

Abstract
Although opioids are highly efficacious analgesics, their abuse potential and other untoward side effects diminish their therapeutic utility. The addition of non-opioid analgesics offers a promising strategy to reduce required antinociceptive opioid doses that concomitantly reduce opioid-related side effects. Inhibitors of the primary endocannabinoid catabolic enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) show opioid-sparing effects in preclinical models of pain. As simultaneous inhibition of these enzymes elicits enhanced antinociceptive effects compared with single enzyme inhibition, the present study tested whether the dual FAAH-MAGL inhibitor SA-57 [4-[2-(4-chlorophenyl)ethyl]-1-piperidinecarboxylic acid 2-(methylamino)-2-oxoethyl ester] produces morphine-sparing antinociceptive effects, without major side effects associated with either drug class. SA-57 dose-dependently reversed mechanical allodynia in the constriction injury (CCI) of the sciatic nerve model of neuropathic pain and carrageenan inflammatory pain model. As previously reported, SA-57 was considerably more potent in elevating anandamide (AEA) than 2-arachidonyl glycerol (2-AG) in brain. Its anti-allodynic effects required cannabinoid (CB)1 and CB2 receptors; however, only CB2 receptors were necessary for the anti-edematous effects in the carrageenan assay. Although high doses of SA-57 alone were required to produce antinociception, low doses of this compound, which elevated AEA and did not affect 2-AG brain levels, augmented the antinociceptive effects of morphine, but lacked cannabimimetic side effects. Because of the high abuse liability of opioids and implication of the endocannabinoid system in the reinforcing effects of opioids, the final experiment tested whether SA-57 would alter heroin seeking behavior. Strikingly, SA-57 reduced heroin-reinforced nose poke behavior and the progressive ratio break point for heroin. In conclusion, the results of the present study suggest that inhibition of endocannabinoid degradative enzymes represents a promising therapeutic approach to decrease effective doses of opioids needed for clinical pain control, and may also possess therapeutic potential to reduce opioid abuse.
Keywords: 2-arachidonyl glycerol (2-AG), anandamide (AEA), cannabinoid, pain, fatty acid amide hydrolase (FAAH), monoacylglycerol lipase (MAGL), heroin, morphine, self-administration
1. Introduction
Opioids represent a front line effective analgesic for the treatment of chronic pathological pain (Ballantyne and Mao, 2003), but their side effects, including constipation, pruritus, and respiratory depression, pose serious clinical limitations (Campbell et al., 2015). Additionally, the clinical use of opioids for the treatment of pain carries a high abuse potential (Thomas et al., 2014), with prescription opioid misuse preceding approximately 80% of new heroin abusers in the United States (Hedegaard et al., 2015). Thus, an urgent need exists to provide pain patients effective analgesics with reduced abuse risk. The combination of opioids and other classes of analgesics represents a promising strategy to accomplish this goal.
Cannabinoid receptor agonists reliably augment the antinociceptive effects of opioids in preclinical studies (Cichewicz and Welch, 2003; Cox et al., 2007; Miller et al., 2012). Interestingly, co-infusion of morphine and the potent cannabinoid receptor agonist HU-210 into the ventrolateral periaqueductal gray (PAG) produced augmented antinociceptive effects in the rat hot plate test (Wilson-Poe et al., 2013). Moreover, initial clinical studies suggest that the primary constituents of cannabis, Δ9-tetrahydrocannabinol (THC) and cannabidiol may be opioid-sparing in pain patients (Abrams et al., 2011; Johnson et al., 2010; 2012). Likewise, inhibitors of fatty acid amide hydrolase (FAAH) (Cravatt et al., 1996; 2001; Kathuria et al., 2003) and monoacylglycerol lipase (MAGL) (Dinh et al., 2002; Long et al., 2009a), responsible for the degradation of the respective endogenous cannabinoids N-arachidonoylethanolamine (anandamide; AEA) (Felder et al., 1996; Di Marzo et al., 1999) and 2-arachidonyl glycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995), not only produce antinociceptive action when administered alone, but also augment morphine-induced antinociception. Specifically, combination of the FAAH inhibitor URB597 and morphine produces additive antinociceptive effects in the acetic acid model of visceral nociception (Miller et al., 2012). Additionally, co-administration of the MAGL inhibitor MJN110 and morphine produces synergistic anti-allodynic effects in the chronic constrictive injury (CCI) of the sciatic nerve neuropathic pain model (Wilkerson et al., 2016). Given the national crisis of clinical opioid misuse and abuse, therapeutic pain options that lessen the total opioid prescription burden, and limiting the overt abuse liability of opioids (e.g., morphine, oxycodone and heroin) are needed. Modulation of the endocannabinoid system through inhibitors targeting endogenous cannabinoid regulating enzymes represents a promising opioid sparing therapeutic option.
Selective FAAH or MAGL inhibitors produces antinociceptive effects in acute thermal (Long et al., 2009b), visceral (Naidu et al., 2009), inflammatory (Kinsey et al., 2011; Guindon et al., 2011), and neuropathic (Kinsey et al., 2009; 2010; Guindon et al., 2013; Ignatowska-Jankowska et al., 2015) models of pain. Interestingly, combined inhibition of FAAH and MAGL results in enhanced antinociceptive effects compared with single inhibition of these enzymes. One such dual inhibitor, JZL195 produces augmented antinociceptive effects in mouse models of acute thermal, visceral (Sakin et al., 2015), inflammatory (Long et al., 2009b; Anderson et al., 2014) and neuropathic (Adamson Barnes et al., 2016) pain. Additionally, the combination of a high dose of the FAAH inhibitor PF3845 and a low dose of the MAGL inhibitor JZL184 produces augmented antinociceptive effects in inflammatory and neuropathic pain assays (Ghosh et al., 2015). The dual FAAH/MAGL inhibitor [4-[2-(4-chlorophenyl)ethyl]-1-piperidinecarboxylic acid 2-(methylamino)-2-oxoethyl ester], SA-57 (Niphakis et al., 2012), which reduces opioid withdrawal signs (Ramesh et al., 2013; Gamage et al., 2015) and serves as a discriminative stimulus in the drug discrimination paradigm (Owens et al., 2016) in mice, has yet to be tested in pain models. Thus, a major objective of the present study was to determine if SA-57 elicits efficacious antinociceptive effects. In initial experiments, we quantified whole brain endocannabinoid and arachidonic acid levels following SA-57 administration, as well as assessed this compound in the carrageenan inflammatory pain assay and the CCI model of neuropathic pain.
Because serious clinical liabilities are associated with opioid prescriptions for pain control, a second major objective of the present study was to determine whether SA-57 would produce morphine sparing effects in the CCI model. Additionally, we examined SA-57 alone or SA-57 in combination with morphine in the tetrad assay. The tetrad assay consists of measures of locomotor activity, thermal antinociception, catalepsy, and hypothermia, and is highly sensitive to THC and other CB1 receptor agonists (Little et al., 1988). Finally, because of the large abuse liability of prescription opioids that can lead to heroin addiction, the final goal of this work was to determine whether SA-57 alters heroin self-administration behavior.
