

Abstract
Deubiquitinases (DUBs) are increasingly linked to the regulation of fundamental processes in normal and cancer cells, including DNA replication and repair, programmed cell death, and oncogenes and tumor suppressors signaling. Here evidence is presented that the deubiquitinase OTUD6B regulates protein synthesis in non-small cell lung cancer (NSCLC) cells, operating downstream from mTORC1. OTUD6B associates with the protein synthesis initiation complex and modifies components of the 48S preinitiation complex. The two main OTUD6B splicing isoforms seem to regulate protein synthesis in opposing fashions: the long OTUD6B-1 isoform is inhibitory, while the short OTUD6B-2 isoform stimulates protein synthesis. These properties affect NSCLC cell proliferation, since OTUD6B-1 represses DNA synthesis while OTUD6B-2 promotes it. Mutational analysis and downstream mediators suggest that the two OTUD6B isoforms modify different cellular targets. OTUD6B-2 influences the expression of cyclin D1 by promoting its translation while regulating (directly or indirectly) c-Myc protein stability. This phenomenon appears to have clinical relevance as NSCLC cells and human tumor specimens have a reduced OTUD6B-1/OTUD6B-2 mRNA ratio compared to normal samples. The global OTUD6B expression level does not change significantly between non-neoplastic and malignant tissues, suggesting that modifications of splicing factors during the process of transformation are responsible for this isoform switch.
Implications
Because protein synthesis inhibition is a viable treatment strategy for NSCLC, these data indicate that OTUD6B isoform 2, being specifically linked to NSCLC growth, represents an attractive, novel therapeutic target and potential biomarker for early diagnosis of malignant NSCLC.
Keywords: Deubiquitinase, protein synthesis initiation, eIF4F, c-Myc, CyclinD1
Introduction
Protein synthesis is a fundamental cellular function intimately connected with cell growth and malignant transformation (1). Translation is a highly regulated process, controlled mainly at the stage of initiation (2). Aberrant protein synthesis initiation is a hallmark of sporadic and familial cancer. This is achieved through over-activation of the Akt/mTORC-1/eIF4E axis and altered expression (or mutation) of translation initiation factors (3). The rate-limiting event in cap-dependent translation is the formation of eukaryotic initiation factor (eIF) 4F and the assembly of the 48S preinitiation complex (2). The eIF4E protein binds the cap and promotes assembly of eIF4F, which also includes the scaffolding protein eIF4G, the RNA helicase eIF4A, and the ancillary factors eIF4B and eIF4H. RNA helicases play a fundamental role in solving complex structures at the 5’ untranslated region (UTR) of mRNAs (4). Enhanced initiation efficiency seems to be particularly important for the translation of malignancy-related mRNAs, which are regulated at the level of translation initiation (5).
Proteome-wide screens have revealed that the majority of initiation factors are modified by ubiquitin, including eIF4E, eIF4A, eIF4G, several subunits of eIF3, eIF2s, eIF5s, and poly A binding proteins (PABPs) (6,7). Ubiquitination can potentially alter the composition and functions of the protein synthesis initiation complex. It is generally accepted that Lysine-48 (K-48) and K-11 polyubiquitination targets proteins for proteosomal degradation, hence radically affecting their steady-state expression levels. Furthermore, several additional functional roles are emerging for distinct arrangements of mono- and polyubiquitination modification (8,9). Although the role of ubiquitination of translation initiation factors has yet to be fully elucidated, a few studies support the idea that ubiquitin modification (mono- and polyubiquitination) may affect the function of translation initiation factors (10-12). A growing number of studies underscore that the ubiquitination status of any given protein results from the concerted activities of ubiquitin ligases and deubiquitinases. Deubiquitinating enzymes (or DUBs) are involved in the regulation of central biological processes, including DNA repair, apoptosis, oncogene expression and function, and checkpoint regulation (13). For this reason, DUBs are being actively explored as anticancer targets (14). DUBs are classified in five groups including the ovarian tumor domain-containing DUBs (OTU family), which comprises enzymes that can display linkage-specificity in their recognition and cleavage of diubiquitin moieties (15). OTUD6B does not have linkage specificity towards diubiquitin dimers and it seems unable to operate as an ubiquitin carboxyl-hydrolase (15). However, it is able to deubiquitinate Ub-Met-β-gal fusion protein in vitro (16). Therefore, it seems to be a functioning DUB.
Although new data are clarifying the emerging functions of DUBs, their role in the regulation of protein synthesis, particularly in cancer, still remains unknown. In this study we investigated the role of the isoforms of the deubiquitinase OTUD6B in the regulation of global protein synthesis in NSCLC cells and its consequences on proliferation. Furthermore, the pattern of expression of OTUD6B isoforms has been verified in normal lung and NSCLC tissues and cells.
Materials and Methods
Unless otherwise specified, all studies are shown 48 hours after experimental manipulation.
SiRNAs, antibodies and oligonucleotides
All siRNA and oligonucleotides are reported in Supplementary Table ST1. Antibodies used in this study are listed in Supplementary Table ST2.
Cells, tissue culture, and human specimens
All NSCLC cell lines and WI38 lung fibroblasts were purchased from ATCC. ME16 has been previously described (17). HaCaT cells were provided by Dr. Mitchell Denning, Loyola University Chicago. All cells were fingerprinted using the GenePrint fluorescent STR system (Promega) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 incubator. Primary airway epithelial cells were purchased from Cambrex and cultured as previously described (18). Transfection of nucleic acids was performed using electroporation (17). Absence of mycoplasma contamination was monitored using the MycoSensor qPCR assay kit (Stratagene).
Fresh human NSCLC biopsies were collected at Loyola University Chicago Medical Center according to Institutional Review Board supervision. Specimens were flash frozen within 30 minutes from surgical resection. Frozen normal lung specimens were from Addgene or from our archive (17).
