

Abstract
The enzyme cytochrome P450 11A1 cleaves the C20,C22 carbon-carbon bond of cholesterol to form pregnenolone, the first 21-carbon precursor of all steroid hormones. Various reaction mechanisms are possible for the carbon-carbon bond cleavage step of P450 11A1 and most current proposals involve the oxoferryl active species, Compound I (FeO3+). Compound I can either (i) abstract an O-H hydrogen atom or (ii) be attacked by a nucleophilic hydroxy group of its substrate, 20R,22R-dihydroxycholesterol. The mechanism of this carbon-carbon bond cleavage step was tested using 18O-labeled molecular oxygen and purified P450 11A1. P450 11A1 was incubated with 20R,22R-dihydroxycholesterol in the presence of molecular oxygen (18O2), and coupled assays were used to trap the labile 18O atoms in the enzymatic products (i.e., isocaproaldehyde and pregnenolone). The resulting products were derivatized and the 18O content was analyzed by high-resolution mass spectrometry. P450 11A1 showed no incorporation of an 18O-atom into either of its carbon-carbon bond cleavage products, pregnenolone and isocaproaldehyde. The positive control experiments established retention of the carbonyl oxygens in the enzymatic products during the trapping and derivatization processes. These results reveal a mechanism involving an electrophilic Compound I species that reacts with nucleophilic hydroxy groups in the 20R,22R-dihydroxycholesterol intermediate of the P450 11A1 reaction to produce the key steroid pregnenolone.
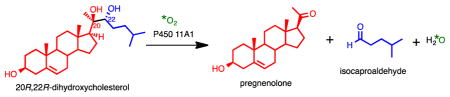
Graphical abstract
INTRODUCTION
Steroid hormone (e.g. progesterone, mineralocorticoids, glucocorticoids, androgens, estrogens) biosynthesis begins with the conversion of cholesterol (Scheme 1, 1) to pregnenolone (Scheme 1, 4), which is catalyzed by cytochrome P450 (P450) 11A1. This reaction proceeds in three steps (Scheme 1). The first two steps involve hydroxylation reactions at the C22- and C20- positions of cholesterol, and the third step is a carbon-carbon (C-C) bond cleavage process. Alternatively, the 20R,22R-dihydroxycholesterol intermediate (Scheme 1, 3) has been proposed to arise from cholesterol through a C20,C22-epoxide intermediate, which in turn is hydrolyzed by water to form the 1,2-diol moiety.1 Another early proposal was that a C20-hydroperoxide is formed and then is converted to 20R,22R-dihydroxycholesterol.2 However, the C20- and C22- hydroxy groups of 20R,22R-dihydroxycholesterol (Scheme 1, 3) both come from molecular oxygen, which was shown from an incubation of cholesterol with adrenal microsomes in the presence of a mixture of 16O2 and 18O2.3 Therefore, the sequential hydroxylation mechanism catalyzed by Compound I (FeO3+) to form 20R,22R-dihydroxycholesterol (Scheme 1, 1 to 3) is favored through the 18O-labeling studies. Moreover, through electron paramagnetic resonance and electron nuclear double resonance (EPR and ENDOR) studies, Compound I has directly been shown to be the active iron species in both the hydroxylation and C-C bond cleavage steps.4,5 (The role in the C-C cleavage is supported by a 50% yield of the product pregnenolone from 20R,22R-dihydroxycholesterol.5) Despite knowledge that Compound I is the active iron intermediate in the C20-C22 bond cleavage step (Scheme 1, 3 to 4 and 5), it is still not clear how Compound I cleaves the side chain of cholesterol.6 Several possible Compound I mechanisms for this C20–C22 lyase step are viable, and we have addressed the issue with 18O-labeling studies.
Scheme 1.

The three steps catalyzed by P450 11A1, which convert cholesterol to pregnenolone, the first steroid precursor found in steroid hormone biosynthesis.
Four mechanistic proposals can potentially explain the carbon-carbon bond cleavage step of P450 11A1 using Compound I as the active iron species (Scheme 2).6–10 The four mechanisms can be divided into two types of reactivity: (i) one involves the nucleophilic attack of the hydroxy group of the substrate (Scheme 2A, 3) on the Compound I species of P450 11A1 (Scheme 2A),11 and (ii) the second type involves the O-H hydrogen atom abstraction of the substrate (Scheme 2B, 3) by Compound I (Scheme 2B).9 In the latter case, where an O-H hydrogen atom abstraction occurs, an oxygen atom (e.g. 18O) from molecular oxygen (e.g. 18O2) would be incorporated into one of the products (Scheme 2B, 4-L or 5-L) after the oxygen rebound step. On the other hand, in the mechanisms involving nucleophilic attack of the substrate on Compound I (i.e., “electrophilic Compound I”) no oxygen atom from molecular oxygen is incorporated into either of the products, pregnenolone or isocaproaldehyde (Scheme 2A, 4 and 5). Assuming that the dehydration step (Scheme 2B) is not stereospecific and that the resulting product isotopologues will be in an equivalent mixture (1:1 of 5:5-L or 4:4-L), we used 18O-labeling experiments to determine whether P450 11A1 undergoes an electrophilic Compound I mechanism (Scheme 2A) or an O-H abstraction mechanism (Scheme 2B) in the C-C bond cleavage step to form pregnenolone and isocaproaldehyde from 20R,22R-dihydroxycholesterol (Scheme 2, 3).
Scheme 2.

