

Abstract
Background and Purpose
H2S protects myocardium against ischaemia/reperfusion injury. This protection may involve the cytosolic reperfusion injury salvage kinase (RISK) pathway, but direct effects on mitochondrial function are possible. Here, we investigated the potential cardioprotective effect of a mitochondria‐specific H2S donor, AP39, at reperfusion against ischaemia/reperfusion injury.
Experimental Approach
Anaesthetized rats underwent myocardial ischaemia (30 min)/reperfusion (120 min) with randomization to receive interventions before reperfusion: vehicle, AP39 (0.01, 0.1, 1 μmol·kg−1), or control compounds AP219 and ADT‐OH (1 μmol·kg−1). LY294002, L‐NAME or ODQ were used to investigate the involvement of the RISK pathway. Myocardial samples harvested 5 min after reperfusion were analysed for RISK protein phosphorylation and isolated cardiac mitochondria were used to examine the direct mitochondrial effects of AP39.
Key Results
AP39, dose‐dependently, reduced infarct size. Inhibition of either PI3K/Akt, eNOS or sGC did not affect this effect of AP39. Western blot analysis confirmed that AP39 did not induce phosphorylation of Akt, eNOS, GSK‐3β or ERK1/2. In isolated subsarcolemmal and interfibrillar mitochondria, AP39 significantly attenuated mitochondrial ROS generation without affecting respiratory complexes I or II. Furthermore, AP39 inhibited mitochondrial permeability transition pore (PTP) opening and co‐incubation of mitochondria with AP39 and cyclosporine A induced an additive inhibitory effect on the PTP.
Conclusion and Implications
AP39 protects against reperfusion injury independently of the cytosolic RISK pathway. This cardioprotective effect could be mediated by inhibiting PTP via a cyclophilin D‐independent mechanism. Thus, selective delivery of H2S to mitochondria may be therapeutically applicable for employing the cardioprotective utility of H2S.
Abbreviations
- AAR
area at risk
- ADT‐OH
5‐(4‐hydroxyphenyl)‐3H‐1, 2‐dithiole‐3‐thione
- AP219
mitochondria‐targeting moiety
- AP39
10‐oxo‐10‐(4‐(3‐thioxo‐3H‐1,2‐dithiol‐5yl)phenoxy)decyl) triphenylphosphonium bromide, mitochondria‐targeting H2S donor
- CsA
cyclosporine A
- eNOS
endothelial NOS
- GSK‐3β
glycogen synthase kinase‐3 β
- IFM
interfibrillar mitochondria
- Mito‐ROS
mitochondrial ROS
- PTP
permeability transition pore
- RISK
reperfusion injury salvage kinase (signalling pathway)
- RNS
reactive nitrogen species
- SSM
subsarcolemmal mitochondria
- TPP+
triphenylphosphonium
Tables of Links
| LIGANDS |
|---|
| LY294002 |
| L‐NAME |
| ODQ |
| Cyclosporine A |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
In myocardial ischaemia/reperfusion injury, rapid pH normalization, Ca2+ overload and overwhelming generation of ROS and reactive nitrogen species (RNS) at reperfusion disturb mitochondrial function and result in the opening of the mitochondrial permeability transition pore (PTP) (Hausenloy and Yellon, 2007). PTP opening leads to collapse of mitochondrial membrane potential, swelling of the mitochondria and the leakage of cytochrome c into the cytoplasm. As a result, ATP production will be impaired, initiating cell apoptosis/necrosis (Murphy et al., 2016; Pell et al., 2016). PTP opening at reperfusion is believed to be the no‐return point of reperfusion injury (Hausenloy et al., 2009). Therapeutic targeting of these processes during the first minutes of reperfusion has been investigated intensively in experimental settings as early reperfusion appears to offer a window of opportunity to prevent PTP opening and ultimately reduce lethal cell injury (Ferdinandy et al., 2014).
The roles of endogenous H2S, in a wide range of physiological systems, has been extensively explored following the discovery that it is produced by several regulated biochemical pathways in mammalian species (Kimura, 2011). In the myocardium, enhanced levels of H2S, whether by H2S supplement or increased endogenous production, have been shown to protect the heart against ischaemia/reperfusion injury (Johansen et al., 2006; Elrod et al., 2007; Karwi et al., 2016). The exact cardioprotective mechanism of H2S has yet to be clarified, but a number of molecular targets have been identified. These include activation of the reperfusion injury salvage kinase (RISK) pathway (Hausenloy, 2013), enhanced cellular and mitochondrial antioxidant defences, and preservation of mitochondrial integrity (Bos et al., 2015). However, these effects have been found to vary in many experimental studies for several reasons including variations in animal species and models, different experimental conditions and inconsistencies in dosing with inorganic sulfide salts (Bos et al., 2015). Inorganic sulfide salts (notably NaHS and Na2S) have been extensively employed to explore the biological activity of H2S. Nevertheless, these salts are impure and generate H2S instantaneously at high (i.e. supraphysiological) concentrations, and there is increasing concern that they are unreliable sources of H2S (Whiteman et al., 2011).
We have examined cardioprotection by a novel mitochondria‐targeting H2S donor, 10‐oxo‐10‐(4‐(3‐thioxo‐3H‐1,2‐dithiol‐5yl)phenoxy)decyl) triphenylphosphonium bromide (AP39) (Le Trionnaire et al., 2014) when given as adjunct to reperfusion and its direct effect on cardiomyocyte mitochondria, namely, subsarcolemmal (SSM) and interfibrillar mitochondria (IFM). The rationale for targeted delivery of H2S to the mitochondria is based on the evidence that H2S can attenuate mitochondrial ROS (mito‐ROS) generation and preserves mitochondrial integrity. There are recent observations that AP39 can successfully deliver H2S into the mitochondria when given at reperfusion and that it reproducibly protects the mitochondria in particular and the cell in general against ischaemia/reperfusion insults in the brain and kidney (Ikeda et al., 2015; Ahmad et al., 2016). We hypothesized that AP39 protects the heart against ischaemia/reperfusion injury when administrated at reperfusion though a cytosolic‐independent mechanism. We also hypothesized that AP39 attenuates mito‐ROS generation and thereby inhibits PTP opening in the SSM and IFM.
Methods
Animals and ethical statement
All animals care and procedures for in vivo studies complied with UK Home Office Guidelines on the Animals (Scientific Procedures) Act 1986, (published by the Stationery Office, London, UK), project licence (PPL30/3032) and was approved by the Animal Welfare and Ethical Review Body at Cardiff University. Studies involved mitochondria isolation was approved by the Animal Welfare Office of the Justus‐Liebig University Giessen. Male Sprague Dawley rats, 300–350 g (9–11 weeks), were obtained for in vivo studies from Harlan, UK. For mitochondria isolation, male Wistar rats, 300–350 g (9–11 weeks), were purchased from Harlan, France. They were housed in polypropylene cages (2–4 rats in each) on wood shavings. Animals acclimatized in the institutional animal house at constant temperature and humidity on a 12 h light/dark cycle for at least 7 days prior to experimentation, with free access to water and a small animal diet at all times. Animals studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilly, 2015).
Acute myocardial infarction model
Myocardial infarction was induced as previously reported (Karwi et al., 2016). Briefly, rats were anaesthetized using thiobutabarbital (Inactin® 200 mg·kg−1, i.p), and anaesthesia was maintained throughout the procedure by supplemental dosing (75 mg kg−1, i.v.) as required. The left jugular vein was cannulated for drug administration while the right common carotid artery was cannulated and connected to a pressure transducer (Powerlab data acquisition system, AD instruments, Abingdon, UK) to monitor the heart rate and the blood pressure throughout the experiment. The trachea was intubated and the animal ventilated with room air using a small animal ventilator (Hugo Sachs Elektronik, March, Germany) at a rate of 75 strokes min−1 and tidal volume of 1.0 to 1.25 mL·100 g−1. The chest was opened by midline sternotomy and the heart exposed using a retractor. The pericardium was incised and a 4/0 braided silk suture (Mersilk, Ethicon Ltd, UK) was placed around the left main coronary artery close to its origin to induce regional ischaemia. ECG was monitored using standard lead lI electrodes inserted s.c. into the limbs and connected to a Powerlab data acquisition system. Rectal temperature was maintained at 37 ± 1°C using a thermal blanket (Harvard Apparatus Ltd, Cambridge, UK). The following inclusion criteria were employed during the stabilization period of 20 min: no ECG or visual signs of ischaemia, steady sinus rhythm without arrhythmia, heart rate ≥250 beats min−1, and diastolic blood pressure ≥50 mmHg.
