompanying AFM images; scale bar = 30 nm. Figures reproduced with permission from ref. [15] © Nature Publishing Group
Prepare a 20 ng/μL RNA solution in TMS buffer (see Note 14).
Cut a ∼1 × 1 cm piece of AP-MICA and glue to an AFM specimen support disk.
Place 5–10 μL of RNA solution on the MICA and incubate for 2–3 min.
Flush the sample thoroughly but gently with 2 mL of HPLC grade water (see Note 15).
Leave the samples to dry on air overnight (see Note 16).
Mount the sample on the AFM stage, load the tip, and align the photodiodes according to the manufacturer's instructions (see Note 17).
Tune the AFM probe to find its resonance frequency and engage the surface (see Note 18).
Slowly reduce the set-point voltage until the surface is visible, optimize the feedback gains and start imaging.
3.3 Flow Cytometry Analysis for In Vitro Detection of Folate-Mediated Human Brain Tumor Cell Targeting (Fig. 2)
Fig. 2.
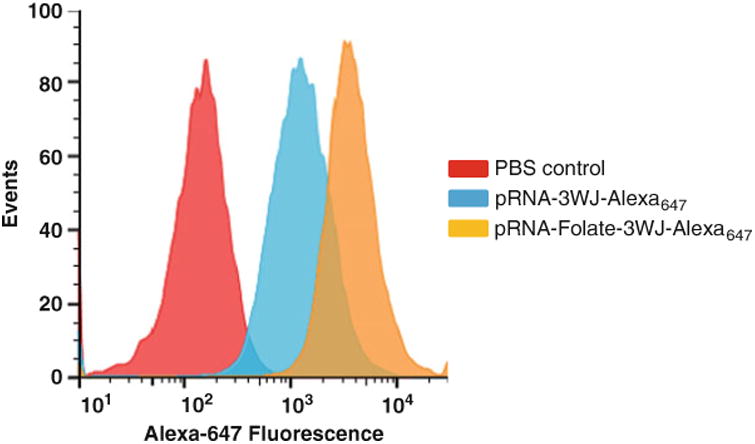
Flow cytometry analysis for in vitro detection of folate-mediated human brain tumor cell targeting. Folate-dependent targeting of human glioma patient-derived GB30 cells by pRNA-3WJ-Folate-Alexa647 or pRNA-3WJ-Alexa647 was analyzed by flow cytometry. PBS was used as gating control (red peak) to locate the cell population targeted by pRNA-3WJ-Folate-Alexa647 (yellow peak) compared to the negative control pRNA-3WJ-Alexa647 (blue peak)
Plate human glioma cells maintained in DMEM/10 % FBS in 6-well 1 day before RNA nanoparticle binding (see Note 19).
Wash the cells with PBS twice.
Incubate the cells with 200 nM of either pRNA-3WJ-Folate-Alexa 647 or pRNA-3WJ-Alexa647 in 200 μL of culture media for 2 h in 37 °C CO2 incubator (see Note 20).
Wash the cells with PBS twice.
Harvest the cells by trypsinization followed by washing with PBS (see Note 21).
Fix the cells in 4 % paraformaldehyde solution for 2 h at 4 °C (see Note 22).
Wash the cells with PBS for 3 times at room temperature (see Note 23).
Resuspend the final PBS washed cells in 200 μL of PBS and transfer into a 5 mL size flow cytometry tube.
Shield the tubes from light until subjected to flow cytometry (see Note 24).
Measure fluorescence from pRNA-3WJ-Folate-Alexa647 associated cells by flow cytometry (comparable to BD FACS Aria- III Cell Sorter).
Analyze the raw data by FlowJo 7.6.1 software to quantify the fluorescence intensity (see Note 25).
3.4 Fluorescence Confocal Microscopy for In Vitro Detection of Folate-Mediated Human Cancer Cell Targeting (Fig. 3)
Fig. 3.
