

Abstract
Medical device related infections are a significant and growing source of morbidity and mortality. Biofilm formation is a common feature of medical device infections that is not effectively prevented or treated by systemic antibiotics. Antimicrobial medical device combination products provide a pathway for local delivery of antimicrobial therapeutics with the ability to achieve high local concentrations while minimizing systemic side effects. In this review, we present considerations for the design of local antimicrobial delivery systems, which can be facilitated by modeling local pharmacokinetics in the context of the target device application. In addition to the need for local delivery, a critical barrier to progress in the field is the need to incorporate agents effective against biofilm. This article aims to review key properties of antimicrobial peptides that make them well suited to meet the demands of the next generation of antimicrobial medical devices, including broad spectrum activity, rapid and biocidal mechanisms of action, and efficacy against biofilm.
Keywords: Antimicrobial, Antimicrobial Peptides, Antibiotic Resistance, Compartment Model, Local Drug Delivery, Medical Device Coatings
Graphical Abstract
1. Introduction
Antimicrobial medical device combination products for local infection prevention is a rapidly growing area of research and product development that holds great promise for improving patient outcomes [1]. Device associated infections (26%) and surgical site infections (22%) together account for nearly half of all US health care–associated infections [2]. The rate of these infections as well as the involvement of multidrug resistant pathogens continues to increase with overall infection rates as high as 25–50% for heart assist devices, 10–30% for bladder catheters, 5–10% for fracture fixation devices and dental implants, 5–8% for central venous catheters (CVC), and 15–20% for patients receiving mechanical ventilation [3–5] [6]. Most implant related infections are resistant to systemic antibiotics and will continue until the implant is removed [7]. In many cases, device removal is associated with substantial morbidity and costs. In addition, removal may not be an option for patients that are dependent on the device for function or that cannot withstand an additional surgery. Infection is also a major contributor to impaired healing in chronic wounds [8–12] and the most expensive complication in acute surgical, trauma, and burn wounds [11].
Perioperative systemic administration of antibiotics is standard practice for preventing infection associated with implanted devices [13–15]. Given the high number of device related infections, perioperative antibiotics are clearly not sufficient and once an infection has developed, systemic antibiotic therapy is largely ineffective. Similarly, there is little evidence to support the use of systemic antibiotics to improve healing of infected chronic wounds [16, 17]. The poor efficacy of systemic antibiotics in these contexts is due to a number of factors including 1) difficulty of achieving effective antibiotic concentrations at the sight of infection [13, 18] 2) high propensity of pathogens to form biofilms on the device surface, peri-device space, or wound bed [7, 19] and 3) continuous development of microbial resistance and loss of susceptibility to existing antibiotics [20, 21]. Successful approaches to prevent device and wound infections remains a pressing clinical challenge.
Local administration of antimicrobials provides an opportunity to overcome some of these challenges. Benefits of local delivery include greater control over the antimicrobial delivery rate, the ability to co-deliver one or more agents, and potential for high local drug concentrations with significantly lower overall systemic exposure [13, 18, 22]. Agents that may be considered for co-administration include those that work synergistically with the primary antimicrobial or agents that improve overall device patency by preventing fouling or promoting healing and tissue integration. Complimentary agents may include for example, biofilm inhibitors and disruptors [23], anti-adhesive materials to reduce protein deposition and microbial attachment [24, 25], anti-inflammatory agents, growth factors and osseointegration promoters [26]. For blood contacting devices, most notably intravascular catheters, there is an association between thrombus formation and infection [27, 28]. For these devices, there is clear need for approaches that provide protection against not only bacteria but also fouling.
Antibiotic resistance is increasing at an alarming rate resulting in many infections that are extremely difficult to treat or untreatable [29, 30]. It is widely recognized that antimicrobial resistance is exacerbated by the fact that the majority of chronic human infectious diseases, including medical device and wound related infections, are associated with biofilm [31–35]. Microbial biofilms comprise polymicrobial communities enclosed within a self-produced extracellular polymeric substance (EPS) adherent to biological tissue and/or synthetic surfaces of medical devices [36, 37]. The susceptibility of bacteria to conventional antibiotics is typically significantly lower in biofilms relative to their planktonic counterparts [38], requiring up to three orders of magnitude higher antibiotic concentrations and longer times to eradicate bacteria in biofilm [39–42]. There is a need to develop antimicrobial approaches for medical devices that rely on alternative antimicrobial agents that are effective against biofilm. This is important to not only ensure efficacy but also to help curb resistance development and preserve the potency of front line clinical antibiotics.
In this review, we present considerations for development of antimicrobial medical devices with a focus on requirements for local drug delivery, strategies for incorporating antimicrobials into devices, and promising approaches for combating device related infections that go beyond the use of conventional antibiotics.
2. Requirements for local antimicrobial delivery systems and combination products
Requirements for local anti-microbial delivery systems depend on the type of device, conditions of use, duration of implantation, and potential pathways and pathogenesis of infection [43, 44]. Design requirements for the duration and strength of the antimicrobial effect should be dictated by the potential routes and duration of microbial invasion for the device’s intended use. Perioperative exposure is widely recognized as the most common route for microbe introduction and device related infections for fully implantable devices. Pathogens may be introduced at the time of surgery from the patient’s skin and clinical environment, including the hands of clinical staff [7, 45–47]. Although infection due to haematogenous seeding is possible, the rate of occurrence is substantially lower for most devices [48–50]. Therefore, the duration of high risk for microbial invasion and device colonization is thought to be limited. Local delivery systems that provide antimicrobial protection of both the device surface and adjacent tissues for a time period sufficient for healing and recovery of host defenses are considered adequate for many such applications [13, 19, 22, 34]. In the case of prosthetic joint devices, however, infections related to haematogenous seeding and late infections can be a significant problem and may justify longer term delivery approaches [51]. A different situation exists with access devices, percutaneous and permucosal implants that breach skin and epithelial barriers thus allowing for continuous microbial invasion during the entire period of device use [43]. For example, continuous invasion of mechanical ventilators and colonization of endotracheal tubes occurs via inhalation, aspiration from the oropharynx, reflux from the stomach and other routes [52]. In the case of intravascular catheters, microbial invasion pathways include the skin at the port of entry, catheter hubs, and infusate [47]. Devices with the risk of continuous microbial invasion require effective protection over the entire period of intended use.
Biofilm is a defining feature of foreign body related infections. In most cases, once a biofilm is established on the surface of a device, it is untreatable and the only viable option is device removal [34, 43, 49, 53]. Therefore, strategies for development of antimicrobial medical devices should focus on preventing attachment of viable bacteria and biofilm formation following implantation. Depending on the specific application, local antimicrobial drug delivery systems may be applied to the site of device placement as a separate dosage form segregated from the device or it might be integrated with the device for example, by impregnation, admixing during device fabrication, or as a coating on the device surface [13, 22]. One example of a segregated implantable antimicrobial delivery system is Collatamp® (EUSA Pharma), a gentamicin loaded, lyophilized resorbable collagen sponge. This product has been shown to reduce postoperative infection rates in patients that have undergone groin hernia repair by insertion of a prosthesis [54] as well as surgical patients with dirty-infected wounds [55]. Short term CVCs impregnated or coated with antiseptics and antibiotics are examples of integrated antimicrobial drug/device combinations that have demonstrated effectiveness in reducing catheter colonization and catheter related blood stream infections [56, 57].
Although segregated antimicrobial implantable dosage forms appear feasible for some applications, the majority of devices will require integration of the antimicrobial delivery system either in bulk or on the surface of the device to cover spatial and overall performance requirements. This is a challenging endeavor that dictates balancing variables needed to achieve successful drug delivery with those required to achieve proper device function [1, 58]. The key challenges include:
Selection of antimicrobials having optimal physicochemical properties (solubility, diffusivity and chemical stability in biologic fluids and tissue)
Selection of antimicrobials that display favorable therapeutic indices for the target application as well as appropriate local pharmacokinetics (PK) and pharmacodynamics (PD) to ensure efficacy while minimizing toxic effects [59];
Selection of a biocompatible delivery carrier that enables controlled drug release without compromising the primary function of the device [43]; and
Development of a manufacturing process, including sterilization, that does not compromise the in process stability or shelf stability of the drug, or the performance properties of the combination product as a whole [1, 58].
2.1. Considerations for selecting an antimicrobial agent
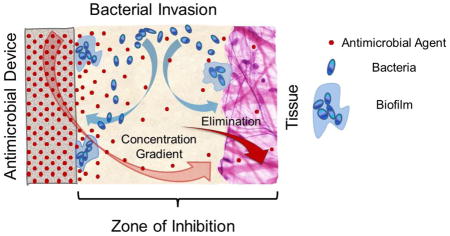
Selection of an appropriate antimicrobial agent is critical for successful prevention of infection. The antimicrobial agent’s potency, therapeutic index, mode of kill, spectrum of antimicrobial activity, and its relevance to the device’s intended use must be considered [60]. Biofilms and device related infections are commonly polymicrobial and selection of a broad spectrum antimicrobial is preferred [60]. When the antimicrobial agent is released from the device surface, a time dependent diffusion driven concentration gradient is created at the device interface with surrounding fluids and tissue. The portion of the gradient above the minimal inhibitory or biocidal concentration is referred to as zone of inhibition (ZOI) or zone of kill [22, 59, 61, 62]. The depth (or volume) of the ZOI decreases and vanishes with time due to depletion of the antimicrobial agent in the carrier, diffusion, and elimination from the sight of potential microbial invasion. Therefore, one needs to understand the rate of kill for a given antimicrobial in the context of the agent’s concentration versus time and distance from the device. It is important to relate these factors to the ZOI size considered suitable for the application and the period of time that it needs to be maintained in vivo in order to design an appropriate antimicrobial drug release mode [59, 63]. At the same time, the prevalence of biofilm forming pathogens found in clinical isolates [31] presents serious challenges; biofilms form rapidly within 24 hours [45] and this process is accompanied by development of tolerance to antibiotics. Tolerance associated with biofilm formation has been shown to increase the concentration of antibiotics required for bacteria inhibition by one to three orders in magnitude [18, 40]. It is also well known that pathogens become more virulent when they reside on foreign body surfaces; the infectious dose (amount of microbes required to cause infection) could be thousands of times lower on a foreign surface as compared to the infectious dose in tissue [64]. Effective antimicrobial treatments capable of decreasing residual pathogens on device surfaces to near zero or well below infectious doses are needed to avoid microbial re-growth and infection development. In this connection, bactericidal antimicrobial agents that are capable of fast and complete or near complete microbial kill offer advantages over bacteriostatic antibiotics that primarily prevent bacterial growth and typically work more slowly [65–67]. Moreover, development of novel anti-microbial agents or novel combination therapeutic approaches that are both rapidly bactericidal and prevent biofilm formation would be ideal [68, 69].
2.2. Compartment model for local drug delivery
The design of local delivery systems could be facilitated by modeling local pharmacokinetics in the context of the target device application. In the case of systemic antibiotic administration, PK/PD modeling and PK/PD characteristic indices are widely used and accepted for interpretation of relationships between antibiotic dosing, concentration in blood circulation, and therapeutic outcomes [70–72]. However, the basis for interpretation of therapeutic effect with systemic administration is usually the time course of drug concentrations in blood rather than concentrations in target tissue. Such indirect modeling of tissue drug levels has been recognized to be flawed and a source of confusion [73, 74]. Unfortunately, development of PK/PD models for local drug administration has not yet been the subject of significant attention, in particular for models relevant to local anti-infective treatment. Experience acquired during development of combination drug/device products such as drug eluting coated stents shows that assessment of local pharmacokinetics both from a theoretical and experimental standpoint is much more difficult relative to systemic PK [75]. Local pharmacokinetics can be significantly modulated by non-uniform conditions at the device/tissue interface such as the presence of fluid flow, liquid pockets, and variations in tissue density. These variable conditions lead to different contributions of diffusion and convection to drug transport as well as drug elimination [75]. However, development of even relatively simple models could be useful for evaluating key factors such as dosing and understanding trends in relationships between kinetics of local delivery and possible pharmacodynamic outcomes. To demonstrate this we adapted a one-compartment PK model for systemic drug administration by substituting the volume of drug distribution Vd, with the volume of zone of inhibition, VZOI [76]. In this model, the input of antimicrobial drug in the VZOI compartment is governed by a first order rate constant for drug release from the carrier, whereas output from the VZOI is controlled by first order elimination rate constant which is a single integral parameter combining such processes as drug diffusion away from the VZOI compartment as well as drug degradation and possible irreversible binding to components of biologic fluid and tissue. Further, we substituted the first order release and elimination rate constants with equivalent terms comprising corresponding half-times (rate constant = ln(2)/half-time). Finally, we normalized the model with respect to unit surface area (per 1 cm2) by dividing VZOI by total surface area of the drug carrier: VZOI/[surface area] = L, which is equal to ZOI depth. The obtained expression for the model is shown in Eq.1 below:
| Eq. 1 |
In Eq.1 C(t) represents the average drug concentration in ZOI compartment at given time, t. A is the initial drug load per unit area of the carrier, L is the minimal desired depth of ZOI (or VZOI per unit surface area of drug carrier), tR and tE represent half times of release into and elimination from the ZOI, respectively.