Chemicals, plasmids and luciferase reporter assay
Torin-1 (1-[4-[4-(1-Oxopropyl)-1-piperazinyl]-3-(trifluoromethyl)phenyl]-9-(3-quinolinyl)-benzo[h]-1,6-naphthyridin-2(1H)-one) (Tocris Bioscience) was dissolved in DMSO and used at a final concentration of 250 nM. Silvestrol (Medchemexpress LLC, Princeton, NJ) was dissolved in DMSO and used at a final concentration of 0.04 μM or 0.1 μM. Homoharringtonine (Tocris) was dissolved in DMSO and used at a final concentration of 0.1 μM. Cycloheximide (Sigma-Aldrich) was dissolved in DMSO and used at a final concentration of 50 μg/ml. Streptavidin-HRP was from ThermoFisher Scientific. pENTR4 and pLENTI4/TO/V5-DEST were from Invitrogen. pcDNA3- was from Addgene. pcDNA3-RLUC-POLIRES-FLUC was a gift from Nahum Sonenberg (McGill University, Montreal). Cells were cotransfected with this plasmid and siRNA to OTUD6B or control siRNA. 48 hours after cotransfection, cells were examined using a dual luciferase reported kit (Promega) following the manufacturer’s instructions. Luciferase units were measured using a Veritas microplate luminometer (Turner Biosystems) and data were normalized for pcDNA3-RLUC-POLIRES-FLUC amounts using qPCR.
OTUD6B isoforms were obtained as synthetic genes (IDT), PCR amplified using primers with extra bases at their 5’-end, digested with Nhe I and Xho I, and cloned into pcDNA3-. OTUD6B-1 C188S and OTUD6B-2 C57S were generated from wild-type counterparts using the GeneArt site-directed mutagenesis kit (Invitrogen) using the manufacturer’s protocols. Human influenza hemagglutinin (HA), six histidines tagged OTUD6B-1 was obtained via PCR amplification with designed primers (Supplementary Table ST1) and Platinum Pfx DNA polymerase (Invitrogen). The PCR product was digested, purified, and ligated into the Nhe I and Xho I sites of pCDNA3.1-.
Protein analysis
Western blot analysis was performed as previously described (18). OTUD6B was detected using a rabbit polyclonal antibody recognizing the OTU domain (Supplementary Table ST2 and Fig. S1). For OTUD6B isoform detection, it is critical to hybridize OTUD6B-1 and -2 separately, most likely due to different accessibility of the epitope in the two isoforms. This was accomplished by cutting nitrocellulose membranes, after transfer, at ~50 and ~25 kDa. This resulted in three membrane strips with the middle and bottom strips hybridized and handled in the same fashion in parallel. Ultimately, the two strips were aligned and exposed to X-ray films. The middle membrane was then stripped of antibodies and rehybridized with an antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for equal loading. As an additional control, all membranes were stained with Ponceau-S, 0.1% (w/v) in 5% acetic acid (Sigma-Aldrich) to confirm equal loading after enhanced chemiluminescence (ECL) development.
For immunoprecipitation, lysates were incubated overnight at 4 °C with specific primary antibodies at concentrations recommended by manufacturers, followed by a 1 hour incubation with Protein A/G Magnetic Beads (Thermo Fisher Scientific Inc.), and subjected to three washes in lysis buffer. After elution, proteins were resolved by SDS-PAGE. 2D gels were performed as follows: the first dimension was resolved on ReadyStrip IPG strips, pH 3-10 (Bio-Rad). Eluates were obtained and equilibrated according to Bio-Rad specifications. The second dimension was then run on 10% SDS PAGE gels and transferred to nitrocellulose membranes.
NSCLC cell lysates were obtained and fractionated through sucrose cushions as previously described (19). Purified fractions were run onto 10% SDS-PAGE gels and either stained with Coomassie blue G-250 (Bio-Rad) or blotted on nitrocellulose membranes.
Cap binding assay, Ni-NTA capture, rate of global protein synthesis, and proliferation
Cap binding assay was performed after transfection with control siRNA or siRNA against OTUD6B as previously described (20) with one modification: initiation complexes were formed using 2’/3’-O-(2-Aminoethyl-carbamoyl)-N7-methyl-guanosine-5’-triphosphate-Biotin, Triethylammonium salt (Jena Bioscience) and captured on streptavidin magnetic beads (Pierce). Complexes were washed and eluted with an excess of m7GTP. As the negative control, cell lysates were incubated with streptavidin magnetic beads only.
For Nickel-conjugated agarose pull down, about 250 μg of total cell lysate was obtained in 400 μl of buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 10mM imidazole, 0.2% NP40, 0.01% Tween 20, 1X Halt™ protease and phosphatase inhibitor cocktail, ThermoFisher Scientific). Lysates were incubated in ice for two minutes, sonicated three times at output 8 using a Microson ultrasonic cell disruptor (Misonix). Cell lysates were centrifuged at 14,000 r.p.m. for 5 minutes in a microfuge, and the supernatants were applied to 100 μl of packed Ni-conjugated agarose (Ni-NTA, Qiagen), and gently rotated for 1 hour at + 4°C. The resin was then collected by centrifugation at 4,000 r.p.m., washed three times with buffer, and eluted in 50 μl of SDS loading gel buffer.
The rate of global protein synthesis was measured using the Click-iT AHA Protein Synthesis kit (Invitrogen) according to the manufacturer’s instruction. Before L-azidohomoalaine (AHA) exposure, cells were incubated for one hour in complete medium lacking methionine, then incubated at 37 °C for two hours in RPMI1640 lacking methionine and FBS, supplemented with 50 μM AHA. Shorter incubation times are reported in figure legends. AHA incorporation was analyzed using a BD FACS Canto II instrument (Becton Dickinson). Flow cytometry data was analyzed by FlowJo software.
For DNA synthesis, bromodeoxyuridine (BrdU) incorporation (FITC BrdU Flow kit, BD Pharmingen)/7-aminoactinomycin D (7-AAD, Sigma-Aldrich) staining was completed according to the manufacturer’s instruction. Incorporation of BrdU was acquired by FACS and analyzed as described above.