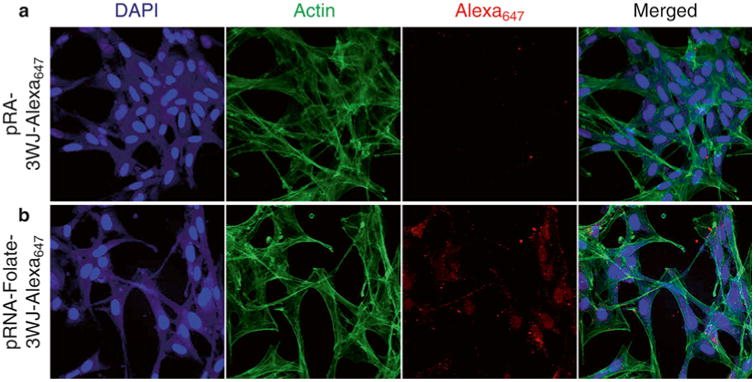
Fluorescence confocal microscopy for in vitro detection of folate-mediated human cancer cell targeting. Human glioblastoma cell U87ΔEGFR treated with 200 nM of pRNA-3WJ-Alexa647 (a) or pRNA-3WJ-Folate-Alexa647 (b) nanoparticles were analyzed immunofl uorescence confocal microscopy. Pseudocolor was used for nuclear (blue), cytoskeleton (green), and Alexa647 (red)
Plate 2 × 103 human glioma cells maintained in 200 μL in DMEM/10 % FBS in Lab-TekII 8-well chamber slide (Nunc).
Next day, wash the cells with 200 μL of PBS twice (see Note 26).
Incubate the cells with 200 nM of either pRNA-3WJ-Folate-Alexa 647 nanoparticles (or controls without Folate) in 200 μL of culture media for 2 h in 37 °C CO 2 incubator (see Note 27).
Aspirate the RNA nanoparticles solution and wash the cells with PBS three times.
Fix the cells in 4 % paraformaldehyde solution for 2 h at 4 °C (see Note 22).
Wash the cells with PBS for three times at room temperature (see Note 26).
Incubate the cells with Alexa488-Phalloidin (Invitrogen) for 30 min at room temperature to stain the cytoskeleton.
Wash with PBS three times.
Incubate the cells with 0.01 % DAPI solution for 10 min at room temperature to counterstain the nucleus (see Note 28).
Rinse the cells with PBS three times (see Note 26).
Mount the cells with PermaFluor Mountant (Thermo Scientific) and cover with a coverslip glass (see Note 29).
Perform fluorescence confocal microscopy.
Detect the cell nucleus stained by DAPI at the wavelengths of 461 nm, the cytoskeleton stained by Alexa488-Phalloidin at 530 nm, and pRNA-3WJ-Alexa647 at 665 nm (see Note 30).
Analyze the images by Olympus FluoView Viewer v4.0 (Olympus). Overlapping fluorescence signals from both fluorophores indicates their physical association in the context.
3.5 Brain Tumor Implantation in Mouse Model System (Fig. 4)
Fig. 4.
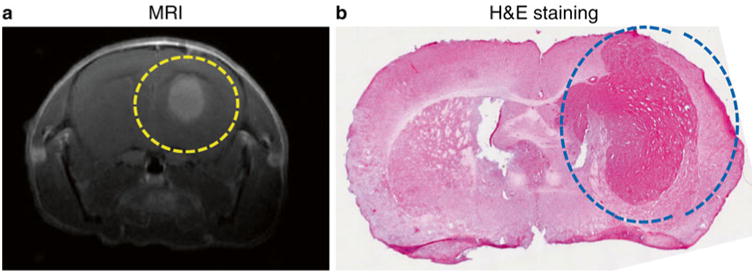
Brain tumor implantation in mouse model system. Brain tumor was induced in immunocompromised mouse by introducing tumorigenic human glioblastoma cell U87ΔEGFR via intracranial surgery only into right hemisphere. The implanted brain tumor (circled dot line) was identified by in vivo magnetic resonance imaging (MRI) (a) and hematoxylin and eosin (H&E) staining on fixed brain tissue (b)
Purchase 6-week-old athymic female nu/nu mice (Jackson Laboratory or comparable supplier). House and handle the experimental animals in accordance with the animal care guidelines approved by the Institutional Review Board and NIH.
Feed the mice with folate-free diet (Harlan) at least for 2 weeks before an injection of folate-harboring RNA nanoparticles to reduce the serum level of free folate in mice blood stream.
Anesthetize the mouse by intraperitoneal injection of ketamine.
Position the anesthetized mouse in a stereotactic apparatus.
Drill a burr hole at 2 mm lateral to the bregma, to a depth of 3 mm.
Inject 1 × 105 human glioma cells per mouse in 2 μL volume resuspended in Hank's buffered salt solution (HBSS) (see Note 31).
Record the date as Day 0.
3.6 Magnetic Resonance Imaging (MRI) for Location and Size of Implanted Brain Tumors in Mice (Fig. 4)
On the indicated day postsurgery of intracranial tumor injection, determine the location and size of the implanted tumors by MRI.
