

Abstract
Background and Purpose
The persistence of lung cancer stem cells (LCSCs) has been proposed to be the main factor responsible for the recurrence of lung cancer as they are highly resistant to conventional chemotherapy. However, the underlying mechanisms are still unclear.
Experimental Approach
We examined the cellular response of a human LCSC line to treatment with cisplatin, a DNA‐damaging anticancer drug that is used extensively in the clinic. We compared the response to cisplatin of LCSCs and differentiated LCSCs (dLCSCs) by determining the viability of these cells, and their ability to accumulate cisplatin and to implement genomic and transcription‐coupled DNA repair. We also investigated the transcription profiles of genes related to drug transport and DNA repair.
Key Results
LCSCs were found to be more stem‐like, and more resistant to cisplatin‐induced cytotoxicity than dLCSCs, confirming their drug resistance properties. LCSCs accumulated less cisplatin intracellularly than dLCSCs and showed less DNA damage, potentially due to their ability to down‐regulate AQP2 and CTR1. The results of the transcription‐coupled repair of cisplatin‐DNA cross‐links indicated a higher level of repair of DNA damage in LCSCs than in dLCSCs. In addition, LCSCs showed a greater ability to repair cisplatin‐DNA interstrand cross‐links than dLCSCs; this involved the activation of various DNA repair pathways.
Conclusions and Implications
Our results further clarify the mechanism of cisplatin resistance in LCSCs in terms of reduced cisplatin uptake and enhanced ability to implement DNA repairs. These findings may aid in the design of the next‐generation of platinum‐based anticancer drugs.
Abbreviations
- dLCSCs
differentiated LCSCs
- ICLs
interstrand cross‐links
- LCSCs
lung cancer stem cells
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Transporters c |
| Notch1 | ATP7B |
| RGS10 | CTR1 |
| Other ion channels b | CTR2 |
| AQP2 | |
| AQP9 |
| LIGANDS |
|---|
| Cisplatin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Lung cancer is estimated to account for the highest number of deaths among all the various types of cancer, and about 224 390 new cases are expected in the United States in 2016 (American Cancer Society, 2016). Lung cancer is known to have a high recurrence rate, since lung cancer cells cannot be completely eliminated by conventional chemotherapeutics due to the development of drug resistance (Chen et al., 2013). A cancer stem cell (CSC) model has been proposed to explain the high rate of recurrence of lung cancer (Bonnet and Dick, 1997). CSCs are known to exist as important subpopulations in tumours and contribute to various functional characteristics such as tumourigenesis and heterogeneity (Singh et al., 2004). Recently, the lung CSC (LCSC) model has been further investigated and a rare population of tumour‐initiating cells was identified and shown to contribute to the self‐renewal and replacement of various heterogeneous cancer cell populations within a tumour in a hierarchical manner (Kubo et al., 2013). LCSCs are the potential cause of relapse because of their high expression of stemness‐related genes, their invasiveness, drug resistance and ability to proliferate, and tumorigenicity (Bonnet and Dick, 1997; Al‐Hajj et al., 2003).
Chemotherapy is still one of the standard treatment regimens for lung cancers and involves the use of different chemotherapeutic agents. Among them, cisplatin [cis‐diamminedichloroplatinum(II)] is a first‐line chemotherapeutic DNA‐damaging drug used to treat lung cancer, which is commonly administered together with other non‐platinum‐based chemotherapeutic drugs to increase the survival rates of lung cancer patients (van Moorsel et al., 1999). The mechanism of action of cisplatin has been extensively investigated (Wang and Lippard, 2005). It is now widely agreed that DNA is the major target of cisplatin. Cisplatin forms adducts of mainly intrastrand platinum (Pt)‐DNA cross‐links, with a minor proportion of interstrand cross‐links (ICLs) (Fichtinger‐Schepman et al., 1985). Pt‐DNA cross‐links induce apoptosis by inhibiting DNA replication and transcription. Complex mechanisms are involved in cisplatin resistance. For example, reduced cellular uptake and elevated efflux are the potential causes of cisplatin resistance (Wang and Lippard, 2005). The resistance can also be attributed to activation of different DNA repair pathways including nucleotide excision repair (NER), based excision repair (BER), mismatch repair (MMR) and single‐strand break repair (SSBR), which are responsible for the removal of Pt‐DNA cross‐links (Graf et al., 2011; Enoiu et al., 2012). Cisplatin resistance has been correlated well with an increased ability to induce DNA repairs (Bartucci et al., 2012), which makes complete elimination of a tumour more difficult.
Although the mode of action and biological properties of cisplatin have been widely described, our knowledge of cisplatin's action in LCSCs is still vague. In addition, how these LCSCs generate drug resistance is still debatable. Cisplatin resistance has been correlated with the stem‐like characteristics of some lung cancer cells (Barr et al., 2013). Recently, cisplatin‐resistant lung cancer cells were generated and these cells were shown to exhibit stemness characterized by the expression of CSC markers, CD133 and CD44, and by an elevated aldehyde dehydrogenase (ALDH) activity (Barr et al., 2013). Another study also demonstrated that a lung tumour cell subpopulation with high ALDH and CD44 co‐expression displayed increased cisplatin resistance and other tumourigenic properties (Liu et al., 2013a). Cisplatin enhances the CD133+ population that exhibits CSC‐like characteristics in lung cancer cells (Bertolini et al., 2009; Liu et al., 2013b). These results indicate that stemness and cisplatin resistance are highly correlated in lung cancer. While some studies claim that the reduced accumulation of cisplatin is the major cause of platinum resistance (Ishida et al., 2002; Safaei and Howell, 2005; Hall et al., 2008), Eljack and colleagues showed that passive diffusion rather than reduced cellular uptake of cisplatin is an important mechanism for drug resistance (Eljack et al., 2014). However, the details of how these stem‐like cancer cells mediate cisplatin resistance are not fully understood.
In the present study, we used a human LCSC line as the model system to study the mechanism of cisplatin resistance and its correlation with cell stemness. We first measured the viability of LCSCs and differentiated LCSCs (dLCSCs) after cisplatin treatment. We next scrutinized the detailed mechanism of cisplatin resistance in LCSCs. The accumulation of platinum in cells as well as the level of platinum on genomic DNA was measured. DNA repair ability in the two cell lines was studied using a transcription‐coupled DNA repair assay. We further investigated the molecular mechanisms of LCSC's increased drug resistance by measuring the transcription profiles of genes related to drug transportation and DNA damage repair. Our study provides further evidence linking the drug resistance of LCSCs to both reduced cellular accumulation of cisplatin and their unique ability to implement DNA repairs of damage induced by this drug.