Understanding the minimal size or depth of ZOI needed for providing adequate antimicrobial protection has a profound impact on the pharmacokinetic requirements of local delivery. Figure 1 presents simulations of concentration-time profiles for four hypothetical cases that involve a relatively thin ZOI (L=0.1 cm). In these examples, the hypothetical delivery system is loaded with 1000 μg/cm2 of a hypothetical antimicrobial agent with known MIC (minimal inhibitory concentration), MBEC (minimal biofilm eradication concentration) and Ctox (maximal tolerated concentration) as shown in the figure. In all simulated cases, the half-time of release is maintained constant at tR = 24 hours, whereas the elimination half-time, tE, is varied. In a thin ZOI, elimination half-times, tE, are in general expected to be short due to the short distances the antimicrobial agent needs to travel before leaving the ZOI. In a thicker ZOI, depending on the physicochemical properties of the antimicrobial molecule, drug degradation and protein/tissue binding [75] will contribute more substantially to elimination. The concentration of an antimicrobial agent in a thin ZOI or in the close vicinity to the surface of a device may be readily maintained well above MIC and MBEC for a prolonged period. However, prolonged exposure to toxic concentrations could result in adverse side effects if elimination is retarded, for example, at locations where the device is in intimate contact with tissue (Example case 4, Figure 1).
Figure 1.

Simulated effects of drug release and elimination half-times on drug concentrations in 0.1 cm deep (L=0.1cm) ZOI compartment using Eq. 1.
A=1000 μg/cm2 (initial drug load); C(t) is the average drug concentration in ZOI compartment. Ctox is the maximum tolerated concentration. MIC – minimum inhibitory concentration, MBEC – minimum biofilm eradication concentration. tR and tE are half-times of release and elimination, respectively.
In Figure 2, the targeted thickness of the ZOI has been increased 10 times to L= 1 cm, while the initial loading of anti-microbial agent was kept at the same level of 1000 μg/cm2. Increasing the ZOI volume leads to drug dilution and a significant drop in average drug concentration within the ZOI as is seen from comparison of case 2 (Fig. 1) with case 5 (Fig. 2), which have the same characteristic release and elimination half-times. In case 5 (Fig. 2) with a thick ZOI, slow release and fast elimination result in sub MIC antimicrobial concentrations rendering the system ineffective even against planktonic bacteria. Moreover, it has been shown that prolonged sub-MIC exposure is not just inefficient but can also select for antibiotic resistance [22, 77]. In addition, there is some evidence that suggests that exposure to sub-MIC concentrations of certain antibiotics can enhance biofilm formation [78–80]. To achieve better outcomes in the thick ZOI the half-times of release (tR) and elimination (tE) were varied in a broader range. For case 6 in Fig. 2, which includes a moderate release half-time and fast elimination half-time, the concentration of antimicrobial agent stays between MIC and MBEC for a relatively short period. Such a system might have efficacy against motile bacteria provided the time of the concentration above MIC is sufficient to kill all microbes, however the system is inefficient against biofilm. A system with fast release accompanied by moderate elimination half-time as shown in the case 7, leads to concentrations in ZOI exceeding maximum tolerable level, Ctox, which might lead to unacceptable side effects. Finally, case 8 in Fig. 2 provides an example of the optimum combination of slow release and elimination half-times that result in antimicrobial concentrations above MBEC and MIC as well as below the toxicity limit for a prolonged period. This type of system would have the highest likelihood for being both efficient against infection and tolerable.
Figure 2.

Simulated effects of drug release and elimination half-times on drug concentrations in 1 cm deep (L=1cm) ZOI compartment using Eq. 1.
A=1000 μg/cm2 (initial drug load); C(t) is the average drug concentration in ZOI compartment. Ctox is the maximum tolerated concentration. MIC – minimum inhibitory concentration, MBEC – minimum biofilm eradication concentration. tR and tE are half-times of release and elimination, respectively.
It should be emphasized that the drug loading in all simulated cases from Figures 1 and 2 was the same. Dramatic differences in the time course of local concentrations resulted exclusively from the key pharmacokinetic properties of the delivery system and the minimal depth of ZOI that was assumed to be needed for effective infection prevention. It should also be noted that antimicrobial agents that are highly effective against biofilm (i.e. the closer the MIC and MBEC are), will provide more flexibility in achieving the necessary release rate.
It is believed that for achieving adequate treatment, in vivo concentrations of antimicrobial drug in the zone of inhibition must be several times higher than the minimal inhibitory concentrations measured in vitro [60]. The depth of an achievable ZOI depends on clinical context. For intravascular devices such as catheters and vascular implants, the ZOI will likely be relatively thin due to dilution of released antimicrobial into the bloodstream. On the contrary, for extravascular devices, implants, and bone cements, wider ZOI might be created provided the drug is not irreversibly binding to surrounding tissues or rapidly degrading in situ.
2.3. Considerations for selecting approaches and materials for coating medical devices
Efficient liberation and full recovery of antimicrobial agents from their carriers in vivo over the desired period is important both for efficacy of treatment and for minimizing the time of exposure to residual sub-MIC antimicrobial concentrations. Recovery of the active agent is dependent on many factors. These include the physicochemical nature of the carrier material and it’s interaction with the active agent, the morphological and phase structure of the delivery composition, and the mode of the antimicrobial agent’s distribution within the carrier material, which could be impacted by the fabrication process. Release of the antimicrobial will also be influenced by the stability of the agent and physicochemical transformations in the system when it is exposed to in vivo conditions and/or during storage and handling prior to use. As an example, polymethylmethacrylate (PMMA) non-biodegradable bone cements loaded with antibiotics demonstrate poor antibiotic recovery, usually less than 10%, over a reasonable therapeutic period [81]. This is followed by very slow flux delivering the drug at sub-MIC concentrations for several years thus contributing to arising drug resistant bacteria such as MRSA [82, 83]. To overcome this problem, efforts have been made to modify PMMA composition by adding water soluble and biodegradable materials that have significantly improved antibiotic recovery and release profiles [84].
Selection and development of materials suitable for combination products must balance the primary functional requirements of the device, namely mechanical properties and biocompatibility [85–87] with the need to achieve an appropriate drug delivery profile, which will depend on both the antimicrobial and the material [44]. Biocompatible materials that display a range of properties are currently available for medical devices and combination products. These include non-biodegradable plastics such as polyethylene, polypropylene and PMMA, non-biodegradable elastomers such as silicones and polyurethanes, synthetic bio-degradable polymers such as poly-lactic, poly-glycolic acids and polycaprolactone, naturally derived biodegradable materials such as collagen, gelatin, hyaluronic acid and demineralized bone, and inorganic fillers such as calcium hydroxyapatite and calcium sulfate [60, 85, 86]. Further, excipients and formulation technologies utilized in implantable [88, 89] and parenteral [90, 91] drug delivery dosage forms might be adapted for combination products to modulate an antimicrobial agent’s release profile.
Integration of antimicrobials with a device might be accomplished either by incorporation of the agent in the bulk of the device material or by coating the device surface with an antimicrobial composition. Another option is creating hollow reservoirs for loading antimicrobials inside the implant with orifices that provide a conduit for the agent to reach the surface [92]. Incorporation of antimicrobials in the bulk of a device by admixing and co-processing with a polymeric device material is usually limited to intrinsically very stable antimicrobial compounds such as inorganic salts, oxides and metals that can withstand common medical device manufacturing conditions that usually involve high temperatures and shear stresses. An example of such a device is the Vantex™ CVC (Edwards Life Sciences, California), which comprises an antimicrobial material extruded from polyurethane combined with silver, platinum metals and carbon black. Some antibiotics might be admixed with curable liquid silicone rubber and co-molded into rubbery silicone devices that can demonstrate long lasting antimicrobial activity [93]. Feasibility of this approach for a broad range of antimicrobials could be hampered by risks of drug degradation and interference with the silicone curing reaction that might lead to leachable chemical by-products of unknown nature and toxicity. However, some drugs such as contraceptive steroids were proven to be suitable for incorporation into silicone rubber matrix to form rods for subcutaneous sustained release dosage forms [94].
Incorporation of antimicrobials in the bulk of a device can also be accomplished by impregnation from a suitable solvent. Efficiency of impregnation strongly depends on the device material and is, for the most part, limited to elastomeric devices, namely silicones [95] and polyurethanes [96] that are capable of swelling in impregnating solutions thus allowing the drug to diffuse deep into the material. The main advantage of incorporating antimicrobials in the bulk is that the whole capacity (volume) of device can be potentially utilized for agent loading thus allowing for higher doses and longer release durations. Although impregnation is relatively simple to apply and offers some advantages, it provides little room to modulate release profiles and commonly yields a relatively high burst release with limited duration of activity. The choice of delivery formulation is substantially limited by the properties of the device material. For example, particulate forms of drugs could be useful formulation tools in modulating release modes but it is rather difficult to impregnate the bulk of device with a suspension. Since impregnation processes are controlled by slow diffusion, achieving uniform concentrations of drug across the thickness of the device could be difficult without employing sufficiently long impregnation times. Thus, due to the nature of the process, impregnated devices have a tendency to accumulate the majority of the drug dose within a relatively thin superficial layer. In turn, such non-uniform impregnation across device depth tends to result in a highly asymmetric release profile with high initial burst followed by sharp drop in the drug flux after a relatively short period of time.
When device impregnation is not a viable option various coating approaches might be employed. For example, ureteral stents made of an olefinic block copolymer were coated with a hydrogel that was subsequently impregnated with antibiotics [97]. These stents demonstrated in vitro antimicrobial activity for 3–16 days against common ureteral pathogens. Antimicrobial agents can be incorporated into a polymer coating by solvent casting methods [98]. For example, silver sulfadiazine has been dispersed in a polyurethane solution in N,N-dimethylacetamide and then dip coated on endotracheal tubes [99], which demonstrated efficacy in prevention of tube colonization for 72 hours in a sheep model. Coating from solvent systems offers a lot of flexibility in the types of tools that can be implemented to tailor release profiles. Coating from solvent allows flexibility in the coating composition as well as the option to utilize multiple layers having different release properties, including top coats that can serve as release controlling membranes. One potential disadvantage associated with coating is that there may be a limit to the allowable thickness, which in turn, will limit the capacity for antimicrobial loading. Another disadvantage is that it could be difficult to apply a uniform coat on devices with intricate shapes. Depending on the device composition and coating materials, poor adhesion could be a problem as well, especially in the case of metal substrates such as titanium alloys that are widely used in implants. Metal substrates might require the use of tie layers or surface modification with primers and chemical grafting to achieve sufficient adhesion. As an example, Morra and coworkers treated titanium screws with propylene plasma followed by acrylic acid grafting to enable coupling of collagen to the surface. The chemically grafted collagen layer then was impregnated with antiseptics from aqueous solutions [100]. Using an anodization process, the surface of titanium can be modified to create titanium nanotubes [101] or titanium oxide nanotubes [102] that have been used as reservoirs for loading antimicrobials. Unfortunately, release from such structures is typically too rapid [101, 102]. However, the rate of release from such structures can be controlled to some degree by coating the active loaded nanotubes with a polymer [103] or by formulating the agent with suitable excipients, for example by co-precipitation [104].
Design considerations for development of antimicrobial medical device combination products are summarized in Table 1.
Table 1.
Summary of factors to consider in the development of antimicrobial medical device combination products.