Anesthetize the mouse with 2.5 % isoflurane mixed with 1 L/min carbogen (95 % O2 with 5 % CO2), then maintain with 1 % isoflurane thereafter (see Note 32).
Maintain core temperature using a warm water bath.
Image using a Bruker 94/30 magnet (Bruker) or a comparable model (see Note 33).
Inject the mouse via intraperitoneal administration with 0.5 mmol/kg Magnevist, gadolinium-based contrast agent (Bayer Health Care Pharmaceuticals).
Insert the mouse into the MRI detection chamber.
Collect T2-weighted RARE (Rapid Acquisition with Refocused Echoes) imaging using a sequence (TR = 3524 ms, TE = 36 ms, rare factor = 8, navgs = 2, FOV = 20 × 20 mm, 0.5 mm slice thickness).
Collect T1-weighted RARE imaging using a sequence (TR = 1224 ms, TE = 12 ms, rare factor = 4, navgs = 4, FOV = 20 × 20 mm, 0.5 mm slice thickness) about 20 min postcontrast injection.
Manually outline region of interest (ROI) based on contrast in signal intensity between brain and tumor tissue (see Note 34).
3.7 Ex vivo Fluorescence Imaging on Brain Tumor-Bearing Mice (Fig. 5)
Fig. 5.
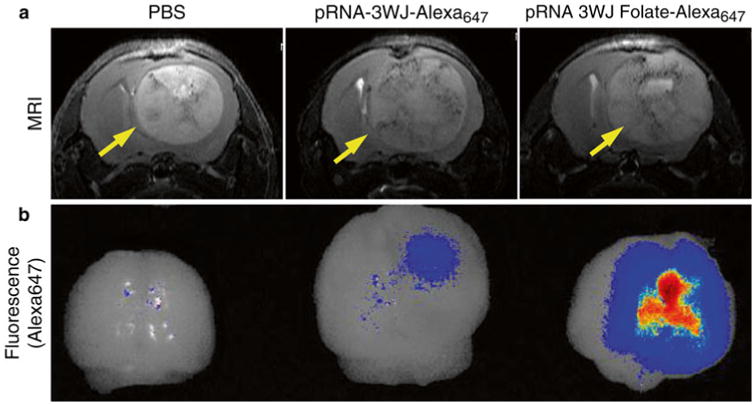
Ex vivo fluorescence imaging on tumor-bearing mice brain. Mouse brain tumor induced by human glioblastoma cell U87ΔEGFR was systemically targeted by pRNA-3WJ-Folate-Alexa647 in comparison to pRNA-3WJ- Alexa647 and PBS as negative controls. RNA nanoparticles with tumor size and RNA dose-dependent manner. The intracranial tumors were located by in vivo MRI (top). The Folate-mediated targeting by pRNA-3WJ nanoparticle was evaluated at 15 h after systemic injection of RNA nanoparticles by ex vivo fluorescence imaging (bottom)
Implant brain tumor as described in Subheading 3.5.
At 15 days posttumor implantation, inject pRNA-3WJ nanoparticles dissolved in 100 μL of PBS through tail vein (see Note 35).
Sacrifice the mouse at 15 h postinjection by cervical dislocation under anesthesia in CO2 chamber.
Dissect out the brain in the dark place (see Note 36).
Transfer the brain to IVIS Lumina Series III Pre-clinical In Vivo Imaging System (Perkin Elmer) or comparable system.
Position the mouse brain on the stage inside the dark chamber.
Preset the focus on the brain by using bright field CCD camera.
Acquire fl uorescence imaging on the positioned brains with excitation at 640 nm and emission at 660 nm for 2 min exposure (see Note 37).
Convert the raw data to express the fluorescence intensity as Mean Radiant Efficiency [p/s/cm2/sr]/[μW/cm2], and further normalize by tumor volume (mm3).
3.8 In vivo Bioluminescence Whole Body Imaging for Detection of siRNA Delivery into Brain Tumor (Fig. 6)
Fig. 6.