2. Methods
2. 1. Animals
Adult male C57BL/6J mice (18–35 gram, Jackson Laboratory, Bar Harbor, ME) served as subjects in the carrageenan, CCI, and tetrad assays. Transgenic mice lacking functional cannabinoid CB1 receptors (CB1) or cannabinoid CB2 receptors (CB2) were bred at Virginia Commonwealth University. Mice lacking either CB1 (Zimmer et al., 1999) or CB2 (Jackson Laboratories, Bar Harbor, ME) receptors were backcrossed onto a C57BL/6J background for more than 15 or 8 generations, respectively. Mice were housed four per cage, in a temperature (20–22 °C), humidity (55 ± 10 %), and light-controlled (12 h light/dark cycle; lights on at 0600) AAALAC-approved facility at Virginia Commonwealth University. Standard rodent chow and water were available ad libitum. Albino male CD-1 mice (8–10 weeks old, 18–25 grams, Charles River, Lyon, France) were used for the heroin self-administration assays. For these experiments standard rodent chow and water were available ad libitum, and mice were tested at the beginning of the dark phase of a reversed light/dark cycle (lights off at 0800 and on at 2000 h), and were conducted at Pompeu Fabra University (Barcelona, Spain). All procedures adhered to the guidelines of the Committee for Research and Issues of the International Association for the Study of Pain and were approved by the Institutional Animal Care and Use Committee (IACUC) of Virginia Commonwealth University and the heroin self-administration studies followed the guidelines of the European Communities Directive 86/609/EEC regulating animal research and were approved by the local ethical committee (CEEA-PRBB).
2.2. Drugs
SA-57 [4-[2-(4-chlorophenyl)ethyl]-1-piperidinecarboxylic acid 2-(methylamino)-2-oxoethyl ester] was synthesized in the Cravatt laboratory at the Scripps Research Institute, as described previously (Niphakis et al., 2012). CP55,940 [(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol] and morphine sulfate were obtained from the National Institute on Drug Abuse (Rockville, MD). Heroin was obtained from Ministerio de Sanidad y Consumo (Spain). All drugs, except morphine and heroin, were dissolved in a vehicle solution consisting of a mixture of ethanol, alkamuls-620 (Sanofi-Aventis, Bridgewater, NJ), and saline (0.9 % NaCl) in a 1:1:18 ratio. Morphine sulfate and heroin were dissolved in sterile 0.9% physiological saline. All injections were given via the intraperitoneal (i.p.) route of administration and were administered in a volume of 10 μl/g body mass, with the exception of the heroin self-administration administration study, which was given via the intravenous (i.v.) route of administration (see 2.8.3 below).
2.3. Carrageenan Model of Inflammatory Pain
Edema was induced by giving an intraplantar injection of 0.3% carrageenan (Sigma, St Louis) in a 20 μl volume using a 30-gauge needle into the hind left paw. Paw thickness was measured with electronic digital calipers (Traceable Calipers, Friendswood, TX), prior to and 5 h following carrageenan administration, which corresponds to peak paw edema (Wise et al. 2008).
2.4. Chronic constriction injury (CCI) surgery
Following baseline (BL) behavioral assessment, the surgical procedure for chronic constriction of the sciatic nerve was performed as previously described (Bennett and Xie, 1988), but modified for the mouse (Ignatowska-Jankowska et al., 2015). In brief, the mice were anesthetized with isoflurane (induction 5% vol. followed by 2.0% in oxygen), and the mid to lower back and the dorsal left thigh were shaved and cleaned with 75% ethanol. Using aseptic procedures, the sciatic nerve was carefully isolated, and loosely ligated with three segments of 4-0 silk suture (Ethicon, Somerville, NJ). The overlying muscle was closed with (1) 4-0 sterile silk suture (Ethicon, Somerville, NJ), and animals recovered from anesthesia within approximately 5 min. In these studies the contralateral paw was used as a within subject non-allodynic control paw, as this procedure produces ipsilateral allodynia, only (Kinsey et al., 2010; Ignatowska-Jankowska et al., 2015). Subjects were tested with drug or vehicle between 5 and 18 days after surgery.
2.5. Assessment of allodynia
The mice were placed inside ventilated polycarbonate chambers on an elevated aluminum mesh table and allowed to acclimate to the apparatus for 60 min before testing. Mechanical allodynia was assessed with von Frey filaments (North Coast Medical, Morgan Hill, CA), using the “up-down” method (Chaplan et al., 1994) 5 h after carrageenan administration. The plantar surface of each hind paw was stimulated five times with each filament (0.16–6.0 g), at a frequency of approximately 2 Hz, starting with the 0.6-g filament and increasing until the mouse responded by licking and/or lifting the paw off the surface of the test apparatus. Three or more responses out of five stimulations were coded as a positive response. Once a positive response was detected, sequentially lower weight filaments were used to assess the sensory threshold for each paw.
2.6. Extraction and quantification of endocannabinoids by liquid chromatography-tandem mass spectrometry
2-AG, arachidonic acid (AA), and AEA levels were quantified from the whole brain of C57BL/6J mice receiving intraplantar carrageenan injection, after acute i.p. administration of SA-57 (1.25, 2.5, 5, or 12.5 mg/kg) or 1:1:18 vehicle. Brains were collected upon completion of behavioral assessment of edema and allodynia, and processed for quantification of 2-AG, AA, and AEA. Because equivalent doses of SA-57 significantly attenuated CCI-induced allodynia at 2 h after injection, mice were euthanized via rapid decapitation at this time point. Brains were rapidly harvested, snap-frozen in dry ice, and stored at −80°C until the time of processing. Tissues were further processed according to methods described previously (Ramesh et al., 2011; Ignatowska-Jankowska et al., 2014). See supplementary methods for details.
2.7. Tetrad assay
Mice (Latin square within subject design) were housed individually overnight. The behavioral testing was conducted in the following order: locomotor activity, bar test (catalepsy), tail withdrawal test, rectal temperature. Testing was performed according to previously described procedures (Long et al., 2009b; Schlosburg et al., 2010). Briefly, locomotor activity was assessed 120 min after treatment, for a 10 min period in a Plexiglas cage (42.7 × 21.0 × 20.4 cm) and Anymaze (Stoelting, Wood Dale, Illinois) software was used to determine the percentage of time spent immobile, mean speed and distance traveled. Catalepsy was assessed on a bar 0.7 cm in diameter placed 4.5 cm off of the ground. Nociception was then assessed in the tail immersion assay. Rectal temperature was assessed by inserting a thermocouple probe 2 cm into the rectum, and temperature was determined by thermometer (BAT-10 Multipurpose Thermometer, Clifton, NJ, USA).
2.8. Heroin self-administration assay
2.8.1. Surgery
Mice were anaesthetized with a ketamine/xylazine mixture and then implanted with indwelling iv silastic catheters, as previously described (Soria et al., 2005), (see Supplemental Methods). The success rate for maintaining patency of the catheter until the end (16 days) of the heroin self-administration training was 66%.
2.8.2. Heroin self-administration apparatus
Heroin self-administration training was performed in operant chambers (Model ENV-307A-CT, Med Associates, Inc., Georgia, VT, USA) equipped with two holes, one randomly selected as the active hole and the other as the inactive (see Supplemental Methods).
2.8.3. Experimental Design
Mice were divided in the following experimental groups: Group 1: Vehicle, Group 2: SA57 (1 mg/kg), Group 3: SA57 (2.5 mg/kg), and Group 4: SA57 (5 mg/kg). Subjects were given 90 min daily acquisition sessions. All the groups were trained to acquire heroin self-administration (0.025 mg/kg/inj, i.v.) on an FR1 schedule over a time period of seven days. The criteria for acquisition of operant responding were achieved when mice maintained a stable responding with less than 25% deviation from the mean of the total number of infusions earned in three consecutive sessions, with at least 65% responding on the reinforced nose-poke, and a minimum of four reinforcements per session (Martin-Garcia et al., 2011; Soria et al., 2008). From days 8 to 15 (eight sessions), mice received their appropriate drug injection 60 min before each FR1 training session. On day 16, mice received the corresponding treatment and 60 min later were evaluated in a progressive ratio (PR) schedule in which the response requirement to earn infusions escalated according to the following series: 1-2-3-5-12-18-27-40-60-90-135-200-300-450-675-1000. The maximum duration of the PR session was 4 h or until mice did not respond on any manipulandum within 1 h. After PR session, the thiopental test was applied and only mice that showed patency of catheter were moved to the extinction and relapse phases. Briefly, during the thiopental test mice were given an infusion of thiopental, which produces anesthetic effects within 3 s, with correct intravenous catheter placement (Martin-Garcia et al., 2011; Soria et al., 2008). The first extinction session occurred 48 h after the thiopental tests to avoid any possible influence residual drug effects.
