

Abstract
Targeted delivery of chemotherapeutic agents to tumors has been explored as a means to increase the selectivity and potency of cytotoxicity. Most efforts in this area have exploited the molecular recognition of proteins highly expressed on the surface of cancer cells followed by internalization. A related approach that has received less attention is the targeting of intracellular proteins by ligands conjugated to anti-cancer drugs. An attractive target for this approach is the enzyme cyclooxygenase-2 (COX-2), which is highly expressed in a range of malignant tumors. Herein, we describe the synthesis and evaluation of a series of chemotherapeutic agents targeted to COX-2 by conjugation to indomethacin. Detailed characterization of compound 12, a conjugate of indomethacin with podophyllotoxin, revealed highly potent and selective COX-2 inhibition in vitro and in intact cells. Kinetics and X-ray crystallographic studies demonstrated that compound 12 is a slow, tight-binding inhibitor that likely binds to COX-2’s allosteric site with its indomethacin moiety in a conformation similar to that of indomethacin. Compound 12 exhibited cytotoxicity in cell culture similar to that of podophyllotoxin with no evidence of COX-2-dependent selectivity. However, in vivo, compound 12 accumulated selectively in and more effectively inhibited the growth of a COX-2-expressing xenograft compared to a xenograft that did not express COX-2. Compound 12, which we have named chemocoxib A, provides proof-of-concept for the in vivo targeting of chemotherapeutic agents to COX-2, but suggests that COX-2-dependent selectivity may not be evident in cell culture-based assays.
Covalent attachment of anti-cancer drugs to antibodies or receptor ligands has generated conjugates that display enhanced cytotoxicity to cells expressing the relevant molecular targets.1, 2 This approach primarily exploits the molecular recognition of proteins selectively enriched on the surface of cancer cells followed by internalization of the conjugate. A related approach that has received less attention is the direct targeting of intracellular proteins by ligand conjugates to anti-cancer drugs. An attractive target for such an approach is the enzyme cyclooxygenase-2 (COX-2), which is highly expressed in a range of premalignant and malignant tumors and plays a role in promoting the malignant phenotype.3–6 The COX-2 inhibitory nonsteroidal anti-inflammatory drugs (NSAIDs) have been extensively explored as both cancer chemopreventative and adjuvant chemotherapeutic agents for cancers expressing the enzyme.7–13 Our laboratory has demonstrated that conjugates of fluorophores and COX-2 inhibitors selectively accumulate in cancer cells that express COX-2 and are taken up by COX-2-expressing human tumor xenografts or nonmelanoma skin cancers in mice, and transitional cell bladder cancers in dogs. Uptake appears to be direct, and the fluorophores co-localize with COX-2 in perinuclear regions of targeted cells.14–16 These studies demonstrate COX-2-targeted delivery of imaging agents to tumors, suggesting the possibility that this approach can be used to selectively deliver chemotherapeutic agents as well. A recent attempt to test this hypothesis using conjugates of cisplatin analogs with the NSAIDs indomethacin and ibuprofen yielded conjugates that showed no COX-2-dependent cytotoxic selectivity in cultured tumor cells; however some conjugates were notable for their ability to overcome cisplatin-related resistance.17 Here, we describe the synthesis and evaluation of a series of chemotherapeutic agent conjugates, leading to the discovery of compound 12 (chemocoxib A), a podophyllotoxin-indomethacin conjugate that displays COX-2-selective accumulation and anti-tumor activity in vivo.
RESULTS AND DISCUSSION
Discovery of COX-2-Selective Indomethacin Conjugates
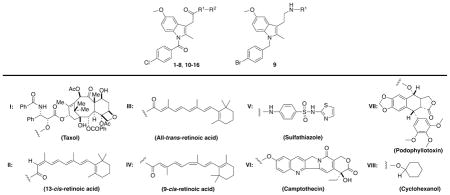
Previous studies have demonstrated that amidation or esterification converts some carboxylic acid-containing NSAIDs that inhibit both the COX-1 and COX-2 isoforms to COX-2-selective inhibitors.18–20 In general, indomethacin derivatives are the most potent and selective, so we synthesized a series of conjugates of chemotherapeutic agents to indomethacin through various amide or ester tethers. Synthetic details and complete chemical characterization of the target molecules are provided in the Supporting Information. Each molecule was evaluated for its ability to inhibit purified mouse COX-2 or ovine COX-1 using a previously described assay.19 Table 1 summarizes these data.
Table 1.