Methods
Cell culture and characterization of human lung cancer cell lines
LCSCs were purchased from Celprogen (USA) and cultured in human parent LCSC media with serum‐free supplement. They are known to express a list of biomarkers, such as CD15 and CD20 (Detail information: http://www.celprogen.com/details.php?pid=6423). This cell line was briefly characterized as described in a previous report (Zou et al., 2015). Third generation LCSCs (P3) were used in this study, and dLCSCs originated from LCSCs after 12 passages (P13) in the culture with an obvious change in cell morphology. dLCSCs were cultured in DMEM/F‐12 medium with 10% FBS and 1% of the antibiotics, penicillin (100 U·mL−1) and streptomycin (100 g·mL−1). The changes in expression of stemness‐related genes in both LCSCs and dLCSCs were characterized by qRT‐PCR. All cells were grown at 37°C in a 5% CO2 incubator.
Immunostaining and FACS analysis
LCSCs and dLCSCs were cultured on 55 cm2 culture dishes. After reaching 90% confluency, cells were detached, washed with PBS and fixed in 4% (v.v‐1) paraformaldehyde for 15 min. Cells were then permeabilized in blocking solution containing 0.5% Triton X100, 1% FBS in PBS. Fluorescein‐conjugated Oct‐3/4 antibody was subsequently added to stain nuclear Oct4 in blocking solution for 1 h. After being washed in PBS containing 0.5% Triton X100, cells were resuspended in PBS and loaded onto a flow cytometer (CantoII, BD Biosciences) for FITC signal detection using a 488 nm laser. Unstained cells were used as negative controls; events that had fluorescent intensities higher than any of the negative controls were considered to be positive; a line segment was drawn to indicate this region on the flow cytometric diagram.
Immunofluorescence microscopy
Ten thousand LCSCs or dLCSCs were seeded onto slides coated with poly‐L‐lysine (Sigma), respectively, and they were allowed to attach onto the surface by overnight incubation at 37°C. Before being stained, the cells were washed with PBS and fixed in 4% (v.v‐1) paraformaldehyde and permeabilized with Triton X100, similar to the above method for immunostaining. After the staining with FITC conjugated Oct4 antibody, Hoechst 33 342 (molecular probes) was used to stain the nucleus. Slides were mounted using 22 mm coverslips (VWR), and cells were imaged using a fluorescence microscope (Leica DMI3000B) with excitation filters of 360/40bp (Hoechst 33 342) and 470/40bp (FITC).
Quantitative real‐time PCR of stemness‐related genes, drug transporters and DNA repair‐related genes
Total RNA of LCSCs and dLCSCs was extracted using TRIzol reagent, and 3 μg of total RNA was reverse transcribed into cDNA by SuperScript™ III reverse transcriptase. Quantitative real‐time PCR was performed by adding SYBR Green Master Mix to cDNA using specific primers for stemness‐ or transporter‐related genes. All primer sequences were listed in the supplementary section. Gene expression levels were analysed by ABI 7500 Fast Real‐Time PCR System (Applied Biosciences) using the following conditions: 1 cycle at 95°C for 10 min, 40 cycles at 95°C for 15 s and 60°C for 1 min, and fold changes of genes were calculated using the formula 2‐(ddCt).
To investigate the expression of DNA repair pathway‐related genes in the two cell lines, the cells were seeded in six‐well plates and treated with 50 μM of cisplatin or cisplatin‐free medium for 12 h. This treatment regime was based on the results from the cell viability, comet and transcription assays. The cisplatin‐treated cells were collected after 12 h and compared with those without treatment for changes in gene expression. Real‐time PCR analysis was performed, as mentioned above. Primer sequences are also included in Table S1.
Tumour sphere formation assay
LCSCs and dLCSCs were seeded at a density of 10 000 cells per well in six‐well non‐adherent plates in DMEM/F12 cell medium, supplemented with human epidermal growth factor (EGF; 10 ng·mL−1), N2 supplement (1% v.v‐1) and human basic fibroblast growth factor (bFGF, 10 ng·mL−1). The medium was freshly prepared and added every other day. On day 14, the cells were inspected under a microscope for sphere formation.
Cell viability assay
LCSCs and dLCSCs were seeded in 96‐well plates at a density of 4000 cells per well. They were treated with cisplatin at 0, 12.5, 25, 50, 75 and 100 μM for 8 and 12 h. Cells without cisplatin treatment were used as the untreated control. For treated cells, the cisplatin‐containing media were then removed and replaced with fresh complete DMEM with 10% FBS and antibiotics. After further incubation, at the endpoint of 48 h, cell viability after each treatment was assessed by an MTT assay. Briefly, 10 μL of MTT solution in PBS (5 mg·mL−1) was added to each well, and the sample plate was incubated at 37°C for 4 h. A total of 100 μL of pure DMSO was added to each well to dissolve the purple formazan crystals. Absorption measurement at 560 nm was subsequently carried out using a 96‐well plate reader.
Whole cell platinum accumulation assay
A hundred thousand each of LCSCs and dLCSCs were seeded and cultured in 55 cm2 culture dishes, allowed to grow to 90% confluence and treated with 50 μM cisplatin for 4 h. Cells were subsequently harvested and washed with PBS three times. Cell numbers were counted using haemocytometer, and the entire cell pellets were digested by 65% nitric acid at 65°C overnight. Pt content in the digestion solutions was determined by an inductively‐coupled plasma‐mass spectrometer, (ICP‐MS, Perkin Elmer, Elan 6100 DRC), which was subsequently divided by the total cell count (106) of the pellet to calculate Pt content per cell.
Immunoslot blot assay
LCSCs and dLCSCs were treated with 50 μM cisplatin for 4 and 12 h. Cells were then harvested, and genomic DNA was extracted using a mammalian genomic DNA miniprep kit (Sigma). After quantification of DNA by a spectrophotometer (Nanodrop, ND‐1000, ThermoFisher), 5 μg of denatured DNA from the control (without cisplatin treatment) and treatment groups was slot‐blotted to nylon membranes (Amersham Hybond‐N+, GE healthcare) and fixed by heating at 80°C for 2 h. Cisplatin‐induced DNA intrastrand cross‐links (Pt‐GG) were detected by an anti‐Pt‐GG primary antibody (oncolyze, German) followed by HRP‐conjugated goat anti‐rat IgG secondary antibody (Life Technologies). The chemiluminescence was recorded on Fujifilm Gel Doc LAS‐4000. The intensity of each band was quantified by use of Multi Gauge V3.2 software.
