

Summary
Regulatory networks play a central role in the relationship between genotype and phenotype in all organisms. However, the mechanisms that underpin the evolutionary plasticity of these networks remain poorly understood. Here, we used experimental selection for enhanced bacterial motility in a porous environment to explore the adaptability of one of the most complex networks known in bacteria. We found that the resulting phenotypic changes are mediated by adaptive mutations in several functionally different proteins, including multiple components of the flagellar motor. Nevertheless, this evolutionary adaptation could be explained by a single mechanism, namely remodeling of the checkpoint regulating flagellar gene expression. Supported by computer simulations, our findings suggest that the specific “bow-tie” topology of the checkpoint facilitates evolutionary tuning of the cost-benefit trade-off between motility and growth. We propose that bow-tie regulatory motifs, which are widespread in cellular networks, play a general role in evolutionary adaptation.
Keywords: evolution, adaptation, microorganisms, bacteria, selection, optimization, network, sigma factor, flagella, bow tie
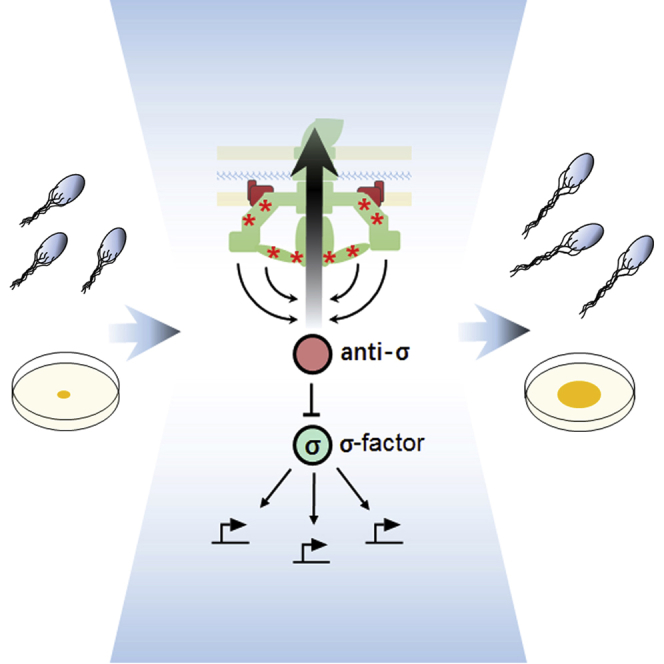
Graphical Abstract
Highlights
-
•
Multiple mutations enhance swimming behavior under selection
-
•
A universal trade-off relationship between motility and growth is observed
-
•
Checkpoint remodeling provides a mechanism of evolutionary adaptation
-
•
Bow-tie topology facilitates evolvability of the motility network
Ni et al. use experimental evolution to investigate remodeling of bacterial motility under selection. Their study reveals a key role of the checkpoint control of flagellar gene expression in motility evolution. Bow-tie topology of the checkpoint control facilitates evolutionary tuning of the trade-off between motility and growth.
Introduction
Global regulatory networks control most cellular decisions, including differentiation, stress response, and environmental adaptation. These networks frequently have a specific “bow-tie” organization where multiple inputs and outputs are connected to a core process or a regulator (Csete and Doyle, 2004). In bacteria, one of the most common global regulatory motifs comprises a sigma factor—a promoter-specific subunit of the bacterial RNA polymerase—and an anti-sigma factor that binds and inhibits the sigma factor (Österberg et al., 2011, Paget, 2015). The multi-input structure of these motifs allows them to integrate a variety of stimuli that control stress responses (Hengge, 2009) and developmental checkpoints (Rudner and Losick, 2001).
One of the best-studied sigma/anti-sigma factor checkpoints is involved in the control of flagellar biosynthesis (Chilcott and Hughes, 2000, Mukherjee and Kearns, 2014). In Escherichia coli and other bacterial species, flagellar genes are organized in a hierarchical regulatory network of three classes based on their order of expression and common transcriptional regulation (Aldridge and Hughes, 2002, Chevance and Hughes, 2008, Chilcott and Hughes, 2000, Kalir et al., 2001) (Figure 1A). Altogether, this network encodes more than 40 proteins that are necessary for flagellar motility and chemotaxis (Minamino and Imada, 2015, Typas and Sourjik, 2015). At the upper level of this regulatory hierarchy is the class I master regulator that is formed by two polypeptides, FlhD and FlhC. It activates class II genes that encode structural components and assembly factors of the flagellar hook-basal body (HBB) complex and of the export apparatus required for biogenesis of flagellar filament. Two other class II genes encode the sigma factor σ28 (FliA) and the anti-σ28 factor (FlgM). At first, both polypeptides are coexpressed, but σ28 is inhibited by binding to FlgM. Upon HBB completion, however, FlgM is secreted from the cell through the flagellar export apparatus to release free σ28. This activates expression of σ28-controlled (class III) genes that encode the subunit of the flagellar filament (FliC), stator components of the flagellar motor (MotA and MotB), as well as the chemotaxis pathway (Hughes et al., 1993). Notably, flgM, fliA, and a number of genes that are required for flagellar assembly possess both class II and class III promoters, and are therefore referred to as class II+III genes (Chevance and Hughes, 2008, Chilcott and Hughes, 2000).
Figure 1.

Experimental Evolution of Bacterial Spreading in Soft Agar
(A) Simplified schematics of the hierarchical regulatory network that controls expression of flagellar genes. Flagellar genes are organized in three classes dependent on the order of their expression, as indicated. Activity of σ28 (FliA) that is required for expression of class III genes (red) is controlled by a checkpoint that involves secretion of inhibitor FlgM (anti-σ28) upon completion of the hook-basal body and export apparatus. Class II+III genes that have both class II and class III promoters are labeled yellow. See text for further details.
(B) Schematic illustration of the experiment. Bacteria are inoculated in the middle of the TB soft agar (TBSA) plate and allowed to spread overnight. Fastest spreaders (∼106 cells) are taken from the edge of spreading colony and used to inoculate a new plate. L1 indicates evolution of line 1, D1 indicates days 1 of evolution, etc. Right: spreading of the parental RP437 and evolved L1-D29 strains.
(C) Time course of enhancement of spreading during evolution of line L1, where spreading was quantified as the diameter of the colony ring on the TBSA plate and normalized to the spreading of RP437.
(D) Spreading of individually evolved lines. All lines spread significantly faster than RP437 according to a two-tailed t test (∗p < 0.05).
(E) Correlation between spreading in TBSA and in minimal medium SA supplemented with 0.1 mM serine. Color code for individual lines here and throughout is the same as in (D). Note that because of the growth defect in minimal medium, line L2 could not be analyzed.
All data here and throughout are represented as mean ± SEM of three replicates. Correlation value is 0.76. Correlation values and p values indicated in panels were calculated as described in the Supplemental Experimental Procedures. See also Figure S1.