The experimental conditions for the extinction phase were similar to the acquisition sessions, except that heroin was not delivered and the cue-light was not presented after active responding. Mice were given 90 min daily extinction sessions, during at least eight consecutive days until the criteria for extinction was achieved (i.e., three consecutive sessions in which mice responded on the active lever less than 30% of the responses reached in the three last acquisition days, and made less than 15 active responses per session).
The experimental sequence was finished in groups 2, 3 and 4 at the end of the extinction training. Once the extinction criteria are reached, mice in group 1 were tested for reinstatement. These mice were initially treated with vehicle, and received infusion of 0.025 mg/kg/inj heroin, previously. For this purpose, cue-induced reinstatement was conducted under the same conditions used in the acquisition phase except that heroin was not delivered. Each response on the active manipulandum in this phase led to the presentation of the cue-light for 2s. The reinstatement criterion was achieved when responding in the nose-poke doubled with respect to extinction responding. Mice from group 1 (vehicle + heroin) were divided in two different subgroups as shown in table 1, below.
Table 1.
Heroin self-administration subgroups and corresponding treatment
| Day 1 Treatment | Second Test Treatment | Third Test Treatment | |||
|---|---|---|---|---|---|
| First Subgroup | Second Subgroup | First Subgroup | Second Subgroup | First Subgroup | Second Subgroup |
| Vehicle + cue-induced reinstatement | SA-57 (1 mg/kg) + cue-induced reinstatement | Vehicle + cue-induced reinstatement | SA-57 (2.5 mg/kg) + cue-induced reinstatement | Vehicle + cue-induced reinstatement | SA-57 (5 mg/kg) + cue-induced reinstatement |
2.10. Data analysis
The ED50 dose, lower equally effective dose values, and 95% confidence limits (Bliss, 1967) were calculated using a standard linear regression analysis of the linear portion of the dose response curve for morphine, SA-57 or the combination of morphine and SA-57 that reversed ipsilateral allodynia. The theoretical additive ED50 value of the combined drugs was calculated from the individual dose-response curves to determine synergistic, additive, or subadditive interactions. The combination was assumed to equal the sum of the effects of each drug. For dose-addition analysis, the ED50 of SA-57 was plotted on the abscissa (x axis) and the isoeffective dose of morphine was plotted on the ordinate (y axis). A line connecting the two points represents the theoretical additive effect of morphine and SA-57 dose combinations. The experimentally derived ED50 values (Zmix) from the dose response curves of the ratios were compared to the predicted additive ED50 values (Zadd). If the empirically derived value and the theoretical value do not differ, the interaction is additive (Tallarida, 2001, 2006). The statistical difference between the theoretical additive ED50 value and the experimental ED50 value was analyzed using a Fisher’s exact test (Naidu et al., 2009). Differences were considered significant at the level of p < 0.05. For the heroin self-administration experiments, analysis of the data obtained during the acquisition phase was conducted using two-way ANOVA with manipulandum (active/inactive) as within-subjects factor and genotype as between-subjects factor. Progressive ratio results were compared using one-way ANOVA, and post-hoc analysis (Newman-Keuls) was performed when required. To evaluate the extinction and cues-induced reinstatement, three-way ANOVA with repeated measures was performed with experimental phase and manipulandum as within-subjects factors, and drug treatment as between-subject factor. Post-hoc analysis (Newman-Keuls) was performed when required. Statistical analysis was performed with either GraphPad Prism version 6.0 (GraphPad Software Inc., San Diego, CA) or the Statistical Package for Social Science program SPSS® 19.0 (SPSS Inc, Chicago, USA).
3. Results
3.1. Pharmacological effects of SA-57 in the carrageenan inflammatory pain model
3.1.1. Dose response effect of SA-57 on carrageenan-induced allodynia and edema
Intraplantar injection of carrageenan produced allodynia and edema at 5 h compared to non-injected control paws (Fig. 1 A, B). Although SA-57 did not alter normal sensory threshold responses to light touch in control paws, it dose-dependently reversed allodynia in the carrageenan-injected paw (F(4,20)=4.71; p < 0.01) and partially reduced the edema (F(4,20)=19.60; p < 0.0001).
Figure 1.

The dual FAAH and MAGL inhibitor SA-57 ameliorates allodynia and inflammation in the carrageenan inflammatory pain model. SA-57 (A) reverses carrageenan-induced allodynia, and (B) partially reverses carrageenan-induced edema. Tests were conducted 2 hr after i.p. administration of SA-57 produces reversal from allodynia. Filled symbols indicate at least p < 0.05 vs. vehicle. Data reflect mean ± SEM, n=6 mice per group.
3.1.2. Dose response effect of SA-57 on brain endocannabinoid levels
Immediately following assessment of paw thickness and von Frey thresholds, the mice were euthanized, brains were collected and endocannabinoid and arachidonic acid levels were quantified. All doses of SA-57 assessed (1.25–12.5 mg/kg) produced approximately 10-fold elevations of AEA (F(4,9) =50.2; p < 0.0001, Fig. 2A). In addition, SA-57 dose-dependently elevated levels of 2-AG (F(4,9)=79.4; p < 0.0001, Figure 2B), while dose-dependently reducing arachidonic acid in whole brain (F(4,9)= 14.9; p < 0.001, Fig. 2C).
Figure 2.
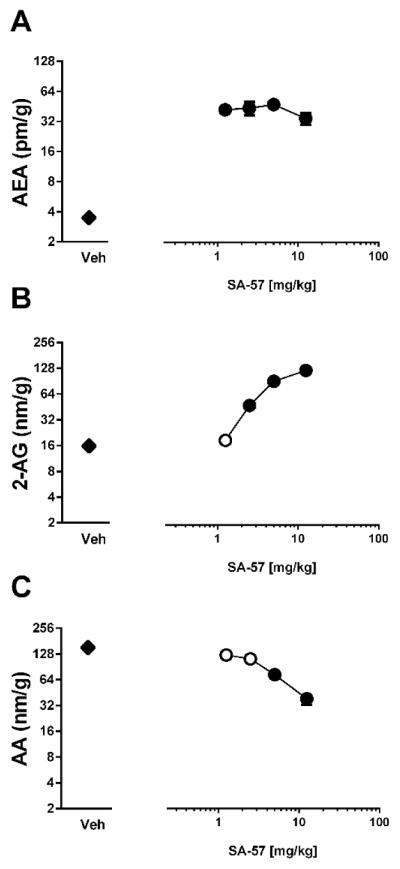
SA-57 alters endocannabinoid levels in whole brain tissue at 2 hr in mice given an intraplantar carrageenan. (A) All doses of SA-57 produce maximal increases AEA compared to vehicle. SA-57 dose-dependently increases 2-AG (B) and (C) decreases AA. *** p<0.0001, ** p<0.001, * p < 0.05 vs. vehicle. Data reflect mean ± SEM, n=6 mice per group.