Comet assay
The extent of cisplatin‐DNA ICLs was assessed by an alkaline comet assay with some modifications according to the literature (Arora et al., 2010). Each type of cell was treated with 25 or 50 μM cisplatin for 12 h in complete media, washed with PBS and incubated in cisplatin‐free fresh medium for another 0 (t = 0) or 12 (t = 12) h. Afterwards, cells were further treated with 100 μM hydrogen peroxide for 15 min to induce DNA strand breaks. Comet assays were then carried out using the OxiSelect 96‐Well comet assay kit (Cell Biolabs, San Diego, CA) according to the manufacturer's instructions. The resulting comets were analysed using a Leica SPE confocal microscope at 200× magnification. Fifty cells per slide were randomly picked and analysed using the Comet Assay IV software. The level of ICLs was calculated by use of the following formula: [1 − (TMpt − TMctl)/(TMH2O2 − TMctl)] × 100, where TMpt is the mean tail moment of the cisplatin + H2O2‐treated sample, TMctl is the mean tail moment of the untreated control sample and TMH2O2 is the mean tail moment of H2O2 treated sample. The data are expressed as a percentage of ICLs that remained at a particular time point where the % at 0 h was normalized to 100%. The tail moment was defined as the products of tail length (the mean distance of migration in the tail) and amount of DNA in the tail region (tail DNA intensity) (Olive et al., 1990).
Transcription‐coupled DNA repair assay
Plasmid construction, preparation and transient transfection into the human lung cancer cells were carried out as previously described (Zhu et al., 2012). Briefly, a mammalian plasmid expressing Gaussia luciferase without the SV40 origin of replication, pGLuc (3983 bp), was constructed. Globally platinated plasmids were prepared accordingly, and Pt contents were determined by ICP‐MS (PerkinElmer, USA). DNA concentrations were measured by absorption measurement at 260 nm on the Nanodrop. The levels of Pt were 15.26 and 30.05 Pt per plasmid. LCSCs and dLCSCs were plated at 5000 cells per well in 96‐well plates and allowed to attach and grow for 48 h. Cells were then washed with antibiotic‐free culture media just before transfection. The experiments were performed in quadruplicate. The plasmids were diluted in OptiMEM (Invitrogen), and Lipofectamine was diluted separately in OptiMEM. After 5 min incubation at room temperature, the two solutions were then mixed and incubated for 20 min at room temperature. The transfection mixture was delivered into each well, and the cells were incubated for 2 h at 37°C. After transfection, cells were washed with PBS and added to antibiotic‐free DMEM/F‐12 medium with 10% FBS. Media after various incubation times were collected for subsequent luminescence analysis, and fresh antibiotic‐free media were added after each collection. The collected media were stored at 4°C in the dark before luminescent measurement of Gaussia luciferase. The inhibitory effect of cisplatin on the transcription of LCSCs and dLCSC was determined by quantification of expressed luciferase using coelenterazine as a substrate.
Group size, randomization and blinding
Each sample was tested in triplicate for each experiment, and five repeat experiments (n = 5) were performed with different samples under the same treatment for each assay. The number of experiments is also stated on the figure legends under each figure. For the comet assay, the data from 50 cell comets for each sample were randomly selected by another researcher who did not participate in the earlier experiment. The method used to measure tail moments was computed by the Comet Assay IV software to exclude human judgement bias. For immunostaining, 10 000 live cell events were randomly collected and analysed by the BD FACSDiva™ software.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All values are expressed as mean ± SEM. Statistical analysis was carried out using one‐way ANOVA followed by an appropriate post hoc test only when the F value from ANOVA reached statistical significance (F < 0.05). As there was no significant variance inhomogeneity in groups of data, ANOVA was carried out using GraphPad PRISM version 5.01 (La Jolla, CA, USA, www.graphpad.com). The threshold of P < 0.05 was designated as statistically significant for all tests. To reduce the effect of other sources of variation on data interpretation, such as different baseline control values of differentiated cells and non‐differentiated cancer stem cells, normalization was performed to better compare the differences after the treatment and to reveal meaningful relevant trends.
Materials
Cisplatin [cis‐diamminedichloroplatinum(II)] was purchased from Sigma–Aldrich (St. Louis, MO) and dissolved in PBS before use. TRIzol reagent and SuperScript™ III reverse transcriptase were obtained from Invitrogen (Life Technologies, Carlsbad, CA). Fluorescein conjugated Oct‐3/4 antibody was purchased from R&D Systems (Minneapolis, MN). Human epidermal growth factor (EGF), N2 supplement and human basic fibroblast growth factor (bFGF) were also purchased from Invitrogen (Life Technologies), while SYBR® Green PCR Master Mix was obtained from Applied Biosystems (Life Technologies). Globally platinated plasmids were prepared as previously described (Zhu et al., 2012). All other reagents and chemicals were from standard suppliers.
Results
LCSCs have distinct cluster morphology and high stemness properties
We first examined the morphological properties of LCSCs and dLCSCs. The dLCSCs originated from LCSCs after 12 passages (P13) in the culture. Figure 1A shows the distinct cell morphologies between LCSCs and dLCSCs. LCSCs grew in clusters, but the cluster morphology had disappeared in dLCSCs. LCSCs had a higher proliferation rate in log phase than dLCSCs (Figure 1B), and tumour sphere formation was observed for LCSCs but not dLCSCs (Figure 1C). Moreover, immunostaining results showed higher Oct4 expression levels in LCSCs after staining with FITC‐conjugated Oct4 antibody, with 76.7% Oct4‐positive LCSCs and 0.9% Oct4‐positive dLCSCs. A strong fluorescent signal for Oct4‐FITC was also observed in fluorescence images of LCSCs (Figure 1D). The stemness of these cells was further characterized by the expression level of a number of stemness‐related genes. Significantly enhanced levels of expression of stemness‐related genes, such as Notch 1, c‐Myc, Klf‐4, Sox2 and Oct4, were found in LCSCs (Figure 1E) when compared with dLCSCs. These results demonstrate the differences between LCSCs and dLCSCs and indicate that LCSCs have enhanced stemness properties and tumourigenicity.
Figure 1.
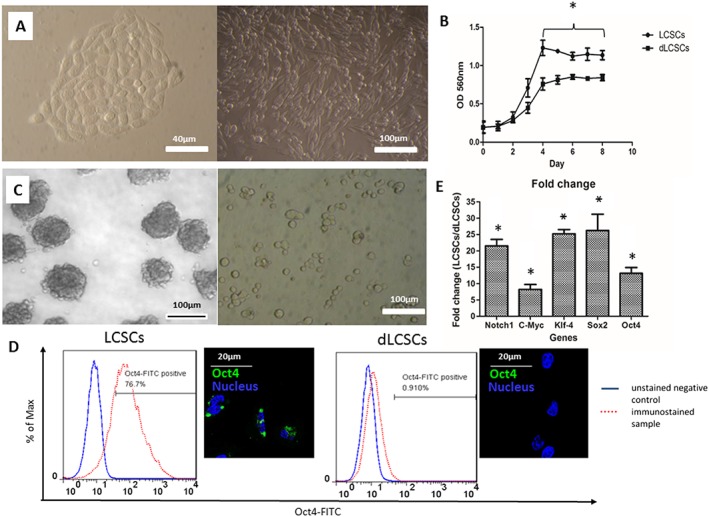
(A) Images showing the difference in cell morphology of the two cell types (left: LCSCs ; right: dLCSCs). (B) Growth curve; (C) ability to form tumour spheres (left: LCSCs ; right: dLCSCs). (D) Difference in Oct4 gene expression of LCSCs and dLCSCs from histograms and fluorescent images. (E) Fold change values of various stemness related genes are expressed as mean ± SEM. Five independent experiments were performed for each assay (n = 5).