Motility and chemotaxis of E. coli are among the most extensively studied biological processes (Sourjik and Wingreen, 2012, Typas and Sourjik, 2015), and the chemotaxis pathway has been used as a model to study evolutionary network optimization (Alon et al., 1999, Barkai and Leibler, 1997, Kollmann et al., 2005, Løvdok et al., 2009, Oleksiuk et al., 2011). Here, we experimentally investigated evolutionary plasticity (evolvability) of motile behavior. Experimental evolution is increasingly applied to investigate predictions of evolutionary theory, particularly in microorganisms (Barrick and Lenski, 2013, Elena and Lenski, 2003, Hindré et al., 2012, Kawecki et al., 2012, Poelwijk et al., 2011, Taute et al., 2014). For example, it has been extensively used to study the evolution of antibiotic and stress resistance (Chait et al., 2007, MacLean et al., 2010) or catabolism of specific nutrients (Blount et al., 2012), typically yielding mutations in a single or a small number of target genes (Blount et al., 2012, González et al., 2015, Toprak et al., 2011, Weinreich et al., 2006). Such experimental evolutionary analyses have helped to better understand several features of the evolutionary process, such as epistatic interactions between multiple mutations, evolutionary trajectories, and evolution in changing environments (Taute et al., 2014). However, understanding the evolution of complex behaviors and of the underlying regulatory networks still remains a challenge (Hindré et al., 2012).
Our results enabled us to characterize the evolvability of the regulatory network that controls bacterial motile behavior. Surprisingly, the observed evolutionary enhancement of motility was achieved through adaptive mutations in a number of functionally very different genes, encoding components of flagellar motor and export apparatus, as well as transcriptional and translational factors. Nevertheless, we showed that most of these mutations acted through a common adaptive mechanism, namely remodeling of the sigma/anti-sigma factor checkpoint, thus leading to common phenotypic changes. This remodeling of the checkpoint apparently adjusts cell motility under selection both during laboratory evolution and in natural isolates of E. coli. Our findings suggest that bow-tie topology can generally enhance evolvability of networks, thus indicating a previously unrecognized evolutionary function of this widespread network motif.
Results
Experimental Evolution Enhances Motility and Chemotaxis
We performed experimental evolution of motile behavior of E. coli using selection for enhanced spreading in tryptone broth (TB) soft agar (TBSA), a porous medium containing a mixture of amino acids that act both as nutrients and as chemoattractants. Consumption of amino acids generates steep local attractant gradients that are subsequently followed by bacteria, which requires motility, chemotaxis, and growth (Berg and Turner, 1979, Wolfe and Berg, 1989). In our experiment, a cell population was allowed to spread for ∼12 hr on a TBSA plate, at a growth rate of approximately one cell generation per hour, and cells from the edge of the outer spreading ring were taken to inoculate a new plate (Figure 1B). We observed that spreading of the evolved strains greatly improved within the first 5–10 rounds of selection (Figure 1C; Figure S1A), although the extent of improvement differed between independently evolved lines (L1–L7; Figure 1D). Consistently, evolved strains had a strong competitive advantage in spreading on TBSA plates compared with the parental strain RP437 (Figure S1B). Evolved E. coli lines showed similarly increased spreading in minimal medium soft agar (SA) supplemented with serine (Figure 1E), confirming that the observed enhancement of spreading is not specific to a particular composition of the medium.
As noted, spreading in SA that initially contains a uniform distribution of attractant requires not only motility and chemotaxis, but also consumption of attractant to create a gradient. To distinguish contributions of motility and metabolism, we tested behavior of evolved strains in gradients of a non-metabolizable analog of aspartate, α-methyl-D,L-aspartate (MeAsp), established either in SA or in liquid (Krembel et al., 2015). Evolved strains showed enhanced chemotactic spreading up the gradient in both assays (Figures S1C and S1D), confirming specific selection for improved motility and chemotaxis in our experiments.
Evolved Strains Show Changes in Motility and Flagellation
To elucidate the mechanisms that underlie the observed enhancement in motility and chemotaxis, we first examined how experimental evolution affected E. coli motility (Figure S2A). The most apparent phenotypic change in all evolved strains was increased swimming velocity in liquid: whereas velocity of the parental strain was comparable with that observed previously (Morehouse et al., 2005, Oleksiuk et al., 2011), it nearly doubled in some of the evolved strains. Moreover, the majority of evolved strains also exhibited an increased frequency of reorientations (tumbling rate). Both of these features were strongly correlated with spreading in TBSA, although the significance of the correlation with increased tumbling rate was weaker. As judged from the chemotactic bias, i.e., chemotactic drift normalized by the swimming velocity, higher velocity alone could mostly account for the increase in the chemotactic drift in liquid.
Our analysis further showed that evolved strains have higher frequency of flagellar motor rotation, as observed for individual motors in immobilized cells, as well as longer and, in the case of L1, also more numerous flagellar filaments (Figure S2B). The statistical analysis of these data suggest that increased flagellar length is the main determinant of higher velocity. In contrast, higher tumbling rate shows only weak correlation with increased length and number of flagella (Figure S2C). Consistent with the observed increase in flagellar length, levels of extracellular flagellin were elevated in all strains (Figure S2D).
Transcription of Class III Flagellar Genes Is Upregulated during Evolution
We next investigated whether the observed enhancement of motility may result from changes in gene expression. For that, we measured transcriptional activity at all three levels of hierarchy of flagellar genes, using reporters for representative class I (flhD), class II (fliE), and class III (fliC) promoters. Interestingly, we observed that the activity of PfliC was upregulated in all evolved strains, whereas the activity of PflhD and PfliE was unchanged or even downregulated (Figure 2A). This increase in PfliC activity showed strong correlation with cell spreading in TBSA (Figure 2B) and with swimming velocity (Figure 2C), indicating that specific upregulation of class III gene expression is a major determinant of the behavioral enhancement. Consistent with this assumption, an increased expression of σ28 (FliA) or titration of anti-σ28 (FlgM) from a plasmid resulted in a similar co-regulation of PfliC activity with spreading in TBSA and with swimming velocity (Figures 2B and 2C). Among the other parameters, flagellar length showed highly significant correlation with PfliC (and anticorrelation with PfliE) activity (Figure 2D; Figure S3A), whereas flagellar number and motor speed did not show significant correlation with gene expression (Figures S3B and S3C). However, overexpression of either flagellin FliC or stator components MotA and MotB was not sufficient to increase spreading (Figure S3D), suggesting that concerted upregulation of multiple class III genes is necessary to enhance motility.
Figure 2.

Upregulation of Class III Flagellar Genes during Evolution
(A) Activity of PflhD, PfliE, and PfliC promoters in evolved strains assayed using plasmid reporters. Color code shows log10 of promoter activity normalized to RP437.
(B–D) Correlation between PfliC promoter activity and spreading in TBSA (B), swimming velocity (C), or flagellar length (D). Gray dot indicates RP437ΔfliA expressing σ28 (FliA) from a plasmid (pBN2; no IPTG induction); dark red dots indicate RP437ΔflgM expressing anti-σ28 (FlgM) from a plasmid (pBN3; 0, 10, and 20 μM IPTG). Other dots indicate RP437 and evolved strains, with colors as in Figure 1D. Correlation values are 0.81 (B), 0.71 (C), and 0.78 (D). Lines indicate hyperbolic fits to the data for visualization.
(E) Changes in genome-wide transcription that correlate with enhanced motility, i.e., showed opposite-sign change upon selection for spreading (L1-L7) or for growth in liquid (R1-R5), as revealed by RNA-seq. Color code shows log2 of promoter activity normalized to RP437.
See also Figures S2 and S3.
These results were further confirmed using genome-wide transcriptome profiling of the evolved strains (Figure 2E). Here, we also considered that during experimental evolution changes in gene expression could result not only from selection for enhanced motility, but also from adaptation to growth in TB at 30°C. In order to distinguish between these two types of selection, we subjected all lines to growth in liquid TB medium at 30°C for another 20 culture passages. This selection indeed either partly or entirely reversed the enhancement of spreading in TBSA (Figure S3E), and these strains were included in the transcriptome analysis.