To induce regional ischaemia in the left ventricle, the left coronary artery was transiently occluded for 30 min by pulling the ligature taut through a plastic snare fixed against the epicardium. Ischaemia was confirmed by colour change of the left ventricle, a drop in the mean arterial pressure (MAP), ST‐segment elevation and arrhythmia developing between 5 and 13 min of ischaemia. The ligature was then released to start reperfusion for 120 min. Successful reperfusion was confirmed by blushing of the previously ischaemic area, reperfusion‐induced arrhythmia and increase in the MAP.
Infarct size determination
At the end of 120 min reperfusion, the heart was harvested and retrogradely perfused with saline through the aorta on a modified Langendorff apparatus. The ligature was re‐occluded and the heart perfused with 2% Evans' blue dye to delineate the ischaemic area at risk (AAR), then quickly frozen at −20°C for 24 h. The heart was transversely sectioned into 5–6 sections of 2 mm thickness and incubated with 1% w.v‐1TTC for 15 min. Sections were then fixed with 4% formalin in PBS for 24 h before being scanned. Sections were scanned using a digital scanner and coded using a random number generator (https://www.random.org), then planimetry was carried out in a blind fashion using the image analysis programme Image J (version 1.47, NIH, Bethesda, USA). The analysis determined the total ventricular area (Evans' blue positive), AAR (TTC positive) and the infarcted area (I, TTC negative), which were converted to volumes by multiplying these areas by 2 mm section thickness. Infarct size was expressed as a percentage of the AAR (% I/AAR).
Treatment protocols
The experimental protocols are summarized in Figure 1. Two series of experiments were carried out. The first series characterized the dose‐dependent infarct‐limiting effect of AP39 along with the control compounds (AP219 and ADT‐OH) to confirm the selective effect of H2S delivery into the mitochondria. The doses of AP39, AP219 and ADT‐OH used in these experiments were derived from in vitro and in vivo studies undertaken by others (Szczesny et al., 2014; Ikeda et al., 2015; Ahmad et al., 2016).
Figure 1.
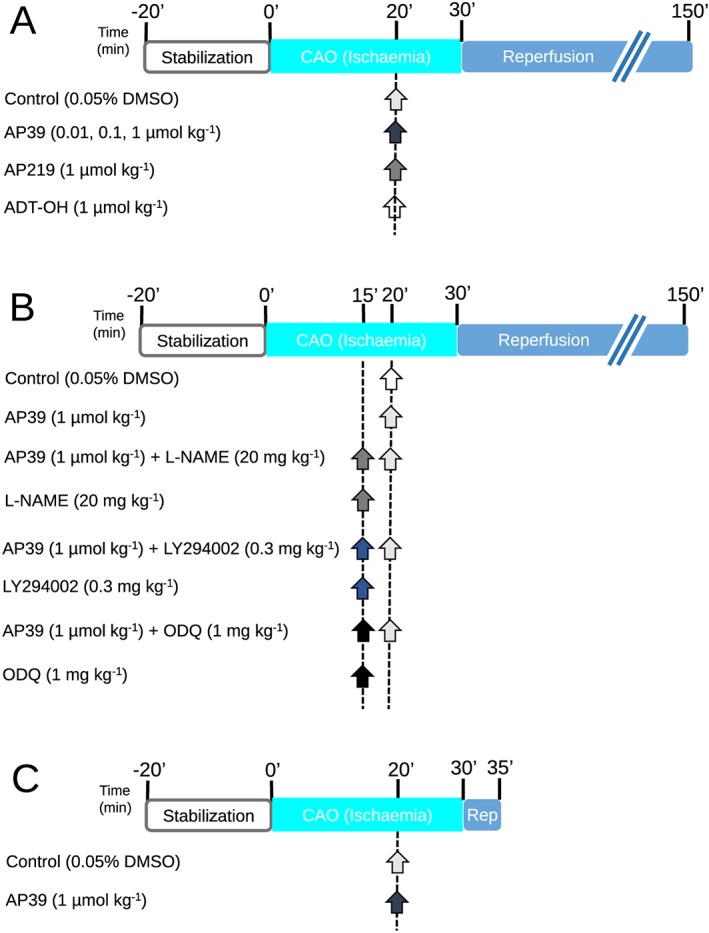
Experimental protocols: Animals underwent 30 min of ischaemia followed by 2 h of reperfusion. Infarct size was determined using Evans' blue/TTC staining technique. Infarction was reported as a percentage of the AAR (I/AAR %). (A) AP39 dose effect on infarct size: Animals were randomly assigned to be treated with either vehicle or AP39 or the controls (AP219 or ADT‐OH) at 10 min before reperfusion. (B) Mechanistic study: Rats were randomised to receive the pharmacological inhibitors, namely LY294002, L‐NAME and ODQ, at 15 min before reperfusion with or without AP39 applied at 10 min before reperfusion. Control group only received the vehicle (0.05% DMSO) 10 min before reperfusion. (C) Myocardium sampling protocol: Rats were randomised to receive either vehicle (0.05% DMSO) or AP39 10 minutes before reperfusion. Myocardial biopsies were harvested at 5 minutes of reperfusion from the left ventricle. Arrows indicate the time of the pharmacological interventions.
Animals were randomised to receive one of six interventions (Figure 1A):
Group 1: Control (n = 10). Animals received a bolus dose of (0.05% DMSO, i.v.) 10 min before reperfusion. DMSO was used as a vehicle for AP39, AP219 and ADT‐OH.
Groups 2–4: Each group (n = 8) received AP39 at (0.01, 0.1 or 1 μmol·kg−1 respectively) as an i.v. bolus 10 min before reperfusion.
Group 5: AP219 (n = 8). Animals received AP219 (1 μmol·kg−1) as an i.v. bolus 10 min before reperfusion.
Group 6: ADT‐OH (n = 8). Animals received ADT‐OH (1 μmol·kg−1) as an i.v. bolus 10 min before reperfusion.
The optimum dose of AP39 (1 μmol·kg−1), selected from the first series of experiments, was used in a second series of experiments that investigated the involvement of the RISK pathway components using inhibitors of Akt phosphorylation (LY294002), eNOS (L‐NAME) or sGC (ODQ). Animals were randomly assigned to one of the following eight treatment groups (Figure 1B).
Group 1: Control (n = 11). Animals received DMSO 0.05% given as an i.v. bolus 15 min before reperfusion. DMSO was used as vehicle for AP39, LY294002 and ODQ.
Group 2: AP39 (n = 8). Animals received AP39 (1 μmol·kg−1) as an i.v. bolus 10 min before reperfusion.
Group 3: AP39 + L‐NAME (n = 8). L‐NAME (20 mg·kg−1) was administered 15 min before reperfusion as an i.v. bolus followed by AP39 (1 μmol·kg−1) 10 min before reperfusion.
Group 4: L‐NAME (n = 8). L‐NAME (20 mg·kg−1) was administered 15 min before reperfusion as an i.v. bolus.
Group 5: AP39 + LY294002 (n = 8). LY294002 (0.3 mg·kg−1) was given 15 min before reperfusion as an i.v. bolus followed by AP39 (1 μmol·kg−1) 10 min before reperfusion.
Group 6: LY294002 (n = 8). LY294002 (0.3 mg·kg−1) was administered 15 min before reperfusion as an i.v. bolus.
Group 7: AP39 + ODQ (n = 8). ODQ (1 mg·kg−1) was given 15 min before reperfusion as an i.v. bolus followed by AP39 (1 μmol·kg−1) 10 min before reperfusion.
Group 8: ODQ (n = 8). ODQ (1 mg·kg−1) was administered 15 min before reperfusion as an i.v. bolus.
In a parallel series prepared for biochemical analysis of RISK pathway components, animals were randomised to receive either vehicle (0.05% DMSO) or AP39 (1 μmol·kg−1) 10 min before reperfusion (Figure 1C). The heart was excised after 5 min of reperfusion and washed with saline to remove any blood residue. Tissue samples were rapidly harvested from the left ventricle, snap frozen with liquid nitrogen then kept at −80°C. These samples were used to investigate the effect of AP39 on the phosphorylation of Akt, eNOS, GSK‐3β and ERK1/2 at the commencement of reperfusion using western blotting.