LCSCs are resistant to the effects of cisplatin treatment
The cytotoxicity of cisplatin on LCSCs and dLCSCs was determined first. Cells were treated with different concentrations of cisplatin for 8 and 12 h, and were further incubated in the fresh media until the endpoint of 48 h. Cell viability was determined by the MTT assay. As shown in Figure 2A–B, LCSCs had significantly higher viability than dLCSCs at both time points. For example, after a 12 h treatment with 25 μM cisplatin, the viability of LCSCs at 48 h was 92.5%, but those of dLCSCs was only 56.8%. Cisplatin displayed higher IC50 values in LCSCs than in dLCSCs under different treatment conditions (Figure 2C). For instance, the corresponding IC50 values of cisplatin in LCSCs and dLCSCs were 57.1 and 37.5 μM, respectively, after a 12 h drug treatment.
Figure 2.
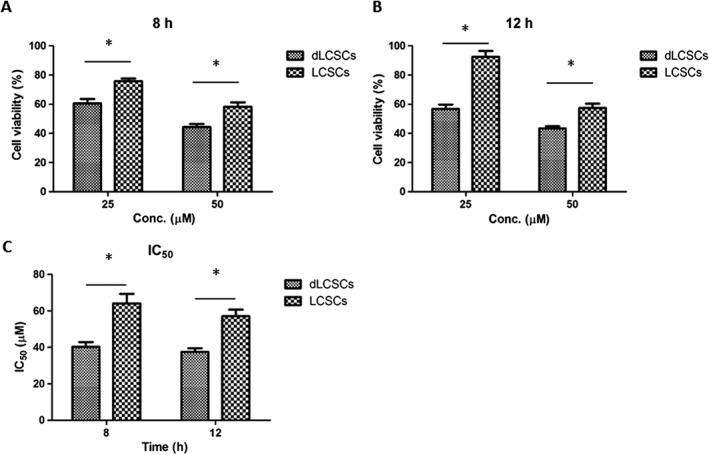
MTT results of the two cell types after (A) 8 h and (B) 12 h of 25 and 50 μM of cisplatin treatment (n = 5). (C) IC50 values of the two cell types at different cisplatin treatment time. Values are expressed as mean ± SEM; *P < 0.05.
LCSCs have reduced cisplatin accumulation at both the cellular and genomic DNA levels
Different mechanisms including reduced cellular uptake and elevated DNA repair contribute to the reduced cytotoxicity of cisplatin in lung cancer cells (Xia and Hui, 2014). We examined the accumulation of cisplatin in LCSCs and dLCSCs. The cells were treated with 50 μM cisplatin for 4 h, and the cellular level of platinum was assessed by inductively coupled plasma‐mass spectrometry (ICP‐MS). The levels of platinum in LCSCs and dLCSCs were 83.69 and 144.83 ng Pt per 106 cells respectively (Figure 3A). Therefore, compared with dLCSCs, LCSCs showed a significantly lower level of platinum inside the cells. We next performed an immunoslot blot assay to determine the platinum levels in genomic DNA (Kang and Leem, 2014). Cells were treated with 50 μM cisplatin for 4 or 12 h, and a fixed amount of DNA was blotted onto the nylon membrane. The level of platinum‐d(GpG) cross‐links, the major adducts formed by cisplatin (Wang and Lippard, 2005), was visualized by using an anti‐Pt‐GG primary antibody followed by an HRP‐conjugated goat anti‐rat IgG secondary antibody. Apparently, higher levels of cisplatin‐DNA intrastrand cross‐links were detected in dLCSCs compared with those in LCSCs (Figure 3B). For example, after a 4 h treatment, the relative band intensities in LCSCs and dLCSCs were 4.04 and 8.94 respectively. Taken together, cisplatin accumulates in LCSCs less efficiently than in dLCSCs, resulting in reduced levels of platinum cross‐linking on genomic DNA, which may be one of the reasons for cisplatin's decreased cytotoxicity in LCSCs.
Figure 3.
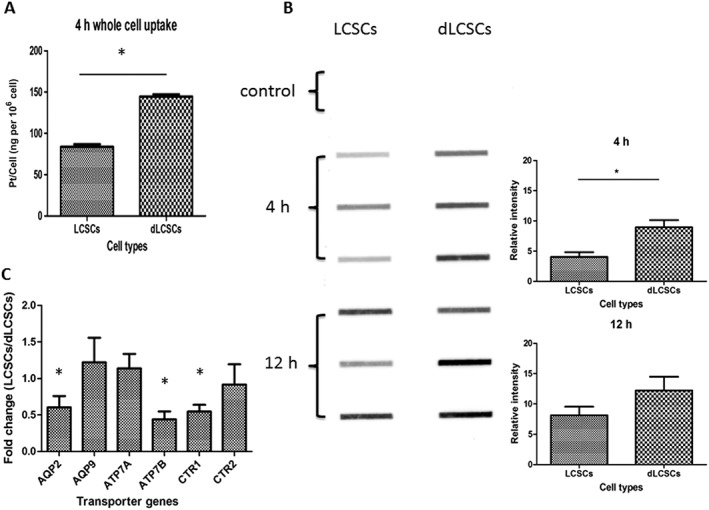
Cisplatin accumulation and expression of transporter genes in LCSCs and dLCSCs. (A) Bar charts showing the whole cell accumulation of cisplatin in LCSCs and dLCSCs after 4 h incubation; expressed as mean ± SEM; *P < 0.05; n = 5. (B) Immunoslot blot assay (left: fixed amount of DNA added for each band; right: bar chart of relative band intensity is shown as mean ± SEM; *P < 0.05) of genomic intrastrand DNA Pt‐GG level of the two cell types. (C) Differential expression of various transporter genes for untreated LCSCs and dLCSCs in terms of fold change (n = 5).
