

Abstract
Lipolysis Stimulated Lipoprotein Receptor (LSR) has been found in the plasma membrane and is believed to function in lipoprotein endocytosis and tight junctions. Given the impact of cellular metabolism and junction signaling pathways on tumor phenotypes and patient outcome, it is important to understand how LSR cellular localization mediates its functions. We conducted localization studies, evaluated DNA binding, and examined the effects of nuclear LSR in cells, xenografts, and clinical specimens. We found LSR within the membrane, cytoplasm, and the nucleus of breast cancer cells representing multiple intrinsic subtypes. Chromatin immunoprecipitation (ChIP) showed direct binding of LSR to DNA, and sequence analysis identified putative functional motifs and post-translational modifications of the LSR protein. While neither overexpression of transcript variants, nor pharmacological manipulation of post-translational modification significantly altered localization, inhibition of nuclear export enhanced nuclear localization, suggesting a mechanism for nuclear retention. Co-immunoprecipitation and proximal ligation assays indicated LSR-pericentrin interactions, presenting potential mechanisms for nuclear-localized LSR. The clinical significance of LSR was evaluated using data from over 1,100 primary breast tumors, which showed high LSR levels in basal-like tumors and tumors from African-Americans. In tumors histosections, nuclear localization was significantly associated with poor outcomes. Finally, in vivo xenograft studies revealed that basal-like breast cancer cells that over-express LSR exhibited both membrane and nuclear localization, and developed tumors with 100% penetrance, while control cells lacking LSR developed no tumors. These results show that nuclear LSR alters gene expression and may promote aggressive cancer phenotypes.
Keywords: Breast cancer, LSR, subcellular localization, pericentrin, tumorigenesis
Introduction
Tumor pathogenesis results from a complex integration of genetic and environmental stimuli. Understating how these oncogenic factors unite to drive the phenotypic characteristics of cancer cells is a critical goal for the development of successful targeted therapies. Recent evidence suggests that the cell surface protein Lipolysis Stimulated Lipoprotein Receptor (LSR) may relay environmental cues to influence breast cancer cell behavior (1-4). LSR has two proposed functions in healthy tissue, hepatic postprandial clearance of triglyceride-rich lipoproteins from blood and mediation of tricellular junctions (tTJ) (5-8).
Experimental studies have shown that LSR plays an important role in lipid metabolism. Complete inactivation of Lsr is embryonic lethal in mice, but removal of a single allele (Lsr-/+) significantly increases plasma triglyceride and cholesterol levels and delays clearance of lipid emulsions after high fat meals (4). LSR is upregulated by leptin and mediates the binding of apoB- and apoE-containing lipoproteins in hepatocytes, leading to their internalization and degradation (4, 5, 9, 10). While the most prominent metabolic alterations in cancer are increased glucose uptake and the use of aerobic metabolism, other metabolic processes including lipid endocytosis and de novo macromolecule synthesis have been observed in cancer-associated metabolic reprogramming (11).
The role of LSR in cell adhesion is similarly well-established. LSR regulates tTJs (8, 12, 13), specialized structural elements within epithelial tricellular contacts. LSR regulates tTJs by recruiting tricellulin to junctions, along with other proteins including ILDR1 and ILDR2 (14). While one would expect cellular junction dissolution/down-regulation as a prerequisite for cellular transformation, and where experimental evidence suggests that TJ/tTJ proteins play dual functional roles in the tumorigenic process (15-17). De-regulation of claudins-3, -4 and -7 in breast cancer is subtype-specific and has been shown to be correlated with aggressive tumor cell behaviors.
Our previous data suggest that LSR's function is cell type- and context-dependent. We have shown that LSR expression is higher in invasive ductal carcinomas compared to invasive lobular carcinomas, and is higher in estrogen receptor α positive (ER+) tumors and cell lines. However, reintroduction of LSR into claudin-low breast cancer cell lines altered gene expression, enhanced cellular proliferation, and increased survival in anchorage independent conditions, highlighting that LSR signaling promoted aggressive/breast cancer stem cell-like behaviors (1). Our previous mechanistic studies complement reports linking LSR to human breast cancer stem cell function and metastatic signatures in two breast cancer models (3, 18). In triple-negative breast cancer cell lines, the CD44hi/CD24lo basal A cells contained high levels of LSR compared to all other populations, and that these cells retained classic cancer stem cell features, such as tumor-initiating capacity in vivo, mammosphere formation and resistance to standard chemotherapy. Further, LSR was identified an integral component of a 31-gene signature capable of predicting distant metastasis in cohorts of ER-negative human breast cancers (3). LSR has also recently been proposed as a poor prognostic plasma biomarker in colon cancer patients (19).
Given the importance of cancer stem cell populations, cell metabolism, and tight junctions in understanding cancer progression, we sought to identify mechanisms of LSR function in breast cancer. Herein, we investigate cellular localization and genomic DNA binding patterns of LSR. We identify downstream functional pathways as well as evaluate transcript variants and post-translational modifications of LSR for effects on cellular localization and function. Finally, we test the effects of LSR expression on tumorigenesis in vivo, and examined the expression and localization of LSR in association with tumor characteristics and patient outcomes.
Materials and Methods
External Dataset analysis
All TCGA data originated from primary breast tumors (N=1097), metastatic tumors (N=7), and adjacent normal tissue (N=114) and were retrieved from the TCGA Data Portal and analyzed using R (V2.13.1). Box and whisker plots were used to visualize differences in expression of LSR by subtype and two-way ANOVAs or t-tests were used to evaluate statistical significance (20, 21). Mutations across TCGA tumor types were identified from the cBioPortal.
GEO accession GSE16997 (22) was downloaded and LSR transcript abundance was compared in luminal progenitor epithelial samples (N=3) with basal/stem cell epithelial samples (N=3) using a linear model. Using the processed hybridization intensities downloaded, we computed expression changes between treatment groups via an empirical Bayes moderated paired t-test (23) using the Bioconductor package limma. Candidate genes for differential expression with a P-value of less than 0.05 were selected. To account for multiple testing, we computed the false discovery rate of the extracted candidate list from the p-value distribution of the corresponding genes using the Bioconductor package qvalue (10% FDR cutoff). Using the same method, we compared the expression of LSR between ALDH+ luminal breast epithelial samples (N=13) and basal breast epithelial tissue samples (N=12) obtained from GEO accession GSE35399 (24).
Immunofluorescence and Proximal Ligation Assay
Tumor biopsies were obtained and analyzed in accordance with the guidelines of the North Carolina Central Institute Review Board, protocol 1201027. Immunofluorescence was performed with appropriate controls as described (1, 2, 25). Anti-LSR antibodies; Atlas Antibodies and Santa Cruz Biotechnology. Imaging was performed on a Nikon Eclipse 50i microscope with digital camera and NIS-Elements 4.11.00. Nuclear localization confirmed via Zeiss 710 laser scanning confocal microscope. Cell culture experiments utilized 8-well chamber slides for cell growth/treatment prior to immunofluorescence. Proximal ligation assays were performed following the Duolink® Starter Kit (Invitrogen) as instructed using either the Atlas Antibodies or the Santa Cruz Biotechnology with the mouse monoclonal pericentrin antibody (Santa Cruz).
Cell culture and treatments
Cells were obtained from American Type Culture Collection (Manassas, VA) or Asterand (Detroit, MI) with the provided authentication documents (2015), and cultured as directed by manufacturers. Primary breast epithelial cells, FMEC2, were previously characterized and cultured as described (25, 26). Overexpression vectors were obtained from Origene (Rockville, MD) and cell transfected as described (1, 2). Proliferating cells were treated with Leptomycin B (37nM) one h prior, or 2-bromopalmitate (27-29) (10 and 100μM) 30min prior to treatment, fixation and imaging as described (1, 2, 25).
Subcellular fractionation, immunoprecipitations, and western analysis
Subcellular fractionation was performed using Qproteome®Cell Compartment kit (Qiagen, Velencia, CA) as instructed. Cells were lysed in mPer Lysis Buffer (Thermo Scientific, Rockford, IL) supplemented with protease and phosphatase inhibitors (Halt™ Thermo Scientific) then subjected to western analyses as described (1, 2, 25). Antibodies: Atlas Antibodies, LSR; Santa Cruz Biotechnology, LSR, vimentin, GAPDH, Sp3, tubulin; Life Technologies (Grand Island, NY), pSeR; Origene (Rockland, MD), VDAC. Immunoprecipitation was performed as described (30).