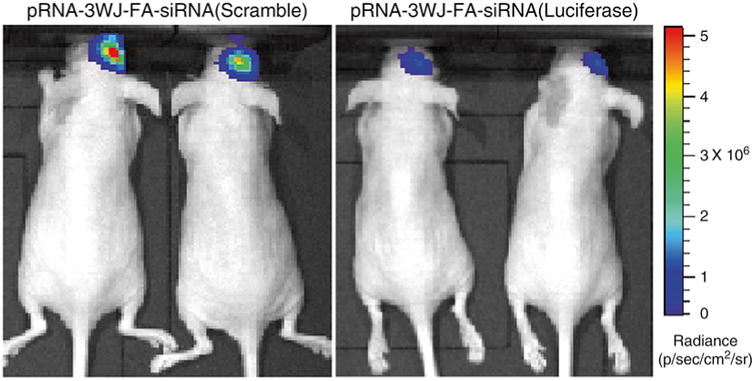
In vivo bioluminescence whole body imaging for detection of siRNA delivery into brain tumor. Gene silencing effect of pRNA-3WJ-Folate-siRNA(Luciferase) nanoparticles in human glioblatoma cell U87ΔEGFR-Luc derived mice brain tumors was evaluated by in vivo bioluminescence whole body imaging after systemic delivery. The change of luciferase activity from the brain tumors was compared to pRNA-3WJ-Folate-siRNA(Scramble) injected mice brains as negative controls. Pseudocolor was used to express the mean radiance (p/s/cm2/sr) of luciferase activity
Implant brain tumor in a mouse as described in Subheading 3.5 (see Note 38).
Inject the brain tumor-bearing mice with RNA nanoparticles harboring siRNA against luciferase gene constituted in total volume of 100 μL of PBS through tail vein (see Note 37).
Before/after the injection, subject the mouse to bioluminescence whole body imaging to detect endogenous luciferase expression level (see Note 39).
Inject the mouse with 75 mg/kg Luciferin (Perkin Elmer) (see Note 40).
Anesthetize the mouse in isoflurane-vapored chamber.
Position the anesthetized mouse on the stage inside the dark chamber. Keep the animal under anesthesia with low flow of isoflurane gas (see Note 41).
Acquire bioluminescence from the anesthetized mice by IVIS Lumina Series III Pre-clinical In Vivo Imaging System (Perkin Elmer) or comparable system.
Convert the raw data to express the luminescence intensity as Mean Radiance [p/s/cm2/sr]/[μW/cm2]. Normalize the Mean Radiance by tumor volume (mm3). Bioluminescence intensity will correspond to the expression level of luciferase gene that a mouse bears.
4 Notes
Sterilize the PBS solutions by autoclaving at 121 °C for 20 min. Cool down to and store at room temperature. Do not freeze or store in a refrigerator since phosphates will irreversibly precipitate.
Sterilize the TMS solution by autoclaving at 121 °C for 20 min.
Aliquot the RNA samples in small volumes and freeze them before use. Thaw the frozen RNA solution on ice. Avoid frequent freezing and thawing of RNA samples to preserve the quality of RNA samples.
Preformulated 4 % PFA solution is also available commercially. Keep refrigerated for long-term storage.
Conventional gel imager is usually equipped with short-wavelength UV lamp, which is strong enough to cause chemical break on phosphodiester bonds on RNA strand. We recommend using a handheld UV lamp with long wavelength in a dark place.
The volume of elution buffer to be used can be determined by measuring the weight of gel piece. Use 1 mL of elution buffer for each gram of gel.
Alternatively, RNA can be precipitated at −80 °C for 20 min. However, overnight precipitation at −20 °C usually gives better results. If isopropyl alcohol is used instead of ethanol, use 1.5 × volume.
After centrifugation, the RNA may be visible as a white pellet if there is a high amount of RNA. However in many cases, the RNA pellet is not visible. To avoid disturbing the RNA pellet, be careful not to touch the area by a pipette tip when adding 70 % ethanol.
Be careful not to overdry the RNA pellet. Medium heat is recommended.
Anything that holds the MICA sheets safely above the surface is feasible, such as mounting the sheets on a rod with paper clips or fixing them on top of a pipette tip box.
Use an overflow gauge to ensure pressure does not build up inside the desiccator due to the vacuum grease seal.
MICA sheets come in different thicknesses. Typically a MICA sheet can be cleaved multiple times to obtain 0.05–0.1 mm thickness. Label the outside surface to ensure only cleaved surfaces are used for sample preparation.
Fresh APTES should be used. If the surface shows heterogeneities after functionalization, the APTES has likely polymerized and needs to be vacuum distilled under Argon atmosphere. Store APTES under Argon.
The surface density varies with substrate functionalization, concentration, size, and shape of the samples. A total of 1–20 ng/μL are typical concentrations but might need to be optimized to ensure samples do not overlap.
The quality of the water plays an important role as Milli-Q or deionized water might still contain nanometer-sized particulates that will accumulate on the substrate and make imaging challenging.