In order to explain the lower cisplatin accumulation in LCSCs, real‐time PCR of various transporter genes was performed for the two cell lines without cisplatin incubation. Figure 3C shows the reduced expression of AQP2, ATP7B and CTR1 influx transporters in LCSCs (fold change: 0.60, 0.44 and 0.55, respectively), while other genes, such as AQP9 and CTR2, did not exhibit any significant difference. The low abundance of influx transporters can reduce the uptake of platinum drugs thus may account for the lower intracellular cisplatin accumulation in LCSCs.
Transcription‐coupled repair of Pt‐DNA cross‐links
Another possible reason for the different cytotoxicity in LCSCs and dLCSCs may result from their different DNA repair capability. We, therefore, carried out a transcription‐coupled DNA repair assay using globally platinated plasmids (Ang et al., 2010). The plasmid encodes secreted Gaussia luciferase that is quantified by a luciferase assay. The presence of Pt‐DNA cross‐links in the plasmids results in transcription inhibition, and the repair of the DNA damage leads to the recovery of transcription activity (Figure 4A). This assay has been used to determine the transcription‐coupled repair mechanism of different types of Pt‐DNA lesions in a variety of mammalian cancer cell lines (Zhu et al., 2012; Zhu et al., 2013). In this study, the cells were transfected with globally platinated plasmids containing 15.26 and 30.05 Pt per plasmid, and the transcription levels were determined at 8, 16, 24, 32 and 44 h. Unmodified plasmid was used as a control, and its expression level was normalized to 100%. In LCSCs, the transcription level recovered from 19.5% at 8 h to 72.9% at 44 h for 15.26 Pt per DNA, while in dLCSCs, the levels were 4.6 and 22.3% at 8 and 44 h, respectively, showing the marked increased repair of Pt‐DNA cross‐links in LCSCs (Figure 4B). Identical effects were observed at 30.05 Pt per DNA (Figure 4C). In addition, D50 values, the number of Pt adducts per plasmid required to reduce the transcription levels to 50% of the control (Wang et al., 2014a), were obtained to quantify the differences in transcription inhibition among the two cell lines. Increased D50 values over time indicated an increased ability of the DNA damage repair on the plasmid. For LCSCs, D50 values increased significantly at different time points, showing the higher DNA repair capability of LCSCs compared to that of dLCSCs (Figure 4D). These data indicate that LCSCs have a higher DNA repair capability than dLCSCs to remove Pt‐DNA damage.
Figure 4.
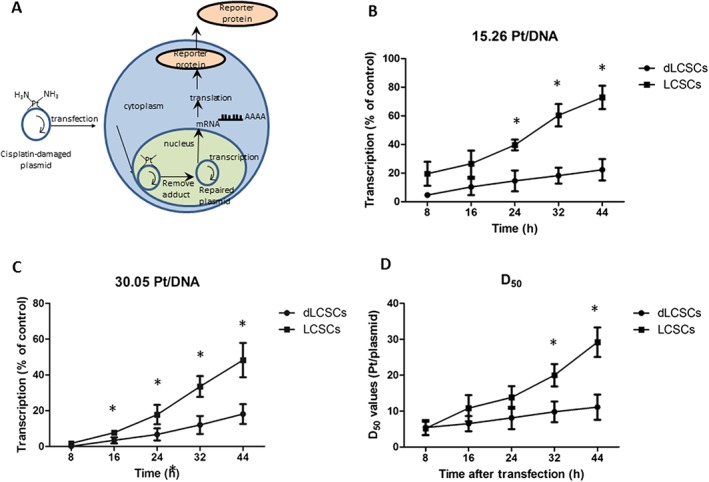
Transcription‐coupled repair profiles of globally platinated plasmids with cisplatin in LCSCs and dLCSCs. (A) Schematics of transcription‐coupled repair. Transcriptional activities of the two cell types using globally platinated plasmids with cisplatin in LCSCs and dLCSCs (n = 5) for (B) 15 Pt/DNA, (C) 30 Pt/DNA plasmids. (D) D50 values of the two cell types are expressed as mean ± SEM.
Repair of cisplatin interstrand cross‐links (ICLs)
We also used the Comet assay to measure the level of cisplatin‐DNA ICLs at the single cell level in the two cell lines (Arora et al., 2010). Cells were treated with 25 or 50 μM cisplatin for 12 h and subsequently incubated in cisplatin‐free media for 12 h. The repair of ICLs was monitored within this period of time. Representative images of gel electrophoresis are shown in Figure 5A, and the remaining images are included in Figure S1 of supporting information. Tables summarizing the tail moments, ICLs and normalized values are provided in Table S2–4 for further elucidating the data. The repair of cisplatin ICLs was evaluated over 12 h after treatment and expressed as the percentage of ICLs remaining at 12 h (Figure 5B). The lower percentage of ICLs remaining indicates a higher degree of repair. LCSCs showed a greater reduction in ICL levels than dLCSCs. For example, after 25 μM cisplatin treatment, the levels of ICLs in LCSCs and dLCSCs were 55.6 and 95.4%, respectively, indicating a higher level of ICL repair in LCSCs. These results show that LCSCs were able to remove Pt‐DNA ICLs in genomic DNA much more efficiently, further proving that LCSCs have a higher DNA repair capability than dLCSCs.
Figure 5.
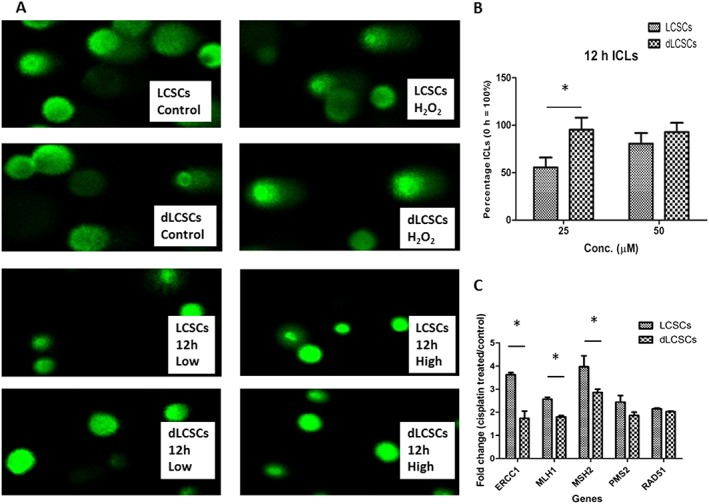
Comet assay and repair of interstrand cross‐links (ICLs) in LCSCs and dLCSCs. (A) Representative images of comet assay after 25 and 50 μM cisplatin treatment at 0 and 12 h. (B) Results of DNA repair in terms of remaining percentage of ICLs after 12 h post‐treatment time. (C) Changes in expression of DNA repair genes after cisplatin damage. Results are shown as mean ± SEM; *P < 0.05; n = 5.