Chip-Seq
Libraries were created from 10ng of LSR ChIP and 10ng of corresponding input from MCF7 cells according to manufacturer's protocol (Illumina, San Diego, CA). Two independent ChIP-seq were performed and analyzed by scientists at the David H. Murdock Research Institute via the Illumina HS2000 sequencing platform, paired-end read, multiplex, 50×. Data were also assessed for ChIP-Seq quality as outlined by the ENCODE and modENCODEconsortia. Reads were trimmed based on a quality limit of 0.05 using CLC Genomics Workbench v 8.0 (http://www.clcbio.com), and mapped to the reference human genome (hg19/GRCh37; Ensembl) in CLC Genomics Workbench (peak calling tool was used to make peak calls using control input data with a maximum P-value<0.05). Sequences were extracted underneath called peaks then passed through TRANSFAC (http://www.biobase-international.com) to create scoring matrices used to predict binding motifs. Predicted transcription factor binding sites are based on 5,000+ positional weight matrices derived from experimentally verified data and 3D homology modeling that is kept in BIOBASE. LSR motif was identified using MEME via Expectation Maximization using one occurrence per sequenced distribution (31). Pathway analyses were performed via Ingenuity IPA (http://www.ingenuity.com/).
Liquid chromatography-tandem mass spectrometry (LC MS/MS)
LSR and LSR-associated proteins were co-immunoprecipitated from MCF cell lysates as described (30). A non-specific antibody served as a control and results were checked for background contaminants via the CRAPome (http://www.crapome.org/). Samples were prepared for bottom-up proteomics using the filter assisted sample preparation protocol (32). Briefly samples digested using trypsin, purified, and analyzed by LC MS/MS using a quadrupole orbitrap mass spectrometer operating in a top 12 data dependent mode. Data was searched against the human Uniprot protein database using Protein Discoverer 1.4. Percolator (33) was used to establish a peptide spectral match q value threshold of <0.01.
Computational Predictions
LSR (ENSG00000105699) transcript variant sequences were compiled from Ensembl, GenBank, and UniProt databases. MWs were calculated using ProtParam (34). Sequences were also analyzed using MOTIF (http://www.genome.jp/tools/motif/). NetPhos was used to predict phosphorylation of serines, threonines, and tyrosines(35), sumoylation sites via GPS-SUMO(36), C-mannosylation sites via NetCGlyc(37), s-nitrosylation via GPS-SNO(38), palmitoylation sites via CSS-Palm(39), and the Sulfinator program was used to predict tyrosine sulfation(40). For evolutionary conservation analysis, sequences were obtained from the NCBI database. Full-length protein sequences of forty animal species were used, including human (Homo sapiens), 37 mammalian species, western painted turtle (Chrysemys picta bellii), and zebrafish (Danio rerio). First, a seed cluster was aligned using Clustal Omega with human, chimpanzee and gorilla sequences. Then, the remaining sequences were iteratively added to the alignment. The final multi-species alignment was visualized and edited using Geneious 7.1 (Biomatters, Aukland, New Zealand). Graphs were prepared using R (V2.13.1).
Xenografts
Animal experiments were conducted in accordance with accepted standards of humane animal care and approved by the Animal Care and Use Committee at North Carolina Central University. Xenografts were generated using 5-week-old female Hsd:Athymic Nude-Foxn1nu mice (Harlan Laboratories, Indianapolis, IN) as described (41) using 5×105 control Hs578t cells or Hs578t LSRδ-overexpressing cells (1) suspended in 30μl of 50/50 PBS:Matrigel. Weekly tumor growth was measured and tumors excised when volume neared 1.0cm3. Tumor fragments were formalin-fixed for immunohistochemical analysis. Three independent experiments were performed to ensure repeatability. Mice that did not form tumors were euthanized five months post-injection.
Results
LSR heterogeneity in breast cancer
LSR has been proposed as a component of breast cancer stem/progenitor cells. This suggests that the functional heterogeneity of LSR we have observed within ER+ and ER- breast cancer cell populations may arise from paths of differential lineages together with microenvironmental cues. Current studies support that both ER+ and ER- tumors arise from ER+ progenitor cells, and others and we have identified LSR in both ER+ and ER- tumors and breast cancer cell lines (22, 24). Thus, we investigated the association of LSR with luminal progenitor cell populations in two prominent reports. Using the data from Lim et al., defining the basal stem/progenitor, luminal progenitor and mature luminal cells isolated from reduction mammoplasty tissue (22), we computed expression changes between treatment groups via an empirical Bayes moderated paired t-test. We found higher expression of LSR in luminal progenitor epithelial cells as compared to the basal/stem cell epithelium (P=0.0002, adjusted P=0.004) using 10% FDR cutoff. Correspondingly, using the data from Shehata et al., (24) we found increased LSR expression in the undifferentiated (EpCAM+CD49f+ALDH+) luminal epithelial progenitors compared to the differentiated and basal cell populations (P=0.004, adjusted P=0.06; 10% FDR cutoff). Last, QPCR evaluation of genes classically associated with luminal progenitor cells and breast cancer stem cell phenotypes (Elf5, MDR1, CD133, FASN, and MUC1) show significantly higher expression in MCF7 cells with high levels of LSR and Hs578t cells overexpressing LSR compared to MCF7 cells with low/undetectable levels and parental control Hs578t cells (Supplementary Fig.S1). These data suggest that the retention or loss of LSR in breast tumor cells may arise from the stage of differentiation at which transformation occurs. Our previous work clearly shows that the functional consequence of LSR expression in claudin-low breast cancers is a highly aggressive, cancer stem cell like phenotype in vitro and in vivo. However, the mechanisms which dictate the subtype-specific, diverse functions of LSR, including regulation of cancer stem cell features, tight junctions, or cell bioenergetics remains to be elucidated.
Subcellular localization
Our previous studies focused on the dominant subcellular localization of LSR on the membrane (1). However, we have also observed LSR localization in the nucleus and other have reported LSR in the nuclear fraction of human epithelial cells (32). As subcellular localization dictates the function of many molecules, we further investigated this observation. We evaluated LSR localization via immunocytofluorescence in breast cancer cell lines (Fig.1A). LSR was observed within the membrane, cytoplasm, and also the nucleus of MCF7 (luminal subtype; ER+/PR+), MDA-MB-468 and SUM1315mo breast cancer cells (basal-like subtype; ER-/PR-), as well as non-transformed breast epithelial cell lines (MCF10A1, FMEC2, HMEC); z-stacks further confirmed nuclear localization (Supplementary Fig.S2), and Hs578t cells served as a negative control for LSR expression. We next performed subcellular fractionation followed by immunoblotting. LSR was expressed in membrane, cytosolic, and nuclear fractions of in all positive cell lines tested (Fig.1B-C). Subcellular-specific proteins SP3 (nuclear), VDAC (membrane), GAPDH (cytosol), and vimentin (cytoskeleton) validated successful separation. These data suggest that LSR may perform a nuclear function in a range of breast epithelial cells.
Figure 1.

Subcellular localization of LSR. A, Representative images of breast cancer cells (MCF7, SUM315mo, MDA-MB-468) and non-transformed normal breast epithelial cells (MCF10AI, FMEC2, HMEC) subjected to immunofluorescence using a LSR specific antibody (DNA stained with DAPI). Immunofluorescence was performed using an anti-LSR primary antibody and an anti-rabbit Alexa Fluor-488 labeled secondary antibody (LSR, Alexa-488/green; DNA, DAPI/blue). Hs578t that do not express LSR were used as a control. Images were obtained at 60X. B, C, Representative western blots of cell lysate separated by cellular compartment (nuclear, membrane, cytosol, cytoskeleton) through subcellular fractionation. Resulting fractions were subjected to western immunoblotting to detect expression of subcellular compartment specific proteins (B, MCF7 representative fractionation) or western immunoblotting for LSR (C, representative cell lines). Data represent a minimum of three independent experiments.
Transcriptome regulation
To investigate a potential nuclear role for LSR, MCF10A1, MCF7, and SUM1315mo cells were treated with formaldehyde to cause DNA-crosslinking to test if LSR bound to DNA, alone or in a complex. We found a significant increase in the amount of LSR bound to DNA in all three cell lines compared to controls (Fig.2A). We then identified specific LSR targets by sequencing (ChIP-seq). Analysis of LSR localization revealed that 90.4% of the sites were in distal intergenic regions (>3000 bp from the transcription start site), 6.3% were within introns, and 0.5% were located in regions <1000 bp from the promoter. Only 0.1% of the sites were within the first 3000 bp 5′ from the start site of genes, the traditional promoter regions (Fig.2B). Examination of the sequences within the peaks using MIME (31) identified a LSR DNA binding sequence motif (E-value of 10-1732, Fig.2C). The LSR-target genes identified were further evaluated via Ingenuity Pathway analysis using the criteria that they contained at least one LSR binding site within all samples evaluated and had a P-value less than 9E-06. The pathways identified to be significantly associated with LSR-target genes included oxidative phosphorylation and mitochondrial dysfunction (Fig.2D). These analyses are complementary to previous findings of a role for LSR in metabolism and further a role in metabolic, developmental and hereditary disorders (Supplementary Figs.S3-S4). Confirmation of LSR target genes was performed via ChIP-QPCR analysis of the top three LSR target genes (Supplementary Fig.S5). These data suggest nuclear LSR function may relate to the reported metabolic functions of LSR while membrane localization relates to aspects of tight junction formation.