Biochemical properties of cytotoxic conjugates. The IC50’s for inhibition of purified ovine COX-1, mouse COX-2, or COX-2 in 1483 HNSCC cell line.
| |||||
|---|---|---|---|---|---|
|
| |||||
| No | R1 | R2 | Purified Enzyme IC50 (μM) |
1483 Cells IC50 (μM) |
|
| COX-1 | COX-2 | COX-2 | |||
| 1 | - | I | > 25 | > 25 | NT |
| 2 |
|
I | > 25 | > 25 | NT |
| 3 |
|
I | > 25 | 2.20 | NT |
| 4 |
|
I | > 25 | 0.25 | 0.42 |
| 5 |
|
II | > 25 | 0.45 | 0.98 |
| 6 |
|
III | > 25 | 0.10 | NT |
| 7 |
|
IV | > 25 | 0.60 | NT |
| 8 |
|
V | > 25 | 0.92 | NT |
| 9 |
|
V | > 25 | > 25 | NT |
| 10 |
|
VI | > 25 | > 25 | NT |
| 11 |
|
VII | > 25 | > 47 | NT |
| 12 |
|
VII | > 25 | 0.29 | 0.20 |
| 13 |
|
VII | > 25 | 0.25 | NT |
| 14 |
|
VII | > 25 | 0.14 | NT |
| 15 |
|
VII | > 25 | 0.18 | NT |
| 16 |
|
VIII | > 25 | 0.48 | 0.20 |
| - | Podophyllotoxin | - | > 25 | > 25 | NT |
| - | Indomethacin | - | 0.05 | 0.75 | NT |
Consistent with prior reports on indomethacin conjugates to bulky fluorophores,20 hydrophobic tethers of moderate length were most effective at enabling selective binding to COX-2. Conjugates to taxol containing no tether (1) or a long, polar tether (2) did not inhibit COX-2 or COX-1, while introduction of a butyl (3) or benzyl (4) tether produced active compounds. Three retinoic acid conjugates, 5–7, were all good inhibitors, while the ethylene diamide conjugate to sulfathiazole, 8, exhibited modest activity. Neither the reverse-indomethacin conjugate to sulfathiazole (9), nor the indomethacin conjugate to camptothecin (10) inhibited COX-2. Each of a series of indomethacin conjugates to podophyllotoxin exhibited good COX-2-selective inhibition (11–15). Podophyllotoxin itself was inactive against both COX enzymes.
Representatives of active classes of inhibitors were assayed for their ability to inhibit COX-2 in intact cells using the 1483 human head and neck squamous cell carcinoma (HNSCC) cell line. Compounds 12 and 16, displayed the highest potency in this assay, with an IC50 value of 200 nM (Table 1).
Kinetics of COX-2 Inhibition by Compound 12
Indomethacin is a slow, tight-binding inhibitor of both COX isoforms. Its inhibition kinetics are consistent with a model that includes an initial rapid equilibrium followed by a slower second step, resulting in a much more tightly bound complex (Eq. 1).
| Eq. 1 |
| Eq. 2 |
Figure 1A displays the time- and concentration-dependent inhibition of mCOX-2 by compound 12. Inhibition proceeded rapidly but plateaued at 20% activity remaining at high inhibitor concentrations. As indicated by Eq. 2, a plot of the observed single exponential rate constant for inhibition as a function of inhibitor concentration (Figure 1B) yields values for the equilibrium constant for initial association (KI = k−1/k1), and the individual rate constants for the second binding step. Compound 12 exhibited greater affinity for initial binding (KI = 2.6 ± 0.4 μM) than indomethacin (KI = 18.6 ± 2.5 μM) and a comparable forward rate constant (compound 12, k2 = 0.0024 s−1; indomethacin, k2 = 0.0052 s−1). However, a measurable second dissociation constant for compound 12 (k−2 = 0.0017 s−1) suggests greater reversibility for the COX-2-compound 12 complex than for the COX-2-indomethacin complex (k−2 ≈ 0 s−1). COX-2 is a structural homodimer that functions as a heterodimer, with one subunit serving as the catalytic site and the second subunit acting in an allosteric role. Prior data indicate that inhibitors may bind to the catalytic site, allosteric site, or both.21–23 Compound 12’s inability to completely inhibit COX-2, even at high concentrations, suggests that it binds in the allosteric site.
Figure 1.

Kinetics of the time-dependent inhibition of COX-2 by compound 12. (A) Purified mCOX-2 was reconstituted with heme and pre-incubated with the indicated concentrations of inhibitor at 37 °C for the indicated times prior to the addition of the substrate (50 μM). Data are the mean ± range of duplicate determinations. (B) Secondary plot of kobs versus inhibitor concentration used to generate values for KI, k2, and k−2.