Isolation of cardiac mitochondria
Isolation of two mitochondrial subpopulations, subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM), was carried out using a modified protocol of that described by Boengler et al. (2009). All the procedures were undertaken at 4°C to maintain mitochondrial integrity. Each rat was anaesthetized with 4% v.v‐1 isoflurane, and the heart was quickly excised and washed with buffer A (100 mM KCl, 50 mM 3‐[N‐morpholino]‐propanesulfonic acid (MOPS), 5 mM MgSO4, 1 mM ATP and 1 mM EGTA, pH 7.4). The ventricles were isolated and weighed. Ventricles were transferred to buffer B (buffer A + 0.04% BSA), finely chopped with scissors then gently minced with six strokes of a teflon pestle in a glass tube. Homogenate was centrifuged at 800 g for 10 min. The supernatant from the first centrifugation was collected and centrifuged for 10 min at 8000 g to isolate the SSM. The sediment of the first centrifugation was resuspended for 1 min in buffer B with protease nargase (8 U·g−1) then gently minced with five strokes of the teflon pestle and glass mortar. The homogenate was centrifuged at 800 g for 10 min, then the supernatant was collected and centrifuged for 10 min at 8000 g to sediment the IFM. The SSM and IFM were washed with buffer A and final pellets were resuspended in buffer A with no ATP. Protein concentration was determined using Lowry assay (BioRad, Hercules, Canada).
Calcium retention capacity
Mitochondrial tolerance to calcium overload, a trigger to PTP opening, was investigated in the presence and absence of AP39 using a modified protocol of Chen et al. (2012). Freshly isolated SSM and IFM (0.1 mg·mL−1) were randomised to be incubated for 4 min in 2 mL (in mM: KCl 125, Tris‐MOPS 10, KH2PO4 1.2, MgCl2 1.2, glutamate 5, malate 2.5). The suspension was supplemented with 8 μL ADP (10 mM), 10 μL EGTA (1 mM), 6 μL CaCl2 (5 mM) and calcium green‐5 N (1 μM, Invitrogen, Carlsbad, Canada). Mitochondria were treated with vehicle (0.003% ethanol) or AP39 (1 μM) during the incubation period. Cyclosporine A (CsA) (1 μM) was used as a positive control as it is a well‐known inhibitor of the PTP opening and increases mitochondrial tolerance to Ca2+ overload by a cyclophilin D‐dependent mechanism. Pulses of Ca2+ (5 μM) were added at 3 min intervals to the solution with stirring at 25°C and mitochondrial calcium tolerance was expressed as μM of Ca2+ mg−1 of protein. Fluorescence was measured with excitation and emission wavelengths 530/530 nm, respectively. Data were coded using a random number generator (https://www.random.org) and blindly analysed.
Mitochondrial oxygen consumption
The respiration of SSM and IFM was measured using a Clark‐type oxygen electrode (Strathkelvin, Glasgow, UK) at 25°C. The concentrations range of AP39 used for the mitochondrial studies were equivalent to the in vivo doses and after assessing the direct effect of AP39 on the mitochondria autoflouresence and membrane potential (data not shown). Basal mitochondrial oxygen consumption was measured in the presence and absence of either the vehicle (0.003% ethanol) or AP39 (0.3, 1, 3, 5 μM). Mitochondria (0.1 mg·mL−1) were randomised to receive one of the treatments and were incubated in two chambers simultaneously, one with complex I substrate (5 mM glutamate and 2.5 mM malate) and the other with complex II substrate (5 mM succinate plus 2 μM of rotenone, to inhibit complex I activity). Respiration was stimulated by addition of 40 μM of ADP, and oxygen consumption was reported as nmol of O2·min‑1·mg‑1 of protein. Oxygraph charts were randomly coded (https://www.random.org) and blindly analysed.
Mito‐ROS generation
Measurement of mito‐ROS generation was carried out as previously descripted by Soetkamp et al. (2014). Freshly isolated SSM or IFM (50 μg) were suspended in incubation buffer (in mM: Tris‐MOPS 10, EGTA 0.02, KCl 125, glutamate 5, malate 2.5, Pi‐Tris 1.2, MgCl2 1.2, pH 7.4). Then 0.1 U·mL−1 HRP (Roche Diagnostic, Grenzach, Germany) and 50 μmol Amplex UltraRed (Invitogen, Eugene, OR) were added to the suspension directly before the measurements were performed. Cardiomyocyte mitochondria were randomly incubated with: (i) no intervention; (ii) vehicle (0.003% ethanol); and (iii) AP39 (0.3, 1, 3 and 5 μM). A second control group with no intervention was employed at the end of the all measurements to ensure that any observed effects are due to AP39 and not because of the decline in the respiratory capacity. SSM and IFM were also incubated with rotenone (2 μM) to induce overproduction of mito‐ROS generation and used as a positive control. Mito‐ROS generation was measured for 4 min at room temperature using Cary Eclipse spectrophotometer (Agilent technologies, Santa Clara, Canada) at excitation/emission wavelengths 565/581 nm. Using a code generator (https://www.random.org), data were coded and the slope of mito‐ROS generation was calculated, as a mean fluorescence per time (a.u.), by an operator blind to the treatments after subtracting the background fluorescence of the incubation buffer.
Western blot analysis
Myocardial samples were homogenized and lysed using a hard tissue lysing kit (Stretton Scientific Ltd, Stretton, UK).Then 30 μg of protein was loaded into each well of a 10% w.v‐1 SDS‐PAGE and separated electrophoretically at 120 mV. Separated proteins were transferred onto nitrocellulose membrane (Amersham, Germany), and the membrane was blocked for non‐specific binding with 5% skimmed milk for 2 h. The membrane was then probed with the primary antibody overnight at 4°C. The membrane was then incubated with secondary antibody (goat anti‐rabbit HRP, 1:15 000, Cell Signalling, UK) for 1 h at room temperature. Super Signal West Dura Extended Duration Substrate (Thermo Scientific, UK) was added on the surface of the membrane to laminate the bands and the bands were visualized on X‐ray film. Films were scanned and coded, and densitometry was carried out in blind fashion using Image J software (1.48v, National Institutes of Health USA). All protein bands were expressed as the relative density of myocardium sample, harvested after 20 min of stabilization (baseline), and then normalised for corresponding GAPDH bands, which served as an internal standard.
Antibodies
The following antibodies were used for Western blotting: Akt (1:1000), phospho‐ ser473Akt (1:1000), endothelial NOS (eNOS 1:500), phospho‐ ser1177eNOS (1:500), GSK‐3β (1:1000), phospho‐ ser9GSK‐3β (1:1000), ERK 1/2 (1:1000), phospho‐ Thr202/Tyr204 ERK 1/2 (1:1000) and GAPDH (1:50 000).
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data passed the Kolmogorov–Smirnov normality test of distribution. Statistical analysis was performed using GraphPad Prism® software (2007, Version 5.01, USA), and data are presented as mean ± SEM. Infarct size data were analysed using one‐way ANOVA with Newman–Keuls post hoc test, and Western blot analysis was performed using Student's unpaired t‐test. Haemodynamic and mitochondrial data were statistically analysed using repeated measures ANOVA supported by Bonferroni's post hoc test. P < 0.05 was considered statistically significant. Post tests were only carried out if P < 0.05 was achieved in the ANOVA.
Materials
AP39 and the control compounds, the mitochondria‐targeting moiety, AP219, and the H2S‐releasing moiety, ADT‐OH (5‐(4‐hydroxyphenyl)‐3H‐1, 2‐dithiole‐3‐thione), were synthesized by us as previously reported (Le Trionnaire et al., 2014; Szczesny et al., 2014; Tomasova et al., 2014). The purity of the compounds was determined by NMR spectroscopy (1H, 31P and 13C). The irreversible haem‐site soluble GC (sGC) inhibitor 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one (ODQ), the constitutive NOS inhibitor L‐nitroarginine methyl ester (L‐NAME), the PI3K inhibitor LY294002, thiobutabarbital sodium salt hydrate (Inactin® hydrate), Evans blue dye, triphenyltetrazolium chloride (TTC) and DMSO were all purchased from Sigma‐Aldrich, Gillingham, UK. Western blotting antibodies were all sourced from Cell Signalling, UK.
Results
For the AP39 dose–response study, 52 rats were used, of which two were excluded: one did not have successful reperfusion, and one rat did not survive ischaemia‐induced ventricular fibrillation. Therefore, data from 50 successfully completed experiments are presented. In the second series, 83 rats were used, of which, four were excluded: two did not complete the ischaemia–reperfusion protocol, one did not have successful TTC staining and one did not survive reperfusion‐induced arrhythmia. Thus, data from 79 rats, 67 infarct size experiments that were successfully completed and 12 tissue sampling experiments were reported. For mitochondria functional studies, data from 20 rats are reported.