Figure 2.

LSR binds to genomic sites in breast cancer cells. A, MCF10A1, MCF7, and SUM1315mo cells were cross-linked with 1% formaldehyde (X-link), or PBS as control (C). Isolation of DNA was performed by ethanol precipitation. Detection of LSR protein was determined by western immunoblotting. Data is expressed as the mean ± SD from three independent experiments. *P<0.05, ***P<0.001. B, ChIP-seq was performed in MCF7 cells for identification of LSR binding to genomic sites. MIME analysis of genomic sequences associated with LSR binding sites in breast cancer cells identified the LSR binding motif. C, Genomic distribution of LSR binding sites relative to genes in breast cancer cells. D, Ingenuity Pathway Analysis of three canonical pathways associated with LSR target genes.
Clinicopathological features of LSR expression in primary breast tumors
We previously reported high LSR expression in less aggressive ER+ tumors and cell lines (1). However, others and we also show LSR expression in ER- breast cancer cells promotes classic cancer stem/progenitor cell features and that LSR is an integral component of a 31-gene signature capable of predicting distant metastasis in cohorts of ER- breast cancers. (1, 3, 18). We sought to determine if these disparate roles were a function of clinicopathological characteristics. Previous studies were limited to cells grown in 2D culture, observations using tissue microarrays, and testing limited datasets of ER- tumors, yet structural, environmental or genetic factors might dictate LSR localization and/or specify function.
To comprehensively examine clinicopathological characteristics associated LSR expression in breast tumors, we analyzed data from 1104 primary and metastatic tumors and 114 adjacent normal tissues within the TCGA breast cancer database. LSR expression was significantly increased in all intrinsic breast cancer subtypes compared to adjacent normal tissues, with the highest expression in basal-like tumors (Fig.3A). We further evaluated the available clinical features and found LSR expression to be significantly higher in African-American patients compared to Caucasians (Fig.3B). No significant correlation was found with other features (age/menopause status, Supplementary Fig.S6). Further examination of race within each subtype shows significant differences for luminal A and Luminal B subtypes (Fig.3C). In contrast, race was not associated with differences in LSR expression among basal-like, Her2-enriched, or normal-like tumors (Supplementary Fig.S7A). Thus, the association between race and LSR expression may be mediated by breast cancer subtype.
Figure 3.
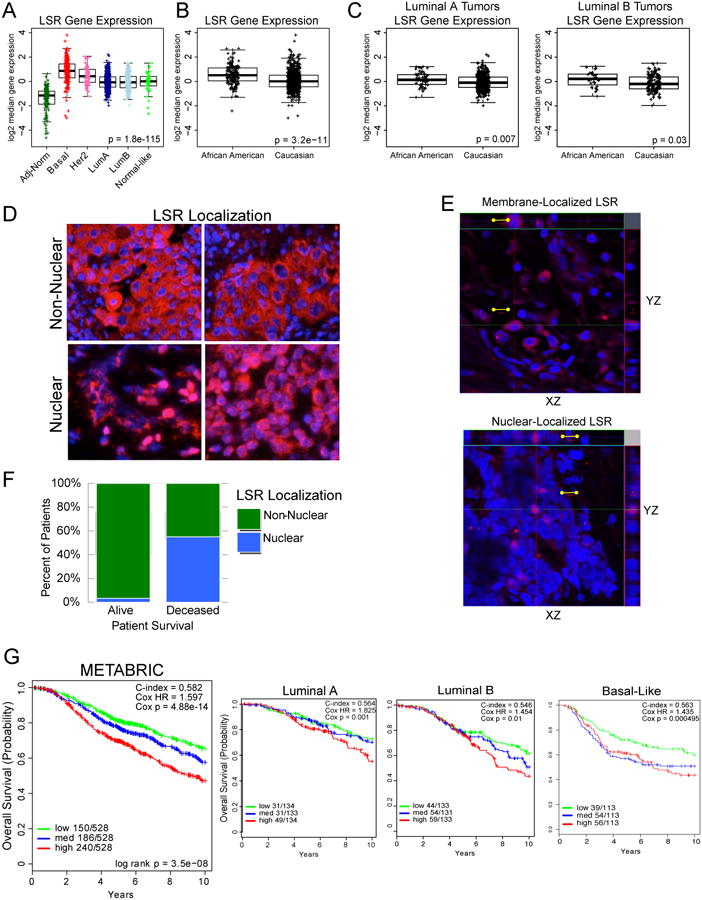
LSR gene expression varies by clinical features in breast cancer. A, Analysis of LSR expression in TCGA breast cancer samples and corresponding adjacent normal breast tissue (N=1,218). B, Data from breast cancer samples were separated by race then evaluated for LSR expression, then data were stratified by breast cancer subtype within each race and LSR expression evaluated (C). Adj-norm, cancer adjacent pathologically normal breast tissue; Basal, basal-like; Her2, Her2-Enriched; LumA, Luminal A; LumB, Luminal B. D, Localization of LSR in breast tumor biopsies and association with patient survival. Histosections of breast tumor samples were examined by immunofluorescence using an LSR-specific antibody and an anti-rabbit Alexa Fluor-594 labeled secondary antibody (LSR, Alexa594/red; DNA, DAPI/blue) and confocal microscopy to establish LSR localization. Top panels show representative images of exclusive membrane and cytoplasmic localization of LSR, bottom panels are representative images of nuclear-localized LSR. All images were obtained at 40×. E, Representative Z-stack projections of primary breast cancer tumors generated from deconvolved slices using the maximum intensity criteria. Top panel image indicates membrane localization of LSR and bottom panel shows nuclear localization. Scale bars, 10 μm. F, Data show percent of patients with ten-year survival data that had nuclear (blue) or non-nuclear (green) LSR expression. (Chi-square, P=5.82x10-8). G, LSR expression associated with overall survival within the METABRIC breast cancer sample datasets. Data was analyzed within all samples and in subsets of Luminal A, Luminal B, and Basal-like subtypes.
Using 80 tumor biopsies from commercially available sources and the Perou lab at the University of North Carolina, Chapel Hill, we evaluated LSR in vivo tumor localization via immunofluorescence (Fig.3D,E). In all samples, LSR was present in the plasma membrane and cytoplasm; however, we also discovered LSR localized to the nucleus in a subset of samples (N=13, nuclear localization further confirmed via z-stacks; Fig.3E). Nuclear localization was scored as positive in each field imaged when a minimum of two cells contained a nucleus foci stained for LSR. Negative staining was determined by the lack of any red fluorescence when the plane of focus was centered on the nucleus. Samples that exhibited distinct nuclear localization were significantly more likely to have died (Fig.3F, N=80; Chi-square, P=5.821×10-8). Fifty-five percent of deceased patients presented nuclear localized LSR compared to only 3.3% of samples from surviving patients. Sample size and the number of clinical details were not sufficient to determine associations with outcome or other clinicopathological features of the tumors imaged. To overcome this limitation we further investigated LSR expression with outcome via a large independent breast cancer dataset using mRNA levels of LSR (METABRIC; Fig.3G). There was a significant correlation of poor survival with high LSR expression (P=3.49E-08). Consistent with TCGA results when stratified by race, a significant difference was observed for overall survival within Luminal A (HR=1.83, cox p=0.001), Luminal B (HR=1.45, cox p=0.01) and Basal-like tumors (HR=1.45, cox p=0.0005). Together with previous studies, these data suggest that that high LSR levels in ER+ tumors potentially indicate a higher proportion of progenitor-like tumor cells that may promote higher rates of aggressive behaviors, drug resistance, metastases, and/or recurrence, leading to the observed poor survival in patients with tumors containing high LSR levels. Overall survival was not associated with differences in LSR expression among Her2-enriched tumors (Supplementary Fig.S7B). Of note, these data are limited to LSR transcript abundance and LSR localization cannot be determined and compared with survival.