Transcriptomics results confirmed strong correlation between enhanced spreading in soft agar and selective upregulation of class III (and class II+III) flagellar genes, consistent with higher activity of σ28. Interestingly, expression of several genes related to cell wall biosynthesis (murD, murE, and murG) showed significant correlation with enhanced motility, whereas genes for ATP synthase (atpA, atpB, atpC, atpD, atpE, atpF, atpG, atpH, and atpI), amino sugar metabolism (nanE, nanK, nagA, nagB, and nagE), and succinyl-coenzyme A (CoA) synthetase (sucC and sucD) were weakly counter-regulated. Furthermore, a small number of genes were consistently up- or downregulated upon selection in both TBSA and liquid TB (Figure S3F), possibly associated with general growth selection.
Evolved Strains Carry Multiple Mutations
Subsequently, we used genome sequencing of the evolved strains to map mutations that emerged during selection for enhanced spreading in TBSA. This analysis yielded point mutations (SNPs) in 14 genes, insertions of insertion sequence 1 (IS1) transposable sequence in 5 genes, as well as 1 gene deletion (Table S1). No mutations were found in non-coding regions, including promoter sequences.
To our surprise, in spite of changes in flagellar gene expression in all evolved strains, only two of these mutations affected known transcriptional regulators of flagellar genes: one in flgM (anti-σ28) (L3) and another in sspA (L1) that encodes a starvation-response factor (Hansen et al., 2005). In addition, strain L1 carries mutation in a tRNA-encoding gene serT that has been previously shown to affect translation of FlgM (Chevance et al., 2006). Instead, multiple mutations mapped to genes that encode functional or structural components of flagellar apparatus. Five strains (L1, L2, L4, L5, and L7) have mutations in FliI, the ATPase component of the flagellar export machinery (Fan and Macnab, 1996). Two of these strains (L1 and L4) have additional mutations in the switch components of the flagellar basal body, FliM and FliG. These proteins are required for rotation of the motor and for its control by the chemotaxis system (Bren and Eisenbach, 1998, Toker and Macnab, 1997), but they are also involved in the function of the export apparatus (González-Pedrajo et al., 2006). Furthermore, strain L3 carries, in addition to a mutation in FlgM, a mutation in CheZ, the phosphatase of the chemotaxis system, and strain L2 has an additional mutation in the aspartate chemoreceptor Tar.
A significant number of mutations mapped to genes that had no previously established relation to the flagellar regulon. These genes encoded a translation factor (yciH), enzymes involved in cell envelope biogenesis (ldtB, mdoH, and idi) or in metabolism (hisA, argH, glpK, and ydfG), transporters (cusS and lacY), and proteins of unknown function (yebO and yceO).
Multiple Adaptive Mutations Upregulate Flagellar Gene Expression and Motility
It is well established that not all mutations arising in evolutionary experiments must be adaptive (Rosenberg, 2001). To distinguish contributions of different mutations to the observed enhancement of motility, they were introduced individually into the wild-type strain and assayed for their effects on spreading in TBSA (Figure 3A). We observed that all mutations in flagellar genes, as well as in genes encoding cell envelope biogenesis enzymes, increased spreading. Moreover, spreading was also enhanced by mutations in genes encoding the transcriptional (SspA) and translational (YciH) regulators, as well as the uncharacterized protein YebO. Other mutations had little or no effect or even decreased spreading.
Figure 3.

Upregulation of Motility and Flagellar Gene Expression by Adaptive Mutations
(A) Spreading in TBSA for RP437 strain carrying individual mutations that were identified in evolved lines. Spreading was normalized to the wild-type RP437. Significant decrease (green asterisks, p < 0.05 according to two-tailed t test) or increase (black asterisks, p < 0.05; red asterisks, p < 0.01) in spreading is indicated. Classification of mutated genes is indicated.
(B–E) Correlation between spreading in TBSA and swimming velocity (B), tumbling rate (C), chemotactic bias in MeAsp gradient in SA (D), or chemotactic drift velocity in MeAsp gradient in liquid (E) for strains carrying single mutations (dark blue) and for evolved strains or strains with varying levels of FliA or FlgM (colors as in Figure 2B). Data for evolved strains in (D) are taken from Figure S1C. Correlation values are 0.82 (B), 0.74 (C), 0.78 (D), and 0.59 (E). Line in (E) indicates an exponential fit.
(F and G) Correlation between PfliC promoter activity and spreading in TBSA or swimming velocity for the same strains. Data for evolved strains are taken from (B) and (C). Correlation values are 0.81 (F) and 0.73 (G). Lines indicate hyperbolic fits.
Despite these very different functionalities of mutated proteins, we observed that the effects of individual mutations on spreading in TBSA are highly correlated with increased swimming velocity and with increased tumbling rate, and similar correlation could be observed for the evolved strains or for varying expression of σ28 or FlgM (Figures 3B and 3C; Figures S2A, S2B, and S4B). This confirms that increased swimming velocity and tumbling rate can account for the enhancement in spreading. Consistent with our analysis for the evolved strains, individual mutations that enhanced spreading in TBSA also conferred an advantage for following MeAsp gradients in soft agar (Figure 3D) or in liquid (Figure 3E).
Finally, essentially all individual adaptive mutations, including those in functional and structural flagellar proteins, led to elevated expression of class III genes (Figure S4A). These changes showed strong correlation with increased spreading in TBSA (Figure 3F) and with swimming velocity (Figure 3G), as observed already for the evolved strains (Figures 2B and 2C), confirming that adaptive changes in class III gene expression are the major determinant of increased motility. Interestingly, the mutation in SspA showed a disproportional enhancement of motility (Figure 3G) and not only longer, but also more numerous flagella (Figure S4C), similar to L1 that carries it.
Checkpoint Remodeling as a Mechanism of Evolutionary Plasticity
How could mutations in flagellar basal body and export apparatus mediate adaptive changes in class III gene expression? A potential mechanism linking these proteins to gene expression is secretion of the anti-σ28 factor FlgM. As discussed above (see Introduction and Figure 1A), FlgM-mediated inhibition ensures that σ28 is activated only upon assembly of the basal body that allows FlgM to be secreted. However, because FlgM is steadily expressed even after the initial checkpoint exit, its cellular levels continue to control expression of class III genes (Kollmann et al., 2005). Therefore, mutations that change the efficiency of FlgM secretion may also affect gene expression. To verify the involvement of this checkpoint control, we deleted flgM in all evolved strains as well as in strains that carry individual mutations. Indeed, deletion of flgM entirely abolished the enhancement of spreading in TBSA in three of the evolved strains (L4, L5, and L7) and reduced it in three other strains (L1, L3, and L6) (Figure 4A; Figure S5A). Similarly, in strains carrying individual mutations in flagellar genes, spreading enhancement was either abolished (for fliI or fliM mutations) or reduced (for fliG mutation) by the deletion of flgM (Figure 4B; Figure S5B). This confirms our hypothesis that increased secretion of the anti-σ28 factor FlgM is the main mechanism of adaptive motility enhancement via mutations in flagellar proteins.
Figure 4.