Pharmacological postconditioning with AP39
Baseline parameters for infarct size studies are shown in (Table 1). There was no difference among the 14 experimental groups in any of the baseline parameters. Risk zone was similar among the experimental groups (50–60% of the total ventricular volume, Figure 2A). Administration of AP39 10 min before reperfusion resulted in a dose‐dependent infarct‐sparing effect compared with vehicle‐treated animals (Figure 2B). The maximum cardioprotection was seen at 1 μmol·kg−1 dose with almost 40% reduction in infarct size compared with vehicle‐treated animals. Postconditioning with AP39 (1 μmol·kg−1) also dose‐dependently increased in the post‐ischaemic functional recovery [% rate pressure product (RPP) recovery as a percentage of pre‐ischaemia RPP] measured at the end of reperfusion (67.2 ± 3.8%) compared with the control hearts (46.2 ± 3.8%, Table 1). The control compounds, namely AP219 and ADT‐OH, did not have a significant effect on either RPP recovery or infarct size, confirming that selective delivery of H2S to the mitochondria mediates AP39's cardioprotection.
Table 1.
Summary of the baseline parameters and haemodynamic data throughout the ischaemia–reperfusion protocol
| Experimental protocol | n | BW (g) | Baseline | 20 min ischaemia | 120 min reperfusion | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HR (beats. min‐1) | RPP (mmHg min−1*103) | MAP (mmHg) | HR (beats. min‐1) | RPP (mmHg min−1*103) | MAP (mmHg) | HR (beats. min‐1) | RPP (mmHg min−1*103) | MAP (mmHg) | |||
| Series 1 | |||||||||||
| Control (0.05% DMSO) | 10 | 342 ± 5 | 342 ± 10 | 36.2 ± 2.1 | 90 ± 6 | 311 ± 14 | 26.0 ± 1.9 | 70 ± 3 | 257 ± 9 | 16.7 ± 2.2 | 55 ± 5 |
| AP39 0.01 μmol·kg−1 | 8 | 347 ± 7 | 351 ± 8 | 38.4 ± 2.8 | 92 ± 4 | 302 ± 10 | 27.1 ± 2.4 | 70 ± 5 | 245 ± 12 | 17.7 ± 1.8 | 58 ± 3 |
| AP39 0.1 μmol·kg−1 | 8 | 339 ± 5 | 349 ± 12 | 40.0 ± 2.0 | 89 ± 6 | 299 ± 14 | 28.7 ± 1.2 | 68 ± 7 | 278 ± 8 | 20.0 ± 1.3 | 54 ± 5 |
| AP39 1 μmol·kg−1 | 8 | 355 ± 5 | 356 ± 9 | 38.5 ± 1.5 | 85 ± 5 | 303 ± 10 | 29.4 ± 2.3 | 67 ± 3 | 332 ± 10 | 25.8 ± 1.2* | 49 ± 4 |
| AP219 1 μmol·kg−1 | 8 | 356 ± 7 | 341 ± 10 | 37.8 ± 2.0 | 87 ± 7 | 315 ± 9 | 27.6 ± 1.6 | 71 ± 5 | 250 ± 9 | 16.2 ± 1.5 | 51 ± 6 |
| ADT‐OH 1 μmol·kg−1 | 8 | 342 ± 6 | 355 ± 12 | 39.4 ± 2.5 | 90 ± 4 | 309 ± 11 | 29.1 ± 2.3 | 68 ± 4 | 243 ± 11 | 17.5 ± 1.8 | 54 ± 5 |
| Series 2 | |||||||||||
| Control (0.05% DMSO) | 11 | 361 ± 5 | 352 ± 12 | 35.5 ± 2.0 | 87 ± 6 | 302 ± 12 | 26.1 ± 1.7 | 65 ± 4 | 255 ± 12 | 15.2 ± 1.8 | 53 ± 5 |
| AP39 1 μmol·kg−1 | 8 | 356 ± 7 | 346 ± 9 | 39.6 ± 3.1 | 92 ± 4 | 289 ± 10 | 31.1 ± 2.0 | 68 ± 5 | 320 ± 9 | 28.2 ± 1.5* | 54 ± 6 |
| AP39 1 μmol·kg−1 + L‐NAME 20 mg·kg−1 | 8 | 365 ± 6 | 345 ± 11 | 36.0 ± 1.9 | 90 ± 7 | 285 ± 15 | 29.5 ± 1.8 | 67 ± 8 | 314 ± 13 | 22.4 ± 2.4* | 53 ± 5 |
| L‐NAME 20 mg·kg−1 | 8 | 371 ± 9 | 343 ± 10 | 39.3 ± 1.6 | 89 ± 5 | 281 ± 12 | 28.6 ± 2.4 | 74 ± 5 | 230 ± 8 | 14.4 ± 2.1 | 56 ± 4 |
| AP39 1 μmol·kg−1 + LY294002 0.3 mg·kg−1 | 8 | 359 ± 10 | 350 ± 7 | 37.4 ± 1.5 | 86 ± 6 | 291 ± 10 | 32.0 ± 2.2 | 70 ± 6 | 306 ± 10 | 24.5 ± 1.6* | 52 ± 5 |
| LY294002 0.3 mg·kg−1 | 8 | 367 ± 9 | 351 ± 11 | 42.3 ± 2.5 | 91 ± 4 | 305 ± 14 | 30.2 ± 1.7 | 69 ± 6 | 235 ± 12 | 16.4 ± 2.0 | 60 ± 7 |
| AP39 1 μmol·kg−1 + ODQ 1 mg·kg−1 | 8 | 365 ± 7 | 350 ± 11 | 39.6 ± 1.5 | 93 ± 5 | 291 ± 11 | 31.5 ± 1.6 | 71 ± 4 | 329 ± 9 | 26.3 ± 1.9* | 55 ± 5 |
| ODQ 1 mg·kg−1 | 8 | 370 ± 8 | 342 ± 9 | 40.3 ± 2.5 | 88 ± 7 | 300 ± 10 | 28.6 ± 1.4 | 66 ± 5 | 228 ± 15 | 15.6 ± 2.0 | 63 ± 4 |
Animals body weight and haemodynamic parameters for infarct size studies at the end of stabilization period (baseline), after 20 min of ischaemia and at the end of reperfusion. BW, body weight; n, number of animals per group. There was no significant difference among experimental groups in the baselines or the application of the pharmacological inhibitors on the haemodynamics. Values expressed as mean ± SEM. (Two‐way ANOVA followed by Bonferroni post hoc test),
P < 0.05 versus control value at the same time point
Figure 2.
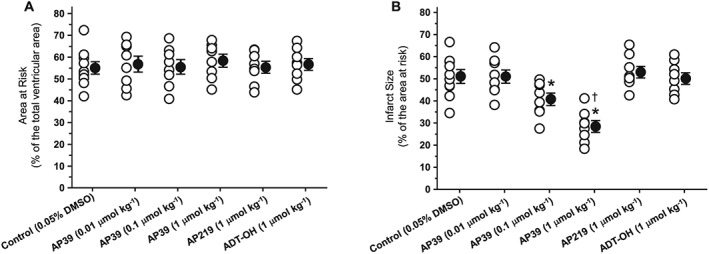
Infarct‐limiting effect of AP39 at reperfusion: (A) area at risk (AAR) reported as a percentage of the total ventricular volume. (B) Infarct size presented as a percentage of the AAR. Data were analysed using one‐way ANOVA with Neuman Keuls post hoc test and presented as mean ± SEM, n = 8 for all groups except the control group (0.05% DMSO) where n = 10. The mean of the infarct size for each group is represented by a filled circle (with error bars) next to the individual values (open circles). * P < 0.05 versus control, † P < 0.05 versus AP39 0.1 μmol·kg−1.
Cytosol‐independent mechanism of postconditioning with AP39
We next investigated the effect of AP39 on the RISK pathway as a relevant protective cytosolic‐signalling pathway using a ‘signal tracing’ technique. Specific pharmacological inhibitors, namely the PI3K inhibitor LY294002 (Jiang et al., 2007), constitutive NOS inhibitor L‐NAME (Fradorf et al., 2010) and sGC inhibitor ODQ (Routhu et al., 2010) were used at doses that have previously been reported to abrogate the activity of their targets in in vivo models.
There was no significant difference in either the baseline characteristics or the risk zone (ischaemic bed) among the groups (Figure 3A). None of the pharmacological inhibitors had a significant effect on infarct size when given alone 15 min before reperfusion compared with the control group (Figure 3B). Blockade of PI3K activity with LY294002 did not abolish the infarct limitation by AP39. Similarly, neither blockade of NO synthesis by L‐NAME nor selective inhibition of its downstream effector, sGC, with ODQ attenuated the protective effect of AP39.
Figure 3.