Four Compound I mechanisms grouped into two categories (A and B) depicting the C-C bond cleavage step of P450 11A1. (A) involves an electrophilic Compound I species while (B) shows an O-H hydrogen atom abstraction by Compound I. (A) shows no incorporation of an oxygen into the products (5 and 4) from molecular oxygen (*O2) and (B) indicates oxygen incorporation from molecular oxygen into products 5-L and 4-L. *O: 18O.
EXPERIMENTAL SECTION
Chemical Synthesis–General
The synthesis of 20R,22R-dihydroxycholesterol was accomplished following a modified route from a previously reported procedure.12 As done previously, compounds were visualized using either ceric ammonium molybdate stain (CAM, Hanessian’s stain: ammonium molybdate (4.8% w/v), ceric ammonium molybdate (0.2% w/v), and H2SO4 (6% v/v) in H2O)13 or a UV lamp (254 nm) on TLC plates with fluorescent indicator (F254). D2O (99.9% enriched) was purchased from Sigma-Aldrich. Bruker NMR spectrometers (600 MHz or 400 MHz) were used to acquire NMR spectra of synthesized compounds in the Vanderbilt facility. The proton and carbon chemical shifts of CDCl3 were referenced to δ 7.26 ppm and 77.16 ppm, respectively, and solvent impurities in the spectra of the purified compounds were identified from previously reported chemical shifts.14 The D2O chemical shift in the proton NMR spectrum was referenced to δ 4.79 ppm. Bruker TopSpin 3.2 software was used to process the NMR spectra. Other experimental details regarding the synthesis of 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol and 4-methylpentan-1-ol picolinoyl ester (e.g. NMR spectra and Rf values of synthetic intermediates) are reported in the Supporting Information. tert-Butyldimethylsilyl (TBDMS) chloride was purchased from Oakwood Chemical (Columbia, SC). Silica gel (SiliaFlash® F60, for purification of compounds) was purchased from Silicycle (Quebec, Canada). The ethylpyridine acetate standard (compound 8d-0) was synthesized according to our previous study.15
Synthesis of Pregnenolone-3-OTBDMS Ether (Compound 6)
Pregnenolone (4, 5.0 g, 16 mmol), tert-butyldimethylsilyl chloride (6.6 g, 44 mmol, 2.8 mol eq), and imidazole (1.0 g, 15 mmol, 0.9 mol eq) were dissolved in CH3CN (100 mL). The reaction was stirred for 17 h at room temperature. The reaction mixture was diluted with H2O (200 mL) and extracted with ethyl acetate (300 mL). The organic extracts were combined and concentrated. The crude material was purified via flash column chromatography (100% hexanes to 20% ethyl acetate in hexanes, v-v) to afford pregnenolone-3-OTBDMS ether 6 (5.7 g, 13 mmol, 84%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 5.32 (broad s, 1H), 3.51–3.45 (m, 1H), 2.53 (apparent t, J = 9.0 Hz, 1H), 2.31-2.23 (m, 1H), 2.21-2.14 (m, 2H), 2.12 (s, 3H), 2.07-2.03 (m, 1H), 2.02-1.96 (m, 1H), 1.84-1.79 (m, 1H), 1.75-1.59 (m, 4H), 1.59-1.52 (m, 3H), 1.52-1.40 (m, 3H), 1.28-1.19 (m, 1H), 1.18-1.11 (m, 1H), 1.06 (td, J1 = 13.1 Hz, J2 = 3.0 Hz, 1H), 1.00 (s, 3H), 0.99-0.94 (m, 1H), 0.89 (broad s, 9H), 0.62 (s, 3H), 0.06 (s, 6H).
Synthesis of Dithiane (Compound 7)
n-Butyl lithium (3.68 mL of a 2.5 M solution in hexanes, 9.2 mmol, 2.0 mol eq) was added dropwise to dithiane (1.11 g, 9.2 mmol, 2.0 mol eq) in THF (15 mL) at −30 °C under an atmosphere of argon. The mixture was stirred for 10 min at the same temperature, and the reaction was cooled to −78 °C. The reaction mixture was stirred for 2 h, and the temperature was gradually warmed to −20 °C. Pregnenolone-3-OTBDMS ether 6 (2.0 g, 4.6 mmol, 1.0 mol eq) in THF (15 mL) was added at −78 °C and the reaction was stirred for 1 h. The reaction was quenched with the addition of H2O (100 mL) and stirred for an additional 10 min at room temperature. The reaction mixture was diluted with ethyl acetate (200 mL) and the organic layer was extracted and concentrated under reduced pressure. The crude material was purified by flash column chromatography (300 mL increments of: 100% hexanes to 10% ethyl acetate in hexanes (v-v) to 20% ethyl acetate/hexanes (v-v)) to afford dithiane 7 (550 mg, 0.64 mmol, 14%) as a white solid. Rf 0.50 (hexanes:ethyl acetate, 4:1, v-v). 1H NMR (600 MHz, CDCl3) δ 5.24 (m, 1H), 4.07 (broad s), 3.45–3.37 (m, 1H), 2.91-2.85 (m, 1H), 2.85-2.73 (m, 2H), 2.21-2.15 (m, 1H), 2.12-2.06 (m, 1H), 2.06-1.99 (m, 1H), 1.94-1.87 (m, 1H), 1.86-1.70 (m, 3H), 1.69-1.61 (m, 1H), 1.61-1.53 (m, 1H), 1.52-1.39 (m, 4H), 1.36 (s, 3H), 1.27-1.15 (m, 1H), 1.15-1.06 (m, 1H), 1.02-0.96 (m, 1H), 0.93 (s, 3H), 0.82 (broad s, 5H), 0.80 (broad s, 7H), −0.02 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 141.3, 120.9, 76.6, 72.4, 61.2, 56.7, 55.0, 50.0, 42.9, 42.7, 40.1, 37.3, 36.5, 32.0, 31.7, 31.4, 31.3, 30.8, 26.0, 25.9, 24.1, 23.7, 21.6, 20.9, 19.4, 18.1, 13.3, −4.6.
Synthesis of Aldehyde (compound 8)
A modified procedure previously reported16 was used. HgCl2 (345 mg, 1.27 mmol, 2.0 mol eq) and HgO (139 mg, 0.64 mmol, 1.0 mol eq) were added to a solution of dithiane 7 (550 mg, 0.64 mmol, 1.0 mol eq) in CH3CN (10 mL) and H2O (0.1 mL). The reaction mixture was heated under reflux for 2.5 h. The reaction mixture was filtered through a short pad of silica gel with ethyl acetate (1 L). The filtrate was concentrated and the crude material was purified via flash column chromatography to afford the aldehyde 8 (90 mg, 0.20 mmol, 31%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 9.56 (s, 1H), 5.31-5.30 (m, 1H), 3.50-3.45 (m, 1H), 2.29-2.23 (m, 1H), 2.18-2.11 (m, 2H), 1.99-1.93 (m, 1H), 1.82-1.75 (m, 2H), 1.74-1.68 (m, 2H), 1.68-1.59 (m, 2H), 1.57-1.41 (m, 7H), 1.35 (s, 3H), 1.33-1.22 (m, 9H), 1.22-1.08 (m, 2H), 1.07-1.01 (m, 2H), 1.00 (s, 3H), 0.98-0.91 (m, 2H), 0.88 (s, 9H), 0.81 (d, J = 6.9 Hz, 3H), 0.79 (s, 3H), 0.05 (s, 6H).
Synthesis of Grignard Adduct (Compound 9)
Isopentylmagnesium bromide (21.7 mL, 2.5 M solution in diethyl ether, 43 mmol, 20 mol eq) was added to a solution of aldehyde 8 (1.0 g, 2.2 mmol, 1.0 mol eq) in THF (100 mL) at −78 °C. The reaction mixture was stirred for 1.5 h. The reaction was diluted with H2O (100 mL) at −78 °C and the reaction mixture was left to warm to room temperature. The reaction mixture was extracted with CH2Cl2 (2 × 250 mL). The combined organic extracts were concentrated and purified by flash column chromatography (300 mL increments of: 100% hexanes to 10% ethyl acetate in hexanes (v-v) to 30% ethyl acetate in hexanes (v-v)) to yield the diol 9 (220 mg, 0.41 mmol, 19%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 5.32-5.31 (m, 1H), 3.51-3.46 (m, 1H), 3.41-3.37 (m, 1H), 2.29-2.24 (m, 1H), 2.21 (d, J = 3.2 Hz, 1H), 2.17 (ddd, J1 = 13.2 Hz, J2 = 5.1 Hz, J3 = 2.3 Hz, 1H), 2.12 (dt, J1 = 12.5 Hz, J2 = 3.6 Hz, 1H), 2.00-1.95 (m, 1H), 1.85-1.79 (m, 2H), 1.75-1.69 (m, 1H), 1.68-1.61 (m, 1H), 1.58-1.40 (m, 11H), 1.26-1.22 (m, 2H), 1.22 (s, 3H), 1.21-1.18 (m, 1H), 1.17-1.11 (m, 1H), 1.06-1.03 (m, 1H), 1.02-0.96 (m, 1H), 1.00 (s, 3H), 0.94-0.92 (m, 1H), 0.90 (s, 3H), 0.88 (broad s, 12H), 0.06 (s, 6H).
Synthesis of 20R,22R-Dihydroxycholesterol (Compound 3)
To a stirring solution of diol 9 (220 mg, 0.41 mmol) in CH3OH:CH2Cl2 (40 mL, 1:1, v-v) was added conc HCl (12 M, 0.02 mL). The reaction was stirred for 15 min and the mixture was concentrated under reduced pressure. The resulting crude material was purified by flash column chromatography (100% hexanes to 50% hexanes/ethyl acetate, v-v) to yield 20R,22R-dihydroxycholesterol (3, 50 mg, 0.12 mmol, 29%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 5.34-5.33 (m, 1H), 3.55-3.46 (m, 1H), 3.39-3.35 (m, 1H), 2.33-2.26 (m, 2H), 2.25-2.19 (m, 1H), 2.12 (dt, J1 = 12.7 Hz, J2 = 3.5 Hz, 1H), 1.99-1.94 (m, 1H), 1.94-1.92 (m, 1H), 1.86-1.79 (m, 3H), 1.67-1.59 (m, 2H), 1.58-1.38 (m, 10H), 1.20 (s, 3H, C-21), 1.19-1.10 (m, 3H), 1.10-1.03 (m, 1H), 1.00 (s, 3H, C-19), 0.90 (d, J = 7 Hz, 6H), 0.89 (s, 3H) (1H NMR characterization matched the previously reported synthesis12); 13C NMR (150 MHz, CDCl3) δ 140.9, 121.7, 77.5, 76.5, 71.8, 56.8, 54.9, 50.2, 43.3, 42.4, 40.3, 37.4, 36.6, 36.5, 31.9, 31.7, 31.4, 29.3, 28.2, 24.1, 23.1, 22.5, 22.1, 20.5, 19.5, 13.7.
Synthesis of [21,21,21-2H3]-Pregnenonlone-3-O-tert-butyldimethylsilyl Ether (Compound 6a)
NaH (0.89 g, 35 mmol, 3.1 mol eq) was added as a solid to a solution of pregnenolone-3-O-tert-butyldimethylsilyl ether 6 (4.9 g, 11 mmol, 1.0 mol eq) in THF (200 mL) at 0 °C. The reaction flask was evacuated and backfilled with argon. D2O (5 mL) was added and the reaction was allowed to stir for 8 days at room temperature. The reaction mixture was concentrated and purified by flash column chromatography (100% hexanes to 10% ethyl acetate in hexanes (v-v) to 30% ethyl acetate in hexanes (v-v)) to afford [21,21,21-2H3]-pregnenonlone-3-O-tert-butyldimethylsilyl ether (6a, 4.0 g, 9.2 mmol, 84%) as a white solid. The proton NMR showed a small peak corresponding to the 21-methyl protons (δ 2.12), which integrated to 0.31 while the 18-methyl protons (δ 0.63) integrated to 3.00 and the 17-proton (δ 2.53) integrated to 0.96. 1H NMR (600 MHz, CDCl3) δ 5.32-5.31 (m, 1H), 3.52-3.44 (m, 1H), 2.53 (apparent t, J = 8.8 Hz, 1H), 2.31-2.21 (m, 1H), 2.21-2.13 (m, 2H), 2.12 (s, 0.3H), 2.11-2.01 (m, 1H), 2.07-1.96 (m, 2H), 1.82 (dt, J1 = 13.2 Hz, J2 = 3.4 Hz, 1H), 1.76-1.40 (m, 8H), 1.29-1.02 (m, 3H), 1.00 (s, 3H), 0.99-0.93 (m, 1H), 0.89 (s, 9H), 0.63 (s, 3H), 0.06 (s, 6H).
Synthesis of Isocaproaldehyde (5) via Swern Oxidation
Dimethyl sulfoxide (20.2 mL, 284 mmol, 2.9 mol eq) was added to oxalyl chloride (12.6 mL, 147 mmol, 1.5 mol eq) in CH2Cl2 (500 mL) at −78 °C. After 10 min, 4-methylpentan-1-ol (12.3 mL, 10 g, 97.9 mmol) was added. Triethylamine (41 mL, 294 mmol, 3.0 mol eq) was added with the use of an addition funnel. After 1 h, the reaction was diluted with H2O (100 mL). The reaction mixture was washed with HCl (1 M, 3 × 300 mL) and the organic layer was dried with anhydrous MgSO4 and concentrated to yield isocaproaldehyde (5) as a clear oil (9 g, 90 mmol, 92%). The purity of the crude material was determined by 1H NMR and used for subsequent experiments (i.e., gem-diol/aldehyde equilibrium in D2O and yeast alcohol dehydrogenase reduction rates). 1H NMR (600 MHz, CDCl3) δ9.77 (t, J = 2.0 Hz, 1H), 2.42 (td, J1 = 7.4 Hz, J2 = 1.9 Hz, 2H), 1.62-1.54 (m, 1H), 1.52 (apparent q, J = 7.4 Hz, 2H), 0.90 (d. J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ203.1 (C1), 42.1 (C2), 30.9 (C3, -CH2-), 27.8 (C4, -CH-), 22.5 (C5 and C6, -CH3).
Determination of the gem-Diol/aldehyde Equilibrium of Isocaproaldehyde
An 1H NMR spectrum of a solution of isocaproaldehyde in potassium phosphate buffer (D2O) was acquired. The following buffer solution was prepared: 100 mM potassium phosphate in H2O (pH = 7.8, 1 mL) was lyophilized and D2O (1 mL) was added (pD 7.4).17–19 A solution (0.07 mL) of isocaproaldehyde (2.7 mg/1 mL of D2O) was added to buffer A (0.63 mL) and the 1H NMR spectrum was acquired on a 600 MHz NMR spectrometer.
Enzyme Incubations
18O2 gas (99% 18O), yeast alcohol dehydrogenase (369 units/mg, one unit converted 1 μmol of ethanol to acetaldehyde min−1 at pH 8.8 and 25 °C, CAS: 9031-72-5), glucose 6-phosphate dehydrogenase (Type XV, 210 units/mg, one unit oxidized 1 μmol of glucose 6-phosphate to 6-phospho-D-gluconate min−1 in the presence of NADP+ at pH 7.4 at 25 °C, CAS: 9001-40-5), and NADH were purchased from Sigma-Aldrich. A 310 mM aqueous solution of (2-hydroxypropyl)-β-cyclodextrin (β-cyclodextrin) was made for solubilization of substrates in enzymatic incubations.20 An OLIS/Aminco DW2 spectrophotometer (On-Line Instrument Systems, Bogart, GA) was used to measure P450 concentrations.21
Human Cytochrome P450 11A1 Expression
A codon-optimized P450 11A1 plasmid was purchased from Genewiz (South Plainfield, NJ) (nucleotide sequence provided in Supporting Information). The P450 11A1 plasmid (pCWori+ vector22) was co-expressed with another plasmid containing the Escherichia coli molecular chaperone GroEL/ES in E. coli JM109 cells in Luria-Bertani (LB) media (100 mL) containing ampicillin (100 μg/mL) and kanamycin (50 μg/mL). This pre-culture was incubated overnight at 37 °C and 220 rpm). The scaled-up expression of P450 11A1 (i.e., bulk culture) was grown in 4.5 L (6 × 750 mL/2.8 L flask) of Terrific Broth (TB) media supplemented with the same concentrations of ampicillin and kanamycin as the pre-culture, along with bactopeptone (2 g/L), trace elements (250 μL/L of culture),23 NaCl (1 mM), thiamine (1 mM), glycerol (4 mL/L), and the pre-culture (dilution of 1:100, v/v). The bulk cultures were incubated at 37 °C and 225 rpm until the OD600 reached 0.7–0.75 (~6 h). P450 expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) (1.0 mM), δ-aminolevulinic acid (1.0 mM), and arabinose (1.0 mM). The induced cultures were incubated for 42 h at 28 °C and 190 rpm in a Multifors incubator. The cells were harvested by centrifugation (3,500 × g for 20 min).
Cytochrome P450 11A1 Purification
All steps were carried out at 4 °C. The cell pellets from bulk cultures (6 L) were resuspended in 300 mL of TES buffer (100 mM Tris-acetate (pH 7.5) containing 0.5 M sucrose and 0.5 mM EDTA) for each pellet derived from 1 L of bulk culture. Lysozyme (2 mg/L of bulk culture) was added and the suspension was incubated on ice for 60 minutes and then centrifuged at 3,500 × g for 20 min. The spheroplasts were resuspended in sonication buffer (50 mL of 100 mM potassium phosphate (pH 7.5) containing 16% glycerol (v/v), 9 mM magnesium acetate, 100 μM dithiothreitol (DTT)), 1.0 mM phenylmethylsulfonyl fluoride (PMSF), and two protease inhibitor tablets (Roche) for each 1 L of bulk culture. After sonication at 0 °C, the contents were centrifuged at 12,000 × g for 15 min. The recovered supernatant was centrifuged at 140,000 × g for 60 min. The pellet was collected, homogenized, and stirred in solubilization buffer (600 mL of 100 mM potassium phosphate buffer (pH 7.5) containing 20% glycerol (v/v), 0.1 mM EDTA, 10 mM β-mercaptoethanol, 0.5 M KCl, and 1% (w/v) sodium 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) overnight at 4 °C. The resulting solution was centrifuged at 140,000 × g for 60 min, and the supernatant was loaded onto a Ni-NTA (nitriloacetic acid) column (1.5 × 8 cm), which had been previously equilibrated with 100 mM potassium phosphate buffer (pH 7.5) containing glycerol (20%, v/v), 10 mM β-mercaptoethanol, 0.5 M KCl (0.5 M), and sodium CHAPS (1% w/v). The column was washed with the same equilibration buffer containing 20 mM imidazole and 2.8 mM pentaethylene glycol monooctyl ether (C8E5) detergent. The concentration of the imidazole was raised to 200 mM in the same buffer to elute P450 11A1. The eluted P450 11A1 fractions (based on A418) were combined and dialyzed against 2 L of dialysis buffer (100 mM potassium phosphate (pH 7.5) containing 20% glycerol (v/v)) 3 × for 24 h. The concentration of P450 11A1 was measured using ferrous vs. ferrous-CO difference spectra,21 and a typical yield of the protein was 135 nmol. The dialyzed protein was divided into 150 μL aliquots and stored at −20 °C until use for incubations.
Adrenodoxin (Adx) Expression
E. coli DH5α cells were transformed with the plasmid pBA1159 for bovine Adx expression.24–27 A pre-culture was prepared by inoculating a single colony into 10 mL of LB media containing 100 μg/mL of ampicillin and grown at 37 °C with gyrorotary shaking at 220 rpm. The pre-culture (5 mL) was then added to bulk culture medium (500 mL of TB media containing ampicillin (100 μg/mL), thiamine (340 μg/mL), and trace elements23 (0.025% v/v). The cultures were incubated at 37 °C with shaking at 220 rpm for 4 h. IPTG (1 mM) was added to induce Adx expression. The induced cultures were incubated for 30 °C for 24 h. The cells were pelleted by centrifugation (3000 × g) for 5 min, and the pelleted cells were sonicated in 10 mM potassium phosphate buffer (pH 7.4). The sonicated material was centrifuged at 100,000 × g for 30 min to remove cell debris, and the supernatant was used for the purification step.
Adrenodoxin (Adx) Purification
The supernatant (vide supra) was loaded onto a DEAE-Sepharose column (2.5 × 20 cm) that had been equilibrated with 50 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA and washed with 1 L of the equilibration buffer. The bound Adx was eluted with the same buffer containing NaCl (170 mM then 300 mM). The fractions that absorbed at 414 nm from the 170 mM pool were highly purified, based on sodium dodecyl sulfate-polyacrylamide electrophresis (15%, w/v, gel). The fractions from the 170 mM pool, (pooled based on A414 readings) were dialyzed twice against 10 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA and applied to a DEAE-Sepharose column (2.5 × 10 cm). The column was washed with the dialysis buffer and eluted with the same buffer containing NaCl (500 mM). Fractions containing adrenodoxin were combined based on A414 measurements. The impure fractions from the DEAE steps were purified further with gel filtration chromatography on a Sephadex G-75 (2.5 × 100 cm) using 10 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA (≤ 20 mL per run). The total yield (from 6 L) was 25.7 μmol of Adx, which was stored in 1 mL aliquots at −70 °C.
Adrenodoxin Reductase (ADR) Expression
E. coli DH5α cells were transformed with the plasmid containing bovine ADR28 in a pCWori+ expression vector22,26,27 with a (His)6 tag added to the C-terminus of the cDNA to facilitate protein purification. LB media (10 mL containing 100 μg/mL ampicillin) was inoculated with a single colony and incubated overnight at 37 °C with gyrorotary shaking at 220 rpm. The overnight culture (5 mL) was used to inoculate 500 mL of TB media containing ampicillin (100 μg/mL), thiamine (340 μg/mL), and trace elements (0.025%, v/v).23 The cultures were incubated at 37 °C and 220 rpm for 4 h. ADR expression was induced with the addition of 1.0 mM IPTG. following induction, the induced cultures were incubated at 30 °C and 220 rpm for another 24 h.
ADR Purification
The cell cultures were harvested by centrifugation (3,000 × g) for 10 min. The cell pellet was sonicated in potassium phosphate buffer (100 mM, pH 7.4) containing EDTA (0.1 mM), glycerol (20% v/v), and protease inhibitor tablets (Roche). The sonicated solution was centrifuged (100,000 × g) for 30 min to remove the cell debris. The supernatant was loaded on a Ni-NTA column, which was then washed with the sonication buffer containing 20 mM imidazole. ADR was eluted with the sonication buffer containing 200 mM imidazole. The eluted ADR was dialyzed against 40 volumes of 100 mM potassium phosphate buffer (pH 7.4) containing glycerol (20% v/v) and 0.1 mM EDTA three times to remove the imidazole and then stored at −80 °C.
Cytochrome P450 17A1
Human P450 17A1 was expressed and purified as described elsewhere.15
Cytochrome b5 and NADPH-P450 Reductase
Recombinant human cytochrome b529 and NADPH P450 rat reductase30 were expressed in E. coli and purified as described previously.
P450 11A1 Coupled Assay with Yeast Alcohol Dehydrogenase (Isocaproaldehyde Analysis)
In preliminary assays, we measured a kcat of 4.5 s−1 and a Km (isocaproaldehyde) of 32 μM for the reduction of isocaproaldehyde by yeast alcohol dehydrogenase (utilizing NADH oxidation at 340 nm). P450 11A1 (450 pmol), Adx (168 pmol), ADR (708 pmol), yeast alcohol dehydrogenase (5 mg) and L-α-1,2-dilauroyl-sn-glycero-3-phosphocholine (50 μg) were added to a Thunberg tube containing 800 μL of 100 mM potassium phosphate buffer (pH 7.4) followed by the addition of either cholesterol (52 nmol) or 20R,22R-dihydroxycholesterol (142 nmol), which was dissolved in a solution of β-cyclodextrin (310 mM). The cap of the Thunberg tube contained the NADPH-regenerating system31 (100 μL of the following mixture: 8.3 μmol of glucose 6-phosphate, 4 μg of glucose 6-phosphate dehydrogenase, 333 nmol of NADP+) plus NADH (100 μL of a 100 mM solution). The Thunberg tube was purged with argon on a gas train by alternate evacuation with reduced pressure and backfilling with argon (repeated 10×). Before injection of 18O2 gas, the tube was evacuated with reduced pressure for 3 minutes. After the 18O2 gas was injected, the contents of the tube and cap were mixed and the incubation was shaken at 50 rpm and 37 °C. After 15 min, the tube was placed on ice and CH2Cl2 (5 mL) was added. The organic layer was extracted and taken to dryness under a stream of N2.
Derivatization with Picolinoyl Chloride
A solution of picolinoyl chloride in THF (200 μL of a 5 mg/mL solution) was added to the dried incubation extracts from the P450 11A1-yeast alcohol dehydrogenase-18O2 reaction (vide supra). The solution was mixed using a vortex device for 5 seconds and dried under a stream of N2. The samples were dissolved in CH3CN (75 μL) for LC-MS analysis.
P450 11A1 Coupled Assay with P450 17A1 (Pregnenolone Analysis)
P450 11A1 (900 pmol), Adx (840 pmol), ADR (900 pmol), P450 17A1 (5.6 nmol), cytochrome b5 (11 nmol), NADPH P450 reductase (6.2 nmol), and L-α-1,2-dilauroyl-sn-glycero-3-phosphocholine (50 μg) were added to a Thunberg tube containing 2 mL of 50 mM potassium phosphate buffer (pH 7.4). Cholesterol (1, 160 nmol in 1.5 μmol β-cyclodextrin) or 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol (3a, 190 nmol in 1.5 μmol β-cyclodextrin) were added to the Thunberg tube. The cap contained the NADPH-regenerating system.31 The Thunberg tube was purged with argon on a gas train by evacuation with reduced pressure and backfilling with argon (repeated 10×). Before injection of 18O2 gas, the tube was evacuated with reduced pressure for 3 minutes. After the 18O2 gas was injected, the contents of the tube and cap were mixed and the incubation was shaken at 50 rpm at 37 °C. After 60 min, the tube was placed on ice and CH2Cl2 (5 mL) was added. HCl (3 M, 1 mL, chilled to 0 °C) was added and the solution was mixed with a vortex device and centrifuged at 1,500 × g for 1 minute. The organic layer was extracted and dried with anhydrous MgSO4 (~50 mg). The MgSO4 was filtered using a Pasteur pipet with a cotton plug. Diazo reagent (vide infra) was added to the CH2Cl2 extract of the reaction mixture and the resulting solution was dried under a stream of nitrogen. The samples were dissolved in CH3CN (75 μL) and analyzed by LC-MS.
Derivatization with Diazo Reagent
The diazo reagent was generated by the addition of NaOH (2 mL of a 30% w/v solution in H2O) to the nitrosourea precursor in diethyl ether (~5 mg in 2 mL) in a borosilicate screw cap vial.15,32 The two layers were mixed with a vortex device and the diethyl ether layer was extracted and dried with anhydrous MgSO4 (~50 mg). The organic layer was filtered through a Pasteur pipet with a cotton plug and was used immediately to treat the incubation extracts prepared above. *Caution: Nitrosourea and diazo compounds are carcinogenic and explosive. Extra care was used when handling the compounds. Double-glove protection and proper personal protective equipment (safety glasses and a laboratory coat) are required. No ground glass joints were used when handling these compounds. A solution of acetic acid (50 mL) chilled on ice was set aside to rinse all of the glassware to react any remaining diazo compound generated. All of the experiments were performed in a fume hood.
Derivatization of P450 11A1/P450 17A1 Incubation Extracts with Picolinoyl Chloride
The CH3CN solution remaining after the LC-MS analyses was added to a solution of picolinoyl chloride in THF (200 μL of a 5 mg/mL solution). The solution was mixed using a vortex device for 5 seconds and dried under a stream of N2. The samples were dissolved in CH3CN (75 μL) for LC-MS analysis. These samples were used to analyze the deuterium content of 17-hydroxypregnenolone picolinoyl esters (Supporting Information).
LC-MS Analysis–General
An Acquity UPLC system was used for the LC-MS analysis of compounds from the enzymatic incubations. Thermo XCalibur Qual Browser 2.2 software (ThermoFisher Scientific) was used to process LC-MS data. An LTQ Orbitrap XL mass spectrometer was used to detect derivatized products in the ESI positive mode with a 100,000 resolution setting. LTQ Tune Plus software was used to calibrate and tune the instrument. The mass window scanned was m/z 50–800.
Calibration of the Mass Spectrometer (LTQ Orbitrap XL)
The mass spectrometer was calibrated before each sequence using Pierce® LTQ ESI Positive Ion Calibration Solution (ThermoFisher) containing a mixture of caffeine, MRFA (the peptide methionine-arginine-phenylalanine-alanine), and Ultramark 1621 in CH3CN, CH3CO2H, and CH3OH by direct infusion (10 μL/min). The mass spectrometer was first tuned for MRFA (m/z 524) and then the tube lens voltage was adjusted to 145 V to fragment caffeine (m/z 195 to 138). The tune conditions were as follows: Sheath Gas Flow Rate: 19, Aux Gas Flow Rate: 3, Sweep Gas Flow Rate: 0, Spray Voltage: 4.00 kV, Capillary Temperature: 300 °C, Capillary Voltage: 4.00 V, Tube Lens: 145.00 V. The exact masses used to calibrate the instrument were (m/z) 138.066190, 195.087652, 524.264964, 1121.997024, 1221.990636, 1321.984249, 1421.977862, 1521.971475, 1621.965088, and 1721.958701.
Mass Spectrometer Tune Conditions
The following tune settings were used to detect the picolinoyl ester of the derivatized isocaproaldehyde product (compound 15) in the APCI positive mode with the following settings (acquired after tuning with the synthetic standard, Supporting Information): Vaporizer Temperature: 450 °C, Sheath Gas Flow Rate: 50, Aux Gas Flow Rate: 5, Sweep Gas Flow Rate: 5, Discharge Current (μA): 5.00, Capillary Temperature: 275 °C, Capillary Voltage: 12.00 V, Tube Lens: 50.00 V.
The tune settings used to detect pregnenolone in the APCI positive mode were as follow: Vaporizer Temperature: 450 °C, Sheath Gas Flow Rate: 50, Aux Gas Flow Rate: 5, Sweep Gas Flow Rate: 5, Discharge Current: 5.00 μA, Capillary Temperature: 275 °C, Capillary Voltage: 43.00 V, Tube Lens: 55.00 V.
The pyridine acetate of the derivatized acetic acid product (compound 18) was detected in ESI positive mode with the following tune settings (acquired after tuning with the synthetic standard): Sheath Gas Flow Rate: 15, Aux Gas Flow Rate: 5, Sweep Gas Flow Rate: 0, Spray Voltage: 4.00 kV, Capillary Temperature: 300 °C, Capillary Voltage: 16.00 V, Tube Lens: 30.00 V.
The tune settings for detecting picolinoylated pregnenolone and mono-picolinoylated-20,22-dihydroxycholesterol (Supporting Information) using ESI positive mode were as follows: Sheath Gas Flow Rate: 8, Aux Gas Flow Rate: 0, Sweep Gas Flow Rate: 0, Spray Voltage: 5.00 kV, Capillary Temperature: 275 °C, Capillary Voltage: 24.00 V, Tube Lens: 70.00 V.
Liquid Chromatography (LC) Methods–General
A Phenomenex Kinetex octylsilane reversed-phase column (2.6 μm, C8, 100 Å, 100 mm × 2.1 mm) was used to separate all of the analytes. For every LC method, the column temperature was held at 40 °C. In order to maintain consistent retention times in each run in the sequence, the column was equilibrated with the initial gradient conditions for at least 10 min until the delta value (i.e. maximum and minimum difference in pressure) was <30 psi. Two to three sequential injections of the standards were made until the retention times remained stable. LC-MS spectra were acquired in full scan mode: m/z 100–1000 (for yeast alcohol dehydrogenase coupled assays), m/z 50–700 or 50–800 (for P450 17A1 coupled assays). ICIS peak algorithm detection was used. Peaks in LC-MS data were acquired in profile mode.
LC Method to Detect Picolinoyl Ester of Isocaproaldehyde Derivative (Compound 15) (Method A)
The LC method used to detect the derivatized isocaproaldehyde product (compound 15) and pregnenolone were run with solvents A (10 mM ammonium acetate, H2O:CH3CN, 95:5, v/v) and B (10 mM ammonium acetate, CH3CN) with the following gradient: 95% A from 0–1.0 min, 95% A to 100% B from 1.0–4.0 min, 100% B from 4.0–5.0 min, 100% B to 95% A from 5.0–5.5 min, 95% A (all v/v) from 5.5–8.0 min with a flow rate of 0.3 mL/min.
LC Method to Detect Pyridine Acetate Derivatives (compound 18) (Method B)
The LC method to detect the derivatized acetic acid product (compound 18) and picolinoylated pregnenolone was run with solvents A (10 mM ammonium formate, H2O) and B (10 mM ammonium formate, CH3CN:H2O, 95:5, v/v) with the following gradient: 100% A from 0–1.0 min, 100% A to 100% B from 1.0–4.0 min, 100% B from 4.0–5.2 min, 100% B to 100% A from 5.2–5.3 min, 100% A (all v/v) from 5.3–8.0 min with a flow rate of 0.3 mL/min.
RESULTS
Overall Strategy
In order to determine if an oxygen atom from molecular oxygen is incorporated into either of the C-C bond cleavage products of P450 11A1 (Scheme 2, 4 and 5), incubations were performed using 20R,22R-dihydroxycholesterol in the presence of purified P450 11A1 and 18O2. Because the carbonyl oxygens in pregnenolone and isocaproaldehyde are readily exchangeable with water, it was necessary to convert the enzymatic products (Scheme 2, 4 and 5) to different chemical entities so that the oxygens would be incapable of exchanging with water. The derivatized C-C bond cleavage products (4 and 5) would be detected by LC-MS for analysis of 18O-content.
Synthesis of Substrates: 20R,22R-dihydroxycholesterol (3) and Its Deuterated Isotopologue, 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol (3a)
The synthesis of 20R,22R-dihydroxycholesterol (3) commenced with pregnenolone as the starting material (Scheme 1). The 3-hydroxy group was protected as a TBDMS ether (Scheme 3, 7). One-carbon homologation of pregnenolone TBDMS ether (7) with a 1,3-dithiane anion nucleophile followed. The resulting dithiane (8) was converted to the aldehyde (9) with mercury-promoted hydrolysis. Grignard addition onto the aldehyde with isopentylmagnesium bromide yielded the 1,2-diol (10). The C3-oxygen was deprotected by removal of the TBDMS group in CH3OH with HCl to furnish 20R,22R-dihydroxycholesterol (3). The 1H NMR spectrum matched a previously reported spectrum.33
Scheme 3.