Sigma Factor Checkpoint Remodeling during Evolution
(A and B) Spreading in TBSA for evolved strains (A) and strains carrying individual mutations (B) upon disruption of flgM gene. Spreading was normalized to RP437ΔflgM. Black (p < 0.05) and red (p < 0.01) asterisks indicate significance of increase in spreading compared with RP437ΔflgM.
(C) FlgM secretion efficiency in indicated strains, assayed by measuring levels of FlgM-HA in supernatant (sup) and pellet (pel) fractions.
(D) Correlation between FlgM secretion efficiency and PfliC promoter activity. Individual mutations that result in highest increase in secretion are indicated. Correlation value is 0.72. Color code is as in Figure 3.
See also Figure S5.
Interestingly, deletion of flgM also reduced the effects of the sspA and yciH mutations, although these gene products have no established connection to the regulation of FlgM secretion. Nevertheless, the effects of yciH, ldtB, mdoH, yebO, and sspA mutations on motility appear to be at least partly independent of the FlgM secretion, which may explain (residual) FlgM-independent enhancement of spreading that is observed in strains L1, L2, L3, and L6 that carry these mutations.
In order to directly confirm the connection between the checkpoint remodeling and enhanced expression of class III genes, we quantified efficiency of FlgM secretion. Consistent with our explanation, in all evolved strains FlgM secretion efficiency was significantly enhanced, leading to a lower cellular level of FlgM relative to its level in the supernatant (Figure 4C). Moreover, there was a strong correlation between the FlgM secretion efficiency and the level of class III gene expression, both in the evolved strains and in strains containing individual mutations that enhanced motility (Figure 4D). Such correlation was not observed for secretion efficiency of FliC (Figure S5C), suggesting that the adaptive enhancement of secretion is specific to FlgM.
Trade-Off between Motility and Growth Fitness
It is well established that evolution frequently needs to optimize different conflicting functions, thus leading to adaptive trade-offs (Schuetz et al., 2012, Shoval et al., 2012). For expression of highly abundant flagellar and chemotaxis proteins, the most likely fitness trade-off may be associated with growth. Indeed, when relative growth fitness of the evolved strains in liquid TB was plotted as a function of PfliC promoter activity, clear anticorrelation between the two was observed (Figure 5A). A similarly apparent trade-off could be observed between the growth fitness and swimming velocity (Figure 5B). Consistent with that, growth rate increased upon disruption of flhC (eliminating expression of the entire flagellar regulon) or fliA (eliminating expression of class III genes), and it was severely reduced by deletion of flgM (upregulation of class III genes). Disruption of fliC gene increased growth rate nearly as much as fliA deletion, confirming that a large part of fitness cost associated with class III gene expression is due to production of flagellin. Indeed, deletion of fliC largely alleviated the fitness burden in strain L1 (Figure 5C). The remaining cost might arise from expression of other class III genes or from the energy expenditure for flagellar rotation. However, mutations in sspA and yciH seem to generally incur disproportionally high cost compared with their fitness increase, likely because they affect not only motility, but also other cellular functions.
Figure 5.

Trade-Off between Motility and Fitness
(A and B) Correlation between relative growth fitness and PfliC promoter activity (A) or swimming velocity (B). Relative growth was determined by co-incubation of tested strain with the wild-type (RP437), and relative fitness was defined as the fraction of the tested strain after 5-day incubation normalized by the fraction at inoculation (0.5). Correlation values are −0.68 (A) and −0.88 (B). Line in (A) indicates a bi-exponential decay fit. Color code is as in Figure 3.
(C) Proportions of indicated strains in competition assay over time.
Checkpoint Activity Correlates with Motility of Natural E. coli Isolates
We hypothesized that the observed mechanism of motility enhancement might also play a role in the evolutionary plasticity of natural isolates of E. coli. Consistent with that, we observed a highly significant correlation between activity of PfliC promoter and spreading of motile natural isolates that were randomly selected from the ECOR collection (Ochman and Selander, 1984) (Figure 6A). In contrast, correlation with the activity of PfliE or PflhD promoters was either much weaker or non-significant (Figures 6B–6D). This confirms our hypothesis that variation in motility of the natural isolates is mainly determined by expression of class III genes rather than of the entire flagellar regulon. The underlying cause again appears to be the plasticity of checkpoint efficiency, because secretion of FlgM is highly correlated with the spreading in TBSA and with the PfliC activity in most natural isolates (Figures 6E and 6F).
Figure 6.

Correlation between Flagellar Gene Expression and Motility in Natural Isolates
(A–D) Spreading in TBSA as a function of PfliC (A), PfliE (B), or PflhD (C) promoter activity in natural E. coli isolates, and significance of correlation based on regression analysis of these data (D).
(E and F) Spreading in TBSA (E) and PfliC promoter activity (F) as a function of FlgM secretion efficiency.
Bow-Tie Topology Accelerates Network Evolution
Taken together, our findings demonstrate that mutations in multiple flagellar proteins modulate motility through a common control of FlgM secretion (Figure 7A). We also obtained similar mutations in flagellar genes in another evolutionary experiment, where selection for motility was performed on minimal medium SA plates (Table S2). We thus hypothesized that the multi-input motif present in the flagellar checkpoint control, but also common in many other cellular networks, might play a general role in evolution. To test this hypothesis, we simulated in silico evolution of a simple bow-tie network where multiple input nodes (X1 to X3) affect activity of a common intermediate node (Y) that in turn activates the output node (Z), and compared it with evolution of linear networks where either one or multiple input nodes act independently (Figure 7B). In these simulations, the fitness increase for the bow-tie network under selection was markedly faster than for the linear networks (Figure 7C), even though the multi-input linear network can in principle attain the same maximal fitness as the bow-tie network (see Supplemental Experimental Procedures). Our analysis thus confirms that the bow-tie topology can accelerate network evolution.
Figure 7.

Multi-input Checkpoint Control Mediates Evolution of Motility
(A) Remodeling of flagellar checkpoint control under selection. Left: flagellar genes are organized in three classes (see Figure 1 and text for details). Checkpoint control couples expression of class III genes (red/orange) to completion of flagellar hook-basal body (HBB) through secretion of FlgM that releases σ28 (FliA). Class I genes (blue) are controlled by a number of upstream transcriptional regulators, only some of which are shown. Right: adaptive mutations in multiple HBB (FliM and FliG) or export apparatus (FliI and FliH) proteins encoded by class II genes (green) enhance export efficiency of FlgM, thereby increasing cellular levels of active FliA and leading to upregulation of class III genes. See also Table S2.
(B and C) In silico evolution of linear and bow-tie networks. Networks have either one (X) or multiple (X1 to X3) inputs that activate the output (Z) through independent or common intermediates (Y), as schematically shown (B). The strength of activation is denoted by W. Networks were subject to simulated evolution as described in the Supplemental Experimental Procedures for 100 generations. Median fitness, assumed to be proportional to Z, was calculated for simulations of each network with 100 randomly selected initial parameter sets (C) and plotted against the number of generations.