Gene Expression Analysis
Total RNA was extracted from cells using the RNeasy Mini kit (Qiagen). Frozen tissues were dissolved in Trizol Plus RNA purification system extraction buffer (ThermoFisher Scientific Inc.) with the help of a polytron. RNA concentrations were determined using a NanoDrop Spectrophotometer (ThermoFisher Scientific Inc.). 2 μg of RNA were reverse transcribed using the iScript Reverse Transcription Supermix RT-qPCR (Bio-Rad). Quantitative real-time PCR was done with SYBR Green PCR Master Mix (Applied Biosystems) in a StepOne thermal cycler (Applied Biosystems). Non–reverse transcription reactions served as negative controls. We used linearity curves calibrations in all experiments, with the relative amounts calculated from these calibration curves in each experiment.
Statistical analysis
We used the GraphPad Prism 5 software for two tailed t-tests, one- and two-way ANOVA. P values are reported in Figures or Figure legends.
Results
OTUD6B affects the rate of cap-dependent protein translation
Depletion of amyloid precursor protein (APP) in NSCLC cells causes a complex phenotype, including increased global protein synthesis (20,21). To determine the critical mediators of APP depletion, we used siRNA to silence mRNAs which were downregulated upon APP depletion in NSCLC. We observed that downregulation of OTUD6B increased the rate of global protein synthesis. To confirm these results, we obtained two siRNAs against the OTUD6B mRNA: siRNA3 and siRNA4 (Table ST1). Both siRNAs efficiently decreased expression of OTUD6B protein (Fig. 1A). Artificial downregulation of OTUD6B caused a 25-30% increase of the methionine surrogate azidohomoalanine (AHA) incorporation in A549 and H1299 cells. Additionally, OTUD6B depletion increased AHA incorporation in H1734 and H1437 cells, and in transformed or primary cells other than NSCLC, specifically the mesothelioma cell line ME16, HaCaT immortalized human keratinocytes, and primary lung fibroblasts WI38 (Fig. 1B). Double-luciferase assays revealed that only cap-dependent translation was affected (Fig. 1C). To assess which step of protein synthesis OTUD6B could affect, we knocked down OTUD6B in the presence of completely inhibitory concentrations of silvestrol (initiation inhibitor, 22), cycloheximide or homoharringtonine (elongation inhibitors, 23). AHA incorporation assays indicated that NSCLC cells with depleted OTUD6B displayed increased AHA incorporation in the presence of silvestrol. siRNAs to OTUD6B were unable to modify AHA incorporation when cells were treated with cycloheximide or homoharringtonine (Fig. 1D, left). OTUD6B depletion in the presence of partially-inhibitory concentrations of silvestrol exhibited increased AHA incorporation over time (Fig. 1D, right). These data suggest that OTUD6B depletion leads to an increased rate of cap-dependent protein synthesis, and that OTUD6B seemed to limit protein synthesis initiation.
Figure 1.

OTUD6B depletion causes increased cap-dependent protein synthesis by affecting the step of initiation. A, immunoblot analysis of the indicated proteins 48 hours after transfection of H1299 cells with the indicated siRNAs. Similar results were obtained in A549 cells. B, AHA incorporation assay/FACS analysis of cells transfected with the indicated siRNAs. Left, representative experiment in H1299 cells. Right, histogram summarizing the results obtained from seven cell lines (one experiment in A549, H1299, H1437, H1734, ME16, HaCaT, WI38 cells). Columns represent averages, bars standard deviations. C, double luciferase assay in cells co-transfected with pcDNA3-RLUC-POLIRES-FLUC (translation of renilla luciferase is cap-dependent, while firefly luciferase translation is dependent on a polio virus IRES) and the indicated siRNAs. Bars represent the averages of a total 12 experiments (four experiments in each A549, H1299, H1347 cell line); bars represent standard deviations”. D, AHA incorporation/FACS analysis during the indicated time points in cells treated with completely inhibitory concentrations of antibiotics (left) and partially inhibitory concentrations (right), and transfected with the indicated siRNAs. On top is reported the silvestrol concentration. Points represent geometric means, while bars represent standard deviations. The graph displays the average of four experiments performed in H1299 cells. Similar results were obtained in A549 cells (two experiments). * represents the overlapping data of no AHA negative control, cell transfected with control siRNA and siRNA to OTUD6B exposed to 10 μg/ml cycloheximide or 100 nM homoharringtonine. No AHA: negative control for autofluorescence.
To assess whether the effect of OTUD6B on protein synthesis was shared among OTU DUBs, we investigated whether depletion of other OTU DUBs resulted in similar effects on protein synthesis. We first analyzed OTUD6A, which shares very high levels of sequence identity with OTUD6B (Supplementary Fig. S1). OTUD6A was not expressed at the mRNA level in five different NSCLC cell lines, as assessed by qPCR. Thus, we focused on two other OTU DUBs, OTUD2 (or YOD1) and OTUD5. In phylogenetic trees, OTUD2 is closely related to OTUD6B, while OTUD5 is more distantly related to OTUD6B (15). A siRNA to OTUD2 or OTUD5 did not affect the rate of AHA incorporation (Supplementary Fig. S2). These data support the notion that the effects of OTUD6B on protein synthesis appear to be specific to this DUB and may not be a general property of OTU DUBs.
OTUD6B operates downstream of mTORC-1
mTORC-1 activity is a master regulator of protein synthesis initiation (24). To elucidate whether depletion of OTUD6B in NSCLC cell lines affects Akt/mTORC-1 signaling, we monitored the phosphorylation status of both p70S6 kinase and Akt after depletion of OTUD6B in NSCLC cells. OTUD6B depletion did not change Akt and S6 kinase phosphorylation at S473 and T385, respectively (Fig. 2A). Furthermore, NSCLC exposure to Torin-1 (an mTOR inhibitor, which target the mTOR catalytic site) abolished OTUD6B depletion-enhanced AHA incorporation in these cells (Fig. 2B). These data strongly indicate that altered OTUD6B expression is unable to affect Akt and mTORC-1 activities. Additionally, complete inhibition of mTOR erased any effect elicited by siRNA to OTUD6B. Taken together, these results suggest that OTUD6B operates downstream from mTORC-1 activation.
Figure 2.

OTUD6B depletion does not affect mTORC-1 activity and does not affect the rate of protein synthesis in the presence of the mTOR inhibitor Torin-1. A, Western blot analysis of the indicated proteins and phosphoproteins in H1299 cells transfected with either control siRNA (c) and siRNA to OTUD6B. Similar results were obtained in A549 cells (one experiment). B, AHA incorporation in H1299 cells transfected with either control siRNA (c) and siRNA to OTUD6B, and exposed to DMSO vehicle (Left) or Torin-1 (Right). These results were reproduced once in A549 and in H1437 cells.