Up‐regulation of DNA repair‐related genes in LCSCs
To further investigate the molecular mechanism of the enhanced DNA repair capability in LCSCs, the expression of DNA repair genes was analysed by real‐time PCR. The two cell lines were incubated with 50 μM cisplatin for 12 h. In the control group, cells were cultured in cisplatin‐free medium for the same amount of time. After treatment, the fold change in DNA repair‐related genes was analysed. It was found that several genes were significantly up‐regulated in LCSCs when compared with dLCSCs. For example, the fold changes in ERCC1, MLH1 and MSH2 in LCSCs were 3.62, 2.56 and 3.97, respectively, while they were 1.74, 1.80 and 2.86 in dLCSCs (P < 0.05). LCSCs also showed higher expression of PMS2 and RAD51 than dLCSCs, but the differences were not significant (LCSCs and dLCSCs =2.44 and 1.87 for PMS2; 2.15 and 2.03 for RAD51). RAD51 and ERCC1 are related to the DSB and NER pathways, respectively (Olaussen et al., 2006; Hashimoto et al., 2012), while MLH1, MSH2 and PMS2 take part in the MMR pathway (Prolla et al., 1994; Kondo et al., 2001). The results show that NER and MMR pathways may play an important role in the enhanced DNA repair capability in LCSCs.
Discussions and conclusion
LCSCs expressed high levels of stemness‐related genes, such as Notch1, c‐Myc, Klf‐4, Sox2 and Oct4. These genes have been commonly investigated in cancer stem cell studies (Shi and Ai, 2013). The high stemness properties have also been correlated with the ability to form tumour spheres (Eramo et al., 2008). When LCSCs differentiated into dLCSCs, the cell cluster morphology of the LCSCs changed to fibroblast‐like and was associated with a loss of stemness and the ability to form tumour spheres. These results indicate that the LCSCs used in this study possessed both stemness properties and tumourigenicity.
Cancer cells may become resistant to DNA‐damaging chemotherapeutics by changing the drug influx or efflux rates, deactivating the drug target by mutation or altering the DNA repair capability (Xia and Hui, 2014). Recently, cisplatin resistance has been correlated with the stem‐like characteristics of lung cancer cells (Barr et al., 2013). Cisplatin‐resistant cells have been found to have an increased ability to repair DNA (Olaussen et al., 2006), which increases the survival of cancer cells. Although the NER pathway was implicated in the removal of cisplatin‐DNA intrastrand cross‐links in lung cancer cells (Graf et al., 2011; Enoiu et al., 2012), the repair pathways that are responsible for the processing of Pt‐DNA cross‐links in LCSCs and the DNA repair capability of LCSCs have not been studied in detail.
We first demonstrated that one mechanism for cisplatin resistance in LCSCs is due to a reduction in the accumulation of cisplatin in the whole cell, which could arise from reduced cisplatin uptake or elevated cisplatin efflux in LCSCs. The immunoslot blot assay showed that the degree of platinum damage in genomic DNA was lower in LDSCs; this may result from both the reduced cellular accumulation of cisplatin and the enhanced ability of these cells to repair DNA damage. The expression of various transporters was also investigated in the present study to determine whether the lower cellular accumulation of cisplatin was due to differences in its influx or efflux. AQP2 and AQP9 are aquaporin transporters, which are mainly responsible for the passage of water molecules (van Lieburg et al., 1995; Ishibashi et al., 1998). Although a connection between aquaporins and cisplatin transport is not clear, reduced AQP2 and AQP9 expression has been correlated with increased cisplatin resistance (Hall et al., 2008; Wang et al., 2014b). Our real‐time PCR results revealed a down‐regulation of AQP2, which may lead to a reduced cellular platinum level thus elevating cisplatin resistance; but there was no significant change in AQP9. The copper efflux transporters, ATP7A and ATP7B, have also been reported to have a role in cisplatin efflux; therefore, increase expression of these transporters could reduce the level of intracellular cisplatin and induce resistance to platinum drugs (Komatsu et al., 2000; Katano et al., 2002). However, we found that the expression ATP7 and ATP7B was either insignificant or even down‐regulated. Hence, they are unlikely to contribute to the reduced platinum level in the cell.
The other copper transporters, CTR1 and CTR2, also regulate cellular copper levels by controlling the cellular intake or efflux of copper (Luk et al., 2003). Studies have shown that CTR1 is responsible for controlling cisplatin uptake and regulating sensitivity to platinum drugs (Ishida et al., 2002; Safaei and Howell, 2005; Larson et al., 2009), while recent evidence demonstrated that a down‐regulation of CTR2 increased cisplatin accumulation (Blair et al., 2010). Our results showed that while CTR2 did not exhibit any significant difference between the two cell lines, the lower expression of CTR1 in LCSCs may contribute to the reduced intracellular accumulation of cisplatin in LCSCs. The down‐regulation of AQP2 and CTR1 may work together to cause a lower platinum level in LCSCs, conferring increased resistance.
The enhanced DNA repair capability in LCSCs was subsequently validated by the transcription inhibition profiles of the two cell lines. The lower recovery of transcription in dLCSCs from 8 to 44 h was attributed to a reduced ability to repair the cisplatin‐damaged plasmid. LCSCs had a much higher transcription recovery than dLCSCs, suggesting the efficient removal of any remaining Pt‐DNA cross‐links and extensive activation of DNA repair genes. The removal of cisplatin ICLs in the genomic DNA of LCSCs was confirmed by the Comet assay. All these results provide solid evidence that the LCSCs are more resistant to cisplatin than dLCSCs because of both a reduction in the cellular accumulation of this drug and an enhanced DNA repair capability.
The molecular mechanisms involved in the DNA repair capability of LCSCs were evaluated based on the expression profiles of various DNA repair genes. The results demonstrated that the NER and MMR pathways were activated in LCSCs by the cisplatin treatment. In the NER pathway, the up‐regulation of endonucleases such as ERCC1 has a major role in cisplatin‐induced DNA damage repair (Arora et al., 2010). Overexpression of ERCC1 is known to correlate with resistance to platinum‐based chemotherapy (Olaussen et al., 2006; Steffensen et al., 2009).
Although the expression change of RAD51 in LCSCs and dLCSCs upon cisplatin treatment was not significantly different (P > 0.05), both cell lines showed an increase in RAD51 expression. This result implies the possible activation of the DSB pathway in both cell lines. Within this pathway, it was reported that MRE11 would interact with RAD51 monomers to form the replisome to facilitate the removal of DNA damage (Hashimoto et al., 2012). The expression of the RAD51 paralog is associated with the overexpression of XRCC3, which also has a role in DNA damage repair (Xu et al., 2005).