3.1.3. Cannabinoid receptors mediate the effects of SA-57 on carrageenan-induced allodynia and edema
Carrageenan-induced allodynia and edema developed comparably to the same magnitude between CB1 (−/−) and (+/+) mice (Fig 3A, B), as well as between CB2 (−/−) and (+/+) mice (Fig. 3C, D). However, SA-57 did not elicit anti-allodynic effects in carrageenan-injected paws of CB1 (−/−) mice (Fig. 3A) or CB2 (−/−) mice (Fig. 3C), indicating that these actions require both cannabinoid receptors. In contrast, these transgenic mice displayed differential effects to the anti-edematous of SA-57. Whereas SA-57 continued to elicit anti-edematous effects in CB1 (−/−) and (+/+) mice (F(1,16)= 16.7; p < 0.001, Fig. 3A, B), the CB2 (−/−) mice were resistant to this action (p = 0.28), (Fig. 3B, D).
Figure 3.

Cannabinoid receptors mediate the anti-edematous and anti-allodynic effects of SA-57 (5 mg/kg, i.p.) in the carrageenan model of inflammatory pain. (A) SA-57 reverses allodynia in CB1 (+/+) mice, but not in CB1 (−/−) mice. (B) SA-57 retains its anti-edematous effects in CB1 (−/−) and (+/+) mice. SA-57 reverses carrageenan-induced allodynia (C) and edema (D) in CB2 (+/+) mice, but not in CB1 (−/−) mice. *** p<0.0001, ** p<0.001, * p < 0.05 vs. vehicle, ## p<0.001, ### p < 0.0001 vs. WT + SA-57. Data reflect mean ± SEM, n=6 mice per group.
3.2. SA-57 produces opioid sparing effects in the CCI model of neuropathic pain
Having established that SA-57 produces anti-allodynic effects in an acute inflammatory pain model, we next examined its effectiveness in the CCI model of neuropathic pain. As depicted in Fig. 4A, 10 mg/kg SA-57, which produced at least 10-fold increases in AEA and 2-AG as well as significant decreases in AA, completely reversed allodynia (F(5,39)=25.4; p < 0.0001), with onset of action at 1 h, peak effects occurred at 2 h, and mice returned to their pre-injection von Frey thresholds by 6 h.
Figure 4. Combination of morphine and SA-57 produces an additive reversal of CCI-induced allodynia.

(A) SA-57 (10 mg/kg, i.p.) produces a time-dependent reversal of CCI-induced allodynia in the ipsilateral paw. In the contralateral paw, which shows pre-surgery paw withdrawal responses, SA-57 does not alter basal mechanical stimulus responses. (B) SA-57 reverses CCI-induced allodynia in a dose-related manner 2 hr after administration. Combination of equally effective doses of (i.e., 1:1) morphine and SA-57 produces a leftward shift of the dose-response curve. (C) Morphine reverses CCI-induced allodynia in a dose-related manner 30 min after administration. A 1:1 equally effective combination of morphine and SA-57 produces a leftward shift of the dose-response curve. (D) The 1:1 equally effective combination of SA-57 and morphine produces an additive effect, as it falls on the line of additivity. The calculated experimental Zmix was approximately equal to than the calculated theoretical Zadd. Data reflect mean ± SEM, n = 5–7 mice/group. *** p<0.0001, ** p<0.001, * p < 0.05 vs. vehicle. Filled symbols indicate at least p < 0.05 vs. vehicle.
We next assessed whether SA-57 would augment the antinociceptive effects of morphine in the CCI model of neuropathic pain. The dose-response relationships of the drugs alone and in combination at equally effective doses are shown in Fig. 4B and C. Both SA-57 (F(2,15) = 18.6; p < 0.0001) and morphine (F(2,15) = 44.9; p < 0.0001) reversed allodynia. An equivalent dose (i.e., 1:1) combination of SA-57 and morphine produced a leftward shift in the dose-response relationship compared with either compound given alone (Fig. 4B and C). Isobolographic analysis revealed an additive interaction between these drugs (Fig. 4D). Specifically, the calculated experimental Zmix (2.89 (2.45–3.33)) mg/kg was approximately equal to than the calculated theoretical Zadd (3.38 (3.33–3.44)) mg/kg (p = 0.08).
Because repeated administration of cannabinoids and opioids often leads to tolerance, in the next experiment we assessed whether the enhanced antinociceptive effects produced by combination of threshold doses of SA-57 and morphine would undergo tolerance following repeated administration. Mice that underwent CCI surgery were given 1.79 mg/kg SA-57, 1.12 mg/kg morphine, or combination of both drugs twice a day for five days, with a final injection on day 6. On day 1 after initial treatment, and again on day 6 after the final treatment, allodynia and thermal hyperalgesia were assessed. The combination of these two threshold doses of SA-57 and morphine fully reversed CCI-induced allodynia (F(6,35) = 12.48; p < 0.0001; Fig. 5A) and thermal hyperalgesia (F(6,35) = 7.13; p < 0.0001; Fig. 5B) following either acute and repeated administration.
Figure 5.

Repeated administration of 1.79 mg/kg SA-57 and 1.12 mg/kg morphine retains its anti-allodynic and anti-thermal hyperalgesic effects. Both acute and repeated administration of the combination of SA-57 and morphine reverse CCI-induced A) allodynia and B) thermal hyperalgesia. *** p<0.0001, * p < 0.05 vs. sham - vehicle. Data reflect mean ± SEM, n=6 mice per group.
3.3. SA-57 produces cannabimimetic effects in the tetrad assay
SA-57 was evaluated in the tetrad assay to infer whether it produced general in vivo cannabimimetic activity. SA-57 produced significant cataleptic (F(5,36)= 64.8; p < 0.0001, Fig. 6A), antinociceptive (F(5,36)= 55.9; p < 0.0001, Fig. 6B), hypothermic (F(5,36)= 82.6; p < 0.0001, Fig. 6C), and hypomotility (F(5,36)=13.6; p < 0.0001, Fig. 6D) effects. Only 12.5 mg/kg SA-57 elicited significant antinociception and catalepsy, while 5 and 12.5 mg/kg SA-57 produced significant hypothermic and locomotor depressive effects.
Figure 6.

High dose SA-57 elicits common cannabimimetic effects in mice. SA-57 (12.5 mg/kg; i.p.) produces increases in (A) catalepsy and (B) antinociception. SA-57 (5, 12.5 mg/kg) produces significant (C) decreases in body temperature and (D) locomotion. *** p<0.0001, ** p<0.001, * p < 0.05 vs. vehicle. Data reflect mean ± SEM, n=6 mice per group.
The equi-effective threshold doses of SA-57 (1.79 mg/kg) and morphine (1.12 mg/kg) in combination were assessed for potential cannabimimetic effects in the tetrad assay. This combination of SA-57 and morphine produced significant antinociceptive effects in the tail withdrawal test (F(2,16)= 23.8; p < 0.0001, Fig. 7B), but did not produce catalepsy (Fig. 7A), hypothermia (Fig. 7C), or locomotor depression (Fig. 7D). In comparison, the potent synthetic cannabinoid agonist CP55,940 (1 mg/kg) produced robust cataleptic, (F(2,16)= 103.8; p < 0.001), antinociceptive, hypothermic (F(2,16)=40.3; p < 0.0001), and hypomotility (F(2,16)=84.4; p < 0.0001) responses.
Figure 7.