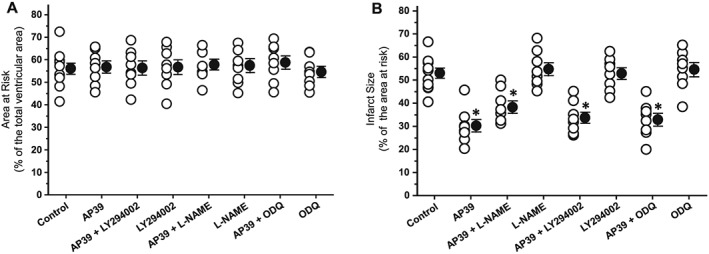
Effect of pharmacological inhibitors of the RISK pathway on infarct‐limitation by AP39: (A) risk zone measurements of experimental groups expressed as a percentage of the total ventricular area. (B) Myocardial infarction data are expressed as a percentage of the risk zone. Individual animal data in each group are represented by empty circles while the mean of infarct size is presented by a full circle. Data were analysed via one‐way ANOVA followed by Newman Keuls post hoc test and reported as mean ± SEM, n = 8 for all groups except the control group where n = 11. * P < 0.05 versus control.
The effect of AP39, used as an adjunct to reperfusion, on the key cytosolic components of the RISK pathway was also evaluated in samples harvested from the left ventricle after 5 min of reperfusion (Figure 4). Immunoblotting was carried out using phospho‐specific antibodies for Akt, eNOS, GSK‐3β and ERK1/2 to outline their role in the cardioprotection. In line with the infarct size data, Western blot analysis showed that administration of AP39 at reperfusion had no significant effect on the phosphorylation of either Akt, eNOS, GSK‐3β or ERK1/2. This confirms that AP39 mediated its cardioprotection independently of these cytosolic components on the RISK pathway.
Figure 4.
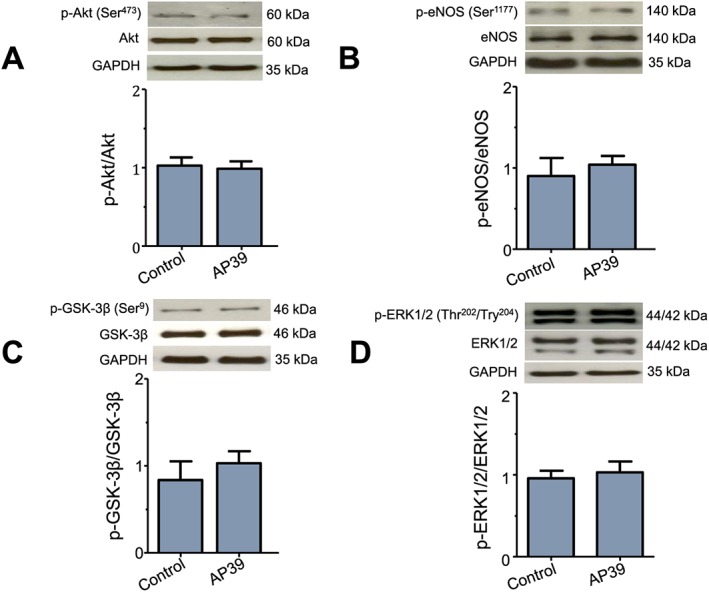
Effect of AP39 on RISK pathway proteins at early reperfusion: representative western blots and densitometry analysis of (A) pAktS473, total Akt and GAPDH; (B) p‐eNOSS1177, total eNOS and GAPDH; (C) p‐GSK‐3βS9, GSK‐3β and GAPDH; (D) p‐ERK1/2Thr202/Tyr204, ERK1/2 and GAPDH. Specific antibodies were used to assess the effect of AP39 on the phosphorylation of the RISK components in myocardial biopsies harvested from the left ventricle at early reperfusion. Histograms show the relative ratio of phosphorylated protein to the total level of protein. GAPDH was used as an internal standard for all quantifications. Data were analysed using Student's t‐test and presented as mean ± SEM, n = 6 per group.
Mitochondrial effects of AP39
We examined the effect of specific‐delivery of H2S into the mitochondria on the susceptibility to PTP opening. We used freshly isolated SSM and IFM and treated them with vehicle or AP39 (1 μM). SSM and IFM were exposed to pulses of Ca2+ in the presence and absence of CsA as a positive control (Figure 5). Untreated IFM showed 30% higher calcium tolerance than untreated SSM. AP39 elicited a significant inhibitory effect on the PTP opening in both SSM and IFM, which represents 30% increase in Ca2+ overload tolerance, compared with vehicle‐treated mitochondria. The inhibitory effect of AP39 on PTP opening was comparable with that observed after CsA in SSM and IFM. Interestingly, AP39 showed 25% additive effect to CsA‐induced inhibition of PTP opening when either SSM or IFM were incubated with both AP39 and CsA before the exposure to Ca2+ pulses, compared with CsA alone.
Figure 5.
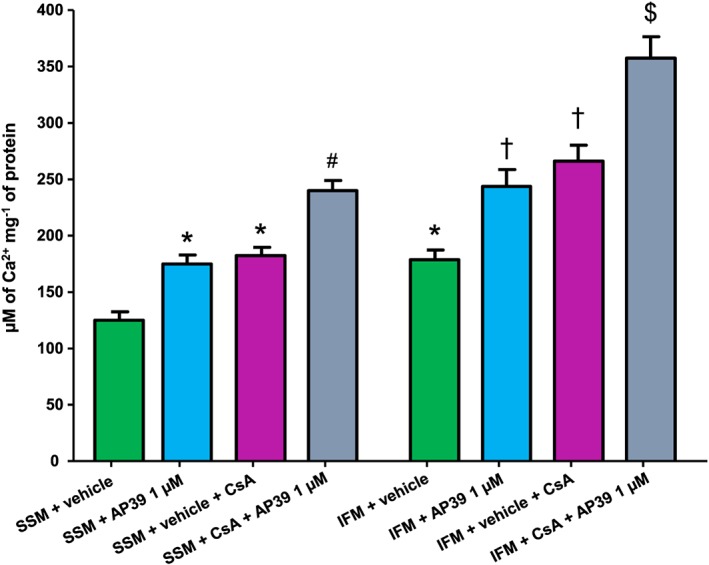
Effect of AP39 on mitochondrial PTP opening: SSM and IFM were incubated individually with vehicle (0.003% ethanol) or different concentrations of AP39 and subjected to pulses of 5 μM of CaCl2 per 3 min at 25°C until the opening of PTP in the presence and absence of CsA. Data expressed as mean ± SEM, n = 10, * P < 0.05 versus SSM + vehicle, # P < 0.05 versus SSM + vehicle + CsA, † P < 0.05 versus IFM + vehicle, $ P < 0.05 versus IFM + vehicle + CsA (two‐way ANOVA followed by Bonferroni post hoc test, n = 10).
Mitochondrial respiration was measured for both SSM and IFM using substrates for complex I (glutamate and malate) and complex II (succinate, in the presence of rotenone to inhibit complex I). There was no difference in the basal respiration of the two subpopulations of mitochondria. ADP‐stimulated respiration was higher in IFM (by 25% and 31% for complex I and II respectively) compared with SSM. Different concentrations of AP39 (0.3, 1, 3, 5 μM) were tested on SSM or IFM; however, none of the concentrations examined significantly influenced mitochondrial oxygen consumption.
Overwhelming mito‐ROS generation at early reperfusion is one of the main determinants of cellular injury. Therefore, we performed in vitro experiments to look at the direct effect of AP39 on H2O2 generation in the isolated rat left ventricle mitochondria (Figure 7A–D). In the control groups, ROS generation was significantly lower by 20% in IFM than SSM. AP39 showed a dose‐dependent inhibition of ROS generation in both SSM and IFM. AP39 (1 μM) exerted the maximum inhibitory effect (38% in SSM and 61% in IFM) compared with control, vehicle‐treated and the second control mitochondria. Interestingly, the inhibitory effect of AP39 on mito‐ROS generation was gradually reduced as the concentration was increased. Rotenone, as a positive control, resulted in overproduction of ROS in both SSM and IFM by 65% and 75%, respectively, compare the basal ROS generation level.
Discussion
The lipophilic triphenylphosphonium (TPP+) scaffold is an attractive moiety for investigating mitochondrial function as it selectively accumulates (100‐ to 500‐fold vs. cytosol) in the mitochondrial matrix (Murphy and Smith, 2007; Smith et al., 2011). Previous work (Prime et al., 2009) demonstrated infarct limitation using a mitochondria‐targeted NO donor (MitoSNO) with an NO‐releasing moiety linked to TPP+. Work by Krieg's group (Methner et al., 2013) showed that MitoSNO works independently of cytosolic protein kinase G that mediates the cardioprotective effect of non‐mitochondrial‐targeted NO donors. AP39 represents the first successful attempt to deliver H2S selectively and at low concentration to the mitochondria (Le Trionnaire et al., 2014; Szczesny et al., 2014; Tomasova et al., 2014; Ikeda et al., 2015; Ahmad et al., 2016).