LSR enhances tumorigenesis in vivo
Only a subset of patient tumor biopsies tested positive for nuclear expression, while a larger portion of cell lines grown in culture showed nuclear localized LSR. We recognize that the subcellular localization of LSR and subsequent downstream physiological effects may be dictated by limitations within 2D culture systems. Accordingly, to examine LSR localization and function in a controlled 3D tissue microenvironment, we utilized breast cancer xenograft models. Others have shown LSR is present in ER- breast tumor initiating cells, but never directly tested the requirement of LSR during tumorigenesis. Thus, we used Hs578t breast cancer cells (i.e. a LSR negative cell line) and transfected the cells with LSRδ or control plasmid and injected into the abdominal mammary glands of female nude mice (Fig.4). In three independent experiments, all mice injected with Hs578t LSRδ-overexpressing cells (variant contains all three complete NES; Fig5A) developed tumors, compared to none of the mice in the control group (Fig4A,B, P<0.05, two-sample proportion test). It is of note that tumors generated from cells overexpressing LSR induced high levels of angiogenesis and the generation of an intra-tumor leaky vasculature. The localization and expression of LSR from the xenografts was evaluated by immunofluorescence and confocal microscopy, as well as immunohistochemistry (Fig4C,D). LSR was readily detectable in the cell membrane, cytoplasmic and nucleus, similar to cells grown in culture (Figure 4C,D). Further, evaluation of MCF7 xenograft tumors with high endogenous LSR levels also showed nuclear, cytoplasmic and nuclear LSR localization (data not shown). Pathological evaluation of the tumors did not reveal any significant correlation of nuclear LSR expression with location within tumor (leading edge vs. tumor core) or phenotypic landmarks (endothelium, stromal cells, necrosis).
Figure 4.
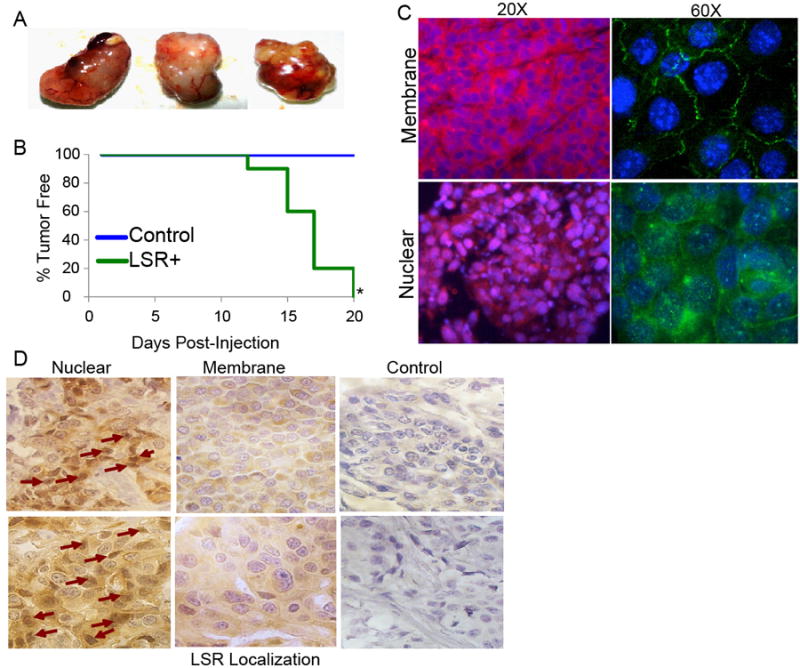
Effect of LSR on tumorigenesis in vivo. A, Representative images of tumor xenografts. Mice were injected into the right abdominal mammary gland with 5.0 × 105 Hs578t cells transfected with control or LSRδ overexpression vectors in 25% Matrigel. B, Tumor incidence, data from one representative experiment of three independent experiments, n=8-10 mice per treatment group. *P<0.001. C, Histosections of xenograft tumor samples were examined by immunofluorescence using an LSR-specific antibody and confocal microscopy to determine LSR localization. Immunofluorescence was performed using an anti-LSR primary antibody and an anti-rabbit Alexa Fluor-594 (left) or Alexa Fluor-488 (right) labeled secondary antibody. (LSR, Red [left] /Green [right]; Blue/DAPI, nucleus/DNA). Top panels show representative images of membrane/cytoplasmic exclusive LSR localization, bottom panels are representative images of nuclear-localized LSR at the two indicated magnifications. D, Histosections of xenograft tumor samples were examined by immunohistochemistry to examine regions of nuclear and membrane localized LSR. (Brown, LSR; Blue, nucleus/DNA). Arrows indicate potential nuclear localized LSR. Images obtained at 40×.
Figure 5.
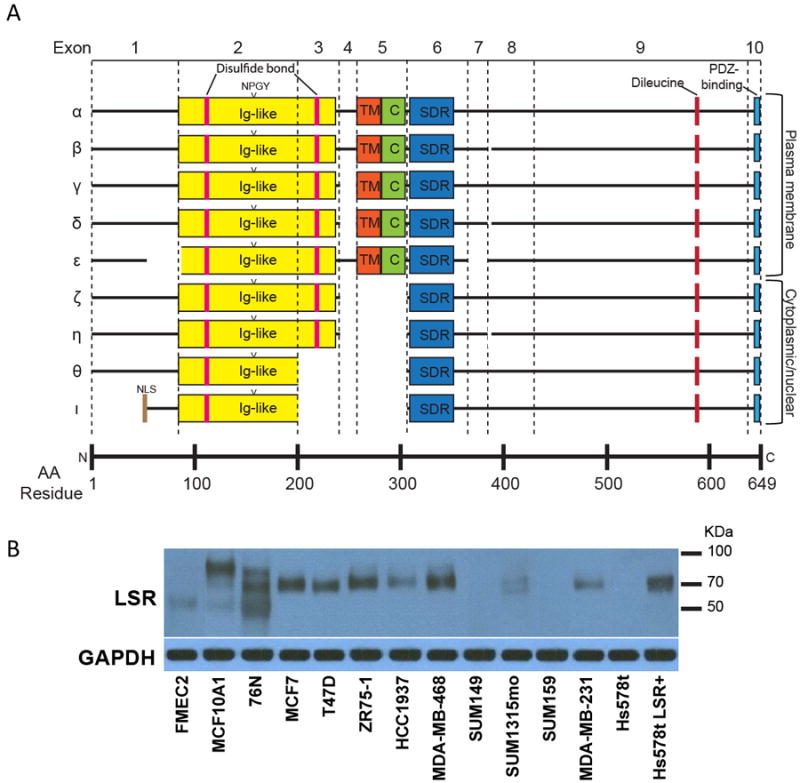
Potential LSR transcript variants. A, LSR transcripts α-ι were identified from online databases and are shown with putative structural and protein-binding domains. Most LSR transcript variants appear to be the result of alternative splicing of particular exons. Ig-like – immunoglobulin-like; NLS – nuclear localization sequence; TM – transmembrane; C – cysteine-rich; SDR – short-chain dehydrogenase/reductase. B, Fourteen cell lines were examined for the expression of LSR (top panel) and a GAPDH loading control (bottom panel) by western blot analysis. Different cell lines expressed LSR at varying levels and at different molecular weights when using a polyclonal antibody that recognizes all predicted isoforms of LSR. Immunoblots are representative of a minimum of three independent experiments.
Analysis of LSR Transcript Variants and Domain Mapping
Similar to breast biopsies, LSR was found in both the membrane and nucleus of cells throughout the xenograft tumors suggesting tissue architecture is not the primary driver of LSR localization. We next investigated autonomous mechanisms that may regulate specific subcellular localization of LSR. The full-length human LSR protein contains 649 amino acids with an estimated MW of 71.4 kDa. Transcript variants of LSR were compiled from Ensembl, GenBank, and UniProt, screened for a complete coding sequence and, where possible, known protein product. In total, nine putative transcript isoforms were identified (Table 1, Fig.5A) and named in order of decreasing protein length using a Greek letter convention, α through ι. Most variants appear to be due to alternative splicing of specific coding exons, notably exons 3, 4, 5, and 7. Multiple transcripts (β, δ, and η) lack a glutamate at position 386 (E386) due to an alternate splice acceptor site. Transcript ε, in addition to omission of exon 7, lacks amino acids 52-88 encoded by exon 1. Lastly, transcript ι features a possible alternative translation initiation codon, beginning with the methionine at position 49 (M49).
Table 1.