The threshold combination of SA-57 and morphine produces antinociception, but no other cannabimimetic effects. (A) CP55,940 (1 mg/kg) produces catalepsy. However, the threshold equi-effective combination of 1.79 mg/kg SA-57 and 1.12 mg/kg morphine does not produce catalepsy. (B) Both 1 mg/kg CP55,940 and the threshold combination of 1.79 mg/kg SA-57 and 1.12 mg/kg morphine produce robust increases in thermal antinociception. CP55,940, but not the threshold combination of 1.79 mg/kg SA-57 and 1.12 mg/kg morphine, produces significant decreases in (C) body temperature and (D) locomotion. *** p<0.0001, * p < 0.05 vs. vehicle. Data reflect mean ± SEM, n=6–7 mice per group.
3.4. SA-57 attenuates heroin drug seeking behavior and self-administration
In the final study, we examined the impact of SA-57 on heroin seeking behavior in mice trained to nose poke for i.v. administration of heroin. As shown in Fig. 8A, vehicle-treated mice developed robust heroin seeking behavior by day 4 and retained this behavior throughout the 15 day period (F(14,1036)= 6.5; p < 0.0001). SA-57 at 1 (time X activity interaction (F(14,448) =1.9; p < 0.05, Fig. 8B), 2.5 (time X activity interaction (F(14,448) = 3.1; p < 0.0001, Fig. 8C), and 5 mg/kg (main effect of activity (F(1,30)=13.2; p < 0.005, Fig. 8D) reduced heroin-reinforced nose poke behavior from days 8 to 15. This effect is clearly shown when the area under each curve is determined (Fig. 8E). Vehicle-treated mice showed a significant increase in nose pokes throughout the test sessions. Each dose of SA-57 prevented increased nose poking behavior from days 8–11 and 12–15 compared with days 1–3, and was significantly less than the responses emitted by the vehicle-treated mice (Fig. 8E). On day 16, mice were assessed in the progressive ratio procedure in which increased ratios of nose-pokes were required to obtain each subsequent infusion of 0.25 mg/kg heroin. Mice in each dose condition of SA-57 showed a significantly reduced break point compared with vehicle-treated mice (F(3,84)=6.3; p < 0.001, Fig. 8F). During the washout period, there were no group differences in extinction rates (Supplemental Fig. 1A–D). To examine the effects of SA-57 on the reinstatement of heroin seeking behaviors, we used mice that had previously received vehicle and heroin infusions of 0.025 mg/kg/inj. SA-57 failed to produce alterations in heroin reinstatement behaviors (Supplemental Fig 1E).
Figure 8.

SA-57 reduces heroin seeking behavior in mice. (A) Mice trained to self-administer heroin display increased drug seeking behavior with vehicle treatment from days 8 to 15, as indicated by increased nose pokes for heroin compared to the corresponding vehicle nose poke hole (n=38 mice per group). (B) Mice trained to self-administer heroin reliably choose the nose poke hole for heroin compared to the vehicle nose poke hole. On day 8, mice receiving 1 mg/kg SA-57 display heroin seeking behavior, but without escalation, as indicated by nose pokes for heroin compared to vehicle (n=17 mice per group). (C) Mice receiving 2.5 mg/kg SA-57 starting on day 8 display no increase in heroin seeking behavior as indicated by nose pokes for heroin compared to the vehicle (n=17 mice per group). (D) Starting on day 8 mice receiving 5 mg/kg SA-57 display no significant heroin seeking behavior overall as indicated by nose pokes for heroin compared to the corresponding vehicle nose poke hole (n=16 mice per group). (E) Area under the curve analysis demonstrates that heroin self-administration significantly increases on time blocks 3 (days 8–11) and 4 (days 12–14) compared with time block 1 (days 1–3) or 2 (days 4–7). All doses of SA-57 (1–5 mg/kg) prevented this increase of heroin-seeking behavior. (F) All doses of SA-57 (1–5 mg/kg) decreased the breaking points of nose pokes for mice to receive infusions of 0.25 mg/kg heroin. **** p < 0.0001 *** p < 0.001, ** p < 0.01, * p < 0.05 vs. respective time-matched vehicle treatment. # p < 0.05 vs. day 1–day 3 vehicle treatment. Data reflect mean ± SEM.
4. Discussion
In the present study, we show that the dual FAAH-MAGL inhibitor, SA-57, fully reversed allodynia in the carrageenan inflammatory and CCI neuropathic pain mouse models, as well as enhanced the antinociceptive effects of morphine in the CCI model. The combination of threshold doses of 1.79 mg/kg SA-57 and 1.12 mg/kg morphine completely reversed CCI-induced allodynia, and this anti-allodynic effect was retained following repeated administration. Moreover, this combination of threshold doses also produced antinociceptive effects in the warm water tail immersion test, but did not elicit common cannabimimetic effects (i.e., hypomotility, catalepsy, and hypothermia). The lack of these effects is likely due to the fact that low dose SA-57 does not raise endocannabinoid brain levels sufficiently to activate CB1 receptor-mediated circuits mediating general cannabimimetic side effects. Thus, the combination of low dose SA-57 and morphine appears not only to enhance antinociceptive effects with some selectively over other cannabimimetic actions, but also its repeated administration may not be sufficient to cause functional changes in mu opioid receptors or CB1 receptors that underlie tolerance. A surprising finding was that SA-57 also reduced the reinforcing effects of heroin in mice. Specifically, 1–5 mg/kg SA-57 decreased heroin seeking behavior and break point. Together, these findings suggest that inhibition of endocannabinoid degradative enzymes may induce pain relief and diminish opioid abuse.
Selective MAGL and FAAH inhibitors reliably produce antinociceptive effects in acute pain models (Kathuria et al., 2003; Long et al., 2009b; Thors et al., 2010; Ignatowska-Jankowska et al., 2015), as well as reverse allodynia to varying degrees in inflammatory (Kinsey et al., 2011; Guindon et al., 2011; Sasso et al., 2012) and neuropathic (Kinsey et al., 2009; 2010; Sasso et al., 2012; Guindon et al., 2013; Ignatowska-Jankowska et al., 2015) pain models. Moreover, simultaneous inhibition of these enzymes produces enhanced antinociceptive effects in these assays (Long et al., 2009b; Adamson Barnes et al., 2016). The present study reports similar findings, as shown by anti-allodynic effects in the carrageenan and CCI assays and thermal antinociception in the tail withdrawal assay, though SA-57 did not elevate paw withdrawal latencies in the contralateral paw of CCI mice. In both the carrageenan and CCI nociceptive models, high doses of SA-57, which substantially elevated both AEA and 2-AG and decreased AA, were required to elicit antinociceptive and anti-edematous effects. However, co-administration of morphine and low doses of SA-57, which elevated AEA brain levels but did not affect 2-AG or AA levels, fully reversed CCI-induced allodynia in an additive fashion. These findings suggest that FAAH inhibition alone sufficiently augments morphine-induced antinociception and are consistent with the results of Miller et al. (2012), who demonstrated that combination of the FAAH inhibitor URB597 and morphine produced additive antinociceptive effects in acetic acid-induced abdominal stretching and depression of wheel running in mice. In contrast combination of morphine and the MAGL inhibitor MJN110 produced synergistic antinociceptive effects in the CCI model (Wilkerson et al., 2016). Taken together, these results suggest that the endocannabinoid hydrolyzing enzyme inhibitors differentially interact with morphine in the CCI model of neuropathic pain, with FAAH inhibition producing additive antinociceptive effects and MAGL inhibition producing synergistic antinociceptive effects. Future studies will ascertain whether other FAAH and MAGL inhibitors interact with morphine in the CCI model in a similar fashion, as well as evaluate whether these respective additive and synergistic interactions extend to other pain models.