Discussion
It is commonly assumed that as a consequence of long-term selection, living organisms have evolved to perform different tasks optimally (de Vos et al., 2013, Flamholz et al., 2013, Ibarra et al., 2002, Kollmann et al., 2005, Noor et al., 2010, Notebaart et al., 2014, Schuetz et al., 2012, Zarrinpar et al., 2003). However, proving this assumption is difficult, not least because details of the past evolutionary selection pressure and the underlying trade-offs between different functions are typically unknown (Hindré et al., 2012). Moreover, establishing the genotype-to-phenotype mapping during evolutionary adaptation remains a challenge, given the difficulty in distinguishing between neutral and adaptive mutations and specifically linking a mutation to one and unlinking it from other traits (Lehner, 2013). Although experimental evolution can address some of these questions by subjecting (micro-)organisms to selection under defined laboratory conditions and monitoring ensuing changes in their phenotype and genotype (Hindré et al., 2012), it is most frequently applied to simple traits that involve only one or a few proteins (Blount et al., 2012, González et al., 2015, Toprak et al., 2011, Weinreich et al., 2006). Here, we used selection for E. coli motility in a porous environment to investigate how adaptive evolution under a specific selection pressure shapes this complex bacterial phenotype and what are the internal constraints and underlying mechanisms of genotype and phenotype remodeling.
We observed that enhanced motility evolved in all our experiments, consistent with high selective pressure at the edge of the spreading colony caused by nutrient depletion. On the phenotypic level, the enhancement of E. coli motility in soft agar was primarily driven by increased swimming velocity, but also by increased tumbling rate. This phenotypic adaptation can be well understood in the context of bacterial chemotaxis between traps created by agar (Wolfe and Berg, 1989). Here, similar to the chemotaxis in liquid, bacteria must swim up the gradient to detect changes in chemical concentration and to bias their motility by reducing tumbling rate (Sourjik and Wingreen, 2012). Higher swimming velocity should thus generally enhance spreading by accelerating both the random movement and chemotaxis. However, in the porous environment, higher swimming velocity alone would also lead to more frequent trapping of bacteria in the agar pores, which would limit spreading. The observed increase in tumbling rate may alleviate this hindrance by enabling bacteria not only to cover distances between pores faster, but also to escape them more efficiently (Wolfe and Berg, 1989).
On the molecular level, this phenotypic adaptation could be accounted for by upregulated expression of class III flagellar genes. This upregulation led to increased flagellar length and motor rotation rate, whereby the former was apparently the main determinant of faster swimming. Such specific upregulation of class III genes in all of the independent evolutionary experiments was surprising, because the established transcriptional regulation of flagellar genes in E. coli occurs primarily at the upper (class I) level of the regulatory hierarchy and affects expression of all three gene classes (Fahrner and Berg, 2015) (Figure 7A). Moreover, this is seemingly in contrast to previous studies showing that poorly motile E. coli strains could activate motility through insertion of insertion sequence 5 (IS5) element into the regulatory region of the flhDC operon (Barker et al., 2004, Wang and Wood, 2011). On the other hand, we observed no transcriptional regulation of individual class III genes or changes in the network topology. Our results thus demonstrate that concerted upregulation of multiple class III genes is required, and also sufficient, to enhance motility, likely because not only FliC, but also other products of class III (and class II+III) genes are required for assembly and energizing of longer flagella, and thereby for faster swimming. Consistent with that explanation, proton channel components MotA and MotB, as well as chaperones (e.g., FliS) that are required for FliC filament assembly (Auvray et al., 2001), are encoded by class III or class II+III genes. We therefore propose that, besides its importance for sequential ordering of expression (Chilcott and Hughes, 2000, Kalir et al., 2001), the topology of flagellar gene regulation may be a consequence of evolutionary selection for high plasticity of flagellar motility. This way, all class III genes that are required for increased motility are under common regulatory control, which enables concerted evolutionary tuning of their levels.
Similarly unexpectedly, we observed that the main mechanism of the adaptive upregulation of class III genes relies on enhanced secretion of the anti-sigma factor FlgM rather than on other possible mechanisms, such as upregulation of the expression or activity of σ28. The role of FlgM secretion as a checkpoint control for the intermediate stage of flagella assembly in E. coli and other bacteria is well established. The observed recurrent utilization of this checkpoint in evolutionary enhancement of cell motility suggests that it also serves another, entirely different adaptive function, namely in enabling bacteria to efficiently adjust motility dependent on environmental selection. This adjustment occurs through modulation of FlgM secretion, and it can be mediated by mutations in a number of proteins that form flagellar secretion machinery or regulate secretion in other ways (Figure 7A). Consistent with its general function in evolutionary plasticity, the σ28/FlgM checkpoint remodeling appears to explain the tuning of motility not only under laboratory selection, but also in natural isolates of E. coli. Interestingly, although the exact mechanisms through which mutations in different motor proteins enhance secretion remain to be investigated, our analysis suggests that this enhancement is specific to FlgM as a substrate. Future studies of these mutations might thus provide further insights into specificity and evolution of the export machinery of type III secretion systems.
We believe that the observed adaptive mechanism may be indicative of a previously unrecognized general role of global checkpoint control mechanisms, and of bacterial sigma/anti-sigma factors in particular, in the evolutionary plasticity of gene expression. Most such global control networks possess a specific bow-tie structure, where multiple regulatory inputs control the activity (or levels) of a key factor that in turn regulates multiple downstream targets, e.g., expression of different genes. Previous adaptive explanations for such network structure suggested that it might enable independent evolution of the input and output functions without affecting the regulatory core (Csete and Doyle, 2004) and also compress cellular input information (Friedlander et al., 2015). Our results strongly suggest that such multi-input design also enhances evolutionary plasticity of the regulatory networks, by substantially increasing the number of possible mutations that affect activity of the common central process (the σ28/FlgM checkpoint in our case), and therefore adjust regulation of the downstream targets. Although it is difficult to assess the relative importance of selection for these individual properties, the enhanced evolvability is likely to be a major factor in the overall evolutionary enrichment of the bow-tie motifs in cellular networks.
Why did phenotype remodeling not occur at the level of class I gene expression, which is controlled by multiple transcriptional inputs (Figure 7C)? For one, the transcription factors involved are not unique to the flagellar regulon, meaning that changes in their activity are likely to affect other cellular functions. Moreover, instead of increasing flagellar length, the upregulation of the entire flagellar regulon might increase flagellar number, which was apparently less critical for the enhancement of motility in our experiments. We thus hypothesize that although previously reported activation of the flhDC operon by IS5 insertion (Barker et al., 2004, Wang and Wood, 2011) can naturally act as a binary ON-switch of motility, subsequent gradual tuning of motility under selection is more efficiently achieved through specific class III gene upregulation. Further supporting the adaptive advantage of such regulation, we observed that even mutations resulting in the FlgM-independent enhancement of motility led to higher expression of class III, but not of other flagellar genes.
Finally, we observed that selection for increased motility carries a cost of reduced growth fitness, meaning that evolution cannot simultaneously maximize both functions. Such conflict between optimization objectives is common in evolution (Garland, 2014, Roff and Fairbairn, 2007), and it might explain why many laboratory E. coli strains, as well as natural isolates, have attenuated motility. Interestingly, recent theoretical analysis has shown that for multiple conflicting optimization tasks, the existing phenotypes fall on a specific Pareto optimality front connecting both extremes of optimization, which in case of two tasks is a line (Shoval et al., 2012). In apparent agreement with this prediction, all strains in our analysis mapped roughly along the same line when growth fitness was plotted against swimming velocity. This was even true for strains that carry individual mutations, which is not trivial because of possible epistatic interactions between multiple mutations in evolved strains. We believe that such universal alignment points to remodeling of FlgM secretion as a general mechanism for optimal tuning of the motility-growth trade-off, so that any change in FlgM export would move the phenotype along the same Pareto optimization line.