Synthesis of 20R,22R-dihydroxycholesterol (3) and 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol (3a) substrates for 18O2-incubation with P450 11A1.
Additionally, 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol (3a) was obtained by synthesizing [21,21,21-2H3]-pregnenolone-3-tert-butyldimethylsilyl ether (6a) and performing the same steps to access 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol from the deuterated silyl ether (Scheme 3). Pregnenolone-3-OTBDMS (6) was deuterated on the C21-position by reacting with NaH and D2O in THF at room temperature for eight days (Scheme 3, 6 to 6a). These reaction conditions resulted in the regioselective deuteration of the C21 position over the C17 position through the formation of the kinetic enolate (see Supporting Information for 1H NMR spectrum).
Gem-Diol/Aldehyde Equilibrium of Isocaproaldehyde (5) in D2O
The equilibrium between the aldehyde and gem-diol forms of isocaproaldehyde in D2O was measured by proton NMR. Similar to the case of 19-oxoandrostenedione,13 the gem-diol and aldehyde were in a 0.9:1.0 ratio in D2O (Figure 1). The fact that isocaproaldehyde is hydrated in water was important in the context of our 18O2 experiments. The purpose of this study is to distinguish between the two types of mechanisms presented in Scheme 2 (i.e. electrophilic Compound I and O-H abstraction). In other words, in order to determine whether or not P450 11A1 incorporates an 18O atom into isocaproaldehyde from molecular oxygen (18O2) (i.e. if Scheme 2Ba supported), the aldehyde oxygen must be trapped at a state where it cannot exchange with H2O.
Figure 1.