Structural Determinants of COX-2 Inhibition
An analysis of the crystal structure of indomethacin complexed with COX-2 reveals that the carboxylic acid group of the inhibitor both hydrogen-bonds and ion pairs with Tyr355 and Arg120, which are located at the base of the active site. Mutation of either Tyr355 to Phe or Arg120 to Ala abolishes inhibition by indomethacin.18 The data in Table 2 demonstrate that compound 12 does not inhibit the Y355F mutant of COX-2, suggesting that, like indomethacin, compound 12 interacts with Tyr355. In contrast, both indomethacin and compound 12 inhibit R120Q COX-2, and compound 12 actually exhibits a 3-fold increase in potency for this mutant. Although the R120Q mutant loses the ability to ion pair with an inhibitor, it retains the ability to hydrogen bond. Thus, these results suggest that hydrogen bonding is important for the interaction of compound 12 with Arg120.
Table 2.
Inhibition of mouse COX-2 mutants by indomethacin or compound 12.
| Enzymes | Indomethacin IC50 (μM)a |
Compound 12 IC50 (μM)a |
|---|---|---|
| wt mCOX-2 | 0.25 | 0.29 |
| wt oCOX-1 | 0.04 | > 4 |
| V349L | 4.0 | > 4 |
| V523I | 0.45 | 1.1 |
| S530A | 0.22 | 1.5 |
| R120Q | 0.30 | 0.09 |
| Y355F | > 4 | > 4 |
IC50 values are average determinations from three experiments. The experimental method is described in the Supporting Information.
Crystal structure data show that the 2′-methyl group of indomethacin inserts into a small hydrophobic pocket comprising Ala527, Val349, Ser530, and Leu531. Indomethacin’s potent, time-dependent inhibition of both COX-1 and COX-2 has been attributed to this interaction.24 Mutation of Val349 to Leu dramatically decreases indomethacin’s potency due to a reduction in the size of the pocket. A similar loss of potency was observed for compound 12 (Table 2). Furthermore, due to its close proximity to the 5-methoxy group of indomethacin, mutation of Val523 to the larger isoleucine results in a 1.8-fold decrease in potency for the inhibitor.25 The reduction in potency against V523I was greater for compound 12 (3.8-fold) (Table 2).
Compound 12:COX-2 Complex Crystal Structure
The parallel pattern of inhibition of COX-2 mutants by indomethacin and compound 12 suggests that they bind to the enzyme in a similar fashion. To test this hypothesis, we crystallized a complex of compound 12 bound to COX-2 and solved its structure by molecular replacement. The complex crystallized in the P21212 space group and diffracted to 2.1 Å (Supplementary Table 1). Two dimers were present in the asymmetric unit. The r.m.s.d. value of any two monomers was 0.12 to 0.16 Å for the backbone atoms. The indomethacin group and the linker of compound 12 were well defined in the electron density, but the podophyllotoxin moiety was not (Supplementary Figure 1). This resulted in a high B-factor for this portion of the ligand (94.8 Å2) compared to that of the indomethacin moiety (37.9 Å2). As a result, the occupancies of the podophyllotxin portion were set to zero in the deposited structure. As predicted, the indomethacin moiety of compound 12 in the crystal structure is positioned in the cyclooxygenase active site in an orientation similar to that previously reported for indomethacin.26 The chlorine atom on the phenyl ring is inserted into the hydrophobic crown made of Leu384, Tyr385, Trp387, Phe518, and Met522, and the carbonyl oxygen forms a hydrogen bond to Ser530 with an effective distance of 3.0 Å. As predicted from the mutation studies, the methoxy group on the indole ring is adjacent to Val523. Interestingly, the carbonyl oxygen of compound 12’s amide group is about 3.6 Å away from Nε of Arg120, suggesting that accommodation of the bridge linker may weaken this interaction as compared to that of indomethacin. Tyr355 is hydrogen-bonded to the nitrogen atom of the amide with a distance of 3.0 Å. The linker region of compound 12 passes through the constriction region at the base of the active site into the channel of the membrane-binding domain of COX-2, placing the podophyllotoxin moiety in the expanded lobby region below the active site. Since only a small region of electron density was observed for the podophyllotoxin moiety, this portion of the structure was modeled into the lobby region (Figure 2). The model predicts that the principal location of the podophyllotoxin portion of the conjugate lies underneath the D-helix that connects the membrane-binding domain (helices A, B, and C) to the catalytic domain.
Figure 2.

Structure of the mCOX-2:compound 12 complex (PDB ID: 4OTJ). Shown is a stereoview of Compound 12 bound to the active site and the lobby region of COX-2. Compound 12 is presented in orange, and COX-2 is shown as a ribbon in grey. Selected key residues are in green.