Although the neuronal circuitry underlying opioid abuse liability differs from that responsible for antinociceptive effects, chronic pain may prime this circuitry to increase abuse liability as well as facilitate the development of addiction (Cahill et al., 2013; Garland et al., 2013; Evans and Cahill, 2016). Case reports and retrospective studies support the proposition that prescription opioid use for pain control may serve as a gateway for heroin addiction, and strategies that limit opioid abuse potential may also be of benefit for the treatment of opioid addiction (Roux et al., 2013; Conroy and Hill, 2014, Mars et al., 2014). Several studies have demonstrated that endocannabinoid hydrolytic inhibitors ameliorate precipitated and spontaneous morphine withdrawal signs in morphine-dependent mice (Ramesh et al., 2011; 2013, Gamage et al., 2015). The present study examined whether SA-57 would alter heroin seeking behavior. All doses of SA-57 (1, 2.5, 5 mg/kg) examined decreased heroin self-administration and decreased the break point of operant responses for heroin reinforcement. The observation that this decreased opioid seeking behavior was seen at 1 mg/kg SA-57, which elevates AEA and does not affect 2-AG or arachidonic acid levels, suggests FAAH inhibition plays a necessary role in this effect. Although the contribution of MAGL cannot be ruled out, the present findings are in line with a previous study reporting that heroin self-administration in rats is associated with increases of AEA, and to a small magnitude decreases of 2-AG, within the nucleus accumbens (Caille et al., 2007). Indeed, CB1 receptors within the nucleus accumbens and prefrontal cortex play a critical role in heroin seeking behavior (Alverez-Jaimes et al., 2008). Thus, inhibitors of endocannabinoid hydrolytic enzymes not only attenuate opioid withdrawal somatic signs (Ramesh et al., 2011; 2013; Gamage et al., 2015), but also reduce the reinforcing effects of heroin.
The experiments examining the effects of SA-57 in CB1 (−/−) and CB2 (−/−) mice in the carrageenan model of inflammatory indicate that these receptors play differential roles in mediating anti-edematous vs. anti-allodynic effects of SA-57. Whereas both cannabinoid receptors were necessary for the anti-allodynic effects of SA-57, the anti-edematous effects of SA-57 required CB2 receptors, while CB1 receptors were not necessary. These receptors are expressed on distinct cell types in which the CB1 receptor is predominately expressed on presynaptic neurons in the brain (Huang et al., 2001; Szabo and Schlicker, 2005), particularly GABAergic interneurons (Katona et al., 2001), and the CB2 receptor is highly expressed on immune cells (Carayon et al., 1998; Galiegue, 1995; Schatz et al., 1997). However, it must be noted that the contribution of other FAAH substrates (e.g., palmitoylethanolamide, oleoylethanolamine; Cravatt et al., 2001) and consequences of MAGL inhibition (i.e., reduced arachidonic acid and prostaglandins; Nomura et al., 2008; 2011) may contribute to the observed antinociceptive effects of SA-57. The underlying mechanisms of SA-57-induced antinociception are likely mediated by activation of both receptors that are expressed on immune cells as well as neuronal signaling pathways. Thus, the antinociceptive effects of SA-57 in this assay required concurrent endocannabinoid activation of CB1 receptors, possibly expressed in the periphery (i.e., sensory nociceptors or dorsal root ganglia) and/or within the CNS (e.g., dorsal horn of the spinal cord or PAG) and CB2 receptors expressed on microglia and other immunological cells.
Likewise, there are multiple potential glial and neuronal mechanisms underlying the opioid modifying actions of SA-57. Given that μ-opioid and cannabinoid receptors are widely expressed in the CNS and periphery, the observed additive antinociceptive effects may be due to activation of these receptors at multiple sites in the neuroaxis implicated in the processing and regulation of nociception, including peripheral nociceptors (Desroches et al., 2014), dorsal root ganglia (Khasabova et al., 2004), dorsal horn of the spinal cord (da Fonseca Pacheco et al., 2008; Desroches et al., 2014), the periaqueductal gray and other brain regions (Paldy et al., 2008; Wilson-Poe et al., 2013). CB2 receptor activation may reduce nociception by increasing the antinflammatory cytokine IL-10, decreasing the proinflammatory cytokine IL-1β, (Wilkerson et al., 2012), decreasing the AKT-Erk1/2 pathway (Merighi et al., 2012) and reducing the mRNA of the critical chemokine MCP1/CCL2 (Deng et al., 2015). Finally, heterodimerization of CB1 receptors and mu-opioid receptors on neurons may also account for these interactions (Rios et al., 2006).
Clinical studies have led to mixed evidence regarding the efficacy of cannabinoid-based medications to treat pain, though a recent meta-analysis concluded that moderate-quality evidence supported the use of these drugs for the treatment of chronic pain (Whiting et al., 2015). Patients taking opioids for non-cancer pain reported that Dronabinol produced additional analgesic effects (Narang et al., 2008). In Phase II clinical trials, combination of Sativex, consisting of THC/cannabidiol, enhanced opioid analgesic effects in patients suffering from advanced cancer pain (Johnson et al., 2010; 2013), though the results of the Phase III trials have yet to be published. In an investigator initiated study, chronic pain patients treated with vaporized cannabis and opioids reported less pain than opioid treatment alone (Abrams et al., 2011). Alternatively, a study from Australia reported that the average chronic non-cancer pain patient using cannabis concomitant with opioids actually used higher doses of opioids over a longer period of time than non-cannabis use patients (Degenhardt et al., 2015). In contrast, there are no reports in the literature testing whether inhibitors of endocannabinoid metabolizing enzymes are beneficial adjutants to opioid therapy in chronic pain patients. The results of the present study taken together with other studies that report individual inhibition of either FAAH or MAGL also produces opioid sparing antinociceptive effects with no added side effects (Miller et al., 2012; Wilkerson et al., 2016) supports the idea to initiate clinical trials to test whether FAAH, MAGL, or simultaneous FAAH and MAGL inhibition is opioid-sparing.
In conclusion, the present study demonstrates that the dual FAAH and MAGL inhibitor SA-57, which increases both AEA and 2-AG levels, interacted in an additive manner with morphine to reverse allodynia in a mouse model of neuropathic pain. In addition, effective doses of these drugs in combination lacked typical cannabis-like side effects, which were seen with higher, analgesic doses of this drug. Importantly, SA-57 also diminished heroin seeking behavior. Overall, these results indicate that inhibition of endocannabinoid degradative enzymes represents a novel therapeutic avenue to decrease doses of opioids needed for clinical pain control, and may additionally be a viable therapeutic in the treatment of opioid addiction.
Supplementary Material
Highlights.
SA-57 inhibits fatty acid amide hydrolase and monoacylglycerol lipase.
SA-57 reverses allodynia in mouse neuropathic and inflammatory pain models.
CB1 and CB2 receptors mediate the antinociceptive effects of SA-57 in the carrageenan model of inflammatory pain.
SA-57 enhances morphine-induced antinociception.
SA-57 reduces heroin self-administration.
Acknowledgments
Research was supported by NIH grants: DA009789, DA017259, DA032933, DA033934-01A1.