| Variable | Considerations | Sub-considerations |
|---|---|---|
| Implant Type & Location | Tissue or Biological Fluid Exposure | Flow or No Flow |
| Hard or Soft Tissue | ||
| Bacteria Exposure | Pathogens Commonly Associated with Infection | |
| Resistance Patterns | ||
| Fully Implanted or Percutaneous | Routes and Sources of Bacterial Invasion | |
| Duration of Use | Loading of Antimicrobial | |
| Antimicrobial Agent | Potency and Spectrum of Activity | Pathogens Commonly Associated with Infection |
| Resistance Patterns | ||
| Mechanism of Action | Bacteriostatic or Bactericidal | |
| Rate of Bacteria Killing | ||
| Potential to Select for Resistance Traits | ||
| Efficacy Against Biofilm | Kills Bacteria or Inhibits Biofilm Formation | |
| Local Concentrations Time Course | Solubility, Diffusivity and Stability In Vivo | |
| Cytotoxicity | Selectivity in Context of Implant Location | |
| Potential for Additive Cytotoxicity with Systemic Antimicrobial Therapy | ||
| Rate of Degradation/Elimination | ||
| Physicochemical Properties | Solubility | |
| Stability to Heat | ||
| Light Sensitivity | ||
| Sensitivity to Moisture | ||
| Susceptibility to Oxidation | ||
| Stability to Sterilization | ||
| Stability in Biological Fluids | Susceptibility to Non-enzymatic and Enzymatic Degradation | |
| Activity in Biological Fluids (Divalent Cations, pH, Susceptibility to Protein Binding) | ||
| Strategy to Incorporate Antimicrobial with Device | Impact on Device Design Change | Impregnation, Bulk, Coating, Integrated Reservoirs |
| Impregnation | Suitability of Device Material: Thermoplastic PU or Silicone Elastomer | |
| Depth of Impregnation and Release Mode | ||
| Matrix in Bulk Material | Cost of Antimicrobial | |
| Stability of Antimicrobial to Processing | ||
| Recovery of Antimicrobial from Bulk Material In Vivo | ||
| Coating | Degradable or Non-degradable | |
| Thickness Required, Capacity, Release Mode | ||
| Compatibility with Device Complexity | ||
| Fill Pores, Reservoirs, or Micro-channels | Loading Capacity and Rate of Release | |
| Coating Material Selection | Compatibility with Device Material | Good Adhesion |
| Compatibility with Device Function | Impact on Mechanical Properties | |
| Solubility | Safety of Final Device | |
| Safety of Manufacturing | ||
| Compatibility with Antimicrobial Agent | Target Release Profile and Stability | |
| Biocompatibility | Potential Leachables | |
| Compatibility with Select Excipients | Stabilizers, Modifiers of Release and Surface Properties | |
| Resistance to Fouling | Impact of Coating Composition | |
| Manufacturing Process | Cost, Scalability, Throughput, Reproducibility | Impact on Product Design |
| Terminal Sterilization | Impact on Activity and Chemical Stability of Antimicrobial Agent | |
| Packaging | Shelf Life at Room Temperature |
3. Biofilms and limitations of antibiotics, antiseptics, and silver
Most bacterial infections can be cleared by either the host immune system or treatment with systemic antibiotics. However, in the presence of a foreign body, bacteria readily bind to the surface and form biofilms where the cells are effectively protected by their self-secreted EPS [105]. EPS constitutes about 80–90% of biofilm and may vary in chemical and physical properties, but it is primarily composed of neutral or polyanionic polysaccharides and other biopolymers such as DNA, proteins and teichoic acids [106]. The EPS matrix provides effective protection to bacteria against both the host defense system and antibiotics. It also causes complex metabolic, genomic and growth mode changes leading to bacterial self-adaptation to external stresses and damage [68]. Susceptibility of bacteria to conventional antimicrobials is usually significantly lower in biofilms as compared to planktonic microbial counterparts [38]. It has been shown in vitro that minimal biofilm eradication concentrations (MBEC) for conventional antibiotics are in the range of 4 – 4000 times higher than the minimal inhibitory concentrations (MIC) for the same bacterial strains in planktonic suspension [39–41] and longer times are often needed to eradicate established biofilm [39, 42]. Most antibiotics were developed against planktonic bacteria and target functions of metabolically active bacteria. The metabolism of bacteria within a biofilm differs significantly from their planktonic counterparts [107]. Even within the biofilm, bacteria near the surface may be more metabolically active than those deep within the biofilm. Antibiotic therapy whether delivered systemically or locally may be effective at reducing symptoms of a biofilm related infection, however, due to tolerance of the more dormant bacteria deep within the biofilm, some bacteria may persist and result in a recurrent infection. As mentioned above in Section 1, this is commonly the case for medical device related infections, necessitating device removal in order the clear the infection.
Many device and wound related biofilms are polymicrobial in nature. They may involve both gram positive and gram negative bacteria as well as fungi, underscoring the need for a broad spectrum approach. One example that highlights the importance of a broad spectrum approach involves the minocycline/rifampin coated CVCs. A significant increase in the clinical rate of Candida colonization has been reported for these catheters by more than one group [108, 109]. Although the minocycline/rifampin catheters reduce colonization by coagulase negative staphylococci, their limited spectrum of activity appears to open the door for opportunistic pathogens which are arguably more virulent. In fact, the rate of mortality for CVC infections involving Candia is approximately 39%, higher than any other CVC related blood stream infection [110].
The majority of antimicrobial modified medical devices are based on the use of antiseptics such as chlorhexidine and/or metals such as silver. Although clinical efficacy has been demonstrated for intravascular catheters impregnated with chlorhexidine and silver sulfadiazine [111], high quality evidence that supports a clinical benefit for the majority of antiseptic and/or silver coated devices, including wound care products, is lacking [112, 113]. In fact, several of these products have been associated with poorer outcomes [112]. Similar to antibiotics, chlorhexidine, as well as a number of antiseptic and silver based dressings, display poor efficacy against bacteria in biofilms [114, 115]. Combined with the fact that most antiseptics and silver ions display toxicity against host cells at levels associated with antimicrobial efficacy, [116, 117] it is clear why it has been difficult to demonstrate a clinical benefit for many antiseptic and silver coated products.
4. Antimicrobial peptides (AMPs) – promising therapeutics for medical device combination products
Antimicrobial peptides (AMPs) are a class of peptides that evolved to provide nonspecific immune protection in organisms ranging from prokaryotes to humans. The most well studied AMPs commonly display an amphipathic conformation having positively charged and hydrophobic groups segregated onto opposite faces of an alpha helix, beta–sheet, or other tertiary structure [118]. This structure confers affinity and selectivity for the outer leaflet of bacteria membranes resulting in binding, membrane disruption, and ultimately death. Most AMP sequences do not exceed 50 amino acids and more than 2600 such peptides either obtained from natural sources or de-novo synthesized are known to date [119]. AMPs are typically classified based on their structure, source, and mechanism of activity which is discussed in a number of excellent reviews and is beyond the scope of this article [120–123]. Numerous AMPs have demonstrated activity against multi-drug resistant bacteria as well as a low rate in selecting resistant bacteria phenotypes leading to a strong interest in these molecules as a potentially vast source of novel anti-infective therapeutic agents to combat antibiotic resistant infections and biofilms [124–129].
4.1. Desirable properties of AMPs for medical device and wound applications
AMPs commonly display broad-spectrum, biocidal activity with multiple modes of action. These include direct kill in many cases and indirect antimicrobial effects including immunomodulatory, chemotactic, and anti-endotoxin activities as well as stimulation of wound healing and angiogenesis [130]. The most prominent mechanism of action of cationic amphiphilic AMPs involves binding to bacteria and permeabilizing or forming pores within their membranes resulting in destabilization and lysis. This mechanism of action is rapid and is expected to remain effective against slow growing or sessile bacteria within biofilms, which are the primary underlying cause of difficult to treat medical device associated infections. Several AMPs have been reported to inhibit biofilm formation and/or the ability to effectively kill bacteria in existing biofilms [125, 131, 132]. In addition, very low concentrations of some AMPs have been shown to inhibit biofilm formation [133]. This is an important contrast to antibiotics where exposure to sub-MIC concentrations has been reported to stimulate biofilm formation [134, 135]. In the case of medical device coatings, where the concentration of an antimicrobial will decrease with distance from the device and will diminish overtime, this could prove to be a significant advantage.
AMPs have co-existed with microbial pathogens over millions of years and remained surprisingly effective. Nevertheless, it is well recognized that some pathogens have acquired mechanisms to reduce their susceptibility to host defense peptides. [136–139]. Mechanisms that have been reported include production of proteases that cleave AMPs, production of proteins that bind host defense peptides, modification of the bacteria’s surface charge resulting in reduced AMP binding, changes to bacterial membrane fluidity, and in some limited cases, expulsion of peptides by multi-drug resistance pumps [140]. It is unlikely that any antimicrobial will be completely immune to the emergence of resistance and there is some concern that extensive use of AMPs could lead to selection of bacteria with reduced susceptibility. Most cationic AMPs display rapid killing kinetics and function by affecting multiple low-affinity targets that create stresses at multiple sites, making it more difficult for microbes to acquire strong resistance mechanisms [130, 141, 142]. In contrast, modern conventional antibiotics are designed to block specific high affinity targets thus allowing pathogens to focus on developing strong antibiotic resistance determinants against the specific target. Deslouches and coworkers recently evaluated the propensity for Pseudomonas aeruginosa resistance development to two de novo designed cationic AMPs relative to rifampin, LL37 and colistin in vitro. Serial passaging of three P. aeruginosa strains in the presence of subinhibitory antimicrobial concentrations (0.5X the MIC) lead to phenotypes resistant (fold MIC, >10) to rifampin within 3 days, to LL37 within 9 days, and to colistin within 13 days. Phenotypes resistant to the de novo designed antimicrobial peptides not did not emerge until 25 to 30 days. These results underscore the fact that not all antimicrobials are created equal and highlight the potential for engineered AMPs to demonstrate longer times to develop experimentally induced resistance when compared both to their natural AMP counterparts and clinical antibiotics.
In addition to a lower propensity to select for resistant bacteria based on mechanism of action, it is important to recognize that relatively rapid elimination of AMPs plays a significant role in limiting exposure and selective pressures for resistance. Rapid elimination and toxicity are two factors that have limited the development of AMPs as systemic antimicrobials [143]. However, the “on demand” localized biosynthesis and fast biodegradation of host defense AMPs is one of the factors that has contributed to their evolutionary sustainability as well as host tolerability [141]. In the same vein, rapid elimination by protein binding and degradation could be a beneficial feature for application of AMPs in medical device coatings. Successful coatings will necessitate formulations that protect peptides from the surrounding environment until release, while maintaining an adequate flux to prevent biofilm formation. Once released, relatively rapid elimination will reduce potential for resistance development as well as toxicity. Perhaps most importantly, AMPs offer a mechanism of action that is distinct from that of conventional antibiotics. As such, use of AMPs to protect medical devices is unlikely to confer resistance to front line clinical antibiotics, and may help preserve their efficacy by reducing infection rates. Moreover, because resistance traits to conventional antibiotics do not appear to impact the efficacy of many AMPs, the efficacy of AMP coated devices are not likely to be lessened by the growing number of antibiotic resistant bacteria.
4.1.2. Challenges associated with applying AMPs to medical devices
The main obstacles to implementing antimicrobial peptides as therapeutics to date have been delivery, stability, and toxicity (particularly hemolysis) which all depend on the application and mode of delivery [144–146]. In addition to safety and technical challenges, regulatory approval of products based on this new class of antimicrobials has proven to be a significant ordeal [147]. With the exception of gramicidin, which is used for topical administration, no AMPs have received US Food and Drug Administration (FDA) approval. In spite of these challenges, medical devices and topical treatments represent a unique opportunity to utilize the benefits of AMPs while minimizing issues that have limited their use for systemic therapies. This will only be possible if stability issues can be overcome. Stability of labile therapeutics depends on their intrinsic stability, availability of stabilization approaches for the entity, overall delivery formulation, conditions required for processing, and packaging. All of these factors must be studied before a successful product can be developed.
Achieving acceptable stability and purity of AMPs in combination products during manufacturing and after terminal sterilization poses significant challenges. Even though many parenteral solid dosage forms comprising peptide drugs and other labile drugs have been successfully developed, these products are usually manufactured in lyophilized forms containing bulking agents such as protective sugars by freezing-drying from liquid formulations that are usually sterilized by filtration and then aseptically filled in final closure containers thus avoiding terminal sterilization [148, 149]. Moreover, cold storage and transportation is an acceptable practice for labile drugs. In contrast, for the large majority of medical devices, which entail more sophisticated fabrication processes, aseptic manufacturing and cold storage are not practical. As a result, the majority of peptide coated devices need to be compatible with terminal sterilization. Room temperature shelf stability post sterilization will also be mandatory in most cases. Standard device sterilization methods namely, gamma irradiation and ethylene oxide gas are known to cause significant degradation of peptides. Gamma irradiation induces formation of radicals that cause oxidation of peptide amino acids via a multitude of reactions [150]. Alternative less common radiation sterilization methods such as UV light are also detrimental for peptides [151]. Due to high reactivity of ethylene oxide, it easily forms hydroxyethylated adducts with many amino acids, in particular such adducts were identified with cysteine, methionine, histidine and N-terminal valine [152, 153].
There is a lack of publications on the impact of process conditions, sterilization methods and storage conditions on the stabilities of AMPs in combination devices. However, some experience acquired during development of implantable peptide drug solid dosage forms could be very valuable. For example A. Rothen-Weinhold et. al. extensively studied biodegradable polylactic acid (PLA) based rods prepared by melt extrusion as sustained delivery implantable depot loaded with vapreotide, a cyclic octapeptide which is a shorter analog of somatostatin, an inhibitor of growth hormone secretion [154]. Vapreotide is a highly hydrophobic peptide which was used in the form of the pamoate - a counter-ion derived from hydrophobic pamoic acid. When extruded into PLA rods veprolide pamoate could withstand temperatures 80–90 °C for up to 1 hour without significant degradation. The veprolide/PLA rods were sterilized with gamma irradiation on dry ice at −78 °C to minimize degradation, and no significant increase in impurities was observed immediately after exposure to a 25 kGy irradiation dose which is considered adequate for sterility assurance [155]. However, 10-months post-irradiation, a stability study showed that the peptide in the samples sterilized and stored in air atmosphere degraded by about 15% at 40 °C, whereas the peptide in the samples sterilized and stored under nitrogen or argon atmosphere underwent about 7% degradation. At the same time, samples stored for 9 months at 4 °C had less than 1% peptide degraded with any single degradant entity not exceeding 0.5%. It has also been shown that the purity of the PLA grade had a significant impact on vapreotide degradation, in particular, residual amounts of lactic acid present in PLA at less than 1% caused more than 5% peptide degradation due to formation of a lactoyl-lactyl-vapreotide conjugate via the N-terminus amine group [156]. It follows from the PLA/vapreotide rod example, that successful development of AMP combination devices with acceptable shelf stability might require exploration and implementation of several strategies related to selection of appropriate materials, formulations and AMP forms that facilitate peptide in-process and shelf stability, optimization of process conditions as well as customization of sterilization and packaging. The cost of AMPs and their associated manufacturing challenges could prove to be a significant barrier to implementation of AMP based coatings for low margin devices.