1H NMR spectrum of isocaproaldehyde (5) in potassium phosphate buffer in D2O (pD 7.4). The aldehyde (δ 9.68) and the gem-dihydroxymethine (δ 5.01, triplet, J = 5.4 Hz) protons are shown (600 MHz).
Design of Coupled Assays to Avoid Exchange of Carbonyl Oxygen Atoms of Isocaproaldehyde and Pregnenolone with H2O
The carbonyl oxygens in steroids can be exchanged readily with H2O.13 As shown in the proton NMR spectrum of isocaproaldehyde (one of the lyase products of P450 11A1) in D2O, the aldehyde exists in equilibrium in the gem-diol form (Figure 1). Moreover, exchange kinetics of ketones and aldehydes with water have also been determined experimentally to occur on a time scale from seconds to minutes (i.e. less than one hour)34,35 while the incubations in this study were run for 15 to 60 minutes in this study. Therefore, coupled enzyme assays were necessary to trap the carbonyl oxygens of the reaction products of P450 11A1, pregnenolone and isocaproaldehyde (Scheme 4, 4 and 5), so that no exchange would occur with the water medium. Two types of coupled assays (yeast alcohol dehydrogenase and P450 17A1) were performed depending on the product (i.e. isocaproaldehyde or pregnenolone) analyzed.
Scheme 4.