Summary information of LSR splice variants.
| Splice Variant | Ensembl Name Transcript ID Protein ID | GenBank Name Protein ID | UniProt Name Identifier | Variant Description* | AA Missing† | Size (AA) | Predicted Molecular Weight (kDa)‡ |
|---|---|---|---|---|---|---|---|
| α | LSR-003 ENST00000361790 ENSP00000354575 |
Isoform 2 NP_991403.1 |
Isoform 1 Q86X29-1 |
Full-length | - | 649 | 71.4 |
| β | Isoform X1 XP_005259037.1 |
Alternate splice acceptor site for intron 7/exon 8 | 386 | 648 | 71.3 | ||
| γ | LSR-012 ENST00000354900 ENSP00000346976 |
Isoform 1 NP_057009.3 |
Isoform 4 Q86X29-4 |
Exon 4 spliced out | 240-258 | 630 | 69.6 |
| δ | LSR-001 ENST00000602122 ENSP00000472569 |
Isoform 4 NP_001247418.1 |
Isoform 3 Q86X29-3 |
Exon 4 spliced out; alternate splice acceptor site for intron 7/exon 8 | 240-258, 386 | 629 | 69.4 |
| ε | LSR-002 ENST00000347609 ENSP00000262627 |
Isoform 2 Q86X29-2 |
Missing AA within exons 1 and 2; exon 7 spliced out | 52-88, 366-386 | 591 | 65.6 | |
| ζ | LSR-004 ENST00000360798 ENSP00000354034 |
Isoform 3 NP_991404.1 |
Isoform 5 Q86X29-5 |
Exons 4 and 5 spliced out | 240-308 | 581 | 64.1 |
| η | Isoform X3 XP_005259039.1 |
Exons 4 and 5 spliced out; alternate splice acceptor site for intron 7/exon 8 | 240-308, 386 | 580 | 63.9 | ||
| θ | Isoform 5 NP_001247419.1 |
Exons 3, 4, and 5 spliced out | 200-308 | 541 | 59.8 | ||
| ι | LSR-201 ENST00000427250 ENSP00000394479 |
Alternate start codon; Exons 3, 4, and 5 spliced out | 1-48, 200-308 | 493 | 54.5 |
AA – amino acids;
numbering the 10 coding exons of LSR as 1-10;
compared to full-length splice variant (649 AA);
calculated using ProtParam.
We quantified LSR variant expression via protein molecular weight in a panel of normal breast and breast cancer cell lines using a polyclonal LSR antibody that binds an epitope common to all LSR variants. We were unable to definitively decipher individual isoforms, however, three distinct bands were observed; approx. 70 kDa, 65 kDa, and in MCF10AI and 76N cells a third near 55 kDa (Fig.3B). In support, RNA-seq data from MCF7, MDA-MB-231, SUM149, and SUM159 suggest the presence of six isoforms (Supplementary Fig.S8; uc002nyn.2, uc010xsr.1, uc002nym.2, uc002nyp.2, uc002nyl.2, uc002nyo that corresponds to LSR variants α/β, θ, γ, ε, α/β, δ, respectively). The projected molecular weight of these isoforms (59.8 to 71.4 KDa, Table 1) corresponded to the molecular weights observed by western analysis in the MCF7 and MDA-MDB-231 cell lines (Fig.5B). While LSR transcript variants were present in SUM149 and SUM159 cells, no detectable protein levels could be detected via western immunoblotting (Fig.5B). Collectively, these data suggest the presence of multiple LSR mRNA/protein isoforms, however, the potential also exists for post-translational modifications of the protein.
As either post-translational modifications or unique variants may provide insight in the diverse functions of LSR, we further investigated LSR for potential structural domains. The UniProt database was used to identify potential structural domains (Table 2). To evaluate domains of particular functional significance, we aligned the full-length human LSR protein sequence with the full-length sequences from 39 other vertebrate species; the subsequent BLAST search did not reveal any more distantly related species, suggesting that LSR evolved in vertebrates with an overall 77.4% sequence conservation. The Ig-like, cysteine-rich, short-chain dehydrogenase/reductase (SDR), and PDZ-binding domains showed high conservation across species (Table 2, Supplementary Fig.9A). Regions without known function in the cytoplasmic domain also displayed high levels of evolutionary conservation.
Table 2. Predicted Functional Domains of LSR Transcription Variants.
| Domain | AA Positions | Avg. Evolutionary Conservation (%) | Present in Transcript Variant | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| α | β | γ | δ | ε | ζ | η | θ | ι | |||
| Extracellular | 1-259 | 71.2 | C | C | C | C | P | P | P | P | P |
| Signal peptide | 49-90 | 53.9 | A | A | A | A | A | A | A | A | C |
| Nuclear export | 77-155 | 86.8 | C | C | C | C | P | C | C | C | C |
| Ig-like | 86-234 | 93.6 | C | C | C | C | P | C | C | P | P |
| SUMO-interaction | 100-104 | 99.5 | C | C | C | C | C | C | C | C | C |
| Disulfide bond | 111 | 92.5 | C | C | C | C | C | C | C | C | C |
| SUMO-interaction | 123-127 | 96.5 | C | C | C | C | C | C | C | C | C |
| NPXY | 157-160 | 98.1 | C | C | C | C | C | C | C | C | C |
| Disulfide bond | 218 | 100 | C | C | C | C | C | C | C | A | A |
| Transmembrane | 260-280 | 84.6 | C | C | C | C | C | A | A | A | A |
| SUMO-interaction | 273-277 | 75.0 | C | C | C | C | C | A | A | A | A |
| Nuclear export | 276-277 | 92.5 | C | C | C | C | C | A | A | A | A |
| Cys-rich | 280-304 | 93.1 | C | C | C | C | C | A | A | A | A |
| SDR active site | 309-313 | 95.0 | C | C | C | C | C | C | C | C | C |
| SDR cofactor binding | 348-354 | 54.3 | C | C | C | C | C | C | C | C | C |
| Dileucine | 587-591 | 93.5 | C | C | C | C | C | C | C | C | C |
| Nuclear export | 640-642 | 89.2 | C | C | C | C | C | C | C | C | C |
| PDZ-binding | 647-649 | 93.3 | C | C | C | C | C | C | C | C | C |
AA – amino acid; C – complete; P – partial; A – absent; SDR – short-chain dehydrogenase/reductase
We next mapped LSR mutations found in human cancers from The Cancer Genome Atlas (TCGA) data using cBioPortal. Forty-seven unique mutations were identified in 54 samples (Supplementary Table 1). Cancer subtype showed no clear correlation with mutation type or distribution across the protein. Missense and in-frame insertion/deletion mutations were examined for domain-specific clustering. Sliding windows of 40 amino acids (42) were used to calculate the frequency of mutations (Supplementary Fig.9B). Of the seven clusters of mutations identified, the largest cluster (20.4% of samples) falls within the SDR domain. The clustering of mutations within this SDR region further underscores the potential importance of this domain for LSR function, either as an enzyme or as a redox sensor.
No robust nuclear localization sequence was identified; therefore we focused on nuclear export signals (NES, Table 2) to substantiate the ability of LSR to translocate between compartments. NucPred (43) identified amino acids 77-155, present in all human LSR transcript variants, as containing a weak to moderate NES. NetNES (44) predicted a NES at leucines 276 and 277 (L276 and L277) and a weaker NES at amino acids 640-LAL-642. Not all LSR transcript variants contain these sequences, so the potential exists for these sequences to affect subcellular localization. Immunofluorescence was used to identify LSR localization in breast cancer cells engineered to express only a transcript isoform that contains all three complete NES (LSR δ) or only the transcript isoform that lacked one of the NES (LSR ι). We found that isoform expression did not alter nuclear localization in the cell lines tested (Fig.6A). However to support nuclear transport, we established that nuclear levels of LSR could be altered via manipulation of the nucleocytoplasmic transport system; that is, chromosome region maintenance 1 (CRM1)-mediated nuclear export was blocked using the nuclear export inhibitor leptomycin B (45-47), resulting in nuclear accumulation of LSR in all cell lines assayed (Fig.6B). Thus, LSR contains an active NES, has the ability to translocate between compartments, and interaction with CRM1 is essential for exportation, but isoform specificity does not regulate subcellular localization.
Figure 6.