OTUD6B interacts with and modifies components of the protein synthesis initiation complex
OTUD6B depletion caused an increased rate of cap-dependent translation. However, OTUD6B downregulation seemed unable to affect Akt or mTORC-1 activity, and the effects of its downregulation were abolished upon Torin-1 treatment. We hypothesized that OTUD6B may exert its functions on either the initiation complex or the ribosome. To determine this, we first performed cell fractionations to identify whether OTUD6B is ribosome-associated. We determined that OTUD6B was detectable in postmitochondrial and postribosomal fractions, but it was not detectable alongside ribosomes and polysomes (Fig. 3A). Although preliminary, this evidence suggested that OTUD6B does not interact with the ribosome. To elucidate whether OTUD6B interacts with the initiation complex, we immunoprecipitated eIF4G and blotted the immunoprecipitates for components of eIF4F and OTUD6B. OTUD6B co-immunoprecipitated alongside eIF4G, eIF4A, eIF4B, and eIF4E (Fig. 3B). To confirm that OTUD6B interacts with the initiation complex, we performed m7GTP (cap) pull-down assays. The results indicated that OTUD6B interacts with the initiation complex (Fig. 3C). Additionally, upon OTUD6B depletion, the amounts of initiation factors associated with eIF4F were quantitatively higher compared to the same experiments performed on lysates obtained from cells transfected with control siRNA. This data may suggest that OTUD6B destabilizes the eIF4F complex. To verify interaction of OTUD6B with the protein synthesis initiation complex, we generated an OTUD6B harboring a C-terminal human influenza hemagglutinin (HA) tag followed by six histidines. This mutant was transfected in NSCLC cells (Fig. 3D, Left). Total cell lysates obtained from cells transfected with this fusion, or with a control plasmid, were purified with Nickel-conjugated resin, washed and eluted with SDS gel loading buffer. The resin incubated with lysates obtained from cells overexpressing OTUD6B-HA-6His retained components of the eIF4F complex (Fig. 3D, Right).
Figure 3.

OTUD6B binds to and deubiquitinates the protein synthesis initiation complex. A, Sucrose fractionation of NSCLC cells extracts followed by immunoblotting. Lanes: 1, total cell lysate, 2, nuclear fraction, 3, postmitochondrial fraction, 4, postribosomal fraction, 5 ribosomes (see Supplementary Fig. S3 for Coomassie staining of representative gel). Ribosomal protein S6 has been used as a marker of ribosomes. B, Western blot analysis in immunoprecipitates obtained using an antibody against eIF4G and 50 μg of total cell lysates (indicated). A549 cells were transfected with either control siRNA or siRNA to OTUD6B. Please note that eIF4A appears as a single band or 2-3 bands whether it is run in minigels, or 20 cm long gels (respectively). OTUD6B also coimmunoprecipitated with eIF4G in H1299 cells (Supplementary Fig. S4). C, Western blot analysis of the indicated proteins in a representative 7mGTP pull-down assay or total cell lysate obtained from H1299 cells transfected with either control siRNA or siRNA to OTUD6B. Similar results were obtained in two additional experiments in H1299 and in A549 cells (one experiment). D, Left, Western blot analysis of H1299 cells transfected with a control plasmid (c) or with a plasmid encoding an OTUD6B fused at its C-terminal portion with an AH tag followed by 6 histidines at the indicated time points after transfection (24 and 48 hours). Right, Ni-NTA pull down of 200 μg of total cell lysates obtained from (24) and (c). E, immunoblots of 2D gels hybridized with an antibody against ubiquitin. Eluates from 7mGTP pull-down assays were run in 3 to 10 pH gradients (arrow), and subsequently run on a 10% SDS page gel, then transferred to nitrocellulose membranes.
We asked whether OTUD6B modifies components of the translation initiation complex. To address this, we transfected NSCLC cells with control siRNA and siRNA to OTUD6B and captured initiation complexes with cap-insoluble carrier. We then ran the complexes on 2D gels, blotted and hybridized 2D-Western membranes with an antibody against ubiquitin (Fig. 3E). Initiation complexes isolated from OTUD6B-depleted NSCLC cells demonstrated an enhancement of at least three strong signals, which likely correspond to poly-ubiquitinated proteins, and one possibly representing a mono-ubiquitinated protein (at a single or at multiple sites). Collectively, these experiments support the interpretation that OTUD6B binds to and deubiquitinates components of the protein synthesis initiation complex in NSCLC cells.
OTUD6B splice isoforms have opposing effects on global protein synthesis and require their catalytic cysteine for function
OTUD6B primary transcripts can be alternatively spliced to yield three splicing variants (Supplementary Fig. S5). Splice variant 1 (OTUD6B-1) contains an N-terminal coiled coil domain, which suggests oligomerization. Splice variant 2 (OTUD6B-2) lacks such a domain, while variant 3 (OTUD6B-3) has a short N-terminal deletion. OTUD6B-1 and -2 are expressed in NSCLC cells. We had no means to specifically measure OTUD6B-3 using specific primers for its mRNA. Western blot analyses indicated that OTUD6B-3 is either poorly expressed or not expressed in NSCLC cells (Fig. 4A). OTUD6B-2 levels of expression varied between cell types (see below). Differences in primary sequence between isoforms could reflect diverse biological properties. To investigate this, we cloned each OTUD6B splice variant in pcDNA3-. In overexpression experiments, OTUD6B-1 depressed the rate of AHA incorporation (Fig. 4B). This result was complementary to experiments performed using siRNA to OTUD6B. Surprisingly, OTUD6B-2 overexpression caused a marked increase in AHA incorporation (Fig. 4C). OTUD6B-3 overexpression decreased methionine surrogate incorporation in a similar fashion compared to the OTUD6B-1 isoform (Fig. 4D). These results indicated that the deletion of the coiled coil domain in OTUD6B-1 yielded an isoform with opposite effects on global protein synthesis, while the minor amino acid changes between OTUD6B-1 and -3 did not result in functional changes between these two isoforms.