Coupled-Enzyme Assays for P450 11A1. The oxygen trapping experiments used to avoid exchange of the carbonyl oxygens with water in (A) isocaproaldehyde (5) and (B) pregnenolone (4), the carbon-carbon bond cleavage products of P450 11A1.
Yeast Alcohol Dehydrogenase Trapping of the Oxygen in Isocaproaldehyde
In order to trap the oxygen in the isocaproaldehyde product (Scheme 2, 5), we used yeast alcohol dehydrogenase to convert the aldehyde to the resulting alcohol (Scheme 4A, 5 to 10),36 which was then derivatized as the picolinoyl ester and analyzed by mass spectrometry. The picolinoyl ester products would either have an 18O- atom or 16O-atom, depending on whether or not P450 11A1 incorporated an oxygen atom into isocaproaldehyde. Derivatization of the alcohol to the picolinoyl ester enhanced the ionization of the compounds through protonation of the pyridine nitrogen in the ESI positive mode. The presence of an 18O-atom in the derivatized product would support the O-H abstraction mechanism (Scheme 2Bb), while the absence of an 18O-atom would support the electrophilic Compound I mechanism (Scheme 2A).
Use of P450 17A1 to Trap the C20-Oxygen in Pregnenolone
Conversely, in order to analyze the carbonyl oxygen in the pregnenolone product of P450 11A1 (Scheme 2, 4), a coupled assay with P450 17A1 was performed to convert pregnenolone into acetic acid and dehydroepiandrosterone (DHEA) (Scheme 4B). The carbon-carbon bond cleavage product of P450 11A1 is pregnenolone, which is a substrate for P450 17A1. The transformation catalyzed by P450 17A1 occurs in two steps: (i) hydroxylation at the C17-position of pregnenolone and (ii) cleavage of the 17,20-carbon-carbon bond of 17α-hydroxypregnenolone to form acetic acid and DHEA (Scheme 4B, 4a to 12a and 13). We have shown that P450 17A1 incorporates a single oxygen (18O) atom from molecular oxygen (18O2) into the acetic acid product in the lyase step (ii) when 17α-hydroxy-[21,21,21-2H3]-pregnenolone is used as the substrate.15 Therefore, in order to determine 18O-incorporation into pregnenolone by P450 11A1, the derivatized acetic acid product was analyzed for incorporation of one or two 18O atoms. If the derivatized acetate product contained two 18O atoms, then the mechanism shown in Scheme 2Ba would be supported. One of the 18O atoms would have come from P450 11A1 and the second 18O atom would have come from P450 17A1. However, if there were only one 18O atom incorporated into the acetate product, then the electrophilic Compound I mechanism (Scheme 2A) is supported. In this case, the 18O atom would only come from P450 17A1.
Positive Control Experiments Using Cholesterol Indicated Retention of the Oxygen Atom in the Carbonyl-Containing Products
Two separate positive control experiments were performed to verify minimal exchange of the carbonyl oxygens in either the isocaproaldehyde (Scheme 5, 5a to 5b) or pregnenolone (Scheme 6, 4a to 4b or 14a to 14b) product with the water medium. If the last step does not incorporate 18O-label into either of the P450 11A1 products (i.e., if Scheme 2A is supported by the results), then the possibility of H2O exchanging with the carbonyl groups of pregnenolone and isocaproaldehyde (Scheme 2B, 4-L and 5-L) to yield products lacking an 18O label must be ruled out. Therefore, these positive control experiments are especially crucial in the case that the P450 11A1 incubation with 20R,22R-dihydroxycholesterol and 18O2 does not incorporate an 18O-label into either pregnenolone or isocaproaldehyde (i.e., the experimental results support the scenario in Scheme 2A).
Scheme 5.

Positive control assay for monitoring the 18O-atom content in the derivatized isocaproaldehyde product (15).
Scheme 6.

Positive control assay for monitoring the 18O-atom content in the pregnenolone product (4a).
Both positive control experiments involve the use of cholesterol as the substrate for P450 11A1 instead of 20R,22R-dihydroxycholesterol (3) (Schemes 5 and 6). These experiments are critical in establishing oxygen retention in the products during the enzyme incubation and derivatization conditions. In previous mechanistic studies with P450 19A1, we showed that the aldehyde oxygen of 19-[18O]-labeled 19-oxoandrostenedione readily exchanges with water in pH 7.4 phosphate buffer (estimated t1/2 < 1 second).13 The use of cholesterol guarantees incorporation of 18O-atoms into isocaproaldehyde (Scheme 5) and pregnenolone (Scheme 6).
Positive Control–Yeast Alcohol Dehydrogenase
The positive control experiment for 18O incorporation into isocaproaldehyde involved the use of cholesterol (Scheme 5, 1) as the starting material with P450 11A1 coupled with yeast alcohol dehydrogenase in the presence of 18O2. The resulting alcohol product was derivatized with picolinoyl chloride and analyzed by LC-MS (Scheme 5, 10 to 15). The detection of an 18O-labeled picolinoyl ester product would show that the oxygen atom from isocaproaldehyde does not exchange completely with the water medium (Scheme 5, 5a to 5b) during the conversion from the aldehyde to alcohol by yeast alcohol dehydrogenase (Scheme 5, 5a or 5b to 10).
LC-MS analysis of 4-methylpentanol picolinoyl ester (Scheme 5, 15) derived from the yeast alcohol dehydrogenase-coupled incubation of P450 11A1 with cholesterol and 18O2 confirmed the presence of an 18O atom in the product (Figure 2B). This observation was important in establishing that the yeast alcohol dehydrogenase reaction was efficient enough to trap the 18O atom in the initial isocaproaldehyde product (Scheme 5, 5a) before exchange with the water medium could occur (Scheme 5, 5a to 5b).
Figure 2.