Effects of Compound 12 in Cells
The anti-proliferative effects of compound 12 were evaluated in 1483 HNSCC cells. Compound 12 induced a dramatic reduction in 1483 cell number at 48 h (EC50 = 62 nM), which was the same as the reduction induced by podophyllotoxin (EC50 = 62 nM) (Figure 3A). The inability to reduce cell number below ~ 35% of the number of cells observed in vehicle-treated cultures suggests that both compounds are cytostatic, not cytotoxic, in 1483 cells. The anti-proliferative effects of 12 were evaluated against other neoplastic cell types, including RKO, HCT116, MDA MB231, HCA-7, IMR-5, NCI-H292, RAW264.7, NHDF, and HEK293T cells, and potent inhibition was observed in all, regardless of COX-2 expression level (Table S2). In contrast, no reduction in cell number was observed at 48 h when nontumorigenic primary human mammary epithelial cell (HMEC) cultures were treated with compound 12, despite the strong cytostatic effect of podophyllotoxin in these cells (EC50 = 360 nM) (Figure 3B).
Figure 3.

Effect of compound 12 on cell viability. Shown are the viabilities of 1483 HNSCC cells (A) or primary human mammary epithelial cells (B) in the presence of Compound 12 (dashed line, ●) or podophyllotoxin (solid line, □) after a 48 h treatment. Data are the mean ± standard deviation (n = 6).
The reversibility of inhibition of 1483 cell growth was evaluated using a high concentration of compound 12. Compound 12 (10 μM) was added to cultured 1483 cells, and cell number was evaluated after 72 h (Figure 4A). As anticipated, both compound 12 and podophyllotoxin reduced total cell number by ~ 80%. In parallel experiments, cells were treated with the compounds for 5 h, then the medium was replaced, and the cells were incubated without compound for an additional 72 h (Figure 4b). The washout had no effect on the reduction in 1483 cell number by compound 12, but there was a significant recovery of cell number in cultures treated with podophyllotoxin and washed after 5 h. The differential cytostatic effects of compound 12 and podophyllotoxin on 1483 and HMEC cells along with the partial reversibility of the effects of podophyllotoxin on 1483 cells suggest differences in their modes of action or retention in the cells after washing. However, these differences could not be attributed to an interaction of compound 12 with COX-2.
Figure 4.

Reversibility of compound 12-mediated cytotoxicity. (A) Viability of 1483 HNSCC cells following incubation in the presence of the vehicle dimethylsulfoxide (DMSO), 10 μM compound 12, or 10 μM podophyllotoxin for 72 h. (B) Viability of 1483 HNSCC cells following incubation in the presence of DMSO, 10 μM compound 12, or 10 μM podophyllotoxin for 5 h followed by washout and further incubation for 72 h. Data are the mean ± standard deviation (n = 6).
Effects of Compound 12 in Vivo
The cell culture studies suggested that compound 12 is selectively cytostatic against tumorigenic as opposed to nontumorigenic cells but fails to discriminate on the basis of COX-2 expression. Thus, COX-2 targeting appears to have no selective value ex vivo. To determine if this conclusion also applies in vivo, we evaluated the ability of compound 12 to affect the growth of human tumor xenografts in nude mice. Treatment of 1483 xenograft-bearing mice with compound 12 for 14 days reduced tumor growth by approximately 50% compared to treatment with vehicle, while parallel experiments conducted with podophyllotoxin demonstrated no effect on tumor size (Figure 5A). A decrease in animal weight was observed in podophylotoxin-treated animals, suggesting toxicity, whereas compound 12 had no effect on animal weight. The importance of COX-2 expression for inhibition of tumor growth was demonstrated by compound 12’s lack of efficacy against COX-2-negative HCT116 human colon cancer cell xenografts (Figure 5B).
Figure 5.

In vivo tumor growth inhibition by compound 12. (A) Female nude mice bearing 1483 HNSCC xenografts were treated with vehicle (solid line, ●), podophyllotoxin (dotted line, □) or compound 12 (dashed line, ▲) for the indicated number of days. Data are the mean ± standard deviation (n = 8). (B) Female nude mice bearing HCT116 cell xenografts were treated with vehicle (solid line, ●) or compound 12 (dashed line, ▲). Data are the mean ± standard deviation (n = 7). (C) Mass spectrometric analysis of extracts of a 1483 cell xenograft 3 h post treatment with compound 12. (D) Levels of compound 12 in 1483 and HCT116 cell xenografts 24 h after completion of a 14 day course of treatment. Data are the mean ± standard deviation (n = 5). (The asterisk denotes that the data are significantly different at p = 0.048).