In the present study, AP39 significantly limited infarct size (Figure 2B) and improved the post‐ischaemic functional recovery (Table 1), both in a dose‐dependent manner, when administered prior to reperfusion. Consistent with these data, Ahmad et al. (2016) reported that AP39 also exerted attenuation in renal damage, oxidative stress and renal inflammation when applied at reperfusion in an in vivo renal ischaemia/reperfusion injury model. We observed that the TPP+ scaffold molecule (AP219) and the H2S‐generating moiety (ADT‐OH), which were used as controls, had no effect on myocardial injury. This is in agreement with other reports where these controls lacked biologically activity when used at nanomolar or micromolar concentrations (Le Trionnaire et al., 2014; Szczesny et al., 2014; Ahmad et al., 2016).
At the time of finalizing this manuscript, recent work with AP39 by Papapetropoulos's group (Chatzianastasiou et al., 2016) has appeared. The main focus of Chatzianastasiou's work is ‘head‐to‐head’ comparison of infarct limitation by different H2S donors (Na2S, GYY4137, thiovaline and AP39) and elucidation of the role of NO in mediating protection. Intriguingly, all donors had the same infarct‐limiting effect in a mouse model of ischaemia/reperfusion injury. It is noteworthy that the optimum cardioprotective doses of GYY4137 and AP39 used were 26.6 and 0.25 μmol·kg−1 respectively. Very recently, we reported that 26.6 μmol·kg−1 GYY4137 was not cardioprotective in an in vivo rat model of ischaemia/reperfusion injury with the optimum cardioprotective dose being 10‐fold higher (Karwi et al., 2016). Similarly, in the present study, we demonstrated that AP39 exerts an infarct‐sparing effect with an optimum cardioprotective dose of 1 μmol·kg−1, fourfold higher than the effective dose in mouse reported by Chatzianastasiou et al. (2016). No haemodynamic data are available to compare AP39‐induced dose‐dependent improvement in post‐ischaemic functional recovery with Chatzianastasiou's paper.
Our present study provides important mechanistic insight into AP39's cardioprotective action in vivo. We found that selective blockade of PI3K, which is known to mediate the cardioprotective effect of non‐mitochondrial H2S donors (Andreadou et al., 2015; Karwi et al., 2016), did not abolish cardioprotection (Figure 3B). Crosstalk/interaction between H2S and NO has different patterns depending on organs/tissues, experimental conditions and species (King et al., 2014; Bibli et al., 2015; Karwi et al., 2016). To test if NO/sGC pathway mediates the effect of AP39, we blocked the endogenous NO synthesis pathway and also inhibited the activity of its end effector (sGC) using L‐NAME and ODQ respectively. We found that cardioprotection by AP39 was still observed in the presence of L‐NAME or ODQ, supported by analysis of protein phosphorylation during early reperfusion. We observed that AP39 did not induce phosphorylation of Akt, eNOS, GSK‐3β, a downstream effector of the RISK pathway, or ERK1/2, a parallel arm of the RISK pathway at 5 min reperfusion (Figure 4A–D). Chatzianastasiou et al. (2016) reported that AP39 did not phosphorylate either eNOS ser1176 or VASP ser239 after 10 min of reperfusion and its cardioprotection was not abolished by either L‐NAME or DT2, indicating a cGMP/PKG‐independent mechanism in the murine model. Their data are complementary to the fuller characterization of the potential cytosolic signalling targets of AP39 that we present here. Viewed together, our data and those of Chatzianastasiou et al. (2016) provide persuasive evidence that, unlike other H2S donors, AP39 mediates its cardioprotection by a mechanism that is independent of activation of the cytosolic components of the RISK signalling cascade.
Intra‐mitochondrial H2S is essential for normal function of the citric acid cycle. Levels are disturbed during oxidative stress due to increased H2S degradation and reduced production (Geng et al., 2004; Doeller et al., 2005; Whiteman et al., 2011; Vandiver and Snyder, 2012). H2S supplements or overexpression of endogenous synthetic enzymes have been shown to protect against ischaemia/reperfusion injury by mitigating oxidative stress and preserving mitochondrial integrity (Andreadou et al., 2015). Interestingly, Kai et al. (2012) found that NaHS‐induced protection was abolished in mitochondria‐free cells. Nevertheless, whether H2S directly interacts with mitochondria or triggers cytosolic signalling pathways that converge on the mitochondria may depend on the intracellular level of H2S. Therefore, we have characterized for the first time the direct effect of AP39 on the most relevant subpopulations of cardiomyocyte mitochondria, namely SSM and IFM. Inhibition of the PTP opening in the first minutes of reperfusion has been extensively reported to protect against reperfusion injury (Halestrap, 2010; Ong et al., 2014). It has been shown that many cardioprotective interventions act to maintain PTP in a closed state (Hausenloy et al., 2009). With this in mind, we investigated the influence of AP39 on the opening of PTP, as a result of Ca2+ overload, in cardiac SSM and IFM. We found that AP39 inhibited PTP opening in SSM and IFM (Figure 5) with no significant difference from the inhibitory effect of the positive control, CsA, which can protect myocardium against ischaemia/reperfusion injury (Hausenloy et al., 2012). We observed that AP39 and CsA in combination increased mitochondrial tolerance to Ca2+ overload and resulted in an additive effect compared with either compound alone. CsA prevents the opening of PTP by desensitizing cyclophilin‐D, a component of the multiprotein complex spanning the inner and outer mitochondrial membranes, which is a modulator of PTP located in the mitochondrial matrix (Bernardi and Di Lisa, 2015). Having an additive effect to CsA suggests that AP39 may inhibit PTP opening via a cyclophilin‐D independent mechanism. Chatzianastasiou et al. (2016) reported that AP39 (0.3 vs. 1 μM in our study), exerted an additive effect to CsA in mouse mitochondria isolated from the whole heart. However, isolating mitochondria from whole heart tissue is potentially problematic as a number of cell types contribute to the isolated mitochondrial fraction, for example endothelial cells, fibroblasts and other local resident cells. Even more important, cardiomyocyte mitochondria, namely SSM and IFM, themselves significantly differ in their main characteristics including oxygen consumption, mito‐ROS generation and calcium retention capacity (Palmer et al., 1977; Palmer et al., 1986). Our present study confirms for the first time the effect of AP39 in both IFM and SSM subpopulations.
It has been reported that H2S can stimulate mitochondrial ATP production by acting as an electron donor for the electron transport chain (Modis et al., 2014; Szabo et al., 2014). Accordingly, we explored the influence of AP39 on mitochondrial respiration through complexes I and II in both SSM and IFM. The respiration control ratio (RCR), an index for the coupling between mitochondrial respiration and oxidative phosphorylation, for isolated mitochondria fractions in this study was around 2.5. Although this result is comparable with our previously data (Boengler et al. 2009), others have reported higher RCR ratios (Chen et al., 2008; Asemu et al., 2013; Gao et al., 2013). This could be due to either measuring oxygen consumption at 30°C instead of 25°C, using trypsin instead of nargase to release the IFM or stimulating the mitochondria with higher concentration of ADP than what was used in this study. We did not detect any significant effect of AP39 on the oxygen consumption of these complexes in either mitochondrial subpopulation (Figure 6). These data suggest the safety margin of the applied concentration range. More importantly and in line with others, these results show that electron supply by H2S (at low concentration) to the electron transport chain occurs at the level of coenzyme Q where sulfide quinone reductase activity is involved. Following that, electrons will flow forward toward Complex III and Complex IV without affecting either Complexes I or II (Goubern et al., 2007; Szabo et al., 2014). It has been demonstrated that inhibition of Complex I using rotenone did not affect sulfide oxidation while inhibition of Complex III or VI by antimycin or cyanide, respectively, impeded it (Volkel & Grieshaber, 1996; Yong & Searcy, 2001; Goubern et al., 2007). Investigating the effect of AP39 on mitochondrial respiration at 37°C also needs further investigation in future work.
Figure 6.