18O2 incubation of P450 11A1 coupled with yeast alcohol dehydrogenase to reduce isocaproaldehyde to the alcohol followed by derivatization with picolinoyl chloride to the ester (positive control with cholesterol). Schemes showing the incubation/derivatization conditions for (A) the cholesterol substrate. Part (B) is the selected ion chromatograms of the derivatized picolinoyl ester containing one 18O atom (m/z 210.1375, 15-1) and no 18O atom (m/z 208.1332, 15-0). Chromatogram information – mass tolerance window: 4 ppm, full scan MS: m/z 100–1000 range. APCI corona positive mode. (C) is the mass spectrum of the detected peak at tR 4.05–4.17 min. The peak of interest eluted at tR 4.09 minutes.
Positive Control–P450 17A1
In order to verify the retention of the oxygen in the C20-position of pregnenolone, a similar control experiment was performed using cholesterol (Scheme 6, 1) as the starting material. Additionally, analysis of the C20-oxygen in the pregnenolone product formed from P450 11A1 was achieved by P450 17A1-mediated conversion of pregnenolone into acetic acid in situ (Scheme 6). The presence of two 18O atoms in the acetic acid product (Scheme 6, 13a) would confirm that at least some, if not all, of the oxygen atoms are retained during the P450 11A1/P450 17A1 coupled assay (Scheme 6).
LC-MS analysis of the pyridine acetate isotopologues from the positive control experiment showed the detection of an acetate containing two 18O atoms (Figure 3B, 18d-2). There was also a pyridine acetate isotopologue containing only one 18O atom in the positive control experiment. The ratio of the acetate isotopologues containing two 18O atoms and one 18O atom was 0.7 to 1.0 (Figure 3B, the ratio of 18d-2:18d-1). The detection of the acetate with a single 18O atom indicates the possibility of some exchange of the C20-oxygen in the 21-carbon steroid products (Scheme 4, 4a to 4b or 16a to 16b) during the enzyme-coupled assay. However, the LC-MS analysis of the synthetic pyridine acetate standard with no 18O label (Supporting Information), which was prepared by treating acetic anhydride with the corresponding alcohol,15 did show the presence of some acetate containing one 18O atom (0.003% 18O). Therefore, the source of the mono-18O-containing acetate detected in the positive control experiment (Figures 3B and 3C, 18d-1) may be from the acetate derived from background acetic acid. Nevertheless, the positive control experiment confirmed that we were still able to detect the acetate isotopologue containing two 18O atoms (Figure 3B), indicating that the P450 17A1 reactions (i.e. 17α-hydroxylation and C17,C20-lyase) are fast enough to trap the original C20-oxygen in pregnenolone generated from P450 11A1 (Scheme 6, 4a). In other words, the positive control experiment verified that the transformation from 4a to 4b (Scheme 6) does not completely occur before P450 17A1 converts pregnenolone to acetic acid (Scheme 6, 4a to 13a).
Figure 3.


Positive control experiment of P450 11A1/P450 17A1 coupled assay. (A) Scheme showing positive control incubation of cholesterol with P450 11A1 in the presence of 18O2 followed by derivatization with the pyridine-containing diazo reagent. (B) Selected ion chromatograms of the pyridine acetate isotopologues (i–iv). (C) Mass spectra of the pyridine acetate isotopologues of the positive control incubation showing the presence of two 18O atoms (18d-2) and confirming retention of 18O atoms in the coupled assay. The expanded box shows the peaks detected (18d-1 and 18f-0) in the m/z 168.06–168.11 range. Information for the chromatogram – full scan MS: m/z 50–700 range, mass tolerance window: 6 ppm. ESI positive mode. The peak of interest eluted at tR 3.15 minutes.
With the results from both positive control experiments using cholesterol as the substrate, we have established that our enzyme coupled assay systems are efficient enough to trap the carbonyl oxygens in the C20-C22 bond cleavage products of P450 11A1 (i.e. isocaproaldehyde and pregnenolone). We now present the results from the 18O-labeling experiments using the direct lyase substrate, 20R,22R-dihydroxycholesterol.
P450 11A1 Incubation Coupled with Yeast Alcohol Dehydrogenase– Analysis of 18O Incorporation from Molecular Oxygen (18O2)
The yeast alcohol dehydrogenase-P450 11A1 coupled incubation with 20R,22R-dihydroxycholesterol in the presence of 18O2 showed no incorporation of an 18O atom derived from molecular oxygen (18O2) (Figure 4B). The lack of 18O incorporation in this experiment, combined with the knowledge that the yeast alcohol dehydrogenase enzyme traps the initial carbonyl oxygen in isocaproaldehyde as the alcohol (Scheme 5, 5a to 10) before it can exchange with water, rules out the C20 O-H abstraction mechanism that is followed by oxygen rebound onto the cleaved side chain (Scheme 2Ba).
Figure 4.


P450 11A1/yeast alcohol dehydrogenase incubation of 20R,22R-dihydroxycholesterol (3) in the presence of 18O2 followed by derivatization. (A) Scheme showing incubation followed by derivatization with picolinoyl chloride (14). (B) Selected ion chromatograms monitoring for picolinoyl esters containing one 18O atom (15-1) and no 18O atom (15-0). (C) Mass spectrum showing the absence of 18O-incorporated picolinoyl ester isotopologue (15-1). The peak of interest is at tR 4.10 minutes.
P450 11A1 Incubation Coupled with P450 17A1–Analysis of 18O-Incorporation from Molecular Oxygen (18O2)
Because of the background acetic acid problem revealed in the positive control experiment regarding the detection of 18O-containing acetate (Figure 3, 18d-1), we used 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol in the P450 11A1 incubation (Figure 5A, 3a) when probing the C20,C22-lyase reaction. The synthesis did not yield a completely (i.e., 100%) 21,21,21-d3-incorporated 20R,22R-dihydroxycholesterol, and analysis of the substrate by LC-MS revealed a mixture of isotopologues containing three, two, and one deuteriums (Supporting Information, compound 3a). This isotopic mixture was inconsequential in our analysis because we employed HRMS (i.e., resolution of the LTQ Orbitrap XL mass spectrometer set at 100,000). With such high resolution, we were able to clearly distinguish between the mass differences of a pyridine acetate isotopologue with two deuteriums (18b-0, m/z 168.0988) vs. one 18O atom (18d-1, m/z 168.0905) vs. two 13C atoms (18f-0, m/z 168.0924), with a difference in mass of 49 ppm (18b-0 vs. 18d-1) and 11 ppm (18d-1 vs. 18f-0) (i.e., a minimum resolving power (m/Δm)37,38 of 20,000 and 88,500 are required for the first two isotopologues (18b-0 and 18d-1) and the last two isotopologues (18d-1 and 18f-0), respectively). As shown in Figure 3C (and in the inset), the two isotopologues, 18d-1 and 18f-0, were cleanly resolved when the resolving power was set to 100,000. The LC-MS analysis of the P450 11A1-P450 17A1 coupled incubation with 21,21,21-d3-incorporated 20R,22R-dihydrocholesterol (Figure 5A, 3a) in the presence of 18O2 revealed that none of the pyridine acetate isotopologues contained two 18O atoms (Figures 5B and 5C, 18). This observation, combined with the fact that the positive control experiment (Figure 3) confirmed the retention of the C20-carbonyl oxygen in pregnenolone through the trapping process (Scheme 6, 4a to 16a), eliminates the C20-C22 bond cleavage mechanism involving the C22 O-H abstraction and oxygen rebound into pregnenolone (Scheme 2Bb).
Figure 5.



P450 11A1/P450 17A1 incubation of 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol (3a) in the presence of 18O2 followed by derivatization. (A) Scheme showing the incubation of P450 11A1 with 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol coupled with P450 17A1 in the presence of 18O2 followed by derivatization with the pyridine-containing diazo reagent. (B) Chromatogram showing different pyridine isotopologues generated from the incubation (i–viii). (C) Mass spectral analysis of pyridine acetate isotopologues from P450 11A1 incubation of 20R,22R-dihydroxy-[21,21,21-2H3]-cholesterol with P450 17A1 in 18O2. No isotopologues with two 18O atoms were detected (i, iii, v, vii). Full scan MS: m/z 50–700 range, mass tolerance: 6 ppm; ESI positive mode. The peak of interest is at tR 3.15 minutes.
The results from both sets of coupled assays (i.e., 18O2 incubation of P450 11A1 with either yeast alcohol dehydrogenase or P450 17A1, Figures 4B and 5B) showed that an 18O atom from molecular oxygen (18O2) was not incorporated into the C20-C22 bond cleavage products (i.e. pregnenolone or isocaproaldehyde). Thus, P450 11A1 cleaves the carbon-carbon bond of 20R,22R-dihydroxycholesterol through an electrophilic Compound I mechanism (Scheme 2A).
DISCUSSION
The incubations with P450 11A1 and its substrate 20R,22R-dihydroxycholesterol in the presence of 18O2 revealed that an oxygen atom from molecular oxygen was not incorporated into either pregnenolone (Figure 5) or isocaproaldehyde (Figure 4), supporting an electrophilic Compound I mechanism (Scheme 2A). The positive control experiments (Figures 2 and 3) confirmed retention of the oxygen atom in the carbonyl functional groups of the products during the coupled assays (i.e. yeast alcohol dehydrogenase to reduce isocaproaldehyde and P450 17A1 to analyze pregnenolone). These observations rule out a carbon-carbon bond cleavage mechanism that involves O-H abstraction followed by oxygen rebound (Scheme 2B) and support an electrophilic Compound I mechanism (Scheme 2A). Nonetheless, it is difficult to determine whether it is the 22R-hydroxy group (Scheme 2Aa) or 20R-hydroxy group (Scheme 2Ab) of 20R,22R-dihydroxycholesterol that attacks Compound I.
A Potential Additional C-H Abstraction Mechanism
P450 11A1 has been shown to oxygenate at different sites on other sterol substrates besides the C20- and C22- positions. For example, ergosterol is hydroxylated on the C23-position by P450 11A1 after epoxidation of the C20-C22 double bond.39 Additionally, vitamin D2 is a substrate for P450 11A1, where it catalyzes the hydroxylation of the C20- and C17- carbons.40 The possibility of P450 11A1 reacting at adjacent positions (i.e., C17- and C23-) on its substrate suggested another possible mechanism for C20-C22 bond cleavage, as shown in Scheme 7. In this scenario, the C17α-H hydrogen atom is abstracted and an electron transfer occurs that forms a tertiary carbocation, which can undergo a 1,2-alkyl shift to form a β-hydroxy ketone (Scheme 7, 19). A subsequent retro-Aldol reaction would then result in the formation of the final products, pregnenolone and isocaproaldehyde (Scheme 7, 19 to 4 and 5). Despite this mechanism being consistent with the 18O-labeling results, the possibility is ruled out by the detection of a tetradeuterated pregnenolone product (Supporting Information) when 20R,22R-dihydroxy-[17,21,21,21-[2H4]-cholesterol was incubated with P450 11A1. Another piece of evidence discounting this proposed mechanism (Scheme 7) is that only the hydroxycholesterol intermediates (Scheme 1, 2 and 3) have been isolated from a reaction with P450 11A1 and cholesterol.33 However, another possible mechanism initiated by abstraction of the C23-hydrogen atom cannot be eliminated.
Scheme 7.