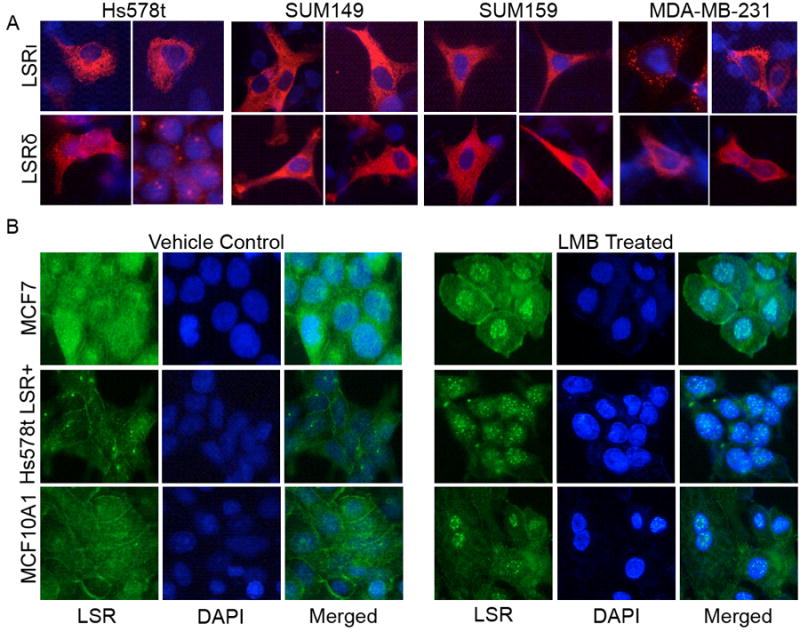
Localization of LSR variants and regulation of export via Leptomycin B. A, Hs57st, SUM149 and SUM159 cells that do not express detectable levels of LSR via western analysis, and MDA-MB-231 with below detectable levels via immunofluorescence, were transfected with a plasmid containing the full length gene for LSR variant δ that contains all nuclear export signal sequences, or transfected with a plasmid containing the full length gene for LSR variant ι that lacks one of the nuclear export signal sequences. All cell lines were subjected to immunofluorescence to identify LSR localization. B, Leptomycin B (LMB) treatment. Cells growing in log phase were treated with either vehicle or 37 nM Leptomycin B (LMB) for one hour prior to fixation and immunofluorescence. Immunofluorescence was performed using an anti-LSR primary antibody and an anti-rabbit Alexa Fluor-594 or -488 labeled secondary antibody (LSR, Alexa594/red top panels and Alexa-488/green on lower panel; DNA, DAPI/blue). All images were obtained at 40×.
Post-Translational Modifications of LSR
Post-translational modifications often promote subcellular localization; therefore we computationally predicted post-translational modifications that may influence LSR function (Supplementary Table 2). Multiple palmitoylation and phosphorylation sites were identified (43 serines, 8 threonines, 8 tyrosines; Supplementary Fig.S10). The ability of LSR to undergo these post-translational modifications was tested using pharmacological inhibitors (staurosporine for phosphorylation and 2-bromopalmitate for palmitoylation) and/or immunoblotting, however, altering these post-translational modifications did not appreciably alter nuclear localization in the cell lines examined under standard culture conditions (Supplementary Fig.S11A-C). These data suggest that palmitoylation and phosphorylation may not be primarily involved in regulating LSR nuclear localization.
Of note, co-immunoprecipitation studies with LSR and phospho-serine identified several bands suggesting phospho-proteins co-immunoprecipiated with LSR (Fig7A). To identify putative LSR interacting protein, co-immunoprecipitation studies using MCF7 lysates were performed followed by LC MS/MS. Pericentrin was one potential LSR interacting protein identified via LC MS/MS that co-immunoprecipitated at one of the MW bands identified on the immunoblot (75KDa), translocates between the cytoplasm and nucleus, contains multiple serine phosphorylation sites, is leptomycin sensitive and subject to CRM-1 mediated nuclear export (similar to LSR Fig.7B) and is often deregulated in cancers (48, 49). We confirmed pericentrin co-immunoprecipitation using LSR, pericentrin, or non-specific antibodies with breast cancer cell lysates. Western analysis shows LSR immunoprecipitated with pericentrin, and that pericentrin was detectable when anti-LSR was used for the immunoprecipitation (Fig.7B). The interaction between LSR and pericentrin was further confirmed using a proximal ligation assay (PLA, Fig.7C), where each species-specific antibody with a unique short DNA strand attached binds either the LSR or pericentrin antibody. When the PLA probes are in close proximity (<40 nm), the DNA strands can interact through a subsequent addition of two other circle-forming DNA oligonucleotides. Several-hundredfold replication of the DNA circle can occur after the amplification reaction, and a fluorescent signal is generated by labeled complementary oligonucleotide probes, visualized by a fluorescent particle. In further support of pericentrin-LSR interactions, data from our two independent, paired-end read ChIP-seq studies were visualized in UCSC Genome Browser. Overlapped peaks from the two datasets were retained and peaks within 10kb of each other were merged into larger peaks, leaving 377 total peaks. Of these, 178 (a striking 47.2%) fall within 5Mb of centromeres (Supplementary Fig.S12). These data suggest LSR and pericentrin interact and present a novel mechanism for subcellular regulation/function of LSR.
Figure 7.
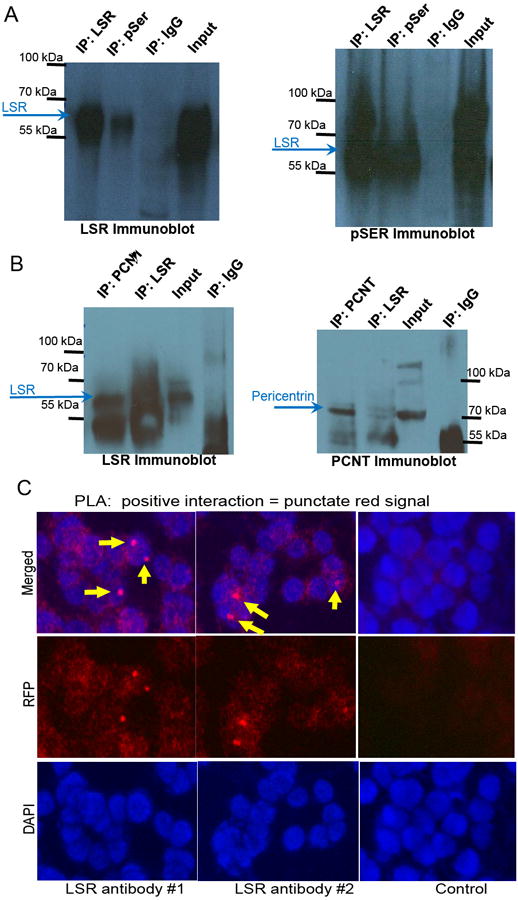
Post-translational modifications of LSR. Representative western blot of immunoprecipitation studies. A, Serine phosphorylation of LSR. T47D cells were serum starved overnight then treated with medium containing 10% fetal bovine serum, 20 ng/mL recombinant human leptin, and 0.8 mM oleic acid. Immunoprecipitation was performed on cell lysates using either an anti-LSR, anti-pSer, or non-specific antibody followed by western blot analysis. B, Co-immunoprecipitation of LSR and Pericentrin. MCF7 cells were treated as stated above. Immunoprecipitation was performed on cell lysates using either an anti-LSR, anti-pericentrin (PCNT), or non-specific antibody followed by western blot analysis. C, Proximal Ligation Assay using MCF7 cells with an anti-LSR antibody (#1 Santa Cruz or #2 Atlas) together with an anti-pericentrin antibody. A positive interaction (<40 nm) emits a punctate red signal. Control images obtained from MCF7 cells with LSR knocked-down below detectable levels and subjected to the PLA. IP, immunoprecipitation; input, cell lysates. Images at 40×.
Discussion
Together with our previous functional studies in breast cancer cell lines (1, 2), the current data support LSR as a novel transcriptional regulator that is responsive to microenvironmental cues. LSR is capable of nuclear localization and DNA binding, and its nuclear functions may be associated with poor patient outcome. LSR is upregulated in all molecular breast cancer subtypes, with highest expression in basal-like tumors, and is significantly associated with African-American race as well as poor outcome in Luminal A, Luminal B and Basal-like tumors. Further, orthotopic xenograft studies demonstrate LSR enhances tumorigenesis in vivo. Our data are consistent with previous observations linking LSR to metastatic signatures in two breast cancer models (3, 18).
LSR is reported to have two main effects: metabolic regulation and cell adhesion. The biological function of LSR may reflect roles of distinct transcript isoforms. Indeed, earlier studies have also found evidence that transcript isoforms of LSR may be expressed in specific cell types (e.g. prostate) or in certain types of cancer, such as lymphoma (50). We identified nine transcript isoforms, multiple unique sequence mutations, and potential post-translational modifications that may be responsible for the differences observed in patient samples. Of the 47 unique mutations identified in TCGA, the majority was missense (83%), and may indicate LSR protein loss does not provide a selective advantage for developing tumors. The highest mutation frequency in LSR was found in the SDR domain, potentially altering the protein's efficiency at catalyzing reactions, changing the substrates recognized by the protein, or even deregulating its function. LSR is also modified post-translationally with evidence of phosphorylation and palmitoylation. Two studies to date have been published on serine phosphorylation of LSR; one showed LSR bound to 14-3-3 via a phosphorylation site on serine 493 (S493) (51) and the other how phosphorylation of LSR regulates its membrane specific localization (52). Mutating serine 288 of mouse LSR to alanine decreased localization specificity to tTJs, instead appearing along the plasma membrane at bicellular tight junctions. Our data support serine phosphorylation, but further these data to indicate that other factors or cell type specificity contributes to LSR localization.