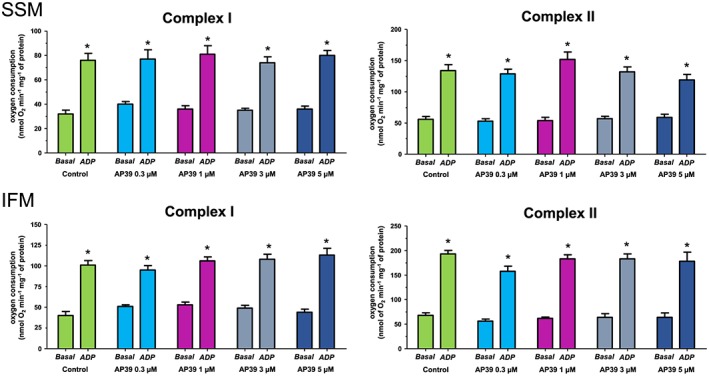
Effect of AP39 on mitochondrial respiration: Respiration of complexes I and II were measured at basal level and after ADP‐stimulation in the presence and absence of the vehicle or different concentrations of AP39 in SSM and IFM mitochondria. Data were analysed by two‐way ANOVA with Bonferroni post hoc test and reported as mean ± SEM, n = 10, * P < 0.05 versus basal respiration.
The detrimental effect of overwhelming mito‐ROS generation, as a result of respiratory chain uncoupling, is one of the hallmarks of ischaemia/reperfusion injury (Venditti et al., 2001; Brown and Griendling, 2015). It is a major contributor to the opening of PTP, initiating cell apoptosis and accelerated necrosis during reperfusion (Hausenloy et al., 2009). Since oxidative stress and the mitochondria play central roles in ischaemia/reperfusion injury, targeting the mitochondria with selective H2S donors is a plausible therapeutic approach to limit ischaemia/reperfusion injury. Here, we have investigated for the first time the influence of AP39 on mito‐ROS generation in both SSM and IFM. We found that AP39 limited mito‐ROS level in both subpopulations (Figure 7), which is in line with other studies of mito‐ROS reduction with AP39 (Le Trionnaire et al., 2014; Szczesny et al., 2014; Ikeda et al., 2015; Ahmad et al., 2016). We also observed an attenuation in AP39's activity as its concentration was increased, consistent with the findings of others (Szczesny et al., 2014; Ahmad et al., 2016).
Figure 7.
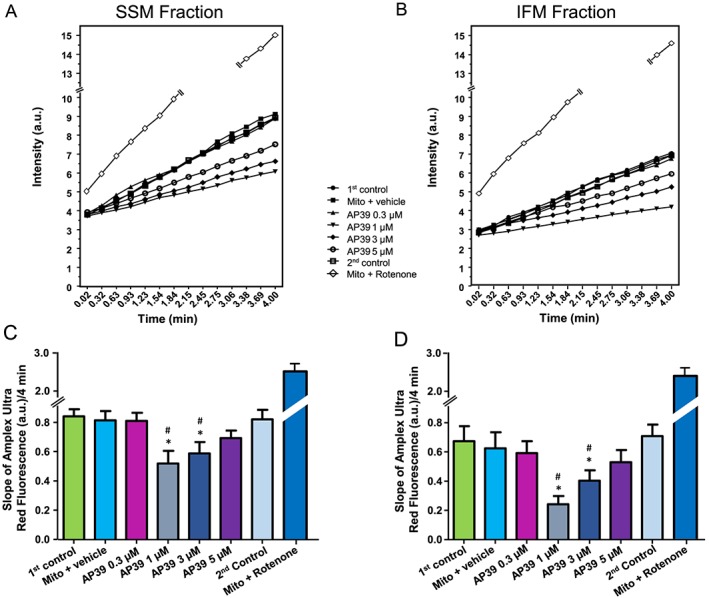
Effect of AP39 on mitochondrial‐ROS generation: Mitochondria were incubated with either vehicle (0.05% DMSO) or different concentrations of AP39. (A) and (B) are representative charts for the ROS generation of SSM and IFM, respectively, and error bars were removed for clarity. The slope of ROS generation was measured continuously for 4 min with the fluorescence indicator Amplex Ultrared both in (C) SSM and (D) IFM mitochondria. Data are expressed as mean ± SEM, n = 10, * P < 0.05 versus first control, # P < 0.05 versus second control (two‐way ANOVA with Bonferroni post hoc test, n = 10).
Although we have provided novel mechanistic insights into how AP39 could mediate cardioprotection, there are several caveats and current limitations. Assessing the extent of mitochondrial H2S level increase following AP39 application remains a challenge due to the lack of sensitive probes. Ikeda et al. (2015) reported that AP39 did not influence the expression of cystathionine γ lyase (CSE), cystathionine β synthase (CBS) or 3‐mercaptopyruvate sulphurtransferase (3‐MST) in the brain. This suggests that AP39 probably elevates mitochondrial H2S level without interfering with the endogenous synthesis of H2S. The focus of ongoing work is to investigate whether AP39 supresses oxidative stress by increasing GSH production or by up‐regulating mito‐ROS scavenging pathways or by directly scavenging ROS. The exact mechanism/target whereby AP39 inhibits PTP opening also remains to be determined. It may be interesting to identify what happens to these subpopulations in the animal treated with AP39. Both subpopulations play significant roles in mediating cardioprotection, although it is possible this includes persulfidation (also called S‐sulfhydration) of mitochondrial proteins such as ATP synthase (Modis et al., 2016; Wedmann et al., 2016). Very recent work by Murphy's group (Sun et al., 2016) also proposed that postconditioning with NaHS protected against myocardial infarction via an increase in S‐nitrosylation and most of the S‐nitrosylated proteins were mitochondrial proteins. This further emphasizes the physiological importance of post‐translation modifications of H2S and its interaction with NO in the mitochondria, a phenomenon that we are now seeking to characterize.
In conclusion, our results confirm that AP39 can protect the heart against myocardial infarction when given at reperfusion in a manner that is independent of classical cytosolic signalling mechanisms. We also report for the first time that AP39 inhibits mito‐ROS generation and PTP opening in both SSM and IFM, probably in a cyclophilin D‐independent manner without affecting mitochondrial respiration. These findings provide proof‐of‐concept that direct delivery of H2S to mitochondria by mitochondria‐targeting H2S donors, of which AP39 is a prototype of several compounds in this class under development, represents a novel and effective adjunctive intervention to mitigate the irreversible myocardial injury associated with reperfusion.
Author contributions
Q.G.K. and G.F.B. conceived and designed the studies and wrote the first draft of the manuscript. Q.K. performed all the in vivo studies, biochemical studies and mitochondrial studies. J.B., K.B. and R.S. designed the mitochondrial studies and assisted Q.K. with performing mitochondrial experiments and analysis of mitochondrial function data. M.E.W., R.T. and M.W. designed, synthesized and characterized AP39 and related compounds. All authors contributed to revision of the manuscript prior to finalization for submission by Q.K. and G.F.B.
Conflict of interest
M.W., M.E.W. and the University of Exeter have intellectual property (patent filings) on AP39, related compounds and their use.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommendation by funding agencies, publishers and other organizations engaged with supporting research.
Acknowledgements
Q.K. acknowledges the generous support of the Iraqi Ministry of Higher Education and Scientific Research. R.T. is the recipient of The Brian Ridge Scholarship. M.W. and M.E.W. would like to thank the Medical Research Council (UK) MR/M022706/1 for their generous support. Authors also thank Elvira Ungefug, Anna Reis, Sabrina Bohme and Christine Hirschhauser for their support.
Karwi, Q. G. , Bornbaum, J. , Boengler, K. , Torregrossa, R. , Whiteman, M. , Wood, M. E. , Schulz, R. , and Baxter, G. F. (2017) AP39, a mitochondria‐targeting hydrogen sulfide (H2S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. British Journal of Pharmacology, 174: 287–301. doi: 10.1111/bph.13688.