Figure 4.

The two main OTUD6B splice variants have opposite effects on global protein synthesis; their activity appears to require the catalytic cysteine. A, Western blot analysis of the OTUD6B isoforms. Left; immunoblots in four NSCLC cell lines (1, A549; 2, H1755; 3, H1437; 4, H1650). For OTUD6B isoforms detection, please see Materials and Methods. Right; representative immunoblots of H1299 cells 48 hours after transient transfection with the specified OTUD6B isoforms cloned in pcDNA3-. Arrowheads indicate the three OTUD6B isoforms. B, C, D, representative AHA incorporations in H1299 after transfection with the indicated plasmids; c, control. E, summary of AHA incorporation in cell lines A549, H1299 and H1437 (two experiments in each cell line) after transfection with the indicated plasmids; c, pCDNA3-; -1, OTUD6B-1; -2, OTUD6B-2; -3, OTUD6B-3. P values refer to t-test performed on all data considered as independent values. F, summary of eight independent AHA experiments performed in H1299 cells after transfection with the indicated OTUD6B isoforms and their mutants at the catalytic cysteine. C, control; -1, OTUD6B-1; -1MUT, OTUD6B-1 C188S; -2, OTUD6B-2; -2MUT, OTUD6B-2 C57S. Statistics: one-way ANOVA; asterisk on left histogram, OTUD6B-1 is statistically significant in Friedman/Dunn's tests to both control (p=0.008) and OTUD6B-1 C188S. Control and OTUD6B-1 C188S are not significantly different. Asterisk on right histogram, OTUD6B-2 is statistically significant in Friedman/Dunn's tests compared to control (p=0.018).
To address whether the catalytic site of the OTUD6B isoforms was responsible for the observed phenotypes on protein synthesis, we mutated the catalytic cysteine (C188, OTUD6B-1 numbering) into serine within the OTUD6B-1 and -2 isoforms. In transient transfection experiments, C→S mutations in the catalytic site caused OTUD6B-1 and -2 to lose their phenotype (Fig. 4E). This would suggest that the catalytic site (hence, DUB activity) is required for these isoforms to regulate protein synthesis. Because mutations of either OTUD6B-1 or OTUD6B-2 did not produce dominant-negative proteins, it stands to reason that the two isoforms process different cellular targets. However, transient transfection experiments performed with mutant isoforms showed an unusually high experimental variation (Fig. 4F). This suggests that OTUD6B isoforms with mutated catalytic sites can interfere with their wild type counterparts, in a fashion that is most likely dose-dependent. To ascertain whether mutations in the catalytic cysteine were present in NSCLC cell lines, we PCR amplified and sequenced the OTU domain from genomic DNA of two primary bronchoepithelial cultures, one primary small airway epithelial culture, and 11 NSCLC cell lines. Only HCC827 cells showed evidence of an allelic mutation at C188 (Supplementary Fig. S6). These results support the interpretation that the catalytic cysteine of OTUD6B (and the OTUD6B gene in general) is rarely mutated in cancer, in accordance with data available in TCGA and COSMIC databases with a large number of primary tumors and cell lines (25,26).
OTUD6B splice isoforms have opposing effects on NSCLC proliferation
Our results indicate that OTUD6B-1 likely acts as a repressor of general protein synthesis. The OTUD6B-2 splice isoform seems to have opposite effects on protein synthesis, since its overexpression almost doubled the rate of methionine analog incorporation in transfected cells. It is generally accepted that deregulated protein synthesis can lead to increased proliferation (3). One mechanism linking these two biological phenomena is efficient translation of malignancy-related mRNAs regulated at the level of translational initiation, including c-Myc and Cyclin-D1 (5). To assess whether overexpression of the two main OTUD6B isoforms affected proliferation, we performed bromodeoxyuridine (BrdU) incorporation and proliferation assays after transient transfection of OTUD6B-1 and -2 in NSCLC. The results revealed that OTUD6B-1 overexpression decreased BrdU incorporation, while OTUD6B-2 overexpression increased BrdU incorporation (Fig. 5). Both OTUD6B-1 and OTUD6B-2 overexpression also causes cell death (Supplementary Fig. S7). This event, which is common when overexpressing proteins that affect growth and proliferation (27, 28), did not allow us to generate reliable growth curves.
Figure 5.

The two main OTUD6B splice variants have opposite effects on DNA synthesis. Bromodeoxyuridine incorporation/FACS analysis of the indicated cells transfected with the specified plasmids. Histogram, summary of six experiments (three in A549 cells and three in H1299 cells). One way ANOVA with Bonferroni post test: R square = 0.0078, F = 0.1431, P = 0.987. 95% CI of diff.: α, 6.248 to 16.92; β, −24.15 to −13.48; γ, −12.57 to −1.891
Taken together, these results indicate that OTUD6B-1, by acting to limit protein synthesis, may have anti-proliferative effects, while OTUD6B-2, by acting to enhance protein synthesis, may favor cell cycle progression and oncogenesis.