Whole genome ChIP-seq identified the LSR binding motif and associated significant pathways/networks and biological functions altered by LSR-DNA interactions. Our analysis was performed using MCF7 cells and given the diverse functional differences between breast cancer subtypes (and intricacies of tissue specificity) different subtypes may dictate different LSR binding targets based on the alterations in DNA such as methylation or histone modification. Indeed, distinct patterns of CpG island methylation according to molecular subtype has been reported (53), as well as specific methylation profiles and frequencies between the five subtypes (54). However, LSR target genes identified pathways including metabolic disease, oxidative phosphorylation and mitochondrial dysfunction are consistent with reports of LSR function regarding activation by free fatty acids (FA) subsequent LDL/VLDL endocytosis (4). Each of these processes/pathways is intimately related to cellular bioenergetics. Given the disparity of increased obesity/metabolic syndrome, cardiovascular disease, and breast cancer mortality in African Americans (55, 56), metabolic factors that can link these comorbidities with survivorship are important. Our observation that LSR target genes are associated with metabolic function/disorder, and that LSR expression is higher in African American and basal-like breast tumors, present an exciting potential of LSR as a molecular link between metabolic status and breast cancer cell behavior via both transcriptome regulation and lipoprotein endocytosis to alter cellular bioenergetics.
We identified pericentrin as a potential LSR interacting protein that may promote nuclear localization and/or function. Pericentrin is an integral component of the centrosome that serves as a multifunctional scaffold for anchoring protein complexes (57). The nuclear import of pericentrin is mediated by importin α/β, regulated by Ran GTPase cycle, and similar to LSR, is sensitive Crm1 mediated export (58). Deregulation of pericentrin would cause alterations in centrosome number, structure, and function thereby altering mitotic spindle organization and function, leading to chromosome missegregation. The resulting losses and gains of chromosomes after cell division could generate aneuploidy or chromosomal instability, potentially selecting for cells with accumulations of chromosomes with activated oncogenes or inactivated tumor suppressors. Rapid proliferation accompanied with changes in chromosomes distribution may account for the phenotypic changes observed in particular breast cancer subtypes with increased LSR levels. Indeed, our data present the potential that LSR influences chromatin organization via pericentrin interaction, and not direct the transcriptional regulation of genes. Alternatively, LSR may serve as a communication link between extracellular microenvironmental cues and nuclear events to induce proliferation and gene expression.
While the role of pericentrin has not been studied in breast cancer, breast cancer susceptibility, survival, and high-risk subtypes have been linked to chromosome defects (59, 60). Further, increased pericentrin expression in cultured prostate cells reproduces many features of aggressive prostate cancer, including centrosome defects, abnormal spindles, chromosome instability, and enhanced anchorage-independent growth (57). However, pericentrin also plays a role in directing cilia assembly, and given the role of LSR in membrane organization and cell junctions, the potential for LSR-pericentrin to have a membrane function exists. The precise mechanisms and functional outcomes of LSR-pericentrin interactions are beyond the scope of the present report, but hold future promise in understanding the role of these two complex molecules in cancer behaviors and patient outcome.
While much is to be learned about the multifaceted functions of LSR, our data establish LSR as a potential transcriptional regulator with important implications for cell behavior. Future studies should determine the specific functions of LSR transcript variants and microenvironmental/cell autonomous cues that regulate its expression. However, given the significant increase in LSR expression in basal-like breast tumors, its cell surface expression, and regulation by dietary factors, LSR is an important regulator of breast cancer.
Supplementary Material
Implications.
LSR functions in the promotion of aggressive breast cancer phenotypes and poor patient outcome via differential subcellular localization to alter cell signaling, bioenergetics, and gene expression.
Acknowledgments
This work was supported by funding from the National Cancer Institute (U54 CA156735, R21 CA175783, and SC2 CA176585), the Howard Hughes Medical Institute (52007553), and the Center for Human Health and the Environment (P30ES025128).
Footnotes
The authors have no conflicts of interest to disclose.
- Conception and design: J. Fleming
- Development of methodology: J. Fleming, D. Reaves, K. Hoadley, D. Jima
- Acquisition of data: D. Reaves, K. Fagan-Solis, M. Bereman, L. Thorpe, J. Hicks, D. McDonald
- Analysis and interpretation of data: J. Fleming, K. Hoadley, D. Jima, C. Perou
- Writing, review/revision of the manuscript: J. Fleming, K. Hoadley, M. Troester, C. Perou
- Study supervision J Fleming, K. Hoadley, M. Troester, C. Perou
References
- 1.Reaves DK, Fagan-Solis KD, Dunphy K, Oliver SD, Scott DW, Fleming JM. The role of lipolysis stimulated lipoprotein receptor in breast cancer and directing breast cancer cell behavior. PloS one. 2014;9:e91747. doi: 10.1371/journal.pone.0091747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fagan-Solis KD, Reaves DK, Rangel MC, Popoff MR, Stiles BG, Fleming JM. Challenging the roles of CD44 and lipolysis stimulated lipoprotein receptor in conveying Clostridium perfringens iota toxin cytotoxicity in breast cancer. Molecular cancer. 2014;13:163. doi: 10.1186/1476-4598-13-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leth-Larsen R, Terp MG, Christensen AG, Elias D, Kuhlwein T, Jensen ON, et al. Functional heterogeneity within the CD44 high human breast cancer stem cell-like compartment reveals a gene signature predictive of distant metastasis. Mol Med. 2012;18:1109–21. doi: 10.2119/molmed.2012.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yen FT, Roitel O, Bonnard L, Notet V, Pratte D, Stenger C, et al. Lipolysis stimulated lipoprotein receptor: a novel molecular link between hyperlipidemia, weight gain, and atherosclerosis in mice. The Journal of biological chemistry. 2008;283:25650–9. doi: 10.1074/jbc.M801027200. [DOI] [PubMed] [Google Scholar]
- 5.Stenger C, Hanse M, Pratte D, Mbala ML, Akbar S, Koziel V, et al. Up-regulation of hepatic lipolysis stimulated lipoprotein receptor by leptin: a potential lever for controlling lipid clearance during the postprandial phase. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:4218–28. doi: 10.1096/fj.10-160440. [DOI] [PubMed] [Google Scholar]
- 6.Bihain BE, Yen FT. Free fatty acids activate a high-affinity saturable pathway for degradation of low-density lipoproteins in fibroblasts from a subject homozygous for familial hypercholesterolemia. Biochemistry. 1992;31:4628–36. doi: 10.1021/bi00134a013. [DOI] [PubMed] [Google Scholar]
- 7.Mann CJ, Khallou J, Chevreuil O, Troussard AA, Guermani LM, Launay K, et al. Mechanism of activation and functional significance of the lipolysis-stimulated receptor. Evidence for a role as chylomicron remnant receptor Biochemistry. 1995;34:10421–31. doi: 10.1021/bi00033a014. [DOI] [PubMed] [Google Scholar]
- 8.Masuda S, Oda Y, Sasaki H, Ikenouchi J, Higashi T, Akashi M, et al. LSR defines cell corners for tricellular tight junction formation in epithelial cells. Journal of cell science. 2011;124:548–55. doi: 10.1242/jcs.072058. [DOI] [PubMed] [Google Scholar]
- 9.Yen FT, Mann CJ, Guermani LM, Hannouche NF, Hubert N, Hornick CA, et al. Identification of a lipolysis-stimulated receptor that is distinct from the LDL receptor and the LDL receptor-related protein. Biochemistry. 1994;33:1172–80. doi: 10.1021/bi00171a017. [DOI] [PubMed] [Google Scholar]
- 10.Yen FT, Masson M, Clossais-Besnard N, Andre P, Grosset JM, Bougueleret L, et al. Molecular cloning of a lipolysis-stimulated remnant receptor expressed in the liver. The Journal of biological chemistry. 1999;274:13390–8. doi: 10.1074/jbc.274.19.13390. [DOI] [PubMed] [Google Scholar]
- 11.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Disease models & mechanisms. 2013;6:1353–63. doi: 10.1242/dmm.011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuse M, Oda Y, Higashi T, Iwamoto N, Masuda S. Lipolysis-stimulated lipoprotein receptor: a novel membrane protein of tricellular tight junctions. Annals of the New York Academy of Sciences. 2012;1257:54–8. doi: 10.1111/j.1749-6632.2012.06486.x. [DOI] [PubMed] [Google Scholar]