In conclusion, experimental evolution demonstrated that bacteria can rapidly adjust such a complex trait as motility under selection pressure, following a defined trade-off line with conflicting optimization for growth. This high phenotypic plasticity appears to be inherent in the design of the hierarchical flagellar gene regulatory network, where genes modulating motility and chemotaxis are under the common control of a sigma/anti-sigma factor checkpoint that can be rapidly tuned under selection. Thus, besides its well-established function in the control of timing of flagellar gene expression, this checkpoint plays a key role in the evolutionary plasticity of E. coli motility. This latter function seems to be aided by its bow-tie architecture, and we propose that the similar design of other global regulatory networks might generally promote evolutionary adaptation.
Experimental Procedures
Strains and Plasmids
All strains used in this study were derived from E. coli K12 strain RP437 that is commonly used as a wild-type for chemotaxis and motility studies (Parkinson, 1978) and has constitutively activated motility because of IS5 insertion in the promoter region of flhDC operon (Barker et al., 2004). Details of strain and plasmid construction can be found in the Supplemental Experimental Procedures. Cells were grown at 30°C in TB medium [1% tryptone and 0.5% sodium chloride (NaCl)] supplemented with appropriate antibiotics (ampicillin: 100 μg⋅mL−1, kanamycin: 50 μg⋅mL−1).
Experimental Microevolution
Microevolution of E. coli strain RP437 was performed in 0.3% soft agar plates supplemented with TB. After an overnight incubation at 30°C, cells from the edge of the spreading ring were collected and re-inoculated onto a new soft agar plate. The same procedure was repeated 20–30 times, as indicated. For reverse evolution, evolved strains were repeatedly grown overnight in 3 mL liquid TB at 30°C.
Motility Analyses
For motility analysis, E. coli cells were grown in 10 mL TB medium at 34°C in a rotary shaker at 180 rpm. Cells (1 mL) at mid-log phase (OD600 [optical density at 600 nm] = 0.6) were collected and re-suspended in 1 mL tethering buffer (10 mM K2HPO4, 10 mM KH2PO4, 100 μM EDTA, 1 μM L-methionine, 10 mM lactic acid [pH 7.0]). Bacterial spreading was measured by inoculating 2 μL cell suspension at the top of 0.3% soft agar containing either TB or Minimal A medium supplemented with 0.1 mM aspartate or serine as indicated, after 7–8 hr incubation in TB or 20–22 hr incubation in Minimal A soft agar at 30°C. For measurements of chemotactic bias in soft agar, gradient of MeAsp was established in Minimal A soft agar containing glucose for 14 hr at 4°C before cell inoculation as described previously (Krembel et al., 2015). Swimming velocity and tumbling rate were measured by cell tracking in a glass chamber using phase-contrast microscopy (Nikon TI Eclipse, 10× objective, NA = 0.3, CMOS camera EoSens 4CXP). Drift velocity and chemotaxis efficiency in MeAsp gradient were tested in a microchamber made of poly-dimethylsiloxane (PDMS) using differential dynamic microscopy (DDM) (Wilson et al., 2011) and phase differential microscopy (φDM) (Colin et al., 2014). The cells were subjected to a linear gradient of MeAsp of 100 μM per 2 mm, and average concentration at the measurement segment was 50 μM. All data were analyzed using ImageJ (https://imagej.nih.gov/ij/) with custom-written plugins for swimming velocity, drift velocity, and tumbling rate analysis (Colin et al., 2014).
Genome and Transcriptome Analyses
E. coli cells were grown in TB as described above and harvested at mid-log phase (OD600 = 0.6). DNA samples were prepared using commercial kits (QIAGEN), and RNA samples were prepared using hot acid/phenol RNA extraction method (Maniatis et al., 1982). Genome sequencing was performed at GATC Biotech; RNA sequencing was performed at the BioQuant sequencing facility (University of Heidelberg). All sequencing data were analyzed using DNASTAR Lasergene software with E. coli strain MG1655 as reference template.
Promoter Activity Analysis
For promoter activity assays, E. coli strains transformed with reporter plasmids were grown in TB supplemented with kanamycin in 96-well plates at 30°C in a rotary shaker at 180 rpm. Cell fluorescence was measured using flow cytometry on BD LSRFortessa SORP cell analyzer (BD Biosciences).
Growth Fitness Analysis
For growth fitness measurements, equal amounts of a tested strain expressing CFP (pVS129) and the wild-type RP437 strain labeled with yellow fluorescent protein (YFP) (pVS132) were inoculated into 1 mL of TB medium supplemented with ampicillin and 20 μM isopropyl-β-D-thiogalactopyranosid (IPTG). After growth in a shaker at 30°C for 20 hr, 10 μL of cell suspensions was transferred into 1 mL PBS (pH 7.4), and the numbers of CFP- and YFP-labeled cells were measured by flow cytometry as described above. The culture was then reinoculated and the dynamics of the two cell populations was so monitored for 5 days.
Secretion Assay
Protein secretion was assayed using C-terminal fusions of a hemagglutinin (HA) tag to FlgM or to 175-amino-acid N-terminal fragment of FliC. The latter construct additionally contained a 71-bp 5′ untranslated region upstream of fliC. Cells were grown in TB as described above. FlgM-HA expression from a plasmid pBN4 was induced by 100 μM IPTG; FliC1-175-HA expression from a plasmid pBN5 was induced by 50 μM IPTG. Protein levels in the pellet and supernatant, respectively, were analyzed by immunoblotting as described in the Supplemental Experimental Procedures.
Quantification of Extracellular and Intracellular Flagellin
For measurements of FliC levels, density of cell culture was adjusted to OD600 = 1, and extracellular FliC filaments were sheared by Precellys homogenizer (Bertin Technologies) at 4,500 rpm for 20 s. Afterward, bacterial cells were collected by centrifugation (13,000 rpm for 1 min) at 4°C. Supernatants and pellets were collected separately for SDS-PAGE and immunoblotting. Anti-FliC antibody at a 1:10,000 dilution and secondary anti-rabbit IgG antibody labeled with peroxidase at a 1:5,000 dilution were used for detection.
Motor Rotation Assay
Motor rotation assay was conducted as described previously (Ryu et al., 2000) with minor modifications. Cells grown in TB medium were collected, re-suspended in tethering buffer, and flagella were sheared by 60 passages through 26 G syringe needle. Cells were allowed to settle on glass coverslips treated with poly-L-lysine (Sigma) and incubated for 5 min with a suspension of 1.1 μm polystyrene beads (Sigma). Unattached beads were washed away using tethering buffer supplemented with 20% TB. Rotation of beads was monitored using phase-contrast microscopy (40× magnification) at 1,000 Hz for 20 s. The beads were tracked using ImageJ with a custom-written plugin, and the traces were analyzed using MATLAB to extract the frequency of rotation.
Electron Microscopy
Carbon-coated copper grids (400 mesh) were hydrophilized by glow discharging (PELCO easiGlow; Ted Pella). Five microliters of a cell suspension (grown to OD600 = 0.5–0.6 as described above) was applied onto the hydrophilized grids and stained with 2% uranyl acetate after a short washing step with water. Samples were analyzed using a JEOL JEM-2100 transmission electron microscope with an acceleration voltage of either 80 or 200 kV. F214 FastScan CCD camera (TVIPS; Gauting) was used for image acquisition.
Statistical Analysis and Simulations
Details of statistical data analysis and computational simulations are described in the Supplemental Experimental Procedures.