4.1.3. AMPs – De novo designed, modified structures, and mimetics
Most natural AMPs evolved to function in a specific environment and their efficacy is limited to a narrow range of conditions. They are also susceptible to proteolytic degradation and exhibit limited selectivity, resulting in issues with hemolysis and cytotoxicity. These features effectively result in a relatively low therapeutic window relative to conventional antibiotics. To overcome these limitations, several groups have developed de novo designed peptide sequences [157–160]. These sequences incorporate characteristics common to natural AMPs with changes designed to increase stability and function within a range of biological media. Deslouches and coworkers have designed de novo AMPs that display improved activity over a broad range of conditions including physiologic NaCl, in the presence of divalent cations, and in serum [161–163]. These peptides have also shown good selectivity for Pseudomonas aeruginosa over mammalian cells within a co-culture system [157] and excellent activity against both gram positive and gram negative multidrug resistant organisms [164].
Life times of AMPs can be extended without losing full biodegradability by employing approaches in peptide design known to improve in vivo stability, such as including cysteine disulfide bonds to increase structure rigidity, introduction of proline residues, and C-terminal amidation [141]. For example, Kim with colleagues used a rational approach in synthesizing new short, lower cost 15-mer peptides comprising all natural amino acids with improved proteolytic stability and selectivity indices by avoiding protease-scissile peptide bonds in the sequences to the extent possible, at the same time maintaining an amphiphilic structure, wherein the hydrophobic face was split with one cationic lysine residue to reduce mammalian cell toxicity [165]. Two peptides were identified in this work to possess significantly improved proteolytic stability and broad-spectrum potency that was only moderately reduced in the presence of saline and human serum. The peptides also demonstrated very low cytotoxicities against human red blood cells, HaCaT keratinocytes, and human foreskin fibroblasts at concentrations about 10–20 times above MICs. Since the activity of AMPs may vary widely over different pathogens, optimization of efficacy and safety of AMP/device combination products can be also facilitated by utilizing a case by case principle of choosing peptides with the best selectivity and activity sub-spectrum relevant to a particular device application. Designing high quality controlled release systems with reproducible delivery of AMPs within the desired therapeutic concentration windows will also contribute to improved combination product safety and efficacy.
High production costs, low stability and low selectivity of AMPs has stimulated an extensive search for AMP mimetics that would be devoid of these weaknesses [166]. AMPs comprising natural L-amino acids are susceptible to relatively fast enzymatic proteolytic degradation. To overcome short life-times a number of approaches to modify or mimic AMP sequences have been explored [167, 168]. In particular, the impact of partial or full substitution of natural L-amino acids with their D- enantiomeric forms on antimicrobial activity, hemolytic toxicity, and proteolytic stability have been studied [169–171]. Proteases and peptidases target peptide bonds comprising L-amino acids, therefore D-amino acid diastereomers possess good proteolytic stability because they are not readily metabolized in humans. Substitution with D-amino acids might lead to either an increase or decrease in antimicrobial potency, as well as changes in toxicity and selectivity indices. However, the most potent examples of D- amino acid diastereomers in general display broad-spectrum antimicrobial bactericidal activity similar to that of their most potent L-amino acid AMPs.
Fluorination of AMPs is another possible pathway of modulating antimicrobial activity, enhancing proteolytic stability, improving selectivity against pathogens, and efficacy against biofilms [172, 173]. Modification of peptide sequences can be accomplished by incorporating fluorinated amino acids such as tri uoroleucine, tri uoroisoleucine, hexa uoroisoleucine and hexa uorovaline or introducing other fluorine containing moieties. Gimenez and Andreu with co-workers explored the effect of introducing fluorine atoms or trifluoromethyl groups within the chain or at the C-terminus of three short 5-mer cationic peptides and found that fluorination enhanced anti-microbial activity against Escherichia coli and Staphylococcus aureus in some cases more than 10–20 times [174].
Large classes of amphiphilic AMP inspired mimetics comprise α-peptoids [175, 176], β-peptides [177, 178] and β-peptoids [179] (Figure 3). Natural peptides are built of α-peptide bonds (structure I in the Figure 3) that are susceptible to enzymatic cleavage, whereas structures II – IV in the Figure 3 are not attacked by proteases and peptidases. Ryge et.al. investigated 20 sequences of lysine- α-peptoid hybrids and found that many of these molecules have high potency and broad-spectrum activity against clinically relevant gram-positive and gram-negative bacteria, including antibiotic resistant strains as well as anti-fungal activity [180]. Activity against gram-negative bacteria in general was lower compared to that for gram-positive bacteria. Toxicity of the mimetics was evaluated against red blood cells (RBC) at lysine-peptoid concentrations of 50 μg/mL and several molecules were found to be slightly hemolytic or non-hemolytic at concentrations 2–30 times above MICs. Huang et al. investigated more than 30 linear and cyclic α-peptoid oligomers against gram-positive and gram-negative bacteria and found potent antimicrobials with relatively low hemolytic activity [175]. Gellman et al. studied structure-property relationships in β-peptides and discovered molecules with potent antibacterial activity at the same time displaying non-hemolytic properties at concentrations up to 10 times above their MICs [177, 178]. C.A. Olsen et.al. designed 17 different β-peptoid-peptide hybrid oligomers and evaluated their antimicrobial activity, hemolytic activity against human red blood cells (hRBC) and cytotoxicity against HeLa cells [179]. A number of the oligomers were potent against methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecium (VRE), Pseudomonas aeruginosa, Escherichia coli and Candida albicans. For a number of the oligomers, 10% hRBC lysis occurred at concentrations (HC10) exceeding 10 times MIC, however in some cases critical pre-hemolytic membrane alterations in hRBCs were observed at lower, close to MIC concentrations. For the majority of the β-peptoid-peptide mimetics investigated 50% viability (IC50) of HeLa cells was observed at near MIC concentrations. The least toxic oligomer had IC50 = 258 μg/mL, HC10> 500 μg/mL and MICs in the range 9 – 143 μg/mL against the panel of pathogens tested.
Figure 3.

Structures of α-peptide, α-peptoid, β-peptide and β-peptoid. R is side chain moiety.
Lipopeptides, in particular conjugates of fatty acids or fatty alkyl groups with cationic peptide motifs or with peptide mimetics are another class of molecules that have been explored as potential antimicrobials. Tirrell et al. took 12-mer and 18-mer lysine rich peptides with poor antimicrobial activity and conjugated them to lauric acid to obtain bactericidal lipopeptides that were potent against planktonic E.coli and S. epidermidis [181]. Gillmor et al. studied activity against planktonic and sessile bacteria of C6 – C16 fatty acid conjugates with the tetrapeptide Orn-Orn-Trp-Trp-NH2 comprising two unnatural amino acids (Orn) [182]. The resulting C10 and C12 lipopeptides demonstrated the highest biocidal activity against planktonic gram-positive and gram-negative bacteria with MICs ranging from 2–16 μg/mL, however poor efficacy was observed against most biofilms tested. Makovitzki with colleagues explored conjugates of C12, C14 and C16 fatty acids with ultra-short cationic di-, tri- and tetra-peptides comprising both L- and D- enantiomeric forms of amino acids [183, 184]. A number of conjugates were discovered to have broad-spectrum activity against planktonic gram-positive and gram-negative bacteria and fungi with no or slight hemolytic activity at concentrations about 2–10 times above MICs. Efficacy of three lead palmitoyl (C16)-peptide conjugates were studied in a fungal infection model in mice. Animals were infected subcutaneously (SC) with C. albicans complexed to Cytodex beads and treated at the site of the infection with SC injection 1 hour after the infection followed by once daily SC treatment over the next 3 days [184]. After 5 days of infection the biopsied skin of the mice treated with C16-KKK lipopeptide was devoid of fungal contamination, only slight fungal contamination was obtained in the skin of mice treated with the other two lipopeptides and no damage to the skin tissue was observed, whereas the skin treated with vehicle control showed severe fungal contamination. Barron et al. explored short α-peptoids alkylated with C5 – C13 fatty tails to mimic lipopeptides [185]. Several molecules were identified with potent broad-spectrum activity against 16 bacterial strains and C. albicans. Hemolytic concentrations yielding lysis of 10% RBC were 2–65 times above MICs for candidates with the best selectivity indices.
Ceragenins recently gained attention as another type of amphiphilic cationic compound designed to mimic the activities of antimicrobial peptides [186, 187]. Ceragenins have been synthesized from a steroid scaffolding of cholic acid derivatized with amine groups. In particular ceragenin CSA-13 has potent bactericidal activity against clinical strains of multidrug resistant Staphylococcus aureus (MIC≈MBC≈1 μg/mL) [188] and clinical biofilm forming isolates of Pseudomonas aeruginosa (MIC≈MBC in the range of 3–25 μg/mL) [189]. Nagant et al. studied activity of ceragenin CSA-13 against young and mature Pseudomonas aeruginosa biofilms including clinical isolates as well as the effect of CSA-13 on the integrity of human umbilical vain endothelial cells (HUVEC) [189]. The anti-biofilm activity was strain dependent and about 3 to 30 times higher (20 – 200 μg/mL) concentrations of CSA-13 were needed to eradicate the biofilms including those formed by tobramycin resistant strains, at the same time eradication of 12-days mature biofilms required 9 days exposure to the drug and some biofilm strains appeared to be fully resistant. CSA-13 concentrations causing significant permeabilization of HUVEC eukaryotic cells were estimated to be in the range of 20 – 50 μg/mL, which is in the lower end of the concentration range effective against biofilms, underscoring the challenges in developing new antimicrobials with high selectivity against biofilms.
4.1.4. Examples of AMP coated devices
C.K. Bower et.al. [190] studied antimicrobial properties of fluorinated ethylene propylene CVCs and polyvinyl chloride tracheotomy tubes treated with nisin, a cationic peptide belonging to the class of lantibiotics produced by Lactococcus lactis and used as a food preservative [191]. The catheters and tubes were immersed in phosphate buffer solutions containing 1 mg/mL nisin immediately before placement in animal models, samples treated with saline were used as controls. Intravenous catheters were placed in jugular vein of sheep, whereas the tracheotomy tubes were inserted in the mid-cervical region of the trachea in ponies. Residual nisin activity in the samples was tested post-implantation against a nisin-sensitive strain of Pediococcus pentosaceus. It was found that the catheters retained residual activity for 5 hours while the tracheotomy tubes only for 1–2 hours. There was no evidence of any adverse effects from placement of nisin-treated samples in veins and trachea. The short lived antimicrobial effect observed in this study is likely associated with the method employed for coating the devices that neither allowed for deposition of significant doses nor led to prolonged nisin release.
The ability of AMP mimetic, cationic steroid ceragenin CSA-13 to prevent peri-operative device related infections was studied using an in vivo sheep model infected with MRSA [192]. Approximately 1/3 of the surface of a titanium plug bone implant was made porous and coated with curable liquid medical grade silicone rubber admixed with 18% CSA-13 and cured at room temperature after coating the implant. The coat average thickness was 150 ±75 microns with average CSA-13 loading 5.25 ± 0.50 mg per implant, and CSA-13 release level established in vitro was 14.84 ± 1.21 μg/mL/24hrs, well above MIC of 2.5 μg/mL. The CSA-13/silicone coated implants were placed in the femoral condyle of sheep bone and were found to be effective in preventing MRSA infection as was evidenced by radiographic, histologic observations and microbiologic samplings from the bone, soft tissues and implant itself. However, one animal (out of five) in the CSA-13/silicone coated group acquired a severe soft tissue infection which was attributed to a spill of about 1/3 of MRSA inoculum in the soft tissue about 3 cm away from the CSA-13 coated implant during the surgical procedure. The distance of the bacterial inoculum spillage in this case proved to be greater than the depth of zone of kill estimated to be few millimeters from a separate in vitro dye diffusion experiments in cadaver bone. The CSA-13 releasing silicone coated titanium plug implants did not have adverse effects on bone ingrowth which was similar to the biocompatibility control group suggesting that the pathogen inoculum was killed fast before it had opportunity to erode the peri-implant bone. Thus, the CSA-13 releasing silicone coating demonstrated bactericidal potential without adversely affecting skeletal attachment.
Minardy et.al. investigated silicone ureteral stents impregnated with Tachyplesin III peptide alone and in combination with intraperitoneal injection of piperacillin-tazobactam (TZP) antibiotic for preventing Pseudomonas aeruginosa biofilm formation in a rat infection model [193]. Tachyplesin III is a 17-mer AMP isolated from horseshoe crabs exhibiting broad-spectrum activity against Gram-positive and Gram-negative bacteria as well as strong anti-biofilm activity as was found in preliminary in vitro tests in this study against adherent Pseudomonas aeruginosa. One group of 15 animals received stents impregnated with the peptide solution immediately before subcutaneous implantation. The second group received peptide impregnated stents followed by TZP intraperitoneal injection. The third group received uncoated stents and TZP injection. Both sterility and infection control groups received uncoated stents. All stents were explanted 5 days after placement and inoculation and bacteria recovered from stents were counted. Both groups with coated stents only and TZP injection only showed more than 3 log10 (cfu/ml) reduction in recovered bacteria compared to the infection controls, whereas the combination of peptide coated stent + TZP injection showed more than 5 log10 (cfu/ml) reduction with residual bacterial counts near the detection limit of 10 cfu/mL. There was no clinical evidence of drug treatment related adverse effects and none of the animals died in any group. Even though the subcutaneous infection model may not fully reflect ureteral conditions the promising results from this study support the potential utility of AMPs for prevention of device related infections.