- 13.Higashi T, Tokuda S, Kitajiri S, Masuda S, Nakamura H, Oda Y, et al. Analysis of the ‘angulin’ proteins LSR, ILDR1 and ILDR2--tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. Journal of cell science. 2013;126:966–77. doi: 10.1242/jcs.116442. [DOI] [PubMed] [Google Scholar]
- 14.Iwamoto N, Higashi T, Furuse M. Localization of angulin-1/LSR and tricellulin at tricellular contacts of brain and retinal endothelial cells in vivo. Cell structure and function. 2014;39:1–8. doi: 10.1247/csf.13015. [DOI] [PubMed] [Google Scholar]
- 15.Kwong RW, Perry SF. The tight junction protein claudin-b regulates epithelial permeability and sodium handling in larval zebrafish, Danio rerio. Am J Physiol Regul Integr Comp Physiol. 2013;304:R504–13. doi: 10.1152/ajpregu.00385.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Runkle EA, Mu D. Tight junction proteins: from barrier to tumorigenesis. Cancer Lett. 2013;337:41–8. doi: 10.1016/j.canlet.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh AB, Sharma A, Dhawan P. Claudin family of proteins and cancer: an overview. J Oncol. 2010;2010:541957. doi: 10.1155/2010/541957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Garcia JM, Pena C, Garcia V, Dominguez G, Munoz C, Silva J, et al. Prognostic value of LISCH7 mRNA in plasma and tumor of colon cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:6351–8. doi: 10.1158/1078-0432.CCR-07-0882. [DOI] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163:506–19. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nature medicine. 2009;15:907–13. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 23.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- 24.Shehata M, Teschendorff A, Sharp G, Novcic N, Russell IA, Avril S, et al. Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast cancer research : BCR. 2012;14:R134. doi: 10.1186/bcr3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleming JM, Miller TC, Kidacki M, Ginsburg E, Stuelten CH, Stewart DA, et al. Paracrine interactions between primary human macrophages and human fibroblasts enhance murine mammary gland humanization in vivo. Breast cancer research : BCR. 2012;14:R97. doi: 10.1186/bcr3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleming JM, Miller TC, Quinones M, Xiao Z, Xu X, Meyer MJ, et al. The normal breast microenvironment of premenopausal women differentially influences the behavior of breast cancer cells in vitro and in vivo. BMC Medicine. 2010;8:27. doi: 10.1186/1741-7015-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drisdel RC, Manzana E, Green WN. The role of palmitoylation in functional expression of nicotinic alpha7 receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:10502–10. doi: 10.1523/JNEUROSCI.3315-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Percherancier Y, Planchenault T, Valenzuela-Fernandez A, Virelizier JL, Arenzana-Seisdedos F, Bachelerie F. Palmitoylation-dependent control of degradation, life span, and membrane expression of the CCR5 receptor. The Journal of biological chemistry. 2001;276:31936–44. doi: 10.1074/jbc.M104013200. [DOI] [PubMed] [Google Scholar]
- 29.Webb Y, Hermida-Matsumoto L, Resh MD. Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. The Journal of biological chemistry. 2000;275:261–70. doi: 10.1074/jbc.275.1.261. [DOI] [PubMed] [Google Scholar]
- 30.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179:4053–64. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 31.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 32.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 33.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–5. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 34.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tools on the ExPasy Server. Humana Press; 2005. [DOI] [PubMed] [Google Scholar]
- 35.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. Journal of molecular biology. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 36.Ren J, Gao X, Jin C, Zhu M, Wang X, Shaw A, et al. Systematic study of protein sumoylation: Development of a site-specific predictor of SUMOsp 2. 0 Proteomics. 2009;9:3409–12. doi: 10.1002/pmic.200800646. [DOI] [PubMed] [Google Scholar]
- 37.Julenius K. NetCGlyc 1.0: prediction of mammalian C-mannosylation sites. Glycobiology. 2007;17:868–76. doi: 10.1093/glycob/cwm050. [DOI] [PubMed] [Google Scholar]
- 38.Xue Y, Liu Z, Gao X, Jin C, Wen L, Yao X, et al. GPS-SNO: computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PloS one. 2010;5:e11290. doi: 10.1371/journal.pone.0011290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein engineering, design & selection : PEDS. 2008;21:639–44. doi: 10.1093/protein/gzn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monigatti F, Gasteiger E, Bairoch A, Jung E. The Sulfinator: predicting tyrosine sulfation sites in protein sequences. Bioinformatics. 2002;18:769–70. doi: 10.1093/bioinformatics/18.5.769. [DOI] [PubMed] [Google Scholar]
- 41.Fleming JM, Miller TC, Meyer MJ, Ginsburg E, Vonderhaar BK. Local regulation of human breast xenograft models. Journal of cellular physiology. 2010;224:795–806. doi: 10.1002/jcp.22190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hudson RR, Kaplan NL. The coalescent process in models with selection and recombination. Genetics. 1988;120:831–40. doi: 10.1093/genetics/120.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brameier M, Krings A, MacCallum RM. NucPred--predicting nuclear localization of proteins. Bioinformatics. 2007;23:1159–60. doi: 10.1093/bioinformatics/btm066. [DOI] [PubMed] [Google Scholar]
- 44.la Cour T, Kiemer L, Molgaard A, Gupta R, Skriver K, Brunak S. Analysis and prediction of leucine-rich nuclear export signals. Protein engineering, design & selection : PEDS. 2004;17:527–36. doi: 10.1093/protein/gzh062. [DOI] [PubMed] [Google Scholar]
- 45.Wolff B, Sanglier JJ, Wang Y. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol. 1997;4:139–47. doi: 10.1016/s1074-5521(97)90257-x. [DOI] [PubMed] [Google Scholar]
- 46.Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, et al. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A. 2013;110:1303–8. doi: 10.1073/pnas.1217203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–60. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 48.Lee K, Rhee K. PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J Cell Biol. 2011;195:1093–101. doi: 10.1083/jcb.201106093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Q, Jiang Q, Zhang C. A fraction of Crm1 locates at centrosomes by its CRIME domain and regulates the centrosomal localization of pericentrin. Biochemical and biophysical research communications. 2009;384:383–8. doi: 10.1016/j.bbrc.2009.04.154. [DOI] [PubMed] [Google Scholar]
- 50.Hauge H, Patzke S, Delabie J, Aasheim HC. Characterization of a novel immunoglobulin-like domain containing receptor. Biochemical and biophysical research communications. 2004;323:970–8. doi: 10.1016/j.bbrc.2004.08.188. [DOI] [PubMed] [Google Scholar]
- 51.Dubois F, Vandermoere F, Gernez A, Murphy J, Toth R, Chen S, et al. Differential 14-3-3 affinity capture reveals new downstream targets of phosphatidylinositol 3-kinase signaling. Molecular & cellular proteomics : MCP. 2009;8:2487–99. doi: 10.1074/mcp.M800544-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakatsu D, Kano F, Taguchi Y, Sugawara T, Nishizono T, Nishikawa K, et al. JNK1/2-dependent phosphorylation of angulin-1/LSR is required for the exclusive localization of angulin-1/LSR and tricellulin at tricellular contacts in EpH4 epithelial sheet. Genes to cells : devoted to molecular & cellular mechanisms. 2014 doi: 10.1111/gtc.12158. [DOI] [PubMed] [Google Scholar]
- 53.Park SY, Kwon HJ, Choi Y, Lee HE, Kim SW, Kim JH, et al. Distinct patterns of promoter CpG island methylation of breast cancer subtypes are associated with stem cell phenotypes. Mod Pathol. 2012;25:185–96. doi: 10.1038/modpathol.2011.160. [DOI] [PubMed] [Google Scholar]
- 54.Holm K, Hegardt C, Staaf J, Vallon-Christersson J, Jonsson G, Olsson H, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12:R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA: a cancer journal for clinicians. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 57.Delaval B, Doxsey SJ. Pericentrin in cellular function and disease. J Cell Biol. 2010;188:181–90. doi: 10.1083/jcb.200908114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Q, Yu J, Zhuo X, Jiang Q, Zhang C. Pericentrin contains five NESs and an NLS essential for its nucleocytoplasmic trafficking during the cell cycle. Cell Res. 2010;20:948–62. doi: 10.1038/cr.2010.89. [DOI] [PubMed] [Google Scholar]
- 59.Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39:1338–49. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 60.Denu RA, Zasadil LM, Kanugh C, Laffin J, Weaver BA, Burkard ME. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC cancer. 2016;16:47. doi: 10.1186/s12885-016-2083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.