OTUD6B-2 affects the expression of c-Myc and Cyclin-D1
OTUD6B-1 cannot be specifically targeted using siRNA. OTUD6B-2, instead, contains a unique exon (about 200 bases long), which can be exploited for specific RNA silencing. This region (the 5’ untranslated portion) of the mRNA is not optimal for RNA silencing. Nevertheless, we constructed three custom siRNAs targeting this exon, plus two exon-skipping, modified oligonucleotides (to prevent the generation of the OTUD6B-2 mRNA; Supplementary Table ST1). We tested the above reagents in transient transfection experiments and we found that one siRNA (siRNA6) was effective in specifically downregulating OTUD6B-2 24 hours after transfection (Fig. 6A). A second siRNA (siRNA9) was effective only at very high concentrations, while the exon-skipping primers were not effective in reducing OTUD6B-2 expression (data not shown). We first asked whether OTUD6B-2 specific downregulation could have produced effects on NSCLC cell proliferation. Cells with depleted OTUD6B-2 displayed a small, but statistically significant reduction in BrdU incorporation 48 hours after transfection with siRNA6 (Fig. 6B). Next, we investigated whether the expression of proteins regulated at the level of translation initiation could have been affected by specific OTUD6B-2 depletion. We found that siRNA to OTUD6B-2 reduced the expression of both c-Myc and Cyclin-D1 while OTUD6B-2 overexpression caused increased c-Myc and Cyclin-D1 expression (Fig. 6C). To establish whether these differences were due to altered translation of the c-Myc and Cyclin-D1 mRNAs, we measured the expression of these mRNAs 24 hours after siRNAs transfection, finding that siRNA6 did not alter the expression of the c-Myc or Cyclin-D1 mRNAs (Fig. 6D). To rule out differences in degradation rates of these two proteins, we treated siRNA-transfected cells with the proteasomal inhibitor MG132. Upon proteasome inhibition, siRNA6 to OTUD6B-2 still repressed Cyclin-D1 expression. However, c-Myc expression was no longer affected by siRNA6 to OTUD6B-2 (Fig. 6E). To understand whether OTUD6B-1 may have opposing effects on c-Myc and Cyclin-D1 expression, we transfected NSCLC cells with a plasmid encoding OTUD6B-1, and measured no differential expression of these oncogenic proteins (Fig. 6F). Collectively, these data suggest that OTUD6B-2 favors the expression of c-Myc and Cyclin-D1. Instead, OTUD6B-1 does not appear to affect the expression of these proteins, further supporting the idea that these two OTUD6B isoforms target different cellular substrates. OTUD6B-2 likely facilitates translation of Cyclin-D1, while affecting the stability of c-Myc. Because DUBs are highly pleiotropic enzymes, the issue of whether OTUD6B-2 directly or indirectly regulates c-Myc stability must be clarified in future studies.
Figure 6.

OTUD6B-2 favors the expression of c-Myc and Cyclin-D1; siRNA to OTUD6B-2 reduces DNA synthesis. A, representative immunoblots of the indicated proteins in H1299 cells 24 hours after transfection with the indicated siRNAs. Similar results were obtained in A549 cells (one experiment). B, representative BrdU incorporation in H1299 cells 48 hours after transfection with the indicated siRNAs. Similar results were obtained in A549 cells (two experiments in each cell line). C, immunoblots of the indicated proteins in H1299 cells 24 hours after transfection of the indicated siRNAs (left), or 48 hours after transfection of the indicated plasmids (right). Similar results were obtained in A549 cells (one experiment). D, qPCR results of the indicated mRNAs in A549 and H1299 cells (three experiments/cell line) 24 hours after transfection with the indicated siRNAs. Results were normalized for the β-globin mRNA. E, immunoblots of the indicated proteins in H1299 24 hours after transfection of the indicated siRNAs or 48 hours after transfection of the indicated plasmids. H1299 cells were treated with MG132. Lanes, 1 and 2, Immunoblot for ubiquitin of cell lysates obtained from control cells in the absence (1) or presence (2) of MG132. 3 and 4, Ponceau-S staining of the nitrocellulose membrane shown in 1 and 2 after ECL development (equal loading control). Please note that the molecular weight ladder does not represent the molecular weight of the bands shown on the right side of the panel. F, immunoblots of the indicated proteins in cell extracts obtained from H1299 cells after transfection of the indicated plasmids.
NSCLC cells and tumors upregulate OTUD6B-2 expression compared to normal counterparts
The data gathered thus far indicate that OTUD6B-1 curtails growth and proliferation, while OTUD6B-2 seems to promote growth and proliferation. These opposing properties would suggest that the ratio between OTUD6B-1 and OTUD6B-2 could be decreased in transformed lung cells and tissues as compared to their normal counterparts. Given the technical challenges posed by the primary antibody (see Materials and Methods), we tested this hypothesis quantitatively at the mRNA level using qPCR and primers able to discriminate the two isoforms. The results showed that NSCLC cells had significantly lower OTUD6B-1/OTUD6B-2 mRNAs ratio compared to primary bronchial and small airway epithelial cells using three internal gene normalizers (Fig. 7A). This ratio falls from an average of about 3.9 in normal cells to an average of 1.3 in NSCLC cells. Immunoblots (although not quantitatively accurate) seemed to support the idea that NSCLC cells upregulate OTUD6B-2 expression when compared to primary cells (Fig. 7B).
Figure 7.
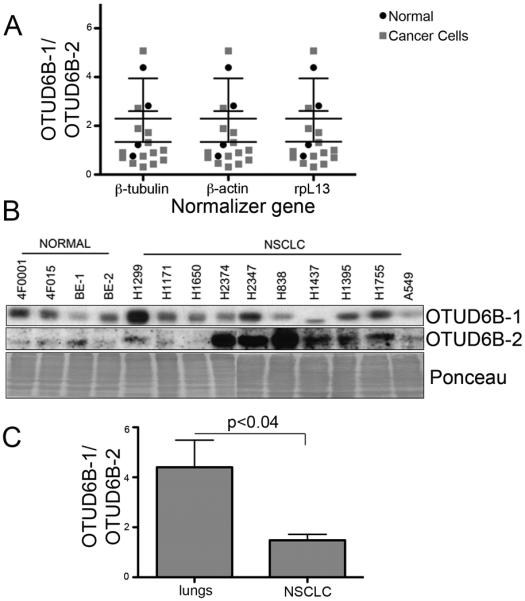
The ratio of OTUD6B-1/OTUD6B-2 expression is reduced in malignant NSCLC cells and tissue compared to their normal counterparts. A, measurements in NSCLC cell lines and primary airway epithelial cells; qPCR results. The expression has been normalized for three internal controls (specified). The two-way ANOVA results are as follows: the ratio normal/cancer affects the results (p=0.0351); the ratio normal/cancer does not have the same effect at all values of normalization gene (p>0.99); the normalization gene does not affect the results (p>0.99). B, immunoblots for the indicated OTUD6B isoform in the indicated primary cell culture and NSCLC cell lines. Staining the nitrocellulose membrane with Ponceau-S after ECL development assessed equal loading. BE-1, 4F0439; BE-2, 4F0624 (primary bronchial epithelial cells, Cambrex). C, summary of the RT-qPCR results obtained in lungs and NSCLC specimens.