Currently bone cements loaded with antibiotics such as gentamicin are used for treatment of osteomyelitis. However, developing resistance against gentamicin is prompting for search of alternative anti-microbial agents. H.P. Stallman et.al. investigated the efficacy of the anti-microbial peptide hLF1-11 in calcium phosphate cement on a rabbit model of femur infection [194]. hLF1-11 is a short AMP with broad-spectrum activity comprising the eleven N-terminal amino acids 1–11 of human lactoferrin. The AMP/cement efficacy was tested against Staphylococcus aureus strain susceptible to gentamicin and compared to gentamicin/cement as well as untreated infection and sterility control groups. Cements loaded with 50mg/1g of hLF1-11 or gentamicin were prepared immediately before bacterial inoculation of drilled bone canals. After 21 days of implantation, animals were sacrificed and bacterial loads (cfu/g) in the excised bone samples with the cements were measured. The hLF1-11group showed 2 log10 (cfu/g) reduction and gentamicin group 2.5 log10 (cfu/g) reduction relative to untreated infected controls. Residual bacterial load was 4.2 log10 (cfu/g) in the AMP group and 3.7 log10 (cfu/g) in the gentamicin group. It is known that in general antibiotic recoveries from setting bone cements are poor and it was previously found by the authors of this study that hLF1-11 release significantly slows down due to peptide binding to the cement [194] underscoring the need for cements with better release properties in particular for more expensive anti-microbial agents such as AMPs.
5. Summary and Conclusions
Medical device related infections are a significant and growing problem that is not adequately addressed through the use of systemic antibiotics. Antimicrobial medical device combination products promise to reduce infection risks by providing high antimicrobial concentrations at the target site while minimizing systemic side effects. With proper formulation, combination products have potential to provide prolonged, consistent antimicrobial delivery at the device surface to prevent device colonization. The design of local antimicrobial delivery systems could be facilitated by modeling local pharmacokinetics in the context of the target device application. A simple compartment model was presented for this purpose along with a variety of materials and strategies that may be implemented to achieve suitable release profiles.
When it comes to device coatings, there is no one size fits all. The type of device, its composition, site of implantation, bacteria exposure and expected healing times should be factored into the antimicrobial delivery strategy. The type of antimicrobial agent, formulation composition, coating strategy, and release profile need to align with the device’s intended use and must not interfere with its primary function or potential for tissue integration.
In addition to the need for controlled local delivery, critical barriers to progress in the field are (1) the need to prevent biofilm formation and eradicate existing biofilms, and (2) the need for an antimicrobial approach that will not impart toxicity to cells required for healing progression. AMPs display several key properties that make them well suited to meet the demands of the next generation of antimicrobial medical devices. They are broad spectrum, rapidly biocidal, and certain structures appear to have excellent activity against biofilm. They function by a different mechanism of action relative to traditional antibiotics and many are effective against multidrug resistant organisms. Application of AMPs is also one way to reduce the burden on conventional antibiotics, thus prolonging their life. De novo designed and mimetic AMPs overcome some of the limitations of natural AMPs to provide improved stability and function within a range of physiological media. Design of high quality controlled release systems capable of maintaining AMP stability both on the shelf and in use will also be critical to success. Well-designed formulations that provide reproducible delivery of AMPs within therapeutic concentration windows will further contribute to improved combination product safety and efficacy.
Acknowledgments
This work is supported by the National Institutes of Health under Grant No. 5R44DK072560-06 and the US Army Medical Research and Materiel Command under Contract No. W81XWH-15-C-0198. The views, opinions and/or findings contained in this report are those of the author(s) and should not be construed as an official NIH or Department of the Army position, policy or decision unless so designated by other documentation.
Abbreviations
- AMP
Antimicrobial Peptide
- Ctox
Maximal Tolerated Concentration
- CVC
Central Venous Catheters
- EPS
Extracellular Polymeric Substance
- hRBC
Human Red Blood Cell
- MBC
Minimum Biocidal Concentration
- MBEC
Minimum Biofilm Eradication Concentration
- MIC
Minimum Inhibitory Concentration
- MRSA
Methicillin-resistant Staphylococcus Aureus
- PD
Pharmacodynamics
- PK
Pharmacokinetics
- PLA
Polylactic Acid
- PMMA
Polymethylmethacrylate
- tE
Half-time of Elimination
- tR
Half-time of Release
- ZOI
Zone of Inhibition
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu P, Grainger DW. Drug/device combinations for local drug therapies and infection prophylaxis. Biomaterials. 2006;27:2450–2467. doi: 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 2.Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, Lynfield R, Maloney M, McAllister-Hollod L, Nadle J, Ray SM, Thompson DL, Wilson LE, Fridkin SK I. Emerging Infections Program Healthcare-Associated, T. Antimicrobial Use Prevalence Survey. Multistate point-prevalence survey of health care-associated infections. N Engl J Med. 2014;370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.S. American Thoracic, A. Infectious Diseases Society of. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171:388–416. doi: 10.1164/rccm.200405-644ST. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DJ, Kirkland KB, Kaye KS, Thacker PA, 2nd, Kanafani ZA, Auten G, Sexton DJ. Underresourced hospital infection control and prevention programs: penny wise, pound foolish? Infect Control Hosp Epidemiol. 2007;28:767–773. doi: 10.1086/518518. [DOI] [PubMed] [Google Scholar]
- 5.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK T. National Healthcare Safety Network, F. Participating National Healthcare Safety Network. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol. 2008;29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 6.Darouiche RO. Device-associated infections: a macroproblem that starts with microadherence. Clin Infect Dis. 2001;33:1567–1572. doi: 10.1086/323130. [DOI] [PubMed] [Google Scholar]
- 7.Schierholz JM, Beuth J. Implant infections: a haven for opportunistic bacteria. J Hosp Infect. 2001;49:87–93. doi: 10.1053/jhin.2001.1052. [DOI] [PubMed] [Google Scholar]
- 8.James GA, Swogger E, Wolcott R, Pulcini E, Secor P, Sestrich J, Costerton JW, Stewart PS. Biofilms in chronic wounds. Wound Repair Regen. 2008;16:37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhao G, Usui ML, Lippman SI, James GA, Stewart PS, Fleckman P, Olerud JE. Biofilms and Inflammation in Chronic Wounds. Advances in wound care. 2013;2:389–399. doi: 10.1089/wound.2012.0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao G, Usui ML, Underwood RA, Singh PK, James GA, Stewart PS, Fleckman P, Olerud JE. Time course study of delayed wound healing in a biofilm-challenged diabetic mouse model. Wound Repair Regen. 2012;20:342–352. doi: 10.1111/j.1524-475X.2012.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763–771. doi: 10.1111/j.1524-475X.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ammons MC. Anti-biofilm strategies and the need for innovations in wound care. Recent Pat Antiinfect Drug Discov. 2010;5:10–17. doi: 10.2174/157489110790112581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darouiche RO. Antimicrobial approaches for preventing infections associated with surgical implants. Clin Infect Dis. 2003;36:1284–1289. doi: 10.1086/374842. [DOI] [PubMed] [Google Scholar]
- 14.Schmidmaier G, Lucke M, Wildemann B, Haas NP, Raschke M. Prophylaxis and treatment of implant-related infections by antibiotic-coated implants: a review. Injury. 2006;37(Suppl 2):S105–112. doi: 10.1016/j.injury.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Uckay I, Hoffmeyer P, Lew D, Pittet D. Prevention of surgical site infections in orthopaedic surgery and bone trauma: state-of-the-art update. J Hosp Infect. 2013;84:5–12. doi: 10.1016/j.jhin.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 16.O’Meara S, Al-Kurdi D, Ologun Y, Ovington LG, Martyn-St James M, Richardson R. Antibiotics and antiseptics for venous leg ulcers. Cochrane Database Syst Rev. 2013;12:CD003557. doi: 10.1002/14651858.CD003557.pub4. [DOI] [PubMed] [Google Scholar]
- 17.O’Meara S, Cullum N, Majid M, Sheldon T. Systematic reviews of wound care management: (3) antimicrobial agents for chronic wounds; (4) diabetic foot ulceration. Health Technol Assess. 2000;4:1–237. [PubMed] [Google Scholar]
- 18.Zilberman M, Elsner JJ. Antibiotic-eluting medical devices for various applications. J Control Release. 2008;130:202–215. doi: 10.1016/j.jconrel.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 19.Costerton JW. Biofilm theory can guide the treatment of device-related orthopaedic infections. Clin Orthop Relat Res. 2005:7–11. doi: 10.1097/00003086-200508000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Harbarth S, Samore MH. Antimicrobial resistance determinants and future control. Emerg Infect Dis. 2005;11:794–801. doi: 10.3201/eid1106.050167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.May J, Shannon K, King A, French G. Glycopeptide tolerance in Staphylococcus aureus. J Antimicrob Chemother. 1998;42:189–197. doi: 10.1093/jac/42.2.189. [DOI] [PubMed] [Google Scholar]
- 22.Richards RG, Moriarty TF, Miclau T, McClellan RT, Grainger DW. Advances in biomaterials and surface technologies. J Orthop Trauma. 2012;26:703–707. doi: 10.1097/BOT.0b013e31826e37a2. [DOI] [PubMed] [Google Scholar]
- 23.Chen M, Yu Q, Sun H. Novel strategies for the prevention and treatment of biofilm related infections. Int J Mol Sci. 2013;14:18488–18501. doi: 10.3390/ijms140918488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Eiff C, Kohnen W, Becker K, Jansen B. Modern strategies in the prevention of implant-associated infections. Int J Artif Organs. 2005;28:1146–1156. doi: 10.1177/039139880502801112. [DOI] [PubMed] [Google Scholar]
- 25.Hasan J, Crawford RJ, Ivanova EP. Antibacterial surfaces: the quest for a new generation of biomaterials. Trends Biotechnol. 2013;31:295–304. doi: 10.1016/j.tibtech.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Goodman SB, Yao Z, Keeney M, Yang F. The future of biologic coatings for orthopaedic implants. Biomaterials. 2013;34:3174–3183. doi: 10.1016/j.biomaterials.2013.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boersma RS, Jie KS, Verbon A, van Pampus EC, Schouten HC. Thrombotic and infectious complications of central venous catheters in patients with hematological malignancies. Ann Oncol. 2008;19:433–442. doi: 10.1093/annonc/mdm350. [DOI] [PubMed] [Google Scholar]
- 28.Thornburg CD, Smith PB, Smithwick ML, Cotten CM, Benjamin DK., Jr Association between thrombosis and bloodstream infection in neonates with peripherally inserted catheters. Thromb Res. 2008;122:782–785. doi: 10.1016/j.thromres.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J, Jr A. Infectious Diseases Society of. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 30.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 31.Sanchez CJ, Jr, Mende K, Beckius ML, Akers KS, Romano DR, Wenke JC, Murray CK. Biofilm formation by clinical isolates and the implications in chronic infections. BMC Infect Dis. 2013;13:47. doi: 10.1186/1471-2334-13-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francolini I, Donelli G. Prevention and control of biofilm-based medical-device-related infections. FEMS Immunol Med Microbiol. 2010;59:227–238. doi: 10.1111/j.1574-695X.2010.00665.x. [DOI] [PubMed] [Google Scholar]
- 33.Gbejuade HO, Lovering AM, Webb JC. The role of microbial biofilms in prosthetic joint infections. Acta Orthop. 2015;86:147–158. doi: 10.3109/17453674.2014.966290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McConoughey SJ, Howlin R, Granger JF, Manring MM, Calhoun JH, Shirtliff M, Kathju S, Stoodley P. Biofilms in periprosthetic orthopedic infections. Future Microbiol. 2014;9:987–1007. doi: 10.2217/fmb.14.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhoads DD, Wolcott RD, Percival SL. Biofilms in wounds: management strategies. J Wound Care. 2008;17:502–508. doi: 10.12968/jowc.2008.17.11.31479. [DOI] [PubMed] [Google Scholar]