The correlation of tumor growth inhibition by compound 12 with COX-2 expression suggests that association with COX-2 may increase compound 12’s concentration in tumors that express this enzyme. Mass spectrometric analysis of 1483 tumor tissue extracts 3 h after injection confirmed the presence of compound 12 at a concentration of 31.6 pmol g−1 tissue (Figure 5C). Compound 12 was the only indomethacin-containing species detected in the tumors, suggesting that it has the in vivo metabolic stability required to circulate and accumulate in the tumor. Comparison of levels of compound 12 in a COX-2-expressing tumor (1483) compared to a non-COX-2-expressing tumor (HCT116) 24 h following chronic dosing in xenograft-bearing nude mice revealed a 3.7-fold higher concentration of compound 12 in the 1483 tumors than the HCT116 tumors (p = 0.048) (Figure 5D). The strong correlation between the COX-2 status of the tumor and the levels of compound 12 that accumulated on chronic administration suggests that COX-2-targeting contributes to the compound’s in vivo anti-tumor activity. We further tested this hypothesis by treating mice bearing 1483 xenografts with doses of both indomethacin and podophyllotoxin comparable to the quantities of each drug present in a 2 mg kg−1 dose of compound 12. This treatment had no effect on tumor growth (Supplementary Figure S2A), indicating that the efficacy of compound 12 requires that the indomethacin-podophyllotoxin conjugate be intact.
COX-2 inhibitors such as indomethacin have been reported to inhibit the growth of certain human tumor xenografts in nude mice, so we conducted additional experiments to determine if the reduction in tumor growth required the presence of the podophyllotoxin moiety. An indomethacin conjugate that contained the same tether as compound 12 but with a cyclohexanol group instead of podophyllotoxin (compound 16) was administered to 1483 tumor-bearing mice in parallel with compound 12. Although compound 16 was an efficient and selective COX-2 inhibitor (IC50 = 480 nM), it did not inhibit tumor growth (Figure S2B).
To our knowledge, compound 12, which we have named chemocoxib A, is the first example of an NSAID-chemotherapeutic agent conjugate that selectively inhibits COX-2 and targets chemotherapeutic activity to a COX-2-expressing tumor in vivo. Our results provide proof-of-concept that targeting an anti-tumor agent to an intracellular protein (in this case, COX-2) may be a useful strategy to increase anti-tumor selectivity and improve the therapeutic window for the treatment of naturally occurring cancers. However, our results also indicate that these effects may not be evident in cell culture. Thus, the benefits of intracellular protein targeting will most likely be maximized in the in vivo environment in which accumulation of inhibitor in the tumor is the result of a dynamic balance between compound administration and delivery, versus metabolism and excretion. Given the diversity of structure and function of cancer chemotherapeutic agents, the range of potential COX-2-targeted chemotherapeutic conjugates is wide, offering many opportunities to exploit this novel approach.
METHODS
COX Enzyme Inhibition Assays
We evaluated the ability of test compounds to inhibit purified ovine COX-1 or murine COX-2 utilizing previously published protocols.14
COX-2 Inhibition Assay in Cells
Mycoplasma negative human 1483 HNSCC cells (Dr. Peter Sacks, New York University School of Dentistry, New York, NY) were cultured in Dulbecco’s Modified Eagle Medium:Nutrient Mixture F12 (Life Technologies) supplemented with 10% (v/v) fetal bovine serum (Atlas Biologicals) and antibiotic/antimycotic (Life Technologies) in six-well plates. Cells (passage 8 to 20) at 60% confluence were overlaid with serum-free medium (2 mL) containing inhibitor dissolved in dimethyl sulfoxide (0–5 μM, final concentration) and preincubated for 30 min at 37 °C. Then, [1-14C]-arachidonic acid (10 μM, ~55 mCi mmol−1) was added, and the cells were incubated for an additional 20 min at 37 °C. Reactions were terminated, and the samples were processed as previously described.14
X-Ray Co-crystallography
Samples for crystallization were prepared as previously described27 except that 10% (v/v) dimethyl sulfoxide was added along with Compound 12 to aid complex formation. Crystallization was achieved using the drop vapor diffusion method as described previously.28 Crystals were mounted after growth to full size in 2–3 weeks and flash frozen in liquid nitrogen for shipment and data collection.
Data were acquired at the Northeastern Collaborative Access Team beam line 24-ID-C at the Advance Photon Source at the Argonne National Laboratory. The data were processed with XDS,29 revealing compound 12:COX-2 cocrystals belonging to space group P21212. Phaser30 was employed to determine initial phases by molecular replacement based on a search model (PDB id: 3NT1). Coot and PHENIX31, 32 were utilized for several rounds of model refinement that including application of global non-crystallographic symmetry. Ligand restraints were built using the PRODRG Server.33 Addition of water molecules during last cycles of refinement preceded TLS refinement, which was applied in the final cycle. Simulated annealing using PHENIX34 excluded the potential of phase bias. The PHENIX Suite also provided the values of the Ramachandran plot for the final structure. The structure factor and atomic coordinates have been deposited in the Protein Data Bank under the access id (http://www.rcsb.org/). The monomers exhibited no significant structural differences, and all illustrations were prepared using the coordinates of monomer A with Pymol (Schrödinger, LLC).