We then analyzed the expression of the OTUD6B isoforms in frozen lung specimens obtained from non-cancerous donors and in NSCLC biopsies (11 lungs and 21 NSCLCs, see Supplementary Fig. S8 for representative specimens). Similarly to NSCLC cells, lung cancer specimens expressed significantly less OTUD6B-1 to OTUD6B-2 mRNA ratio (from about 4.4 to about 1.5; Fig. 7C). These results indicate that malignant lung cells and tissues overexpress the OTUD6B isoform that favors increased protein synthesis and proliferation.
Additionally, the relative expression of the two OTUD6B isoforms explains why siRNAs to the OTU domain (siRNA3 and siRNA4; Figure 1 and Supplementary Figure S5) causes a net decrease in protein synthesis: since OTUD6B-1 is quantitatively more expressed than OTUD6B-2 in all cell lines tested, genetic depletion of both isoforms is expected to determine a phenotype predominantly influenced by OTUD6B-1 loss-of-function.
Discussion
Here we report, to our knowledge for the first time, a role for OTUD6B in the regulation of protein synthesis. OTUD6B-1 appears to interact with the protein synthesis initiation complex, and seems to modify it at the level of ubiquitination. Depletion of OTUD6B, although modifying the rate of the methionine surrogate AHA incorporation, was unable to alter critical indicators of the Akt/mTORC-1 axis activity status (29), thus indicating that OTUD6B operates downstream from mTORC-1. On the other hand, complete mTORC-1 inhibition using Torin-1 abolished the effects that OTUD6B depletion elicits on protein synthesis. These observations are consistent with a model whereby mTORC-1 activity is required for the formation of the initiation complex. OTUD6B, through binding and/or modification of some initiation complex components, most likely alters the composition and/or function of the complex itself either after or during its formation.
The two main OTUD6B isoforms seem to have opposing effects on protein synthesis and proliferation, but both require their catalytic activity for function. In normal and transformed cells, the expression of the OTUD6B isoform that represses protein synthesis and proliferation is quantitatively higher. This explains why siRNA to UTUD6B, which is unable to distinguish between the two isoforms, results in enhanced AHA incorporation in many cell lines. These results support a recent study in which cytokine-induced proliferation of B lymphocytes required the loss of OTUD6B expression (16).
Here we show that transformed lung tissues and cells upregulate OTUD6B-2 expression. The decreased OTUD6B-1/OTUD6B-2 ratio seems to promote proliferation in NSCLC cells. NSCLC cells transfected with a siRNA that specifically targets OTUD6B-2 showed reduced DNA incorporation and expression (at the protein level) of c-Myc and Cyclin-D1. The expression of these oncogenes is also regulated at the level of mRNA translation (30, 31). OTUD6B-2 depletion led to reduced expression of both c-Myc and CyclinD-1 proteins without alteration of the expression of their mRNAs, with no indication of augmented Cyclin-D1 degradation. This observation suggests that increased OTUD6B-2 may contribute to qualitative modifications in translation efficiency linked to malignant transformation (5). However, overexpression of OTUD6B-1 did not alter the relative expression of c-Myc or Cyclin-D1. This can be explained by the assumption that the two OTUD6B isoforms may target different substrates. This is supported by results obtained after overexpression of the two isoforms and their mutants in the catalytic core. Neither C188S OTUD6B-1 yielded results similar to OTUD6B-2, nor C57S OTUD6B-2 gave phenotypes similar to OTUD6B-1. Therefore, it is likely that the two isoforms do not function as dominant-negative to each other. Deubiquitinases discriminate their substrates in complex with other cellular proteins (8,9,32). Because of their primary structure, OTUD6B-1 and OTUD6B-2 are likely expressed in different oligomerization statuses. For this reason, the two isoforms most likely segregate into non-identical protein complexes. OTUD6B-2 forced expression in NSCLC cells failed to further increase the relative expression of c-Myc and Cyclin-D1. This may suggest that the abundance of OTUD6B-2 in NSCLC cells favors a qualitative selection of these mRNAs for translation, and any further increase in OTUD6B-2 expression affects quantitative protein synthesis alone. To clarify this issue, and to determine whether OTUD6B-2 may directly or indirectly target c-Myc, future studies in non-transformed cells and tissues will be required.
Isoform switching is a common phenomenon in cancer. For example, the Bcl-x primary transcript can produce two splicing isoforms with opposing effects on apoptosis (namely, a pro-survival isoform, or Bcl-xL, and a shorter Bcl-xS, that promotes cell death; 33). The induction of the transcription factor E2F-1 in NSCLC cells regulates transcription of pro-survival genes, but also determines pre-mRNA splicing events that lead to increased Bcl-xL/Bcl-xS ratio (34). Similarly, enhanced expression of MDM-2 splice variants that promote transformation are found in a variety of human cancers (35, 36).
In conclusion, it appears that the OTUD6B isoforms play important roles in the maintenance of NSCLC proliferation and may be a contributing factor in NSCLC development at the level of protein synthesis deregulation. OTUD6B appears to favor transformation due to the preferential expression of a pro-oncogenic isoform rather than gene mutations or altered net expression of its gene. Data on knock out mice do not rule out such interpretation. OTUD6B does not appear to be required for normal development or survival (37). This is somewhat reminiscent of that observed in E-type Cyclins (E1 and E2) and p53 knock out mice, which do not display gross phenotypic abnormalities (38,39), while these proteins play fundamental roles in oncogenesis. Additional investigations on the possible overlapping function of OTUD6A, which shares a very high level of sequence identity with OTUD6B, is required to clarify whether OTUD6B mirrors a situation similar to that of type E Cyclins.
Currently, protein synthesis initiation inhibitors are being explored as anticancer therapeutics. The experience gathered thus far indicates that complete inhibitors of protein synthesis, albeit efficient, have unacceptable toxicities. This is why most attention is presently focused on compounds able to modulate protein synthesis (e.g., rapamycin-like molecules), rather than completely inhibiting protein synthesis. For this reason, OTUD6B-2 could be a very valuable therapeutic target as its specific inhibition over OTUD6B-1 could restore physiologic protein translation while curtailing malignancy-associated protein synthesis.