Author Contributions
V.S., B.N., and F.S.P. designed experiments. B.N., F.S.P., R.C., and T.H. performed experiments. B.N., B.G., and R.C. analyzed data. B.G. performed computer simulations. V.S. and B.N. wrote the manuscript.
Acknowledgments
We thank Howard C. Berg, Ned S. Wingreen, Sean Murray, and Kelly Hughes for insightful discussions and comments on the manuscript; Lars Velten for help with experiments; and Karen A. Fahrner for providing pKAF131. This work was supported by grant 294761-MicRobE from the European Research Council and grant R01 GM082938 from the NIH.
Published: January 24, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.12.088.
Accession Numbers
The accession number for the original sequencing data reported in this paper is NIH SRA: SRP095233.
Supplemental Information
References
- Aldridge P., Hughes K.T. Regulation of flagellar assembly. Curr. Opin. Microbiol. 2002;5:160–165. doi: 10.1016/s1369-5274(02)00302-8. [DOI] [PubMed] [Google Scholar]
- Alon U., Surette M.G., Barkai N., Leibler S. Robustness in bacterial chemotaxis. Nature. 1999;397:168–171. doi: 10.1038/16483. [DOI] [PubMed] [Google Scholar]
- Auvray F., Thomas J., Fraser G.M., Hughes C. Flagellin polymerisation control by a cytosolic export chaperone. J. Mol. Biol. 2001;308:221–229. doi: 10.1006/jmbi.2001.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkai N., Leibler S. Robustness in simple biochemical networks. Nature. 1997;387:913–917. doi: 10.1038/43199. [DOI] [PubMed] [Google Scholar]
- Barker C.S., Prüss B.M., Matsumura P. Increased motility of Escherichia coli by insertion sequence element integration into the regulatory region of the flhD operon. J. Bacteriol. 2004;186:7529–7537. doi: 10.1128/JB.186.22.7529-7537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick J.E., Lenski R.E. Genome dynamics during experimental evolution. Nat. Rev. Genet. 2013;14:827–839. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg H.C., Turner L. Movement of microorganisms in viscous environments. Nature. 1979;278:349–351. doi: 10.1038/278349a0. [DOI] [PubMed] [Google Scholar]
- Blount Z.D., Barrick J.E., Davidson C.J., Lenski R.E. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature. 2012;489:513–518. doi: 10.1038/nature11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bren A., Eisenbach M. The N terminus of the flagellar switch protein, FliM, is the binding domain for the chemotactic response regulator, CheY. J. Mol. Biol. 1998;278:507–514. doi: 10.1006/jmbi.1998.1730. [DOI] [PubMed] [Google Scholar]
- Chait R., Craney A., Kishony R. Antibiotic interactions that select against resistance. Nature. 2007;446:668–671. doi: 10.1038/nature05685. [DOI] [PubMed] [Google Scholar]
- Chevance F.F.V., Hughes K.T. Coordinating assembly of a bacterial macromolecular machine. Nat. Rev. Microbiol. 2008;6:455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevance F.F., Karlinsey J.E., Wozniak C.E., Hughes K.T. A little gene with big effects: a serT mutant is defective in flgM gene translation. J. Bacteriol. 2006;188:297–304. doi: 10.1128/JB.188.1.297-304.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilcott G.S., Hughes K.T. Coupling of flagellar gene expression to flagellar assembly in Salmonella enterica serovar typhimurium and Escherichia coli. Microbiol. Mol. Biol. Rev. 2000;64:694–708. doi: 10.1128/mmbr.64.4.694-708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin R., Zhang R., Wilson L.G. Fast, high-throughput measurement of collective behaviour in a bacterial population. J. R. Soc. Interface. 2014;11:20140486. doi: 10.1098/rsif.2014.0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csete M., Doyle J. Bow ties, metabolism and disease. Trends Biotechnol. 2004;22:446–450. doi: 10.1016/j.tibtech.2004.07.007. [DOI] [PubMed] [Google Scholar]
- de Vos M.G.J., Poelwijk F.J., Tans S.J. Optimality in evolution: new insights from synthetic biology. Curr. Opin. Biotechnol. 2013;24:797–802. doi: 10.1016/j.copbio.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Elena S.F., Lenski R.E. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 2003;4:457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- Fahrner K.A., Berg H.C. Mutations that stimulate flhDC expression in Escherichia coli K-12. J. Bacteriol. 2015;197:3087–3096. doi: 10.1128/JB.00455-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F., Macnab R.M. Enzymatic characterization of FliI. An ATPase involved in flagellar assembly in Salmonella typhimurium. J. Biol. Chem. 1996;271:31981–31988. doi: 10.1074/jbc.271.50.31981. [DOI] [PubMed] [Google Scholar]
- Flamholz A., Noor E., Bar-Even A., Liebermeister W., Milo R. Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc. Natl. Acad. Sci. USA. 2013;110:10039–10044. doi: 10.1073/pnas.1215283110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander T., Mayo A.E., Tlusty T., Alon U. Evolution of bow-tie architectures in biology. PLoS Comput. Biol. 2015;11:e1004055. doi: 10.1371/journal.pcbi.1004055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland T., Jr. Trade-offs. Curr. Biol. 2014;24:R60–R61. doi: 10.1016/j.cub.2013.11.036. [DOI] [PubMed] [Google Scholar]
- González C., Ray J.C.J., Manhart M., Adams R.M., Nevozhay D., Morozov A.V., Balázsi G. Stress-response balance drives the evolution of a network module and its host genome. Mol. Syst. Biol. 2015;11:827. doi: 10.15252/msb.20156185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Pedrajo B., Minamino T., Kihara M., Namba K. Interactions between C ring proteins and export apparatus components: a possible mechanism for facilitating type III protein export. Mol. Microbiol. 2006;60:984–998. doi: 10.1111/j.1365-2958.2006.05149.x. [DOI] [PubMed] [Google Scholar]
- Hansen A.M., Qiu Y., Yeh N., Blattner F.R., Durfee T., Jin D.J. SspA is required for acid resistance in stationary phase by downregulation of H-NS in Escherichia coli. Mol. Microbiol. 2005;56:719–734. doi: 10.1111/j.1365-2958.2005.04567.x. [DOI] [PubMed] [Google Scholar]
- Hengge R. Proteolysis of sigmaS (RpoS) and the general stress response in Escherichia coli. Res. Microbiol. 2009;160:667–676. doi: 10.1016/j.resmic.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Hindré T., Knibbe C., Beslon G., Schneider D. New insights into bacterial adaptation through in vivo and in silico experimental evolution. Nat. Rev. Microbiol. 2012;10:352–365. doi: 10.1038/nrmicro2750. [DOI] [PubMed] [Google Scholar]
- Hughes K.T., Gillen K.L., Semon M.J., Karlinsey J.E. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science. 1993;262:1277–1280. doi: 10.1126/science.8235660. [DOI] [PubMed] [Google Scholar]
- Ibarra R.U., Edwards J.S., Palsson B.O. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature. 2002;420:186–189. doi: 10.1038/nature01149. [DOI] [PubMed] [Google Scholar]
- Kalir S., McClure J., Pabbaraju K., Southward C., Ronen M., Leibler S., Surette M.G., Alon U. Ordering genes in a flagella pathway by analysis of expression kinetics from living bacteria. Science. 2001;292:2080–2083. doi: 10.1126/science.1058758. [DOI] [PubMed] [Google Scholar]