The samples can be dried under an Argon stream or in the desiccator as well. Overnight is preferred as it also ensures that the epoxy support has fully cured.
The size of the RNA nanoparticles makes imaging challenging. Typically, ultrasharp tips are used as narrow as 1 nm with a spring constant of 40 N/m and a resonance frequency of >300 kHz.
Due to the fragility of ultrasharp Silicon tips, great care should be applied when approaching the surface and only low forces should be applied.
We recommend to plate 1 × 105 cells per well, which will reach 70–80 % of confluence next day (about 24 h after the plating). Overplating of cells will reduce RNA binding and immunostaining efficiency as well.
Folate-free culture media is recommended to ensure an efficient recognition of folate receptors by folate ligand conjugated to RNA nanoparticles.
Trypsinization and neutralization can be carried out as general subculture method. Briefl y, incubate the cells in 0.05 % trypsin solution for no more than 5 min in CO2 incubator followed by neutralizing with 10 × volume of culture media. Centrifuge to pellet the cells followed by washing the pellet with PBS, then centrifuge again. Remove the PBS.
Generally, 1 mL of 4 % PFA is enough for the cells harvested from single 6-well.
Once the cells are fixed in 4 % PFA, it becomes more fragile to impact, therefore handle with delicate pipetting.
Usually wrap the tubes with aluminum foil and keep in ice. Briefly agitate right before inserting into flow cytometer stage.
Use tumor cells incubated with pRNA-3WJ-Alexa647 lacking specific targeting ligand (ex. folate) as a negative control to normalize the specific binding.
Squirting PBS directly onto the cells may cause the cells falling off from the surface. Flow the PBS along the side wall of the chamber not to disturb the cell layer.
Minimum volume to cover the surface area is 50 μL.
Alternatively, DAPI solution can be added to the mounting solution.
Leave the mounted chamber at room temperature for at least a half day to dry. Then, the chamber can be wrapped by aluminum foil and stored at 4 °C up to 1 month.
Acquire the fluorescence from DAPI and Phalloidin Alexa488 at minimum intensity to maximize that from Alexa647 conjugated to RNA nanoparticles.
The injection can be carried out either by manually with a Hamilton syringe or an automatic injector by setting the injection speed at 0.4 μL per minute for total volume of 2 μL.
Monitor physiological parameters including respiration and temperature using a small animal monitoring system (Model 1025, Small Animals Instruments, Inc. or comparable system). Respiration can be monitored with pneumatic pillow.
Use 72 mm volume coil as a transistor and a mouse brain coil as a receiver.
Tumor volume can be calculated from the outlined ROI.
Hold the mouse in a mouse restrainer to keep the mouse from wriggling while tail vein injection.
Keep the working place dark to minimize photobleaching of the fluorophores in the tissue.
The acquisition time can be determined experimentally, since the optimal condition can vary depending on the strength of light source.
For an experiment of luminescence detection, human glioma cells transfected with luciferase gene can be used. After brain tumor implantation, we usually detect the luminescence at around 5 days as tumor grows.
Dissecting the brain out is not necessary. Unlike fluorescence, bioluminescence can penetrate the mouse skull.
For the maximal luminescence intensity, the manufacturer recommends to inject up to 300 mg/kg of luciferin. However, 75 mg/kg usually produce enough strength of luminescence intensity from a mouse brain.
We usually reduce the flow rate of isoflurane gas at this step, since a high flow rate may kill the anesthetized mouse while imaging, which usually takes up to 10 min.
Acknowledgments
We thank Dr. Anna Bratasz in the Small Animal Imaging Core (SAIC) of the Ohio State University for helpful advice for this work. The current work was supported by the NIH grants U01CA 151648 (P.G.), R01EB019036 (P.G.), U01CA152758 (C.M.C.), P30NS045758 (B.K.), R01064607 (B.K.), R01CA150153 (B.K.), and P01CA163205 (B.K.). Funding to Peixuan Guo's Endowed Chair in Nanobiotechnology at University of Kentucky is by the William Farish Endowment Fund. P.G. is a cofounder of Kylin Therapeutics, Inc., and Biomotor and RNA Nanotechnology Development Corp. Ltd.