Mismatch repair (MMR) is another important pathway in DNA damage repair. Studies have revealed that MLH1, MSH2 and PMS2 are important components in MMR, and they form the heterodimers complex for the recognition of the mismatched damage (Prolla et al., 1994; Wang et al., 1999; Kondo et al., 2001). We observed greater fold change of MLH1, MSH2 and PMS2 in LCSCs after cisplatin treatment than in dLCSCs, which suggests that an increase in the activity of the MMR pathway in LCSCs may improve cisplatin‐induced repair. Although it remains unclear how the MMR pathway was activated in the cisplatin‐treated LCSCs to induce repair, it is known that ERCC1, which is the mammalian homologue of Rad10, is able to interact with MSH2 (Schrader et al., 2004). When ERCC1 and MSH2 are both highly expressed in LCSCs, the DNA repair might be enhanced. Overall, the results from this study show that while the DSB pathway was activated in both cell lines, the NER and MMR pathways were more strongly activated in LCSCs, which may account for its higher repair capabilities.
In conclusion, we have carried out a detailed study on the mechanism of cisplatin resistance in LCSCs and their differentiated counterpart dLCSCs. LCSCs were found to be more resistant to the cytotoxic effects of cisplatin treatment than dLCSCs. The cellular accumulation of cisplatin and the level of platinum on genomic DNA were lower in LCSCs than in dLCSCs, possibly due to a down‐regulation of the influx transporters AQP2 and CTR1, leading to an elevated cisplatin resistance in LCSCs. The greater DNA repair capability of LCSCs compared to dLCSCs was further validated by the transcription‐coupled repair assay and the Comet assay, and is likely to be the result of the activation of DNA repair pathways such as NER, DSB and MMR. This study provides insights into the mechanism of cisplatin resistance in LCSCs and sheds light on the design of the next‐generation of anticancer agents that may be a more effective treatment against LCSCs. Platinum agents that are able to accumulate in LCSCs, and can form Pt‐DNA adducts in these cells that are difficult to remove will be promising anticancer drug candidates.
Author contributions
M.Y. and G.Z. conceived the study and revised the paper; W.K.Y. performed the research, analysed the data and wrote the paper; Z.W. conducted whole cisplatin accumulation and transcription inhibition assays; C.C.F. designed the experiments; D.L. performed LCSCs characterization assays; T.C.Y and S.K.A. commented on the experiment design. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Raw images of comet assay of LCSCs and dLCSCs for control and H2O2 treatment group.
Table S1 A list of primer sequences used in real time PCR.
Table S2 Average tail moments of various samples from comet assay.
Table S3 The level of remaining interstrand cross‐link of various sample. Values were calculated using the raw values from Table S2 and the formula [1 − (TMpt − TMctl)/(TMH2O2 − TMctl)] × 100.
Table S4 Normalized values of remaining interstrand cross‐link at 12 h. Values of 0 h were normalized to 100%.
Supporting info item
Acknowledgements
This work was supported by grants from the Innovation and Technology Fund, Hong Kong Special Administrative Region, China (ITS/100/14FP), the General Research Fund of Hong Kong Research Grant Council (CityU_11303815) and Knowledge Innovation Programme of Shenzhen Municiple Government (JCYJ20140419115507575). We also thank the National Natural Science Foundation of China (21371145) for funding support. [Correction Note: The final sentence of this section was added during issue compilation, after first publication online Early View.]
Yu, W.‐K. , Wang, Z. , Fong, C.‐C. , Liu, D. , Yip, T.‐C. , Au, S.‐K. , Zhu, G. , and Yang, M. (2017) Chemoresistant lung cancer stem cells display high DNA repair capability to remove cisplatin‐induced DNA damage. British Journal of Pharmacology, 174: 302–313. doi: 10.1111/bph.13690.
Contributor Information
Guangyu Zhu, Email: guangzhu@cityu.edu.hk.
Mengsu Yang, Email: bhmyang@cityu.edu.hk.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Other ion channels. Br J Pharmacol 172: 5942–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF (2003). Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100: 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Cancer Society (2016). Cancer Facts & Figures 2016. American Cancer Society: Atlanta. [Google Scholar]
- Ang WH, Myint M, Lippard SJ (2010). Transcription inhibition by platinum‐DNA cross‐links in live mammalian cells. J Am Chem Soc 132: 7429–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora S, Kothandapani A, Tillison K, Kalman‐Maltese V, Patrick SM (2010). Downregulation of XPF‐ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair (Amst) 9: 745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O'Flaherty JD et al. (2013). Generation and characterisation of cisplatin‐resistant non‐small cell lung cancer cell lines displaying a stem‐like signature. PLoS One 8: e54193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartucci M, Svensson S, Romania P, Dattilo R, Patrizii M, Signore M (2012). Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ 19: 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L et al. (2009). Highly tumorigenic lung cancer CD133(+) cells display stem‐like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A 106: 16281–16286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair BG, Larson CA, Adams PL, Abada PB, Safaei R, Howell SB (2010). Regulation of copper transporter 2 expression by copper and cisplatin in human ovarian carcinoma cells. Mol Pharmacol 77: 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, Dick JE (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730–737. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Zhong W, Zhang L, Zhao J, Li LY, Wang MZ (2013). Recurrence patterns of advanced non‐small cell lung cancer treated with gefitinib. Chin Med J (Engl) 126: 2235–2241. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPH, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP . Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eljack ND, Ma HY, Drucker J, Shen C, Hambley TW, New EJ et al. (2014). Mechanisms of cell uptake and toxicity of the anticancer drug cisplatin. Metallomics 6: 2126–2133. [DOI] [PubMed] [Google Scholar]
- Enoiu M, Jiricny J, Scharer OD (2012). Repair of cisplatin‐induced DNA interstrand crosslinks by a replication‐independent pathway involving transcription‐coupled repair and translesion synthesis. Nucleic Acids Res 40: 8953–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A et al. (2008). Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15: 504–514. [DOI] [PubMed] [Google Scholar]
- Fichtinger‐Schepman AM, van der Veer JL, den Hartog JH, Lohman PH, Reedijk J (1985). Adducts of the antitumor drug cis‐diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry 24: 707–713. [DOI] [PubMed] [Google Scholar]