Cell Viability Assay
Human 1483 HNSCC cells were cultured as described above. Primary HMECs (a gift of Dr. Jennifer Pietenpol, Vanderbilt University) were grown in HMEC medium containing Mammary Epithelial Growth Supplement (Life Technologies). Cells were plated at 8000 to 10000 cells per well in 100 μl medium in 96 well plates (Sarstedt). No cells were plated in the first column of the plate. Following a 24 h incubation, fresh growth medium containing the desired concentrations of test compounds or vehicle (dimethyl sulfoxide, final concentration, 0.1% (v/v)) were placed in each well. After 48 h, cell viability was assessed using the WST-1 Cell Proliferation Reagent (Roche, 11644807001) as described.35
In Vivo Tumor Growth in Nude Mice
In vivo studies were carried out using a protocol (M/10/144) approved by the Vanderbilt University Institutional Animal Use and Care Committee. Female nude mice (Athymic Nude-Foxn 1nu, 5–6 weeks old) were purchased from Harlan Labs. HCT116 cells were trypsinized and resuspended in cold phosphate-buffered saline at a concentration of 2 × 107 cells mL−1. 1483 HNSCC cells were trypsinized and resuspended in cold phosphate-buffered saline containing 30% (v/v) Matrigel (BD Biosciences) at a concentration of 2 × 107 cells mL−1. Each mouse received a subcutaneous injection of the HCT116 or 1483 HNSCC cell suspension (100 μL) on the left flank. Daily intraperitoneal injections (100 μL) of vehicle (dimethyl sulfoxide) or test compound in vehicle (2 mg kg−1) began on the following day (Day 1) and continued for 14 days. Electronic caliper (General Tools) measurements of xenografts and total body weight measurements were obtained on Days 2, 7, 12, and 15. Mice were euthanized on day 15.
Tissue analysis
In one study, three mice were injected with 1483 cells subcutaneously as above, and the xenografts were allowed to grow for 10 days followed by a single intraperitoneal dose of compound 12 (2 mg kg−1). Three hours post-injection, the mice were euthanized for analysis of compound 12 in tumor tissue, which was carried out by a previously described method.36 Quantification of 12 was based on a 5-point standard curve derived from samples that were subjected to the proteinase K digestion and liquid-liquid extraction procedures.
Solvents and Reagents
Fisher was the source for HPLC grade solvents. All reagents, fluorescent dyes, and deuterated solvents were purchased from the Aldrich Chemical Company. These were used without further purification.
Chromatography
Compound purity was assessed using HPLC. The HPLC evaluation of fluorescent compounds (used for all compounds except compound 16) was conducted using a 2996 HPLC system from Waters (C18 column, UV or fluorescence detector). Condition 1 – HPLC fluorescence detection. H2O with 0.5% (v/v) HCO2H (component A), 50% (v/v) MeOH in MeCN with 0.5% (v/v) HCO2H (component B). Condition 2 (used for compound 16) – HPLC UV detection. H2O with 5 mM octanesulfonic acid (pH ~ 3, component A), MeCN with 10% (v/v) component A (component B).
Spectroscopic Analysis
All new compounds were characterized by 1H NMR, 13C NMR, and/or HRMS analyses as described.20
General Synthetic Methods
Method A: Carbodiimide-catalyzed esterification of a secondary alcohol
To a stirred solution of indomethacin or indomethacin-linker-COOH (0.0234 mmol) in toluene (2 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (0.0234 mmol), and the resulting solution was stirred for 15 min at room temperature. Dimethylaminopyridine (0.04 mmol) was added, and the solution was stirred 5 min at room temperature. Then, toxin-OH (0.0234 mmol) was added and stirred 30 h at 60 °C. The reaction mixture was cooled to room temperature and extracted with ethyl acetate. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The product was purified by silica gel column chromatography using n-hexane:ethyl acetate (3:4).
Method B: Carbodiimide-catalyzed esterification of a primary alcohol
To a stirred solution of a toxin-carboxylic acid (10 mmol) in methylene chloride (100 ml) was added the indomethacin-linker-OH (30 mmol), 1-hydroxybenzotriazole hydrate (15 mmol), N,N-dimethylaminopyridine (15 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (11 mmol) at 25 °C. The resultant mixture was stirred for 16 h at 25 °C. Removal of solvent in vacuo afforded a residue, which was dissolved in 100 mL of water and extracted with ethyl acetate. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The product was purified by crystallization or by silica gel column chromatography using n-hexane:ethyl acetate (4:1).
Method C: Carbodiimide-catalyzed amidation
To a stirred solution of a toxin-carboxylic acid (1 mmol) in dimethyl formamide (10 mL) was added indomethacin-linker-amine (3 mmol), 1-hydroxybenzotriazole hydrate (1.5 mmol), N,N-diisopropylethylamine (3 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.1 mmol) at 25 °C. The resultant mixture was stirred for 16 h at 25 °C. Removal of the solvent in vacuo afforded a residue, which was dissolved in 10 mL water and extracted with ethyl acetate. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The amide product was purified by crystallization or by silica gel column chromatography using chloroform:methanol:aqueous ammonia (35:7:1).