References
- 1.Purow B, Schiff D. Advances in the genetics of glioblastoma: are we reaching critical mass? Nat Rev Neurol. 2009;5:419–426. doi: 10.1038/nrneurol.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lesniak MS, Brem H. Targeted therapy for brain tumours. Nat Rev Drug Discov. 2004;3:499–508. doi: 10.1038/nrd1414. [DOI] [PubMed] [Google Scholar]
- 3.Cheng Y, Meyers JD, Agnes RS, et al. Addressing brain tumors with targeted gold nanoparticles: a New gold standard for hydrophobic drug delivery? Small. 2011;7:2301–2306. doi: 10.1002/smll.201100628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He H, Li Y, Jia XR, et al. PEGylated Poly(amidoamine) dendrimer-based dual- targeting carrier for treating brain tumors. Biomaterials. 2011;32:478–487. doi: 10.1016/j.biomaterials.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Kievit FM, Veiseh O, Fang C, et al. Chlorotoxin labeled magnetic nanovectors for targeted gene delivery to glioma. ACS Nano. 2010;4:4587–4594. doi: 10.1021/nn1008512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang HW, Hua MY, Hwang TL, et al. Non-invasive synergistic treatment of brain tumors by targeted chemotherapeutic delivery and amplified focused ultrasound- hyperthermia using magnetic nanographene oxide. Adv Mater. 2013;25:3605–3611. doi: 10.1002/adma.201301046. [DOI] [PubMed] [Google Scholar]
- 7.Brem H, Gabikian P. Biodegradable polymer implants to treat brain tumors. J Control Release. 2001;74:63–67. doi: 10.1016/s0168-3659(01)00311-x. [DOI] [PubMed] [Google Scholar]
- 8.Jensen SA, Day ES, Ko CH, et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci Transl Med. 2013;5:209ra152. doi: 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharpe MA, Marcano DC, Berlin JM, et al. Antibody-targeted nanovectors for the treatment of brain cancers. ACS Nano. 2012;6:3114–3120. doi: 10.1021/nn2048679. [DOI] [PubMed] [Google Scholar]
- 10.Guo P, Zhang C, Chen C, et al. Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Mol Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- 11.Guo P. The emerging fi eld of RNA nanotechnology. Nat Nanotechnol. 2010;5:833–842. doi: 10.1038/nnano.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo P, Haque F, Hallahan B, et al. Uniqueness, advantages, challenges, solutions, and perspectives in therapeutics applying RNA nanotechnology. Nucleic Acid Ther. 2012;22:226–245. doi: 10.1089/nat.2012.0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo P, Erickson S, Anderson D. A small viral RNA is required for in vitro packaging of bacteriophage phi29 DNA. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- 14.Shu D, Moll WD, Deng Z, et al. Bottom-up assembly of RNA arrays and superstructures as potential parts in nanotechnology. Nano Lett. 2004;4:1717–1723. doi: 10.1021/nl0494497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu D, Shu Y, Haque F, et al. Thermodynamically stable RNA three-way junctions for constructing multifuntional nanoparticles for delivery of therapeutics. Nat Nanotechnol. 2011;6:658–667. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haque F, Shu D, Shu Y, et al. Ultrastable synergistic tetravalent RNA nanoparticles for targeting to cancers. Nano Today. 2012;7:245–257. doi: 10.1016/j.nantod.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shu Y, Haque F, Shu D, et al. Fabrication of 14 different RNA nanoparticles for specific tumor targeting without accumulation in normal organs. RNA. 2013;19:766–777. doi: 10.1261/rna.037002.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shu Y, Shu D, Haque F, et al. Fabrication of pRNA nanoparticles to deliver therapeutic RNAs and bioactive compounds into tumor cells. Nat Protoc. 2013;8:1635–1659. doi: 10.1038/nprot.2013.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jasinski D, Khisamutdinov EF, Lyubchenko YL, et al. Physicochemically tunable poly-functionalized RNA square architecture with fluorogenic and ribozymatic properties. ACS Nano. 2014;8:7620–7629. doi: 10.1021/nn502160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khisamutdinov EF, Jasinski DL, Guo P. RNA as a boiling-resistant anionic polymer material to build robust structures with defined shape and stoichiometry. ACS Nano. 2014;8:4771–4781. doi: 10.1021/nn5006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdelmawla S, Guo S, Zhang L, et al. Pharmacological characterization of chemically synthesized monomeric pRNA nanoparticles for systemic delivery. Mol Ther. 2011;19:1312–1322. doi: 10.1038/mt.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee TJ, Haque F, Shu D, et al. RNA nanoparticle as a vector for targeted siRNA delivery into glioblastoma mouse model. Oncotarget. 2015 doi: 10.18632/oncotarget.3632. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]