- Kawecki T.J., Lenski R.E., Ebert D., Hollis B., Olivieri I., Whitlock M.C. Experimental evolution. Trends Ecol. Evol. 2012;27:547–560. doi: 10.1016/j.tree.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Kollmann M., Løvdok L., Bartholomé K., Timmer J., Sourjik V. Design principles of a bacterial signalling network. Nature. 2005;438:504–507. doi: 10.1038/nature04228. [DOI] [PubMed] [Google Scholar]
- Krembel A., Colin R., Sourjik V. Importance of multiple methylation sites in Escherichia coli chemotaxis. PLoS ONE. 2015;10:e0145582. doi: 10.1371/journal.pone.0145582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B. Genotype to phenotype: lessons from model organisms for human genetics. Nat. Rev. Genet. 2013;14:168–178. doi: 10.1038/nrg3404. [DOI] [PubMed] [Google Scholar]
- Løvdok L., Bentele K., Vladimirov N., Müller A., Pop F.S., Lebiedz D., Kollmann M., Sourjik V. Role of translational coupling in robustness of bacterial chemotaxis pathway. PLoS Biol. 2009;7:e1000171. doi: 10.1371/journal.pbio.1000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean R.C., Hall A.R., Perron G.G., Buckling A. The population genetics of antibiotic resistance: integrating molecular mechanisms and treatment contexts. Nat. Rev. Genet. 2010;11:405–414. doi: 10.1038/nrg2778. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E.F., Sambrook J. Cold Spring Harbor Laboratory; 1982. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- Minamino T., Imada K. The bacterial flagellar motor and its structural diversity. Trends Microbiol. 2015;23:267–274. doi: 10.1016/j.tim.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Morehouse K.A., Goodfellow I.G., Sockett R.E. A chimeric N-terminal Escherichia coli--C-terminal Rhodobacter sphaeroides FliG rotor protein supports bidirectional E. coli flagellar rotation and chemotaxis. J. Bacteriol. 2005;187:1695–1701. doi: 10.1128/JB.187.5.1695-1701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S., Kearns D.B. The structure and regulation of flagella in Bacillus subtilis. Annu. Rev. Genet. 2014;48:319–340. doi: 10.1146/annurev-genet-120213-092406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor E., Eden E., Milo R., Alon U. Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol. Cell. 2010;39:809–820. doi: 10.1016/j.molcel.2010.08.031. [DOI] [PubMed] [Google Scholar]
- Notebaart R.A., Szappanos B., Kintses B., Pál F., Györkei Á., Bogos B., Lázár V., Spohn R., Csörgő B., Wagner A. Network-level architecture and the evolutionary potential of underground metabolism. Proc. Natl. Acad. Sci. USA. 2014;111:11762–11767. doi: 10.1073/pnas.1406102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H., Selander R.K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 1984;157:690–693. doi: 10.1128/jb.157.2.690-693.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiuk O., Jakovljevic V., Vladimirov N., Carvalho R., Paster E., Ryu W.S., Meir Y., Wingreen N.S., Kollmann M., Sourjik V. Thermal robustness of signaling in bacterial chemotaxis. Cell. 2011;145:312–321. doi: 10.1016/j.cell.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Österberg S., del Peso-Santos T., Shingler V. Regulation of alternative sigma factor use. Annu. Rev. Microbiol. 2011;65:37–55. doi: 10.1146/annurev.micro.112408.134219. [DOI] [PubMed] [Google Scholar]
- Paget M.S. Bacterial sigma factors and anti-sigma factors: structure, function and distribution. Biomolecules. 2015;5:1245–1265. doi: 10.3390/biom5031245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J.S. Complementation analysis and deletion mapping of Escherichia coli mutants defective in chemotaxis. J. Bacteriol. 1978;135:45–53. doi: 10.1128/jb.135.1.45-53.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelwijk F.J., de Vos M.G.J., Tans S.J. Tradeoffs and optimality in the evolution of gene regulation. Cell. 2011;146:462–470. doi: 10.1016/j.cell.2011.06.035. [DOI] [PubMed] [Google Scholar]
- Roff D.A., Fairbairn D.J. The evolution of trade-offs: where are we? J. Evol. Biol. 2007;20:433–447. doi: 10.1111/j.1420-9101.2006.01255.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg S.M. Evolving responsively: adaptive mutation. Nat. Rev. Genet. 2001;2:504–515. doi: 10.1038/35080556. [DOI] [PubMed] [Google Scholar]
- Rudner D.Z., Losick R. Morphological coupling in development: lessons from prokaryotes. Dev. Cell. 2001;1:733–742. doi: 10.1016/s1534-5807(01)00094-6. [DOI] [PubMed] [Google Scholar]
- Ryu W.S., Berry R.M., Berg H.C. Torque-generating units of the flagellar motor of Escherichia coli have a high duty ratio. Nature. 2000;403:444–447. doi: 10.1038/35000233. [DOI] [PubMed] [Google Scholar]
- Schuetz R., Zamboni N., Zampieri M., Heinemann M., Sauer U. Multidimensional optimality of microbial metabolism. Science. 2012;336:601–604. doi: 10.1126/science.1216882. [DOI] [PubMed] [Google Scholar]
- Shoval O., Sheftel H., Shinar G., Hart Y., Ramote O., Mayo A., Dekel E., Kavanagh K., Alon U. Evolutionary trade-offs, Pareto optimality, and the geometry of phenotype space. Science. 2012;336:1157–1160. doi: 10.1126/science.1217405. [DOI] [PubMed] [Google Scholar]
- Sourjik V., Wingreen N.S. Responding to chemical gradients: bacterial chemotaxis. Curr. Opin. Cell Biol. 2012;24:262–268. doi: 10.1016/j.ceb.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taute K.M., Gude S., Nghe P., Tans S.J. Evolutionary constraints in variable environments, from proteins to networks. Trends Genet. 2014;30:192–198. doi: 10.1016/j.tig.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Toker A.S., Macnab R.M. Distinct regions of bacterial flagellar switch protein FliM interact with FliG, FliN and CheY. J. Mol. Biol. 1997;273:623–634. doi: 10.1006/jmbi.1997.1335. [DOI] [PubMed] [Google Scholar]
- Toprak E., Veres A., Michel J.B., Chait R., Hartl D.L., Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 2011;44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A., Sourjik V. Bacterial protein networks: properties and functions. Nat. Rev. Microbiol. 2015;13:559–572. doi: 10.1038/nrmicro3508. [DOI] [PubMed] [Google Scholar]
- Wang X., Wood T.K. IS5 inserts upstream of the master motility operon flhDC in a quasi-Lamarckian way. ISME J. 2011;5:1517–1525. doi: 10.1038/ismej.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich D.M., Delaney N.F., Depristo M.A., Hartl D.L. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- Wilson L.G., Martinez V.A., Schwarz-Linek J., Tailleur J., Bryant G., Pusey P.N., Poon W.C.K. Differential dynamic microscopy of bacterial motility. Phys. Rev. Lett. 2011;106:018101. doi: 10.1103/PhysRevLett.106.018101. [DOI] [PubMed] [Google Scholar]
- Wolfe A.J., Berg H.C. Migration of bacteria in semisolid agar. Proc. Natl. Acad. Sci. USA. 1989;86:6973–6977. doi: 10.1073/pnas.86.18.6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A., Park S.H., Lim W.A. Optimization of specificity in a cellular protein interaction network by negative selection. Nature. 2003;426:676–680. doi: 10.1038/nature02178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.