- Graf N, Ang WH, Zhu GY, Myint M, Lippard SJ (2011). Role of endonucleases XPF and XPG in nucleotide excision repair of platinated DNA and cisplatin/oxaliplatin cytotoxicity. Chembiochem 12: 1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM (2008). The role of cellular accumulation in determining sensitivity to platinum‐based chemotherapy. Annu Rev Pharmacol Toxicol 48: 495–535. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Puddu F, Costanzo V (2012). RAD51‐and MRE11‐dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol 19: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi K, Kuwahara M, Gu Y, Tanaka Y, Marumo F, Sasaki S (1998). Cloning and functional expression of a new aquaporin (AQP9) abundantly expressed in the peripheral leukocytes permeable to water and urea, but not to glycerol. Biochem Biophys Res Commun 244: 268–274. [DOI] [PubMed] [Google Scholar]
- Ishida S, Lee J, Thiele DJ, Herskowitz I (2002). Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A 99: 14298–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Leem SH (2014). Modulation of ATR‐mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res 42: 4427–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katano K, Kondo A, Safaei R, Holzer A, Samimi G, Mishima M et al. (2002). Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res 62: 6559–6565. [PubMed] [Google Scholar]
- Komatsu M, Sumizawa T, Mutoh M, Chen ZS, Terada K, Furukawa T et al. (2000). Copper‐transporting P‐type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res 60: 1312–1316. [PubMed] [Google Scholar]
- Kondo E, Horii A, Fukushige S (2001). The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2. Nucleic Acids Res 29: 1695–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Takigawa N, Osawa M, Harada D, Ninomiya T, Ochi N et al. (2013). Subpopulation of small‐cell lung cancer cells expressing CD133 and CD87 show resistance to chemotherapy. Cancer Sci 104: 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson CA, Blair BG, Safaei R, Howell SB (2009). The role of the mammalian copper transporter 1 in the cellular accumulation of platinum‐based drugs. Mol Pharmacol 75: 324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xiao ZJ, Wong SKM, Tin VPC, Ho KY, Wang JW et al. (2013a). Lung cancer tumorigenicity and drug resistance are enhanced through ALDH(hi)CD44(hi) tumor initiating cells. Oncotarget 4: 1686–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YP, Yang CJ, Huang MS, Yeh CT, Wu ATH, Lee YC (2013b). Cisplatin selects for multidrug‐resistant CD133(+) cells in lung adenocarcinoma by activating notch signaling. Cancer Res 73: 406–416. [DOI] [PubMed] [Google Scholar]
- Luk E, Jensen LT, Culotta VC (2003). The many highways for intracellular trafficking of metals. J Biol Inorg Chem 8: 803–809. [DOI] [PubMed] [Google Scholar]
- Olaussen KA, Dunant A, Fouret P, Brambilla E, André F, Haddad V et al. (2006). DNA repair by ERCC1 in non‐small‐cell lung cancer and cisplatin‐based adjuvant chemotherapy. N Engl J Med 355: 983–991. [DOI] [PubMed] [Google Scholar]
- Olive PL, Banáth JP, Durand RE (1990). Heterogeneity in radiation‐induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res 122: 86–94. [PubMed] [Google Scholar]
- Prolla TA, Pang Q, Alani E, Kolodner RD, Liskay RM (1994). MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science 265: 1091–1093. [DOI] [PubMed] [Google Scholar]
- Safaei R, Howell SB (2005). Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit Rev Oncol Hematol 53: 13–23. [DOI] [PubMed] [Google Scholar]
- Schrader CE, Vardo J, Linehan E, Twarog MZ, Niedernhofer LJ, Hoeijmakers JH (2004). Deletion of the nucleotide excision repair gene Ercc1 reduces immunoglobulin class switching and alters mutations near switch recombination junctions. J Exp Med 200: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ai W (2013). Function of KLF4 in stem cell biology In: Bhartiya D. (ed). ISBN:Pluripotent Stem Cells. InTech: Rijeka: ISBN:978–953–51‐1192–4. DOI: 10.5772/54370 [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al. (2004). Identification of human brain tumour initiating cells. Nature 432: 396–401. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen KD, Waldstrøm M, Jakobsen A (2009). The relationship of platinum resistance and ERCC1 protein expression in epithelial ovarian cancer. Int J Gynecol Cancer 19: 820–825. [DOI] [PubMed] [Google Scholar]
- van Lieburg AF, Knoers NV, Deen PM (1995). Discovery of aquaporins: a breakthrough in research on renal water transport. Pediatr Nephrol 9: 228–234. [DOI] [PubMed] [Google Scholar]
- van Moorsel CJA, Pinedo HM, Veerman G, Bergman AM, Kuiper CM, Vermorken JB et al. (1999). Mechanisms of synergism between cisplatin and gemcitabine in ovarian and non‐small‐cell lung cancer cell lines. Br J Cancer 80: 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Lippard SJ (2005). Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4: 307–320. [DOI] [PubMed] [Google Scholar]
- Wang B, Wang Z, Ai F, Tang WK, Zhu G (2014a). A monofunctional platinum(II)‐based anticancer agent from a salicylanilide derivative: synthesis, antiproliferative activity, and transcription inhibition. J Inorg Biochem 142C: 118–125. [DOI] [PubMed] [Google Scholar]
- Wang H, Lawrence CW, Li G, Hays JB (1999). Specific binding of human MSH2·MSH6 mismatch‐repair protein heterodimers to DNA incorporating thymine‐ or uracil‐containing UV light photoproducts opposite mismatched bases. J Biol Chem 274: 16894–16900. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yin JY, Li XP, Chen J, Qian CY, Zheng Y et al. (2014b). The association of transporter genes polymorphisms and lung cancer chemotherapy response. PLoS One 9: e91967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia HP, Hui KM (2014). Mechanism of cancer drug resistance and the involvement of noncoding RNAs. Curr Med Chem 21: 3029–3041. [DOI] [PubMed] [Google Scholar]
- Xu ZY, Loignon M, Han FY, Panasci L, Aloyz R (2005). Xrcc3 induces cisplatin resistance by stimulation of Rad51‐related recombinational repair, S‐phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther 314: 495–505. [DOI] [PubMed] [Google Scholar]
- Zhu G, Myint M, Ang WH, Song L, Lippard SJ (2012). Monofunctional platinum‐DNA adducts are strong inhibitors of transcription and substrates for nucleotide excision repair in live mammalian cells. Cancer Res 72: 790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Song L, Lippard SJ (2013). Visualizing inhibition of nucleosome mobility and transcription by cisplatin‐DNA interstrand crosslinks in live mammalian cells. Cancer Res 73: 4451–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Yue W, Yu WK, Liu D, Fong CC, Zhao J et al. (2015). Microfluidic platform for studying chemotaxis of adhesive cells revealed a gradient‐dependent migration and acceleration of cancer stem cells. Anal Chem 87: 7098–7108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Raw images of comet assay of LCSCs and dLCSCs for control and H2O2 treatment group.
Table S1 A list of primer sequences used in real time PCR.
Table S2 Average tail moments of various samples from comet assay.
Table S3 The level of remaining interstrand cross‐link of various sample. Values were calculated using the raw values from Table S2 and the formula [1 − (TMpt − TMctl)/(TMH2O2 − TMctl)] × 100.
Table S4 Normalized values of remaining interstrand cross‐link at 12 h. Values of 0 h were normalized to 100%.
Supporting info item