Supplementary Material
Acknowledgments
We are grateful to National Institutes of Health (R01 CA89450) to support this project and to Northeastern Collaborative Access Team beam lines for X-ray co-crystal diffraction studies funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403) and Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We are also grateful to the Vanderbilt Small Molecule NMR Core and the Vanderbilt Mass Spectroscopy Research Center for compound characterizations.
ABBREVIATIONS
- COX
cyclooxygenase
- NSAID
nonsteroidal anti-inflammatory drug
- HNSCC
head and neck squamous cell carcinoma
- HMEC
human mammary epithelial cells
Footnotes
ACCESSION CODES: PDB ID: 4OTJ
Supporting Information. Supporting Information includes detailed synthetic methods, Tables S1 (X-ray crystallography data collection and statistical information) and S2 (viability of cell lines treated with compound 12), and Figures S1 (full representation of the crystal structure of the mCOX-2:compound 12 complex) and S2 (in vivo tumor growth suppression for indomethacin plus podophyllotoxin and compound 12 versus compound 16). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00480.
References
- 1.Peters C, Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015;35:e00225. doi: 10.1042/BSR20150089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vlahov IR, Leamon CP. Engineering folate-drug conjugates to target cancer: from chemistry to clinic. Bioconjug Chem. 2012;23:1357–1369. doi: 10.1021/bc2005522. [DOI] [PubMed] [Google Scholar]
- 3.Abdalla SI, Lao-Sirieix P, Novelli MR, Lovat LB, Sanderson IR, Fitzgerald RC. Gastrin-induced cyclooxygenase-2 expression in Barrett’s carcinogenesis. Clin Cancer Res. 2004;10:4784–4792. doi: 10.1158/1078-0432.CCR-04-0015. [DOI] [PubMed] [Google Scholar]
- 4.Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, Kimura S, Kato H, Kondo M, Hla T. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995;55:3785–3789. [PubMed] [Google Scholar]
- 5.Shirvani VN, Ouatu-Lascar R, Kaur BS, Omary MB, Triadafilopoulos G. Cyclooxygenase 2 expression in Barrett’s esophagus and adenocarcinoma: Ex vivo induction by bile salts and acid exposure. Gastroenterology. 2000;118:487–496. doi: 10.1016/s0016-5085(00)70254-x. [DOI] [PubMed] [Google Scholar]
- 6.Taketo MM. COX-2 and colon cancer. Inflamm Res. 1998;47(Suppl 2):S112–116. doi: 10.1007/s000110050295. [DOI] [PubMed] [Google Scholar]
- 7.Edelman MJ, Watson D, Wang X, Morrison C, Kratzke RA, Jewell S, Hodgson L, Mauer AM, Gajra A, Masters GA, Bedor M, Vokes EE, Green MJ. Eicosanoid modulation in advanced lung cancer: cyclooxygenase-2 expression is a positive predictive factor for celecoxib + chemotherapy--Cancer and Leukemia Group B Trial 30203. J Clin Oncol. 2008;26:848–855. doi: 10.1200/JCO.2007.13.8081. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62:233–244. doi: 10.1016/s1734-1140(10)70262-0. [DOI] [PubMed] [Google Scholar]
- 9.Kao J, Sikora AT, Fu S. Dual EGFR and COX-2 inhibition as a novel approach to targeting head and neck squamous cell carcinoma. Curr Cancer Drug Targets. 2009;9:931–937. doi: 10.2174/156800909790192437. [DOI] [PubMed] [Google Scholar]
- 10.Khan Z, Khan N, Tiwari RP, Sah NK, Prasad GB, Bisen PS. Biology of Cox-2: an application in cancer therapeutics. Curr Drug Targets. 2011;12:1082–1093. doi: 10.2174/138945011795677764. [DOI] [PubMed] [Google Scholar]
- 11.Moreira L, Castells A. Cyclooxygenase as a target for colorectal cancer chemoprevention. Curr Drug Targets. 2011;12:1888–1894. doi: 10.2174/138945011798184218. [DOI] [PubMed] [Google Scholar]
- 12.Stan SD, Singh SV, Brand RE. Chemoprevention strategies for pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2010;7:347–356. doi: 10.1038/nrgastro.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiel A, Mrena J, Ristimaki A. Cyclooxygenase-2 and gastric cancer. Cancer Metastasis Rev. 2011;30:387–395. doi: 10.1007/s10555-011-9312-1. [DOI] [PubMed] [Google Scholar]