- 36.Arciola CR, Campoccia D, Ravaioli S, Montanaro L. Polysaccharide intercellular adhesin in biofilm: structural and regulatory aspects. Front Cell Infect Microbiol. 2015;5:7. doi: 10.3389/fcimb.2015.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donlan RM. Biofilms: microbial life on surfaces. Emerg Infect Dis. 2002;8:881–890. doi: 10.3201/eid0809.020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stewart PS, William Costerton J. Antibiotic resistance of bacteria in biofilms. The Lancet. 2001;358:135–138. doi: 10.1016/s0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 39.Melchior MB, Fink-Gremmels J, Gaastra W. Extended antimicrobial susceptibility assay for Staphylococcus aureus isolates from bovine mastitis growing in biofilms. Vet Microbiol. 2007;125:141–149. doi: 10.1016/j.vetmic.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 40.Laverty G, Alkawareek MY, Gilmore BF. The In Vitro Susceptibility of Biofilm Forming Medical Device Related Pathogens to Conventional Antibiotics. Dataset Papers in Science. 2014;2014:1–10. [Google Scholar]
- 41.Trafny EA. Susceptibility of adherent organisms from Pseudomonas aeruginosa and Staphylococcus aureus strains isolated from burn wounds to antimicrobial agents. Int J Antimicrob Agents. 1998;10:223–228. doi: 10.1016/s0924-8579(98)00042-9. [DOI] [PubMed] [Google Scholar]
- 42.Hengzhuang W, Wu H, Ciofu O, Song Z, Hoiby N. In vivo pharmacokinetics/pharmacodynamics of colistin and imipenem in Pseudomonas aeruginosa biofilm infection. Antimicrob Agents Chemother. 2012;56:2683–2690. doi: 10.1128/AAC.06486-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campoccia D, Montanaro L, Arciola CR. A review of the biomaterials technologies for infection-resistant surfaces. Biomaterials. 2013;34:8533–8554. doi: 10.1016/j.biomaterials.2013.07.089. [DOI] [PubMed] [Google Scholar]
- 44.Arruebo M, Vilaboa N, Santamaria J. Drug delivery from internally implanted biomedical devices used in traumatology and in orthopedic surgery. Expert Opin Drug Deliv. 2010;7:589–603. doi: 10.1517/17425241003671544. [DOI] [PubMed] [Google Scholar]
- 45.Guggenbichler JP, Assadian O, Boeswald M, Kramer A. Incidence and clinical implication of nosocomial infections associated with implantable biomaterials - catheters, ventilator-associated pneumonia, urinary tract infections. GMS Krankenhhyg Interdiszip. 2011;6:Doc18. doi: 10.3205/dgkh000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Eiff C, Jansen B, Kohnen W, Becker K. Infections associated with medical devices: pathogenesis, management and prophylaxis. Drugs. 2005;65:179–214. doi: 10.2165/00003495-200565020-00003. [DOI] [PubMed] [Google Scholar]
- 47.Pascual A. Pathogenesis of catheter-related infections: lessons for new designs. Clin Microbiol Infect. 2002;8:256–264. doi: 10.1046/j.1469-0691.2002.00418.x. [DOI] [PubMed] [Google Scholar]
- 48.Chiesa R, Astore D, Frigerio S, Garriboli L, Piccolo G, Castellano R, Scalamogna M, Odero A, Pirrelli S, Biasi G, Mingazzini P, Biglioli P, Polvani G, Guarino A, Agrifoglio G, Tori A, Spina G. Vascular prosthetic graft infection: epidemiology, bacteriology, pathogenesis and treatment. Acta Chir Belg. 2002;102:238–247. doi: 10.1080/00015458.2002.11679305. [DOI] [PubMed] [Google Scholar]
- 49.Swain TW, 3rd, Calligaro KD, Dougherty MD. Management of infected aortic prosthetic grafts. Vasc Endovascular Surg. 2004;38:75–82. doi: 10.1177/153857440403800110. [DOI] [PubMed] [Google Scholar]
- 50.Wells DL, Allen JM. Ventriculoperitoneal shunt infections in adult patients. AACN Adv Crit Care. 2013;24:6–12. doi: 10.1097/NCI.0b013e31827be1d1. quiz 13–14. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez D, Pigrau C, Euba G, Cobo J, Garcia-Lechuz J, Palomino J, Riera M, Del Toro MD, Granados A, Ariza X, Group R. Acute haematogenous prosthetic joint infection: prospective evaluation of medical and surgical management. Clin Microbiol Infect. 2010;16:1789–1795. doi: 10.1111/j.1469-0691.2010.03157.x. [DOI] [PubMed] [Google Scholar]
- 52.Safdar N, Crnich CJ, Maki DG. The pathogenesis of ventilator-associated pneumonia: its relevance to developing effective strategies for prevention. Respir Care. 2005;50:725–739. discussion 739–741. [PubMed] [Google Scholar]
- 53.Zimmerli W. Clinical presentation and treatment of orthopaedic implant-associated infection. J Intern Med. 2014;276:111–119. doi: 10.1111/joim.12233. [DOI] [PubMed] [Google Scholar]
- 54.Musella M, Guido A, Musella S. Collagen tampons as aminoglycoside carriers to reduce postoperative infection rate in prosthetic repair of groin hernias. Eur J Surg. 2001;167:130–132. doi: 10.1080/110241501750070592. [DOI] [PubMed] [Google Scholar]
- 55.Chia CL, Shelat VG, Low W, George S, Rao J. The use of Collatamp G, local gentamicin-collagen sponge, in reducing wound infection. Int Surg. 2014;99:565–570. doi: 10.9738/INTSURG-D-13-00171.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramritu P, Halton K, Collignon P, Cook D, Fraenkel D, Battistutta D, Whitby M, Graves N. A systematic review comparing the relative effectiveness of antimicrobial-coated catheters in intensive care units. Am J Infect Control. 2008;36:104–117. doi: 10.1016/j.ajic.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 57.Raad I, Darouiche R, Dupuis J, Abi-Said D, Gabrielli A, Hachem R, Wall M, Harris R, Jones J, Buzaid A, Robertson C, Shenaq S, Curling P, Burke T, Ericsson C. Central venous catheters coated with minocycline and rifampin for the prevention of catheter-related colonization and bloodstream infections. A randomized, double-blind trial. The Texas Medical Center Catheter Study Group. Ann Intern Med. 1997;127:267–274. doi: 10.7326/0003-4819-127-4-199708150-00002. [DOI] [PubMed] [Google Scholar]
- 58.Hupcey MA, Ekins S. Improving the drug selection and development process for combination devices. Drug Discov Today. 2007;12:844–852. doi: 10.1016/j.drudis.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 59.Brooks BD, Davidoff SN, Grainger DW, Brooks AE. Comparisons of Release of Several Antibiotics from Antimicrobial Polymer-Coated Allograft Bone Void Filler. International Journal of Biomedical Materials Research. 2013;1:21–25. [Google Scholar]
- 60.Campoccia D, Montanaro L, Speziale P, Arciola CR. Antibiotic-loaded biomaterials and the risks for the spread of antibiotic resistance following their prophylactic and therapeutic clinical use. Biomaterials. 2010;31:6363–6377. doi: 10.1016/j.biomaterials.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 61.Hanna H, Bahna P, Reitzel R, Dvorak T, Chaiban G, Hachem R, Raad I. Comparative in vitro efficacies and antimicrobial durabilities of novel antimicrobial central venous catheters. Antimicrob Agents Chemother. 2006;50:3283–3288. doi: 10.1128/AAC.01622-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davidoff SN, Sevy JO, Brooks BD, Grainger DW, Brooks AE. Evaluating antibiotic release profiles as a function of polymer coating formulation - biomed 2011. Biomed Sci Instrum. 2011;47:46–51. [PubMed] [Google Scholar]
- 63.Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26:1–10. doi: 10.1086/516284. quiz 11–12. [DOI] [PubMed] [Google Scholar]
- 64.Elek SD, Conen PE. The virulence of Staphylococcus pyogenes for man; a study of the problems of wound infection. Br J Exp Pathol. 1957;38:573–586. [PMC free article] [PubMed] [Google Scholar]
- 65.Levison ME. Pharmacodynamics of antimicrobial drugs. Infect Dis Clin North Am. 2004;18:451–465. vii. doi: 10.1016/j.idc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 66.French GL. Bactericidal agents in the treatment of MRSA infections--the potential role of daptomycin. J Antimicrob Chemother. 2006;58:1107–1117. doi: 10.1093/jac/dkl393. [DOI] [PubMed] [Google Scholar]
- 67.Pankey GA, Sabath LD. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin Infect Dis. 2004;38:864–870. doi: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- 68.Fux CA, Costerton JW, Stewart PS, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 69.Brooks BD, Brooks AE. Therapeutic strategies to combat antibiotic resistance. Adv Drug Deliv Rev. 2014;78:14–27. doi: 10.1016/j.addr.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 70.Derendorf H, Meibohm B. Modeling of pharmacokinetic/pharmacodynamic (PK/PD) relationships: concepts and perspectives. Pharm Res. 1999;16:176–185. doi: 10.1023/a:1011907920641. [DOI] [PubMed] [Google Scholar]
- 71.Mouton JW, Dudley MN, Cars O, Derendorf H, Drusano GL. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: an update. J Antimicrob Chemother. 2005;55:601–607. doi: 10.1093/jac/dki079. [DOI] [PubMed] [Google Scholar]
- 72.Schuck EL, Derendorf H. Pharmacokinetic/pharmacodynamic evaluation of anti-infective agents. Expert Rev Anti Infect Ther. 2005;3:361–373. doi: 10.1586/14787210.3.3.361. [DOI] [PubMed] [Google Scholar]
- 73.Muller M, dela Pena A, Derendorf H. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: distribution in tissue. Antimicrob Agents Chemother. 2004;48:1441–1453. doi: 10.1128/AAC.48.5.1441-1453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu P, Muller M, Derendorf H. Rational dosing of antibiotics: the use of plasma concentrations versus tissue concentrations. Int J Antimicrob Agents. 2002;19:285–290. doi: 10.1016/s0924-8579(02)00024-9. [DOI] [PubMed] [Google Scholar]
- 75.Yang C, Burt HM. Drug-eluting stents: factors governing local pharmacokinetics. Adv Drug Deliv Rev. 2006;58:402–411. doi: 10.1016/j.addr.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 76.Bauer L. Applied Clinical Pharmacokinetics. McGraw-Hill; 2008. Clinical Pharmacokinetic Equations and Calculations; pp. 34–37. [Google Scholar]
- 77.Gullberg E, Cao S, Berg OG, Ilback C, Sandegren L, Hughes D, Andersson DI. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011;7:e1002158. doi: 10.1371/journal.ppat.1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rachid S, Ohlsen K, Witte W, Hacker J, Ziebuhr W. Effect of subinhibitory antibiotic concentrations on polysaccharide intercellular adhesin expression in biofilm-forming Staphylococcus epidermidis. Antimicrob Agents Chemother. 2000;44:3357–3363. doi: 10.1128/aac.44.12.3357-3363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoffman LR, D’Argenio DA, MacCoss MJ, Zhang Z, Jones RA, Miller SI. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature. 2005;436:1171–1175. doi: 10.1038/nature03912. [DOI] [PubMed] [Google Scholar]
- 80.Ahmed NA, Petersen FC, Scheie AA. AI-2/LuxS is involved in increased biofilm formation by Streptococcus intermedius in the presence of antibiotics. Antimicrob Agents Chemother. 2009;53:4258–4263. doi: 10.1128/AAC.00546-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neut D, van de Belt H, van Horn JR, van der Mei HC, Busscher HJ. The effect of mixing on gentamicin release from polymethylmethacrylate bone cements. Acta Orthop Scand. 2003;74:670–676. doi: 10.1080/00016470310018180. [DOI] [PubMed] [Google Scholar]
- 82.Neut D, van de Belt H, van Horn JR, van der Mei HC, Busscher HJ. Residual gentamicin-release from antibiotic-loaded polymethylmethacrylate beads after 5 years of implantation. Biomaterials. 2003;24:1829–1831. doi: 10.1016/s0142-9612(02)00614-2. [DOI] [PubMed] [Google Scholar]
- 83.Schmolders J, Hischebeth GT, Friedrich MJ, Randau TM, Wimmer MD, Kohlhof H, Molitor E, Gravius S. Evidence of MRSE on a gentamicin and vancomycin impregnated polymethyl-methacrylate (PMMA) bone cement spacer after two-stage exchange arthroplasty due to periprosthetic joint infection of the knee. BMC Infect Dis. 2014;14:144. doi: 10.1186/1471-2334-14-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shi M, Kretlow JD, Nguyen A, Young S, Scott Baggett L, Wong ME, Kasper FK, Mikos AG. Antibiotic-releasing porous polymethylmethacrylate constructs for osseous space maintenance and infection control. Biomaterials. 2010;31:4146–4156. doi: 10.1016/j.biomaterials.2010.01.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Helmus MN, Gibbons DF, Cebon D. Biocompatibility: meeting a key functional requirement of next-generation medical devices. Toxicol Pathol. 2008;36:70–80. doi: 10.1177/0192623307310949. [DOI] [PubMed] [Google Scholar]