References
- Ahmad A, Olah G, Szczesny B, Wood ME, Whiteman M, Szabo C (2016). AP39, a mitochondrially targeted hydrogen sulfide donor, exerts protective effects in renal epithelial cells subjected to oxidative stress in vitro and in acute renal injury in vivo. Shock 45: 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadou I, Iliodromitis EK, Rassaf T, Schulz R, Papapetropoulos A, Ferdinandy P (2015). The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol 172: 1587–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asemu G, O'Connell KA, Cox JW, Dabkowski ER, Xu W, Ribeiro RF Jr et al. (2013). Enhanced resistance to permeability transition in interfibrillar cardiac mitochondria in dogs: effects of aging and long‐term aldosterone infusion. Am J Physiol Heart Circ Physiol 304: H514–H528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Di Lisa F (2015). The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78: 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibli SI, Andreadou I, Chatzianastasiou A, Tzimas C, Sanoudou D, Kranias E et al. (2015). Cardioprotection by H2S engages a cGMP‐dependent protein kinase G/phospholamban pathway. Cardiovasc Res 106: 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz‐Meana M, Garcia‐Dorado D et al. (2009). Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104: 141–147. [DOI] [PubMed] [Google Scholar]
- Bos EM, van Goor H, Joles JA, Whiteman M, Leuvenink HG (2015). Hydrogen sulfide: physiological properties and therapeutic potential in ischaemia. Br J Pharmacol 172: 1479–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DI, Griendling KK (2015). Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzianastasiou A, Bibli SI, Andreadou I, Efentakis P, Kaludercic N, Wood ME et al. (2016). Cardioprotection by H2S donors: nitric oxide‐dependent and ‐independent mechanisms. J Pharmacol Exp Ther . doi:10.1124/jpet.116.235119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ (2008). Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294: C460–C466. [DOI] [PubMed] [Google Scholar]
- Chen Q, Paillard M, Gomez L, Li H, Hu Y, Lesnefsky EJ (2012). Postconditioning modulates ischemia‐damaged mitochondria during reperfusion. J Cardiovasc Pharmacol 59: 101–108. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP et al. (2005). Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal Biochem 341: 40–51. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L et al. (2007). Hydrogen sulfide attenuates myocardial ischemia–reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A 104: 15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R (2014). Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev 66: 1142–1174. [DOI] [PubMed] [Google Scholar]
- Fradorf J, Huhn R, Weber NC, Ebel D, Wingert N, Preckel B et al. (2010). Sevoflurane‐induced preconditioning: impact of protocol and aprotinin administration on infarct size and endothelial nitric‐oxide synthase phosphorylation in the rat heart in vivo. Anesthesiology 113: 1289–1298. [DOI] [PubMed] [Google Scholar]
- Gao XH, Qanungo S, Pai HV, Starke DW, Steller KM, Fujioka H et al. (2013). Aging‐dependent changes in rat heart mitochondrial glutaredoxins‐‐Implications for redox regulation. Redox Biol 1: 586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y et al. (2004). Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem Biophys Res Commun 318: 756–763. [DOI] [PubMed] [Google Scholar]
- Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F (2007). Sulfide, the first inorganic substrate for human cells. FASEB J 21: 1699–1706. [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2010). A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans 38: 841–860. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ (2013). Cardioprotection techniques: preconditioning, postconditioning and remote conditioning (basic science). Curr Pharm Des 19: 4544–4563. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Boston‐Griffiths EA, Yellon DM (2012). Cyclosporin A and cardioprotection: from investigative tool to therapeutic agent. Br J Pharmacol 165: 1235–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Ong SB, Yellon DM (2009). The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 104: 189–202. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM (2007). The evolving story of “conditioning” to protect against acute myocardial ischaemia‐reperfusion injury. Heart 93: 649–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Marutani E, Hirai S, Wood ME, Whiteman M, Ichinose F (2015). Mitochondria‐targeted hydrogen sulfide donor AP39 improves neurological outcomes after cardiac arrest in mice. Nitric Oxide 49: 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Zatta A, Kin H, Wang N, Reeves JG, Mykytenko J et al. (2007). PAR‐2 activation at the time of reperfusion salvages myocardium via an ERK1/2 pathway in in vivo rat hearts. Am J Physiol Heart Circ Physiol 293: H2845–H2852. [DOI] [PubMed] [Google Scholar]
- Johansen D, Ytrehus K, Baxter GF (2006). Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia–reperfusion injury‐‐Evidence for a role of K ATP channels. Basic Res Cardiol 101: 53–60. [DOI] [PubMed] [Google Scholar]
- Kai S, Tanaka T, Daijo H, Harada H, Kishimoto S, Suzuki K et al. (2012). Hydrogen sulfide inhibits hypoxia‐ but not anoxia‐induced hypoxia‐inducible factor 1 activation in a von hippel–lindau‐ and mitochondria‐dependent manner. Antioxid Redox Signal 16: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwi QG, Whiteman M, Wood ME, Torregrossa R, Baxter GF (2016). Pharmacological postconditioning against myocardial infarction with a slow‐releasing hydrogen sulfide donor, GYY4137. Pharmacol Res 111: 442–451. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H (2011). Hydrogen sulfide: its production and functions. Exp Physiol 96: 833–835. [DOI] [PubMed] [Google Scholar]
- King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK et al. (2014). Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase‐nitric oxide dependent. Proc Natl Acad Sci U S A 111: 3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Trionnaire S, Perry A, Szczesny B, Szabo C, Winyard PG, Whatmore JL et al. (2014). The synthesis and functional evaluation of a mitochondria‐targeted hydrogen sulfide donor, (10‐oxo‐10‐(4‐(3‐thioxo‐3H‐1,2‐dithiol‐5‐yl)phenoxy)decyl)triphenylphosphonium bromide (AP39). Med Chem Comm 5: 728–736. [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methner C, Lukowski R, Grube K, Loga F, Smith RA, Murphy MP et al. (2013). Protection through postconditioning or a mitochondria‐targeted S‐nitrosothiol is unaffected by cardiomyocyte‐selective ablation of protein kinase G. Basic Res Cardiol 108: 337–343. [DOI] [PubMed] [Google Scholar]
- Modis K, Bos EM, Calzia E, van Goor H, Coletta C, Papapetropoulos A et al. (2014). Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br J Pharmacol 171: 2123–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis K, Ju Y, Ahmad A, Untereiner AA, Altaany Z, Wu L et al. (2016). S‐sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol Res 113: 116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN et al. (2016). Mitochondrial function, biology, and role in disease: a scientific statement from the american heart association. Circ Res 118: 1960–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Smith RA (2007). Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 47: 629–656. [DOI] [PubMed] [Google Scholar]
- Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ (2014). The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol 78: 23–34. [DOI] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252: 8731–8739. [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL (1986). Heterogeneous response of subsarcolemmal heart mitochondria to calcium. Am J Physiol 250: H741–H748. [DOI] [PubMed] [Google Scholar]
- Pell VR, Chouchani ET, Frezza C, Murphy MP, Krieg T (2016). Succinate metabolism: a new therapeutic target for myocardial reperfusion injury. Cardiovasc Res 111: 134–141. [DOI] [PubMed] [Google Scholar]
- Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC et al. (2009). A mitochondria‐targeted S‐nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia–reperfusion injury. Proc Natl Acad Sci U S A 106: 10764–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routhu KV, Tsopanoglou NE, Strande JL (2010). Parstatin (1‐26): the putative signal peptide of protease‐activated receptor 1 confers potent protection from myocardial ischemia–reperfusion injury. J Pharmacol Exp Ther 332: 898–905. [DOI] [PubMed] [Google Scholar]
- Smith RA, Hartley RC, Murphy MP (2011). Mitochondria‐targeted small molecule therapeutics and probes. Antioxid Redox Signal 15: 3021–3038. [DOI] [PubMed] [Google Scholar]
- Soetkamp D, Nguyen TT, Menazza S, Hirschhauser C, Hendgen‐Cotta UB, Rassaf T et al. (2014). S‐nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res Cardiol 109: 433–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Aponte AM, Menazza S, Gucek M, Steenbergen C, Murphy E (2016). Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S‐sulfhydration vs. S‐nitrosylation. Cardiovasc Res 110: 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Ransy C, Modis K, Andriamihaja M, Murghes B, Coletta C et al. (2014). Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol 171: 2099–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Modis K, Yanagi K, Coletta C, Le Trionnaire S, Perry A et al. (2014). AP39, a novel mitochondria‐targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 41: 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasova L, Pavlovicova M, Malekova L, Misak A, Kristek F, Grman M et al. (2014). Effects of AP39, a novel triphenylphosphonium derivatised anethole dithiolethione hydrogen sulfide donor, on rat haemodynamic parameters and chloride and calcium Ca3 and RyR2 channels. Nitric Oxide 46: 131–144. [DOI] [PubMed] [Google Scholar]
- Vandiver M, Snyder SH (2012). Hydrogen sulfide: a gasotransmitter of clinical relevance. J Mol Med 90: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditti P, Masullo P, Di Meo S (2001). Effects of myocardial ischemia and reperfusion on mitochondrial function and susceptibility to oxidative stress. Cell Mol Life Sci 58: 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkel S, Grieshaber MK (1996). Mitochondrial sulfide oxidation in Arenicola marina. Evidence for alternative electron pathways. Eur J Biochem 235: 231–237. [DOI] [PubMed] [Google Scholar]
- Wedmann R, Onderka C, Wei S, Szijártó IA, Miljkovic JL, Mitrovic A et al. (2016). Improved tag‐switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem Sci 7: 3414–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Le Trionnaire S, Chopra M, Fox B, Whatmore J (2011). Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci 121: 459–488. [DOI] [PubMed] [Google Scholar]
- Yong R, Searcy DG (2001). Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp Biochem Physiol B Biochem Mol Biol 129: 129–137. [DOI] [PubMed] [Google Scholar]