Abbreviations
- 2-AG
2-arachidonoylglycerol
- AA
arachidonic acid
- AEA
anandamide
- CB
cannabinoid
- CCI
chronic constriction injury
- CNS
central nervous system
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- THC
Δ9-tetrahydrocannabinol
- TNF-α
tumor necrosis factor alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams DI, Couey P, Shade SB, Kelly ME, Benowitz NL. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90:844–851. doi: 10.1038/clpt.2011.188. [DOI] [PubMed] [Google Scholar]
- Adamson Barnes NS, Mitchell VA, Kazantzis NP, Vaughan CW. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine neuropathic pain model. Br J Pharmacol. 2016;173:77–87. doi: 10.1111/bph.13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WB, Gould MJ, Torres RD, Mitchell VA, Vaughan CW. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine inflammatory pain model. Neuropharmacology. 2014;81:224–230. doi: 10.1016/j.neuropharm.2013.12.018. [DOI] [PubMed] [Google Scholar]
- Ballantyne JC, Mao J. Opioid therapy for chronic pain. N Engl J Med. 2003;349:1943–1953. doi: 10.1056/NEJMra025411. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Xue L, Grenier P, Magnussen C, Lecour S, Olmstead MC. Changes in morphine reward in a model of neuropathic pain. Behav Pharmacol. 2013;24:207–213. doi: 10.1097/FBP.0b013e3283618ac8. [DOI] [PubMed] [Google Scholar]
- Caille S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci. 2007;27:3695–3702. doi: 10.1523/JNEUROSCI.4403-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell G, Nielsen S, Bruno R, Lintzeris N, Cohen M, Hall W, Larance B, Mattick RP, Degenhardt L. The Pain and Opioids IN Treatment study: characteristics of a cohort using opioids to manage chronic non-cancer pain. Pain. 2015;156:231–242. doi: 10.1097/01.j.pain.0000460303.63948.8e. [DOI] [PubMed] [Google Scholar]
- Carayon P, Marchand J, Dussossoy D, Derocq JM, Jbilo O, Bord A, Bouaboula M, Galiegue S, Mondiere P, Penarier G, Fur GL, Defrance T, Casellas P. Modulation and functional involvement of CB2 peripheral cannabinoid receptors during B-cell differentiation. Blood. 1998;92:3605–3615. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Meth. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL, Welch SP. Modulation of oral morphine antinociceptive tolerance and naloxone-precipitated withdrawal signs by oral Delta 9-tetrahydrocannabinol. J Pharmacol Exp Ther. 2003;305:812–817. doi: 10.1124/jpet.102.046870. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Conroy S, Hill D. Failure to identify or effectively manage prescription opioid dependence acted as a gateway to heroin use-buprenorphine/naloxone treatment and recovery in a surgical patient. BMJ Case Rep. 2014:2014. doi: 10.1136/bcr-2014-207458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox ML, Haller VL, Welch SP. Synergy between delta9-tetrahydrocannabinol and morphine in the arthritic rat. Eur J Pharmacol. 2007;567:125–130. doi: 10.1016/j.ejphar.2007.04.010. [DOI] [PubMed] [Google Scholar]
- da Fonseca Pacheco D, Klein A, de Castro Perez A, da Fonseca Pacheco CM, de Francischi JN, Duarte ID. The mu-opioid receptor agonist morphine, but not agonists at delta- or kappa-opioid receptors, induces peripheral antinociception mediated by cannabinoid receptors. Br J Pharmacol. 2008;154:1143–1149. doi: 10.1038/bjp.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt L, Lintzeris N, Campbell G, Bruno R, Cohen M, Farrell M, Hall WD. Experience of adjunctive cannabis use for chronic non-cancer pain: Findings from the Pain and Opioids IN Treatment (POINT) study. Drug Alcohol Depend. 2015;147:144–150. doi: 10.1016/j.drugalcdep.2014.11.031. [DOI] [PubMed] [Google Scholar]
- Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG. Chronic cannabinoid receptor 2 activation reverses Paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol Psychiatry. 2015;77:475–487. doi: 10.1016/j.biopsych.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, De Petrocellis L, Melck D, Martin BR. Cannabimimetic fatty acid derivatives: the anandamide family and other endocannabinoids. Curr Med Chem. 1999;6:721–744. [PubMed] [Google Scholar]
- Evans CJ, Cahill CM. Neurobiology of opioid dependence in creating addiction vulnerability. F1000Res. 2016:5. doi: 10.12688/f1000research.8369.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Nielsen A, Briley EM, Palkovits M, Priller J, Axelrod J, Nguyen DN, Richardson JM, Riggin RM, Koppel GA, Paul SM, Becker GW. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996;393:231–235. doi: 10.1016/0014-5793(96)00891-5. [DOI] [PubMed] [Google Scholar]
- Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Gamage TF, Ignatowska-Jankowska BM, Muldoon PP, Cravatt BF, Damaj MI, Lichtman AH. Differential effects of endocannabinoid catabolic inhibitors on morphine withdrawal in mice. Drug Alcohol Depend. 2015;146:7–16. doi: 10.1016/j.drugalcdep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland EL, Froeliger B, Zeidan F, Partin K, Howard MO. The downward spiral of chronic pain, prescription opioid misuse, and addiction: cognitive, affective, and neuropsychopharmacologic pathways. Neurosci Biobehav Rev. 2013;37:2597–2607. doi: 10.1016/j.neubiorev.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Kinsey SG, Liu QS, Hruba L, McMahon LR, Grim TW, Merritt CR, Wise LE, Abdullah RA, Selley DE, Sim-Selley LJ, Cravatt BF, Lichtman AH. Full Fatty Acid Amide Hydrolase Inhibition Combined with Partial Monoacylglycerol Lipase Inhibition: Augmented and Sustained Antinociceptive Effects with Reduced Cannabimimetic Side Effects in Mice. J Pharmacol Exp Ther. 2015;354:111–120. doi: 10.1124/jpet.115.222851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Guijarro A, Piomelli D, Hohmann AG. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol. 2011;163:1464–1478. doi: 10.1111/j.1476-5381.2010.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG. Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol Res. 2013;67:94–109. doi: 10.1016/j.phrs.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard H, Chen LH, Warner M. Drug-poisoning deaths involving heroin: United States, 2000–2013. NCHS Data Brief. 2015:1–8. [PubMed] [Google Scholar]
- Huang CC, Lo SW, Hsu KS. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol. 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska B, Wilkerson JL, Mustafa M, Abdullah R, Niphakis M, Wiley JL, Cravatt BF, Lichtman AH. Selective Monoacylglycerol Lipase Inhibitors: Antinociceptive versus Cannabimimetic Effects in Mice. J Pharmacol Exp Ther. 2015;353:424–432. doi: 10.1124/jpet.114.222315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JR, Burnell-Nugent M, Lossignol D, Ganae-Motan ED, Potts R, Fallon MT. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer-related pain. J Pain Symptom Manage. 2010;39:167–179. doi: 10.1016/j.jpainsymman.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Johnson JR, Lossignol D, Burnell-Nugent M, Fallon MT. An open-label extension study to investigate the long-term safety and tolerability of THC/CBD oromucosal spray and oromucosal THC spray in patients with terminal cancer-related pain refractory to strong opioid analgesics. J Pain Symptom Manage. 2013;46:207–218. doi: 10.1016/j.jpainsymman.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. 2004;24:1744–1753. doi: 10.1523/JNEUROSCI.4298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. J Pain. 2010;11:1420–1428. doi: 10.1016/j.jpain.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Nomura DK, O’Neal ST, Long JZ, Mahadevan A, Cravatt BF, Grider JR, Lichtman AH. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Wise LE, Ramesh D, Abdullah R, Selley DE, Cravatt BF, Lichtman AH. Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther. 2013;345:492–501. doi: 10.1124/jpet.112.201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little PJ, Compton DR, Johnson MR, Melvin LS, Martin BR. Pharmacology and stereoselectivity of structurally novel cannabinoids in mice. J Pharmacol Exp Ther. 1988;247:1046–1051. [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009a;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny M, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A. 2009b;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mars SG, Bourgois P, Karandinos G, Montero F, Ciccarone D. “Every ‘never’ I ever said came true”: transitions from opioid pills to heroin injecting. Int J Drug Policy. 2014;25:257–266. doi: 10.1016/j.drugpo.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Garcia E, Burokas A, Kostrzewa E, Gieryk A, Korostynski M, Ziolkowska B, Przewlocka B, Przewlocki R, Maldonado R. New operant model of reinstatement of food-seeking behavior in mice. Psychopharmacology (Berl) 2011;215:49–70. doi: 10.1007/s00213-010-2110-6. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Merighi S, Gessi S, Varani K, Fazzi D, Mirandola P, Borea PA. Cannabinoid CB(2) receptor attenuates morphine-induced inflammatory responses in activated microglial cells. Br J Pharmacol. 2012;166:2371–2385. doi: 10.1111/j.1476-5381.2012.01948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LL, Picker MJ, Umberger MD, Schmidt KT, Dykstra LA. Effects of alterations in cannabinoid signaling, alone and in combination with morphine, on pain-elicited and pain-suppressed behavior in mice. J Pharmacol Exp Ther. 2012;342:177–187. doi: 10.1124/jpet.112.191478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narang S, Gibson D, Wasan AD, Ross EL, Michna E, Nedeljkovic SS, Jamison RN. Efficacy of dronabinol as an adjuvant treatment for chronic pain patients on opioid therapy. J Pain. 2008;9:254–264. doi: 10.1016/j.jpain.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Niphakis MJ, Johnson DS, Ballard TE, Stiff C, Cravatt BF. O-hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem Neurosci. 2012;3:418–426. doi: 10.1021/cn200089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Hudak CS, Ward AM, Burston JJ, Issa RS, Fisher KJ, Abood ME, Wiley JL, Lichtman AH, Casida JE. Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorg Med Chem Lett. 2008;18:5875–5878. doi: 10.1016/j.bmcl.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens RA, Ignatowska-Jankowska B, Mustafa M, Beardsley PM, Wiley JL, Jali A, Selley DE, Niphakis MJ, Cravatt BF, Lichtman AH. Discriminative Stimulus Properties of the Endocannabinoid Catabolic Enzyme Inhibitor SA-57 in Mice. J Pharmacol Exp Ther. 2016;358:306–314. doi: 10.1124/jpet.115.229492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paldy E, Bereczki E, Santha M, Wenger T, Borsodi A, Zimmer A, Benyhe S. CB(2) cannabinoid receptor antagonist SR144528 decreases mu-opioid receptor expression and activation in mouse brainstem: role of CB(2) receptor in pain. Neurochem Int. 2008;53:309–316. doi: 10.1016/j.neuint.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Ramesh D, Gamage TF, Vanuytsel T, Owens RA, Abdullah RA, Niphakis MJ, Shea-Donohue T, Cravatt BF, Lichtman AH. Dual inhibition of endocannabinoid catabolic enzymes produces enhanced antiwithdrawal effects in morphine-dependent mice. Neuropsychopharmacology. 2013;38:1039–1049. doi: 10.1038/npp.2012.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, Long JZ, Nomura DK, Sim-Selley LJ, Cravatt BF, Akbarali HI, Lichtman AH. Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J Pharmacol Exp Ther. 2011;339:173–185. doi: 10.1124/jpet.111.181370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA. mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148:387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux P, Sullivan MA, Cohen J, Fugon L, Jones JD, Vosburg SK, Cooper ZD, Manubay JM, Mogali S, Comer SD. Buprenorphine/naloxone as a promising therapeutic option for opioid abusing patients with chronic pain: reduction of pain, opioid withdrawal symptoms, and abuse liability of oral oxycodone. Pain. 2013;154:1442–1448. doi: 10.1016/j.pain.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakin YS, Dogrul A, Ilkaya F, Seyrek M, Ulas UH, Gulsen M, Bagci S. The effect of FAAH, MAGL, and Dual FAAH/MAGL inhibition on inflammatory and colorectal distension-induced visceral pain models in Rodents. Neurogastroenterol Motil. 2015;27:936–944. doi: 10.1111/nmo.12563. [DOI] [PubMed] [Google Scholar]
- Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, Moreno-Sanz G, Reggiani A, Piomelli D. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res. 2012;65:553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz AR, Lee M, Condie RB, Pulaski JT, Kaminski NE. Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol Appl Pharmacol. 1997;142:278–287. doi: 10.1006/taap.1996.8034. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria G, Barbano MF, Maldonado R, Valverde O. A reliable method to study cue-, priming-, and stress-induced reinstatement of cocaine self-administration in mice. Psychopharmacology (Berl) 2008;199:593–603. doi: 10.1007/s00213-008-1184-x. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215(1):89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Szabo B, Schlicker E. Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol. 2005:327–365. doi: 10.1007/3-540-26573-2_11. [DOI] [PubMed] [Google Scholar]
- Tallaria RJ. Drug synergism: Its detection and application. J Pharmacol Exp Ther. 2001;298:865–872. [PubMed] [Google Scholar]
- Tallaria RJ. An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther. 2006;319:1–7. doi: 10.1124/jpet.106.104117. [DOI] [PubMed] [Google Scholar]
- Thomas D, Frascella J, Hall T, Smith W, Compton W, Koroshetz W, Briggs J, Grady P, Somerman M, Volkow N. Reflections on the role of opioids in the treatment of chronic pain: a shared solution for prescription opioid abuse and pain. J Intern Med. 2014 doi: 10.1111/joim.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thors L, Burston JJ, Alter BJ, McKinney MK, Cravatt BF, Ross RA, Pertwee RG, Gereau RWt, Wiley JL, Fowler CJ. Biochanin A, a naturally occurring inhibitor of fatty acid amide hydrolase. Br J Pharmacol. 2010;160:549–560. doi: 10.1111/j.1476-5381.2010.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PF, Wolff RF, Deshpande S, Di Nisio M, Duffy S, Hernandez AV, Keurentjes JC, Lang S, Misso K, Ryder S, Schmidlkofer S, Westwood M, Kleijnen J. Cannabinoids for Medical Use: A Systematic Review and Meta-analysis. JAMA. 2015;313:2456–2473. doi: 10.1001/jama.2015.6358. [DOI] [PubMed] [Google Scholar]
- Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, Kuhn MN, Thakur GA, Makriyannis A, Milligan ED. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain. 2012;153:1091–1106. doi: 10.1016/j.pain.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JL, Niphakis MJ, Grim TW, Mustafa MA, Abdullah RA, Poklis JL, Dewey WL, Akbarali HI, Banks ML, Wise LE, Cravatt BF, Lichtman AH. The selective monoacylglycerol lipase inhibitor MJN110 produces opioid sparing effects in a mouse neuropathic pain model. J Pharmacol Exp Ther. 2016;357:145–156. doi: 10.1124/jpet.115.229971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Poe AR, Pocius E, Herschbach M, Morgan MM. The periaqueductal gray contributes to bidirectional enhancement of antinociception between morphine and cannabinoids. Pharmacol Biochem Behav. 2013;103:444–449. doi: 10.1016/j.pbb.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise LE, Cannavacciulo R, Cravatt BF, Martin BF, Lichtman AH. Evaluation of fatty acid amides in the carrageenan-induced paw edema model. Neuropharmacology. 2008;54:181–188. doi: 10.1016/j.neuropharm.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.