- 86.Morais JM, Papadimitrakopoulos F, Burgess DJ. Biomaterials/tissue interactions: possible solutions to overcome foreign body response. AAPS J. 2010;12:188–196. doi: 10.1208/s12248-010-9175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Onuki Y, Bhardwaj U, Papadimitrakopoulos F, Burgess DJ. A review of the biocompatibility of implantable devices: current challenges to overcome foreign body response. J Diabetes Sci Technol. 2008;2:1003–1015. doi: 10.1177/193229680800200610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Solanki HK, Thakkar JH, Jani GH. Recent advances in implantable drug delivery. International Journal of Pharmaceutical Sciences Review and Research. 2010;4:168–177. [Google Scholar]
- 89.Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm. 2002;54:107–117. doi: 10.1016/s0939-6411(02)00061-9. [DOI] [PubMed] [Google Scholar]
- 90.Akers MJ. Excipient-drug interactions in parenteral formulations. J Pharm Sci. 2002;91:2283–2300. doi: 10.1002/jps.10154. [DOI] [PubMed] [Google Scholar]
- 91.Wu L, Janagam DR, Mandrell TD, Johnson JR, Lowe TL. Long-acting injectable hormonal dosage forms for contraception. Pharm Res. 2015;32:2180–2191. doi: 10.1007/s11095-015-1686-2. [DOI] [PubMed] [Google Scholar]
- 92.Gimeno M, Pinczowski P, Perez M, Giorello A, Martinez MA, Santamaria J, Arruebo M, Lujan L. A controlled antibiotic release system to prevent orthopedic-implant associated infections: An in vitro study. Eur J Pharm Biopharm. 2015;96:264–271. doi: 10.1016/j.ejpb.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tcholakian RK, Raad Durability of anti-infective effect of long-term silicone sheath catheters impregnated with antimicrobial agents. Antimicrob Agents Chemother. 2001;45:1990–1993. doi: 10.1128/AAC.45.7.1990-1993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Benagiano G, Gabelnick H, Farris M. Contraceptive devices: subcutaneous delivery systems. Expert Rev Med Devices. 2008;5:623–637. doi: 10.1586/17434440.5.5.623. [DOI] [PubMed] [Google Scholar]
- 95.Kohnen W, Kolbenschlag C, Teske-Keiser S, Jansen B. Development of a long-lasting ventricular catheter impregnated with a combination of antibiotics. Biomaterials. 2003;24:4865–4869. doi: 10.1016/s0142-9612(03)00379-x. [DOI] [PubMed] [Google Scholar]
- 96.Darouiche RO, Raad, Heard SO, Thornby JI, Wenker OC, Gabrielli A, Berg J, Khardori N, Hanna H, Hachem R, Harris RL, Mayhall G. A comparison of two antimicrobial-impregnated central venous catheters. Catheter Study Group. N Engl J Med. 1999;340:1–8. doi: 10.1056/NEJM199901073400101. [DOI] [PubMed] [Google Scholar]
- 97.John T, Rajpurkar A, Smith G, Fairfax M, Triest J. Antibiotic pretreatment of hydrogel ureteral stent. J Endourol. 2007;21:1211–1216. doi: 10.1089/end.2007.9904. [DOI] [PubMed] [Google Scholar]
- 98.Schierholz JM, Steinhauser H, Rump AF, Berkels R, Pulverer G. Controlled release of antibiotics from biomedical polyurethanes: morphological and structural features. Biomaterials. 1997;18:839–844. doi: 10.1016/s0142-9612(96)00199-8. [DOI] [PubMed] [Google Scholar]
- 99.Berra L, Curto F, Li Bassi G, Laquerriere P, Baccarelli A, Kolobow T. Antibacterial-coated tracheal tubes cleaned with the Mucus Shaver: a novel method to retain long-term bactericidal activity of coated tracheal tubes. Intensive Care Med. 2006;32:888–893. doi: 10.1007/s00134-006-0125-6. [DOI] [PubMed] [Google Scholar]
- 100.Morra M, Cassinelli C, Cascardo G, Carpi A, Fini M, Giavaresi G, Giardino R. Adsorption of cationic antibacterial on collagen-coated titanium implant devices. Biomed Pharmacother. 2004;58:418–422. doi: 10.1016/j.biopha.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 101.Popat KC, Eltgroth M, Latempa TJ, Grimes CA, Desai TA. Decreased Staphylococcus epidermis adhesion and increased osteoblast functionality on antibiotic-loaded titania nanotubes. Biomaterials. 2007;28:4880–4888. doi: 10.1016/j.biomaterials.2007.07.037. [DOI] [PubMed] [Google Scholar]
- 102.Ma M, Kazemzadeh-Narbat M, Hui Y, Lu S, Ding C, Chen DD, Hancock RE, Wang R. Local delivery of antimicrobial peptides using self-organized TiO2 nanotube arrays for peri-implant infections. J Biomed Mater Res A. 2012;100:278–285. doi: 10.1002/jbm.a.33251. [DOI] [PubMed] [Google Scholar]
- 103.Gulati K, Ramakrishnan S, Aw MS, Atkins GJ, Findlay DM, Losic D. Biocompatible polymer coating of titania nanotube arrays for improved drug elution and osteoblast adhesion. Acta Biomater. 2012;8:449–456. doi: 10.1016/j.actbio.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 104.Yao C, Webster TJ. Prolonged antibiotic delivery from anodized nanotubular titanium using a co-precipitation drug loading method. J Biomed Mater Res B Appl Biomater. 2009;91:587–595. doi: 10.1002/jbm.b.31433. [DOI] [PubMed] [Google Scholar]
- 105.Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Annu Rev Microbiol. 1995;49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- 106.Kostakioti M, Hadjifrangiskou M, Hultgren SJ. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med. 2013;3:a010306. doi: 10.1101/cshperspect.a010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Davey ME, O’Toole AG. Microbial biofilms: from ecology to molecular genetics. Microbiol Mol Biol Rev. 2000;64:847–867. doi: 10.1128/mmbr.64.4.847-867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leon C, Ruiz-Santana S, Rello J, de la Torre MV, Valles J, Alvarez-Lerma F, Sierra R, Saavedra P, Alvarez-Salgado F Cabana Study G. Benefits of minocycline and rifampin-impregnated central venous catheters. A prospective, randomized, double-blind, controlled, multicenter trial. Intensive Care Med. 2004;30:1891–1899. doi: 10.1007/s00134-004-2378-2. [DOI] [PubMed] [Google Scholar]
- 109.Wright F, Heyland D, Drover JW, McDonald S, Zoutman D. Antibiotic-coated central lines: do they work in the critical care setting? Clin Intensive Care. 2001;12:21–28. [Google Scholar]
- 110.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 111.Lorente L, Lecuona M, Jimenez A, Lorenzo L, Santacreu R, Ramos S, Hurtado E, Buitrago M, Mora ML. Efficiency of chlorhexidine-silver sulfadiazine-impregnated venous catheters at subclavian sites. Am J Infect Control. 2015;43:711–714. doi: 10.1016/j.ajic.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 112.Wasiak J, Cleland H, Campbell F. Dressings for superficial and partial thickness burns. Cochrane Database Syst Rev. 2008:CD002106. doi: 10.1002/14651858.CD002106.pub3. [DOI] [PubMed] [Google Scholar]
- 113.Meakins JL. Silver and new technology: dressings and devices. Surg Infect (Larchmt) 2009;10:293–296. doi: 10.1089/sur.2008.9942. [DOI] [PubMed] [Google Scholar]
- 114.Vestby LK, Nesse LL. Wound care antiseptics - performance differences against Staphylococcus aureus in biofilm. Acta Vet Scand. 2015;57:22. doi: 10.1186/s13028-015-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Phillips PL, Yang Q, Davis S, Sampson EM, Azeke JI, Hamad A, Schultz GS. Antimicrobial dressing efficacy against mature Pseudomonas aeruginosa biofilm on porcine skin explants. Int Wound J. 2015;12:469–483. doi: 10.1111/iwj.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Poon VK, Burd A. In vitro cytotoxity of silver: implication for clinical wound care. Burns. 2004;30:140–147. doi: 10.1016/j.burns.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 117.de Souza LB, de Aquino SG, de Souza PP, Hebling J, Costa CA. Cytotoxic effects of different concentrations of chlorhexidine. Am J Dent. 2007;20:400–404. [PubMed] [Google Scholar]
- 118.Giangaspero A, Sandri L, Tossi A. Amphipathic alpha helical antimicrobial peptides. Eur J Biochem. 2001;268:5589–5600. doi: 10.1046/j.1432-1033.2001.02494.x. [DOI] [PubMed] [Google Scholar]
- 119.Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44:D1087–1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Koczulla AR, Bals R. Antimicrobial peptides: current status and therapeutic potential. Drugs. 2003;63:389–406. doi: 10.2165/00003495-200363040-00005. [DOI] [PubMed] [Google Scholar]
- 121.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 122.Reddy KV, Yedery RD, Aranha C. Antimicrobial peptides: premises and promises. Int J Antimicrob Agents. 2004;24:536–547. doi: 10.1016/j.ijantimicag.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 123.Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 124.Laverty G, Gorman SP, Gilmore BF. The potential of antimicrobial peptides as biocides. Int J Mol Sci. 2011;12:6566–6596. doi: 10.3390/ijms12106566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Batoni G, Maisetta G, Brancatisano FL, Esin S, Campa M. Use of antimicrobial peptides against microbial biofilms: advantages and limits. Curr Med Chem. 2011;18:256–279. doi: 10.2174/092986711794088399. [DOI] [PubMed] [Google Scholar]
- 126.Park SC, Park Y, Hahm KS. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int J Mol Sci. 2011;12:5971–5992. doi: 10.3390/ijms12095971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xie J, Gou Y, Zhao Q, Wang K, Yang X, Yan J, Zhang W, Zhang B, Ma C, Wang R. Antimicrobial activities and membrane-active mechanism of CPF-C1 against multidrug-resistant bacteria, a novel antimicrobial peptide derived from skin secretions of the tetraploid frog Xenopus clivii. J Pept Sci. 2014;20:876–884. doi: 10.1002/psc.2679. [DOI] [PubMed] [Google Scholar]
- 128.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, Montelaro RC. Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens. Antimicrob Agents Chemother. 2015;59:1329–1333. doi: 10.1128/AAC.03937-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mohamed MF, Hammac GK, Guptill L, Seleem MN. Antibacterial activity of novel cationic peptides against clinical isolates of multi-drug resistant Staphylococcus pseudintermedius from infected dogs. PLoS One. 2014;9:e116259. doi: 10.1371/journal.pone.0116259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Afacan NJ, Yeung AT, Pena OM, Hancock RE. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr Pharm Des. 2012;18:807–819. doi: 10.2174/138161212799277617. [DOI] [PubMed] [Google Scholar]
- 131.Lashua LP, Melvin JA, Deslouches B, Pilewski JM, Montelaro RC, Bomberger JM. Engineered cationic antimicrobial peptide (eCAP) prevents Pseudomonas aeruginosa biofilm growth on airway epithelial cells. J Antimicrob Chemother. 2016 doi: 10.1093/jac/dkw143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bayramov D, Li Z, Patel E, Neff J. Evaluating activities of novel antimicrobial peptides against Staphylococcus aureus biofilms. Front Bioeng Biotechnol; 10th World Biomaterials Congress; Montreal, Canada. 2016. [Google Scholar]
- 133.Overhage J, Campisano A, Bains M, Torfs EC, Rehm BH, Hancock RE. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun. 2008;76:4176–4182. doi: 10.1128/IAI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nucleo E, Steffanoni L, Fugazza G, Migliavacca R, Giacobone E, Navarra A, Pagani L, Landini P. Growth in glucose-based medium and exposure to subinhibitory concentrations of imipenem induce biofilm formation in a multidrug-resistant clinical isolate of Acinetobacter baumannii. BMC Microbiol. 2009;9:270. doi: 10.1186/1471-2180-9-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang Q, Sun FJ, Liu Y, Xiong LR, Xie LL, Xia PY. Enhancement of biofilm formation by subinhibitory concentrations of macrolides in icaADBC-positive and -negative clinical isolates of Staphylococcus epidermidis. Antimicrob Agents Chemother. 2010;54:2707–2711. doi: 10.1128/AAC.01565-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Peschel A. How do bacteria resist human antimicrobial peptides? Trends Microbiol. 2002;10:179–186. doi: 10.1016/s0966-842x(02)02333-8. [DOI] [PubMed] [Google Scholar]
- 137.Nizet V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr Issues Mol Biol. 2006;8:11–26. [PubMed] [Google Scholar]
- 138.LaRock CN, Nizet V. Cationic antimicrobial peptide resistance mechanisms of streptococcal pathogens. Biochim Biophys Acta. 2015;1848:3047–3054. doi: 10.1016/j.bbamem.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 140.Koprivnjak T, Peschel A. Bacterial resistance mechanisms against host defense peptides. Cell Mol Life Sci. 2011;68:2243–2254. doi: 10.1007/s00018-011-0716-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peschel A, Sahl HG. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol. 2006;4:529–536. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- 142.Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2012;11:37–51. doi: 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- 143.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 144.Helmerhorst EJ, Reijnders IM, van ’t Hof W, Veerman EC, Nieuw Amerongen AV. A critical comparison of the hemolytic and fungicidal activities of cationic antimicrobial peptides. FEBS Lett. 1999;449:105–110. doi: 10.1016/s0014-5793(99)00411-1. [DOI] [PubMed] [Google Scholar]