An alternative mechanistic proposal involving C-H abstraction as the initial step for the C20-C22 carbon-carbon bond cleavage step of P450 11A1.
An Electrophilic Compound I Mechanism Involving a Molozonide Intermediate
Interestingly, the substrate for the carbon-carbon bond cleavage reaction contains two adjacent alcohol groups (i.e. a 1,2-diol) at the site of bond breakage (C20- and C22- positions), which suggests another possible mechanism. Furthermore, any “monohydroxylated” substrate such as 22R-hydroxycholesterol (Scheme 1, 2) results in a second hydroxylation by P450 11A1 to form 20R,22R-dihydroxycholesterol (Scheme 1, 3) and not a carbon-carbon bond cleavage reaction. This observation suggests that the second hydroxy group near the site of C-C bond cleavage may play a role in the lyase reaction. In other words, the C-C bond cleavage reaction does not occur with 22R-hydroxycholesterol (Scheme 1, 2) to yield pregnene-3,20-diol and isocaproaldehyde. As shown in Scheme 8, which shows the peroxide intermediate (Scheme 8, 3-P1) after initial reaction between electrophilic Compound I and 20R,22R-dihydroxycholesterol (Scheme 2A), a nucleophilic attack by the second hydroxy group may result in a molozonide intermediate (Scheme 8, 3-O1), which can directly break down to the C-C bond cleavage products (Scheme 8, 4 and 5). This mechanism involving the direct breakdown of the molozonide intermediate (Scheme 8, 3-O1) would be consistent with the observation that the cleaved products (Scheme 8, 3-O1 to 4 and 5) do not contain 18O-labeling from molecular oxygen. In the field of chemical synthesis, ozonolysis is a common way to cleave C-C bonds of alkene functional groups through a molozonide to ozonide transformation.41 However, the decomposition of the ozonide intermediates (Scheme 8, 3-O2 and 3-O3) may result in the incorporation of an 18O label in the products (Scheme 8, 4 and 5). Since we did not observe any incorporation of an 18O atom into the isocaproaldehyde product (Scheme 8, 5), we can rule out the ozonide (Scheme 8, 3-O3 to 5) reaction pathway, but the direct breakdown of the molozonide is still viable (Scheme 8, 3-O1 to 5).
Scheme 8.

(A) An alternative mechanism for C-C bond cleavage involving a molozonide intermediate (3-O1).
Other Alternative Compound I Proposals
Some alternate mechanistic possibilities can be considered (Scheme 9). In parts A–D of Scheme 9, mechanisms are presented in which there is no oxygen rebound but instead a proton-coupled electron transfer (formal removal of a hydrogen atom) occurs to yield the carbonyl products. This mechanism, formally analogous to a carbon desaturation, would be consistent with the 18O labeling results. Such mechanisms have been considered before, for at least 30 years, but no evidence has been obtained to support this possibility. Doing the most relevant experiments has not been possible with P450 11A1, in that the only types of labeling experiments possible with the substrate cholesterol are described here and the ability to catalyze other reactions is limited.27 P450 11A1 does catalyze a number of other reactions, particularly with vitamin D,42,43 but none of these involve the formation of carbonyls. Reactions with other P450s can be considered, e.g. the P450 19A1 reaction that converts its aldehyde substrate to the carboxylic acid (i.e. the reactions in Scheme 9A–D do not occur).13 The 19-oxoandrostenedione substrate yielded a mixture of 19-oic androgen product isotopologues that had one 18O atom and no 18O atom. If the carbonyl forming reaction of P450 19A1 occurred in the fashion shown in Scheme 9A/B, then we would not have seen any 18O-incorporated in the 19-oic androgen products.13
Scheme 9.

Alternate possibilities considered to explain the 18O incorporation results.
Some earlier work with other P450s showed varying results for incorporation of label from 18O2 into carbonyl products.4445–47 However, all of these studies cited were done under incubation conditions extending from minutes to hours, conditions under which carbonyl oxygens are known to rapidly exchange with water,13,34,35,48,49 and none of these results can be considered valid. Surprisingly, the rapid oxygen exchange was already well-established in the P450 field, and an enzymatic trapping procedure had been utilized in 18O incorporation studies in which aldehydes were generated.50 Although we cannot completely discount the alcohol “desaturation” possibility (Scheme 9A–D), we believe that there is currently no real precedent for this in P450s.
Another possibility that can be considered is shown in Scheme 9E/F, in which recombination of the Compound II form of P450 with the carbinol radical occurs to give an iron linked gem-diol (Scheme 9, 4G-1). Base-catalyzed removal of a proton from the hydroxyl not linked to iron (Scheme 9 E/F, path a) would lead to unlabeled carbonyl products. In our Introduction, we started with the assumption that the dehydration step (Scheme 2B) is not stereospecific. We are inclined against this mechanism (Scheme 9E/F), in that formation of P450 iron-bound gem diols does not have precedent.
CONCLUSIONS
In summary, isotope labeling experiments combined with coupled enzyme assays support an electrophilic Compound I mechanism (Scheme 2A, i.e. nucleophilic attack of a hydroxyl on the electrophilic FeO3+), proposed elsewhere6–10 and also suggested in structural 11 and EPR/ENDOR5 studies, and rule out an initial O-H hydrogen atom abstraction mechanism (Scheme 2B) in the carbon-carbon bond cleavage step of P450 11A1 to form pregnenolone from cholesterol. The electrophilic Compound I mechanism is consistent with a mechanism we have proposed in the P450 17A1-catalyzed C17-C20 bond cleavage reaction of 17α-hydroxypregnenolone to form dehydroepiandrosterone and acetic acid (Scheme 10B, 16c to 12 and 13).15 A similar mechanism probably occurs in the P450 1A2 catalyzed carbon-carbon bond cleavage process of nabumetone,51 where an α-hydroxy ketone intermediate comparable to 17α-hydroxypregnenolone (16c) was isolated from the incubation. On the contrary, previous 18O-labeling studies suggested that P450 19A1 does not undergo the electrophilic Compound I mechanism as it catalyzes the cleavage of the C10-C19 bond in androgens to form estrogens and formic acid. Instead, the enzyme abstracts the C1β-H hydrogen atom of its 19-oxo androgen substrate as the initial step.13 Furthermore, the direct C-C bond cleavage substrate of P450 19A1 (e.g. Scheme 10C, 20, 19-oxoandrostenedione) differs from those of P450 11A1 (e.g. Scheme 10A, 3, 20R,22R-dihydroxycholesterol) and P450 17A1 (e.g. Scheme 9B, 16c, 17α-hydroxypregnenolone) in that there is no free hydroxy group present in the case of P450 19A1. However, to remain consistent with the electrophilic Compound I mechanisms proposed for P450s 11A1 and 17A1, we show an alternative mechanism for P450 19A1, which generates a ferric peroxy hemiacetal intermediate (Scheme 10D, 25). This tetrahedral intermediate is similar to the one proposed in the mechanism involving the nucleophilic attack of the ferric peroxide species onto the aldehyde (20)15 except that in Scheme 10D, the 18O label from molecular oxygen (18O2) is not directly attached to the C19-carbon (Scheme 10D, 25). This mechanism for P450 19A1 (Scheme 10D) involves the formation of an alcohol substrate through hydration of the aldehyde (Scheme 10, 20 to 24), and the resulting gem-diol (Scheme 10D, 24) conducts a nucleophilic attack on Compound I. The 18O-labeling studies in this system9 are consistent with the mechanism shown in Scheme 10D (i.e., lack of incorporation of 18O label in the formic acid product (23) from 18O2). The formation of this tetrahedral intermediate (Scheme 10D, 25) has been explored without enzyme present, where the hydroperoxy hemiacetal intermediate was generated in situ by treating 19-oxoandrostenedione with hydrogen peroxide.52 However, an estrogen product was not formed in the study, and instead the Δ4,5-double bond was epoxidized to yield 19-oxo-androst-4β,5β-epoxydione. The authors concluded that the presence of the enzyme drives the reaction towards formation of the estrogen (i.e. Scheme 10, 22) rather than the epoxide.
Scheme 10.