- 14.Uddin MJ, Crews BC, Blobaum AL, Kingsley PJ, Gorden DL, McIntyre JO, Matrisian LM, Subbaramaiah K, Dannenberg AJ, Piston DW, Marnett LJ. Selective visualization of cyclooxygenase-2 in inflammation and cancer by targeted fluorescent imaging agents. Cancer Res. 2010;70:3618–3627. doi: 10.1158/0008-5472.CAN-09-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cekanova M, Uddin MJ, Bartges JW, Callens A, Legendre AM, Rathore K, Wright L, Carter A, Marnett LJ. Molecular imaging of cyclooxygenase-2 in canine transitional cell carcinomas in vitro and in vivo. Cancer Prev Res. 2013;6:466–476. doi: 10.1158/1940-6207.CAPR-12-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ra H, Gonzalez-Gonzalez E, Uddin MJ, King BL, Lee A, Ali-Khan I, Marnett LJ, Tang JY, Contag CH. Detection of non-melanoma skin cancer by in vivo fluorescence imaging with fluorocoxib A. Neoplasia. 2015;17:201–207. doi: 10.1016/j.neo.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann W, Crews BC, Sarosi MB, Daniel CM, Ghebreselasie K, Scholz MS, Marnett LJ, Hey-Hawkins E. Conjugation of cisplatin analogues and cyclooxygenase inhibitors to overcome cisplatin resistance. ChemMedChem. 2015;10:183–192. doi: 10.1002/cmdc.201402353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalgutkar AS, Crews BC, Rowlinson SW, Marnett AB, Kozak KR, Remmel RP, Marnett LJ. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc Natl Acad Sci USA. 2000;97:925–930. doi: 10.1073/pnas.97.2.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalgutkar AS, Marnett AB, Crews BC, Remmel RP, Marnett LJ. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J Med Chem. 2000;43:2860–2870. doi: 10.1021/jm000004e. [DOI] [PubMed] [Google Scholar]
- 20.Uddin MJ, Crews BC, Ghebreselasie K, Marnett LJ. Design, synthesis, and structure-activity relationship studies of fluorescent inhibitors of cycloxygenase-2 as targeted optical imaging agents. Bioconjug Chem. 2013;24:712–723. doi: 10.1021/bc300693w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong L, Vecchio AJ, Sharma NP, Jurban BJ, Malkowski MG, Smith WL. Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J Biol Chem. 2011;286:19035–19046. doi: 10.1074/jbc.M111.231969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchener MM, Hermanson DJ, Shockley EM, Brown HA, Lindsley CW, Reese J, Rouzer CA, Lopez CF, Marnett LJ. Competition and allostery govern substrate selectivity of cyclooxygenase-2. Proc Natl Acad Sci USA. 2015;112:12366–12371. doi: 10.1073/pnas.1507307112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zou H, Yuan C, Dong L, Sidhu RS, Hong YH, Kuklev DV, Smith WL. Human cyclooxygenase-1 activity and its responses to COX inhibitors are allosterically regulated by nonsubstrate fatty acids. J Lipid Res. 2012;53:1336–1347. doi: 10.1194/jlr.M026856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prusakiewicz JJ, Felts AS, Mackenzie BS, Marnett LJ. Molecular basis of the time-dependent inhibition of cyclooxygenases by indomethacin. Biochemistry. 2004;43:15439–15445. doi: 10.1021/bi048534q. [DOI] [PubMed] [Google Scholar]
- 25.Blobaum AL, Uddin MJ, Felts AS, Crews BC, Rouzer CA, Marnett LJ. The 2′-Trifluoromethyl Analogue of Indomethacin Is a Potent and Selective COX-2 Inhibitor. ACS Med Chem Lett. 2013;4:486–490. doi: 10.1021/ml400066a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 27.Windsor MA, Hermanson DJ, Kingsley PJ, Xu S, Crews BC, Ho W, Keenan CM, Banerjee S, Sharkey KA, Marnett LJ. Substrate-Selective Inhibition of Cyclooxygenase-2: Development and Evaluation of Achiral Profen Probes. ACS Med Chem Lett. 2012;3:759–763. doi: 10.1021/ml3001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duggan KC, Walters MJ, Musee J, Harp JM, Kiefer JR, Oates JA, Marnett LJ. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J Biol Chem. 2010;285:34950–34959. doi: 10.1074/jbc.M110.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 34.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uddin MJ, Smithson DC, Brown KM, Crews BC, Connelly M, Zhu F, Marnett LJ, Guy RK. Podophyllotoxin analogues active versus Trypanosoma brucei. Bioorg Med Chem Lett. 2010;20:1787–1791. doi: 10.1016/j.bmcl.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uddin MJ, Crews BC, Ghebreselasie K, Huda I, Kingsley PJ, Ansari MS, Tantawy MN, Reese J, Marnett LJ. Fluorinated COX-2 inhibitors as agents in PET imaging of inflammation and cancer. Cancer Prev Res. 2011;4:1536–1545. doi: 10.1158/1940-6207.CAPR-11-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.