- 145.Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SA, Vogel HJ. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pacor S, Giangaspero A, Bacac M, Sava G, Tossi A. Analysis of the cytotoxicity of synthetic antimicrobial peptides on mouse leucocytes: implications for systemic use. J Antimicrob Chemother. 2002;50:339–348. doi: 10.1093/jac/dkf141. [DOI] [PubMed] [Google Scholar]
- 147.Fox JL. Antimicrobial peptides stage a comeback. Nat Biotechnol. 2013;31:379–382. doi: 10.1038/nbt.2572. [DOI] [PubMed] [Google Scholar]
- 148.Niu CH, Chiu YY. FDA perspective on peptide formulation and stability issues. J Pharm Sci. 1998;87:1331–1334. doi: 10.1021/js9800782. [DOI] [PubMed] [Google Scholar]
- 149.Rathore N, Rajan RS. Current perspectives on stability of protein drug products during formulation, fill and finish operations. Biotechnol Prog. 2008;24:504–514. doi: 10.1021/bp070462h. [DOI] [PubMed] [Google Scholar]
- 150.Garrison WM. Reaction mechanisms in radiolysis of peptides, polypeptides, and proteins. Chem Rev. 1987;87:381–398. [Google Scholar]
- 151.Huebsch N, Gilbert M, Healy KE. Analysis of sterilization protocols for peptide-modified hydrogels. J Biomed Mater Res B Appl Biomater. 2005;74:440–447. doi: 10.1002/jbm.b.30155. [DOI] [PubMed] [Google Scholar]
- 152.Wu KY, Chiang SY, Shih WC, Huang CC, Chen MF, Swenberg JA. The application of mass spectrometry in molecular dosimetry: ethylene oxide as an example. Mass Spectrom Rev. 2011;30:733–756. doi: 10.1002/mas.20299. [DOI] [PubMed] [Google Scholar]
- 153.Chen L, Sloey C, Zhang Z, Bondarenko PV, Kim H, Ren D, Kanapuram S. Chemical modifications of therapeutic proteins induced by residual ethylene oxide. J Pharm Sci. 2015;104:731–739. doi: 10.1002/jps.24257. [DOI] [PubMed] [Google Scholar]
- 154.Rothen-Weinhold A, Besseghir K, De Zelicourt Y, Gurny R. Development and evaluation in vivo of a long-term delivery system for vapreotide, a somatostatin analogue. J Control Release. 1998;52:205–213. doi: 10.1016/s0168-3659(97)00216-2. [DOI] [PubMed] [Google Scholar]
- 155.Rothen-Weinhold A, Besseghir K, Vuaridel E, Sublet E, Oudry N, Gurny R. Stability studies of a somatostatin analogue in biodegradable implants. Int J Pharm. 1999;178:213–221. doi: 10.1016/s0378-5173(98)00376-7. [DOI] [PubMed] [Google Scholar]
- 156.Rothen-Weinhold A, Oudry N, Schwach-Abdellaoui K, Frutiger-Hughes S, Hughes GJ, Jeannerat D, Burger U, Besseghir K, Gurny R. Formation of peptide impurities in polyester matrices during implant manufacturing. Eur J Pharm Biopharm. 2000;49:253–257. doi: 10.1016/s0939-6411(00)00066-7. [DOI] [PubMed] [Google Scholar]
- 157.Deslouches B, Phadke SM, Lazarevic V, Cascio M, Islam K, Montelaro RC, Mietzner TA. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob Agents Chemother. 2005;49:316–322. doi: 10.1128/AAC.49.1.316-322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu D, DeGrado WF. De novo design, synthesis, and characterization of antimicrobial beta-peptides. J Am Chem Soc. 2001;123:7553–7559. doi: 10.1021/ja0107475. [DOI] [PubMed] [Google Scholar]
- 159.Loose C, Jensen K, Rigoutsos I, Stephanopoulos G. A linguistic model for the rational design of antimicrobial peptides. Nature. 2006;443:867–869. doi: 10.1038/nature05233. [DOI] [PubMed] [Google Scholar]
- 160.Muhle SA, Tam JP. Design of Gram-negative selective antimicrobial peptides. Biochemistry. 2001;40:5777–5785. doi: 10.1021/bi0100384. [DOI] [PubMed] [Google Scholar]
- 161.Deslouches B, Islam K, Craigo JK, Paranjape SM, Montelaro RC, Mietzner TA. Activity of the de novo engineered antimicrobial peptide WLBU2 against Pseudomonas aeruginosa in human serum and whole blood: implications for systemic applications. Antimicrob Agents Chemother. 2005;49:3208–3216. doi: 10.1128/AAC.49.8.3208-3216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Deslouches B, Gonzalez IA, DeAlmeida D, Islam K, Steele C, Montelaro RC, Mietzner TA. De novo-derived cationic antimicrobial peptide activity in a murine model of Pseudomonas aeruginosa bacteraemia. The Journal of antimicrobial chemotherapy. 2007;60:669–672. doi: 10.1093/jac/dkm253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Novak KF, Diamond WJ, Kirakodu S, Peyyala R, Anderson KW, Montelaro RC, Mietzner TA. Efficacy of the de novo-derived antimicrobial peptide WLBU2 against oral bacteria. Antimicrobial agents and chemotherapy. 2007;51:1837–1839. doi: 10.1128/AAC.00924-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Mietzner TA, Montelaro RC. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob Agents Chemother. 2013;57:2511–2521. doi: 10.1128/AAC.02218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kim H, Jang JH, Kim SC, Cho JH. De novo generation of short antimicrobial peptides with enhanced stability and cell specificity. J Antimicrob Chemother. 2014;69:121–132. doi: 10.1093/jac/dkt322. [DOI] [PubMed] [Google Scholar]
- 166.Tillotson GS, Theriault N. New and alternative approaches to tackling antibiotic resistance. F1000Prime Rep. 2013;5:51. doi: 10.12703/P5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Findlay B, Zhanel GG, Schweizer F. Cationic amphiphiles, a new generation of antimicrobials inspired by the natural antimicrobial peptide scaffold. Antimicrob Agents Chemother. 2010;54:4049–4058. doi: 10.1128/AAC.00530-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rotem S, Mor A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim Biophys Acta. 2009;1788:1582–1592. doi: 10.1016/j.bbamem.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 169.Pag U, Oedenkoven M, Papo N, Oren Z, Shai Y, Sahl HG. In vitro activity and mode of action of diastereomeric antimicrobial peptides against bacterial clinical isolates. J Antimicrob Chemother. 2004;53:230–239. doi: 10.1093/jac/dkh083. [DOI] [PubMed] [Google Scholar]
- 170.Chen Y, Vasil AI, Rehaume L, Mant CT, Burns JL, Vasil ML, Hancock RE, Hodges RS. Comparison of biophysical and biologic properties of alpha-helical enantiomeric antimicrobial peptides. Chem Biol Drug Des. 2006;67:162–173. doi: 10.1111/j.1747-0285.2006.00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.de la Fuente-Nunez C, Reffuveille F, Mansour SC, Reckseidler-Zenteno SL, Hernandez D, Brackman G, Coenye T, Hancock RE. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem Biol. 2015;22:196–205. doi: 10.1016/j.chembiol.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Marsh EN, Buer BC, Ramamoorthy A. Fluorine--a new element in the design of membrane-active peptides. Mol Biosyst. 2009;5:1143–1147. doi: 10.1039/b909864j. [DOI] [PubMed] [Google Scholar]
- 173.Meng H, Kumar K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J Am Chem Soc. 2007;129:15615–15622. doi: 10.1021/ja075373f. [DOI] [PubMed] [Google Scholar]
- 174.Gimenez D, Andreu C, del Olmo M, Varea T, Diaz D, Asensio G. The introduction of fluorine atoms or trifluoromethyl groups in short cationic peptides enhances their antimicrobial activity. Bioorg Med Chem. 2006;14:6971–6978. doi: 10.1016/j.bmc.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 175.Huang ML, Shin SB, Benson MA, Torres VJ, Kirshenbaum K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem. 2012;7:114–122. doi: 10.1002/cmdc.201100358. [DOI] [PubMed] [Google Scholar]
- 176.Kapoor R, Wadman MW, Dohm MT, Czyzewski AM, Spormann AM, Barron AE. Antimicrobial peptoids are effective against Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother. 2011;55:3054–3057. doi: 10.1128/AAC.01516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Raguse TL, Porter EA, Weisblum B, Gellman SH. Structure-activity studies of 14-helical antimicrobial beta-peptides: probing the relationship between conformational stability and antimicrobial potency. J Am Chem Soc. 2002;124:12774–12785. doi: 10.1021/ja0270423. [DOI] [PubMed] [Google Scholar]
- 178.Porter EA, Weisblum B, Gellman SH. Mimicry of host-defense peptides by unnatural oligomers: antimicrobial beta-peptides. J Am Chem Soc. 2002;124:7324–7330. doi: 10.1021/ja0260871. [DOI] [PubMed] [Google Scholar]
- 179.Olsen CA, Ziegler HL, Nielsen HM, Frimodt-Moller N, Jaroszewski JW, Franzyk H. Antimicrobial, hemolytic, and cytotoxic activities of beta-peptoid-peptide hybrid oligomers: improved properties compared to natural AMPs. Chembiochem. 2010;11:1356–1360. doi: 10.1002/cbic.201000232. [DOI] [PubMed] [Google Scholar]
- 180.Ryge TS, Frimodt-Moller N, Hansen PR. Antimicrobial activities of twenty lysine-peptoid hybrids against clinically relevant bacteria and fungi. Chemotherapy. 2008;54:152–156. doi: 10.1159/000119707. [DOI] [PubMed] [Google Scholar]
- 181.Chu-Kung AF, Bozzelli KN, Lockwood NA, Haseman JR, Mayo KH, Tirrell MV. Promotion of peptide antimicrobial activity by fatty acid conjugation. Bioconjug Chem. 2004;15:530–535. doi: 10.1021/bc0341573. [DOI] [PubMed] [Google Scholar]
- 182.Laverty G, McLaughlin M, Shaw C, Gorman SP, Gilmore BF. Antimicrobial activity of short, synthetic cationic lipopeptides. Chem Biol Drug Des. 2010;75:563–569. doi: 10.1111/j.1747-0285.2010.00973.x. [DOI] [PubMed] [Google Scholar]
- 183.Makovitzki A, Avrahami D, Shai Y. Ultrashort antibacterial and antifungal lipopeptides. Proc Natl Acad Sci U S A. 2006;103:15997–16002. doi: 10.1073/pnas.0606129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Makovitzki A, Baram J, Shai Y. Antimicrobial lipopolypeptides composed of palmitoyl Di- and tricationic peptides: in vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry. 2008;47:10630–10636. doi: 10.1021/bi8011675. [DOI] [PubMed] [Google Scholar]
- 185.Chongsiriwatana NP, Miller TM, Wetzler M, Vakulenko S, Karlsson AJ, Palecek SP, Mobashery S, Barron AE. Short alkylated peptoid mimics of antimicrobial lipopeptides. Antimicrob Agents Chemother. 2011;55:417–420. doi: 10.1128/AAC.01080-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bucki R, Niemirowicz K, Wnorowska U, Byfield FJ, Piktel E, Watek M, Janmey PA, Savage PB. Bactericidal Activity of Ceragenin CSA-13 in Cell Culture and in an Animal Model of Peritoneal Infection. Antimicrob Agents Chemother. 2015;59:6274–6282. doi: 10.1128/AAC.00653-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lai XZ, Feng Y, Pollard J, Chin JN, Rybak MJ, Bucki R, Epand RF, Epand RM, Savage PB. Ceragenins: cholic acid-based mimics of antimicrobial peptides. Acc Chem Res. 2008;41:1233–1240. doi: 10.1021/ar700270t. [DOI] [PubMed] [Google Scholar]
- 188.Chin JN, Rybak MJ, Cheung CM, Savage PB. Antimicrobial activities of ceragenins against clinical isolates of resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51:1268–1273. doi: 10.1128/AAC.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nagant C, Pitts B, Stewart PS, Feng Y, Savage PB, Dehaye JP. Study of the effect of antimicrobial peptide mimic, CSA-13, on an established bio lm formed by Pseudomonas aeruginosa. Microbiologyopen. 2013;2:318–325. doi: 10.1002/mbo3.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bower CK, Parker JE, Higgins AZ, Oest ME, Wilson JT, Valentine BA, Bothwell MK, McGuire J. Protein antimicrobial barriers to bacterial adhesion: in vitro and in vivo evaluation of nisin-treated implantable materials. Colloids and Surfaces B: Biointerfaces. 2002;25:81–90. [Google Scholar]
- 191.Cheigh CI, Pyun YR. Nisin biosynthesis and its properties. Biotechnol Lett. 2005;27:1641–1648. doi: 10.1007/s10529-005-2721-x. [DOI] [PubMed] [Google Scholar]
- 192.Sinclair KD, Pham TX, Williams DL, Farnsworth RW, Loc-Carrillo CM, Bloebaum RD. Model development for determining the efficacy of a combination coating for the prevention of perioperative device related infections: a pilot study. J Biomed Mater Res B Appl Biomater. 2013;101:1143–1153. doi: 10.1002/jbm.b.32924. [DOI] [PubMed] [Google Scholar]
- 193.Minardi D, Ghiselli R, Cirioni O, Giacometti A, Kamysz W, Orlando F, Silvestri C, Parri G, Kamysz E, Scalise G, Saba V, Giovanni M. The antimicrobial peptide tachyplesin III coated alone and in combination with intraperitoneal piperacillin-tazobactam prevents ureteral stent Pseudomonas infection in a rat subcutaneous pouch model. Peptides. 2007;28:2293–2298. doi: 10.1016/j.peptides.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 194.Stallmann HP, Faber C, Bronckers AL, Nieuw Amerongen AV, Wuisman PI. Osteomyelitis prevention in rabbits using antimicrobial peptide hLF1-11- or gentamicin-containing calcium phosphate cement. J Antimicrob Chemother. 2004;54:472–476. doi: 10.1093/jac/dkh346. [DOI] [PubMed] [Google Scholar]