Summary of the mechanisms of three P450 systems supported by the 18O-labeling studies discussed in the text: P450 11A1 (A), P450 17A1 (B), and P450 19A1 (C and D).
The peroxy/molozonide mechanism we propose has some precedent in the work of Hochberg,53,54 as pointed out by Ortiz de Montellano and De Voss.9 In the Hochberg et al.53 work, a crude adrenal mitochondrial preparation efficiently (23% yield) converted the substrate analog (20R)-20-(p-tolyl)-5-pregnene-3β,20-diol to pregnenolone (and its product progesterone), in the presence of the cofactor NADPH. In other work by Hochberg,54 a similar reaction occurred with the 20-cyclopropylcarbinyl analog to yield pregnenolone and cyclopropanecarboxylic acid. Experiments with non-hydroxylated analogs did not yield these steroid products. The results can be explained by the heterolytic addition of FeO3+ to the 20-hydroxyl group to form a Fe–O–O–C bond (as in Scheme 2, 3-P2), which decomposes to yield pregenolone.9
With the experimental evidence disclosed in this report and more data from other systems (i.e., P450s 17A1 and 19A16,13,15,55), we can begin to unambiguously classify how P450 enzymes cleave carbon-carbon bonds. Several questions still remain about the nature of the active oxygenated species involved in catalysis by mammalian P450 51A1, the lanosterol 14α-demethylase, and its microbial orthologues.6,55
Supplementary Material
Acknowledgments
Supported in part by National Institutes of Health grants R37 CA090426 and R01 GM118122. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. I.-J. J. was supported by the Vanderbilt International Scholars Program.
Footnotes
Supporting Information. Codon optimization and construction of expression vector for human P450 11A1, enzyme-catalyzed conversion of isocaproaldehyde to 4-methylpentan-1-ol, detection of naturally abundant mono-18O-isotope incorporated ethylpyridine acetate from synthetic standard, LC-MS analysis of picolinoylated 20,22-dihydroxy-[21,21,21-2H3]-cholesterol, LC-MS analysis of picolinoylated-pregnenolone using 20,22-dihydroxy-[17,21,21,21-2H4]-cholesterol as the starting material, and experimental data for synthesized chemicals.
References
- 1.Kraaipoel RJ, Degenhart HJ, Leferink JG, Van Beek V, De Leeuw-Boon H, Visser HK. FEBS Lett. 1975;50:204–209. doi: 10.1016/0014-5793(75)80489-3. [DOI] [PubMed] [Google Scholar]
- 2.Van Lier JE, Smith LL. Biochim Biophys Acta. 1970;210:153–163. doi: 10.1016/0005-2760(70)90070-6. [DOI] [PubMed] [Google Scholar]
- 3.Burstein S, Middleditch BS. Biochem Biophys Res Commun. 1974;61:692–697. doi: 10.1016/0006-291x(74)91012-2. [DOI] [PubMed] [Google Scholar]
- 4.Davydov R, Gilep AA, Strushkevich NV, Usanov SA, Hoffman BM. J Am Chem Soc. 2012;134:17149–17156. doi: 10.1021/ja3067226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davydov R, Strushkevich N, Smil D, Yantsevich A, Gilep A, Usanov S, Hoffman BM. Biochemistry. 2015;54:7089–7097. doi: 10.1021/acs.biochem.5b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Voss J, Cryle MJ. In: The Ubiquitous Roles of Cytochrome P450 Enzymes. Sigel A, Sigel H, Sigel RKO, editors. Vol. 3. John Wiley & Sons; Chichester, U.K: 2007. pp. 397–435. [Google Scholar]
- 7.Ortiz de Montellano PR. In: Cytochrome P-450. 1. Ortiz de Montellano PR, editor. Plenum Press; New York: 1986. pp. 217–271. [Google Scholar]
- 8.Ortiz de Montellano PR. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. 2. Ortiz de Montellano PR, editor. Plenum Press; New York: 1995. pp. 245–303. [Google Scholar]
- 9.Ortiz de Montellano PR, De Voss JJ. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Ortiz de Montellano PR, editor. Plenum Publishers; New York: 2005. pp. 183–245. [Google Scholar]
- 10.Ortiz de Montellano PR. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. 4. Ortiz de Montellano PR, editor. Springer; New York: 2015. pp. 111–176. [Google Scholar]
- 11.Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, Park HW. Proc Natl Acad Sci U S A. 2011;108:10139–10143. doi: 10.1073/pnas.1019441108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morisaki M, Sato S, Ikekawa N. Chem Pharmaceut Bull. 1977;25:2576–2583. [Google Scholar]
- 13.Yoshimoto FK, Guengerich FP. J Am Chem Soc. 2014;136:15016–15025. doi: 10.1021/ja508185d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottlieb HE, Kotlyar V, Nudelman A. J Org Chem. 1997;62:7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimoto FK, Gonzalez E, Auchus RJ, Guengerich FP. J Biol Chem. 2016;291:17143–17164. doi: 10.1074/jbc.M116.732966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shingate BB, Hazra BG, Pore VS, Gonnade RG, Bhadbhade Mohan M. Tetrahedron. 2007;63:5622–5635. [Google Scholar]
- 17.Glasoe PK, Long FA. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 18.Jencks WP. Catalysis in Chemistry and Enzymology. McGraw-Hill; New York: 1969. p. 276. [Google Scholar]
- 19.Walsh C. Enzymatic Reaction Mechanisms. W. H. Freeman Co; San Francisco: 1979. p. 121. [Google Scholar]
- 20.De Caprio J, Yun J, Javitt NB. J Lipid Res. 1992;33:441–443. [PubMed] [Google Scholar]
- 21.Omura T, Sato R. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 22.Barnes HJ, Arlotto MP, Waterman MR. Proc Natl Acad Sci U S A. 1991;88:5597–5601. doi: 10.1073/pnas.88.13.5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandhu P, Baba T, Guengerich FP. Arch Biochem Biophys. 1993;306:443–450. doi: 10.1006/abbi.1993.1536. [DOI] [PubMed] [Google Scholar]
- 24.Sagara Y, Hara T, Ariyasu Y, Ando F, Tokunaga N, Horiuchi T. FEBS Lett. 1992;300:208–212. doi: 10.1016/0014-5793(92)80847-a. [DOI] [PubMed] [Google Scholar]
- 25.Palin MF, Berthiaume L, Lehoux JG, Waterman MR, Sygusch J. Arch Biochem Biophys. 1992;295:126–131. doi: 10.1016/0003-9861(92)90497-k. [DOI] [PubMed] [Google Scholar]
- 26.Enright JM, Toomey MB, Sato SY, Temple SE, Allen JR, Fujiwara R, Kramlinger VM, Nagy LD, Johnson KM, Xiao Y, How MJ, Johnson SL, Roberts NW, Kefalov VJ, Guengerich FP, Corbo JC. Curr Biol. 2015;25:3048–3057. doi: 10.1016/j.cub.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acimovic J, Goyal S, Kosir R, Golicnik M, Perse M, Belic A, Urlep Z, Guengerich FP, Rozman D. Sci Reports. 2016;6:28462. doi: 10.1038/srep28462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagara Y, Wada A, Takata Y, Waterman MR, Sekimizu K, Horiuchi T. Biol Pharmaceut Bull. 1993;16:627–630. doi: 10.1248/bpb.16.627. [DOI] [PubMed] [Google Scholar]
- 29.Guengerich FP. Arch Biochem Biophys. 2005;440:204–211. doi: 10.1016/j.abb.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 30.Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. Arch Biochem Biophys. 1998;350:324–332. doi: 10.1006/abbi.1997.0534. [DOI] [PubMed] [Google Scholar]
- 31.Guengerich FP. In: Hayes’ Principles and Methods of Toxicology. 6. Hayes AW, Kruger CL, editors. CRC Press-Taylor & Francis; Boca Raton, FL: 2014. pp. 1905–1964. [Google Scholar]
- 32.Yoshimoto FK, Auchus RJ. J Steroid Biochem Molec Biol. 2014;151:52–65. doi: 10.1016/j.jsbmb.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugano S, Miura R, Morishima N. J Biochem (Tokyo) 1996;120:780–787. doi: 10.1093/oxfordjournals.jbchem.a021479. [DOI] [PubMed] [Google Scholar]
- 34.Byrn M, Calvin M. J Am Chem Soc. 1966;88:1916–1922. [Google Scholar]
- 35.Greenzaid P, Luz Z, Samuel D. J Am Chem Soc. 1967;89:749–755. [Google Scholar]
- 36.Guengerich FP, Crawford WM, Jr, Watanabe PG. Biochemistry. 1979;18:5177–5182. doi: 10.1021/bi00590a023. [DOI] [PubMed] [Google Scholar]
- 37.Perry RH, Cooks RG, Noll RJ. Mass Spectrom Rev. 2008;27:661–699. doi: 10.1002/mas.20186. [DOI] [PubMed] [Google Scholar]
- 38.Scigelova M, Hornshaw M, Giannakopulos A, Makarov A. Mol Cell Proteomics. 2011;10:1–19. doi: 10.1074/mcp.M111.009431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuckey RC, Nguyen MN, Chen J, Slominski AT, Baldisseri DM, Tieu EW, Zjawiony JK, Li W. Drug Metab Dispos. 2012;40:436–444. doi: 10.1124/dmd.111.042515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slominski A, Semak I, Wortsman J, Zjawiony J, Li W, Zbytek B, Tuckey RC. FEBS J. 2006;273:2891–2901. doi: 10.1111/j.1742-4658.2006.05302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geletneky C, Berger S. Eur J Org Chem. 1998;1998:1625–1627. [Google Scholar]
- 42.Tuckey RC, Li W, Zjawiony JK, Zmijewski MA, Nguyen MN, Sweatman T, Miller D, Slominski A. FEBS J. 2008;275:2585–2596. doi: 10.1111/j.1742-4658.2008.06406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slominski AT, Li W, Kim TK, Semak I, Wang J, Zjawiony JK, Tuckey RC. J Steroid Biochem Mol Biol. 2015;151:25–37. doi: 10.1016/j.jsbmb.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng KC, Schenkman JB. J Biol Chem. 1983;258:11738–11744. [PubMed] [Google Scholar]
- 45.Wood AW, Swinney DC, Thomas PE, Ryan DE, Hall PF, Levin W, Garland WA. J Biol Chem. 1988;263:17322–17332. [PubMed] [Google Scholar]
- 46.Bellucci G, Chiappe C, Pucci L, Gervasi PG. Chem Res Toxicol. 1996;9:871–874. doi: 10.1021/tx9600053. [DOI] [PubMed] [Google Scholar]
- 47.Matsunaga T, Tanaka H, Higuchi S, Shibayama K, Kishi N, Watanabe K, Yamamoto I. Drug Metab Dispos. 2001;29:1485–1491. [PubMed] [Google Scholar]
- 48.Bell RP. Adv Phys Org Chem. 1966;4:1–29. [Google Scholar]
- 49.Greenzaid P, Luz Z, Samuel D. J Am Chem Soc. 1967;89:756–759. [Google Scholar]
- 50.McMahon RE, Culp HW, Occolowitz JC. J Am Chem Soc. 1969;91:3389–3390. [Google Scholar]
- 51.Varfaj F, Zulkifli SNA, Park H-G, Challinor VL, De Voss JJ, Ortiz de Montellano PR. Drug Metab Dispos. 2014;42:828–838. doi: 10.1124/dmd.114.056903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cole PA, Robinson CH. J Chem Soc, Perk Trans 1. 1990:2119–2125. [Google Scholar]
- 53.Hochberg RB, McDonald PD, Feldman M, Lieberman S. J Biol Chem. 1974;249:1277–1285. [PubMed] [Google Scholar]
- 54.Hoyte RM, Hochberg RB. J Biol Chem. 1979;254:2278–2286. [PubMed] [Google Scholar]
- 55.Fischer RT, Trzaskos JM, Magolda RL, Ko SS, Brosz CS, Larsen B. J Biol Chem. 1991;266:6124–6132. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.