Supplementary Material
Acknowledgements
We thank Nicholas Armijo and Jonathan Eby for their summer rotations in our laboratory, Patricia Simms and Ashley Hess for their help with FACS experiments optimization and interpretation, and Levi Barse for critical reviewing this manuscript.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Rhoads RE. Protein synthesis, cell growth and oncogenesis. Curr Opin Cell Biol. 1991;3:1019–24. doi: 10.1016/0955-0674(91)90123-g. [DOI] [PubMed] [Google Scholar]
- 2.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–27. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruggero D. Translational control in cancer etiology. Cold Spring Harb Perspect Biol. 2013;5:1–27. doi: 10.1101/cshperspect.a012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsyan A, Svitkin Y, Shahbazian D, Gkogkas C, Lasko P, Merrick WC, et al. mRNA helicases: the tacticians of translational control. Nat Rev Mol Cell Biol. 2011;12:235–45. doi: 10.1038/nrm3083. [DOI] [PubMed] [Google Scholar]
- 5.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 6.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.013284. M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 9.Sahtoe DD, Sixma TK. Layers of DUB regulation. Trends Biochem Sci. 2015;40:456–67. doi: 10.1016/j.tibs.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Li WX. A novel function of Drosophila eIF4A as a negative regulator of Dpp/BMP signalling that mediates SMAD degradation. Nat Cell Biol. 2006;8:1407–14. doi: 10.1038/ncb1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yanagiya A, Suyama E, Adachi H, Svitkin YV, Aza-Blanc P, Imataka H, et al. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol Cell. 2012;46:847–58. doi: 10.1016/j.molcel.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh D, Mohr I. Coupling 40S ribosome recruitment to modification of a cap-binding initiation factor by eIF3 subunit e. Genes Dev. 2014;28:835–40. doi: 10.1101/gad.236752.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharya S, Ghosh MK. Cell death and deubiquitinases: perspectives in cancer. Biomed Res Int. 2014;2014:435197. doi: 10.1155/2014/435197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther. 2015;147:32–54. doi: 10.1016/j.pharmthera.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–84. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Z, Zheng Y, Zhu Y, Kong X, Hu L. Evidence for OTUD-6B participation in B lymphocytes cell cycle after cytokine stimulation. PLoS One. 2011;6:e14514. doi: 10.1371/journal.pone.0014514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graziani I, Eliasz S, De Marco MA, Chen Y, Pass HI, De May RM, et al. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008;68:9678–85. doi: 10.1158/0008-5472.CAN-08-0969. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, De Marco MA, Graziani I, Gazdar AF, Strack PR, Miele L, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67:7954–9. doi: 10.1158/0008-5472.CAN-07-1229. [DOI] [PubMed] [Google Scholar]
- 19.Rivera MC, Maguire B, Lake JA. Isolation of ribosomes and polysomes. Cold Spring Harb Protoc. 2015;2015:293–9. doi: 10.1101/pdb.prot081331. [DOI] [PubMed] [Google Scholar]
- 20.Sobol A, Galluzzo P, Liang S, Rambo B, Skucha S, Weber MJ, et al. Amyloid precursor protein (APP) affects global protein synthesis in dividing human cells. J Cell Physiol. 2015;230:1064–74. doi: 10.1002/jcp.24835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sobol A, Galluzzo P, Weber MJ, Alani S, Bocchetta M. Depletion of Amyloid Precursor Protein (APP) causes G0 arrest in non-small cell lung cancer (NSCLC) cells. J Cell Physiol. 2015;230:1332–41. doi: 10.1002/jcp.24875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Nair SJ, Lescarbeau A, Belani J, Peluso S, Conley J, et al. Synthetic silvestrol analogues as potent and selective protein synthesis inhibitors. J Med Chem. 2012;55:8859–78. doi: 10.1021/jm3011542. [DOI] [PubMed] [Google Scholar]
- 23.Gurel G, Blaha G, Moore PB, Steitz TA. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009;389:146–56. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. http://cancer.sanger.ac.uk/cosmic.
- 26. http://cancergenome.nih.gov.
- 27.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 28.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–21. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 29.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–9. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito H, Hayday AC, Wiman K, Hayward WS, Tonegawa S. Activation of the c-myc gene by translocation: a model for translational control. Proc Natl Acad Sci U S A. 1983;80:7476–80. doi: 10.1073/pnas.80.24.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenwald IB, Lazaris-Karatzas A, Sonenberg N, Schmidt EV. Elevated levels of Cyclin D1 protein in response to increased expression of eukaryotic initiation factor 4E. Mol Cell Biol. 1993;13:7358–63. doi: 10.1128/mcb.13.12.7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfoh R, Lacdao IK, Saridakis V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr Relat Cancer. 2015;22:T35–54. doi: 10.1530/ERC-14-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang BS, Kelekar A, Harris MH, Harlan JE, Fesik SW, Thompson CB. The BH3 domain of Bcl-x(S) is required for inhibition of the antiapoptotic function of Bcl-x(L) Mol Cell Biol. 1999;19:6673–81. doi: 10.1128/mcb.19.10.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merdzhanova G, Edmond V, De Seranno S, Van den Broeck A, Corcos L, Brambilla C, et al. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008;15:1815–23. doi: 10.1038/cdd.2008.135. [DOI] [PubMed] [Google Scholar]
- 35.Sigalas I, Calvert AH, Anderson JJ, Neal DE, Lunec J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: transforming ability and frequent detection in human cancer. Nat Med. 1996;2:912–7. doi: 10.1038/nm0896-912. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto R, Tada M, Nozaki M, Zhang CL, Sawamura Y, Abe H. Short alternative splice transcripts of the mdm2 oncogene correlate to malignancy in human astrocytic neoplasms. Cancer Res. 1998;58:609–13. [PubMed] [Google Scholar]
- 37.Gardin A, White J. The Sanger mouse genetics programme: high throughput characterisation of knockout mice. Acta Ophtalmol. 2010;88:s246. [Google Scholar]
- 38.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 39.Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, et al. Cyclin E ablation in the mouse. Cell. 2003;114:431–43. doi: 10.1016/s0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.