

Summary
Intestinal epithelial cells (IECs), an important barrier to gut microbiota, are subject to low oxygen tension, particularly during intestinal inflammation. Hypoxia inducible factor‐1α (HIF‐1α) is expressed highly in the inflamed mucosa of inflammatory bowel disease (IBD) and functions as a key regulator in maintenance of intestinal homeostasis. However, how IEC‐derived HIF‐1α regulates intestinal immune responses in IBD is still not understood completely. We report here that the expression of HIF‐1α and IL‐33 was increased significantly in the inflamed mucosa of IBD patients as well as mice with colitis induced by dextran sulphate sodium (DSS). The levels of interleukin (IL)−33 were correlated positively with that of HIF‐1α. A HIF‐1α‐interacting element was identified in the promoter region of IL‐33, indicating that HIF‐1α activity regulates IL‐33 expression. Furthermore, tumour necrosis factor (TNF) facilitated the HIF‐1α‐dependent IL‐33 expression in IEC. Our data thus demonstrate that HIF‐1α‐dependent IL‐33 in IEC functions as a regulatory cytokine in inflamed mucosa of IBD, thereby regulating the intestinal inflammation and maintaining mucosal homeostasis.
Keywords: HIF‐1α, inflammatory bowel disease, IL‐33
Introduction
Inflammatory bowel diseases (IBD), encompassing Crohn's disease (CD) and ulcerative colitis (UC), are considered as chronic relapsing inflammation of the intestine. The massive inflammatory cell infiltration and excessive immune responses have been shown as the hallmarks in intestinal mucosal immunopathology 1, 2, 3. Imbalance of regulatory T cells (Treg) and effector T cells (Teff), including T helper type 1 (Th1), Th17 and Th2 cells, has been reported to exacerbate inflammatory damage in gut mucosa of CD and UC, respectively 4, 5. Moreover, the changes of intestinal microenvironment also contribute to the development of intestinal inflammation. Such changes may induce intestinal mucosal hypoxia, a reduction in oxygen delivery below tissue demand, which has been demonstrated as one of the pathophysiological characteristics of IBD. Under these conditions, hypoxia regulates a subset of genes that trigger the metabolic or oxygen delivery pathways to adapt the organs to oxygen deprivation. Such hypoxia‐dependent regulation is mediated by the heterodimeric nuclear transcription factor, hypoxia inducible factor (HIF), which consists of an oxygen‐sensitive α‐subunit (including three isoforms HIF‐1α, HIF‐2α, HIF‐3α) and a constitutively expressed β‐subunit 6, 7, 8. This heterodimer then binds to hypoxic response elements (HREs) of target genes, leading to a series of reactions to hypoxia, such as alterations in energy metabolism and angiogenesis 9.
It has been shown that HIF‐1α expression is increased in the inflamed mucosa of IBD, which is regulated by various inflammatory stimuli, such as proinflammatory cytokines and lipopolysaccharide (LPS) 10, 11, 12, 13. Tumour necrosis factor (TNF), one of the critical proinflammatory cytokines in the inflammatory microenvironment in the intestine of IBD, has been observed to induce the expression of HIF‐1α under normal oxygen 14. Recently, the pleiotrophic roles of HIF‐1α in regulating mucosal immune responses in the intestine have also been reported. Interleukin (IL)‐33 is a member of the IL‐1 cytokine family 15, produced mainly by non‐haematopoietic cells, including intestinal epithelial cells (IEC) and fibroblasts. IL‐33 exerts its biological effects by binding its receptor suppression of tumorigenicity 2 (ST2), an IL‐1 receptor‐related protein. The IL‐33–ST2 axis has been implicated in the pathogenesis of several autoimmune diseases, such as rheumatoid arthritis, asthma and IBD 16, 17. Primarily, the functions of IL‐33 in the regulation of intestinal immune responses are to promote the Th2‐associated immune response via driving the production of Th2‐related cytokines (e.g. IL‐5, IL‐13) 18. However, several recent studies have demonstrated that IL‐33 also prevents intestinal inflammation through the promotion of Treg and alternative activated macrophage (i.e. M2) differentiation in mice 19, 20, 21. In addition, IL‐33‐dependent group 2 innate lymphoid cells (ILC2) protect the intestinal barrier and ameliorate intestinal inflammation in an amphiregulin‐dependent manner 22. Taken together, these data suggest that IL‐33 regulates intestinal immune responses and may play an important role in maintaining homeostasis in the intestines.
It has been reported that expression of HIF‐1α and IL‐33 is increased in the inflamed mucosa of patients with IBD 15, 16, 23, and that IL‐33 can be released by necrotic IECs when subjected to inflammation and injury 24. Tissue hypoxia can serve as an initial manifestation of organism injury, particularly in intestinal mucosal inflammation. However, whether or not the increased HIF‐1α directly influences IL‐33 expression in the intestines remains to be elucidated. Furthermore, although TNF up‐regulates both HIF‐1α 14 and IL‐33 25 expression in inflamed mucosa of IBD patients, the mechanisms involved are still largely undefined. In this study, we demonstrated that HIF‐1α targets the IL‐33 gene directly and promotes IL‐33 expression in IEC. TNF also induces IL‐33 expression in inflamed mucosa of IBD in an HIF‐1α‐dependent manner. Our data thus indicate that HIF‐1α maintains the homeostasis of intestinal mucosa via the induction of IL‐33 expression in IEC.
Materials and methods
Patients
Colonic biopsies were collected from patients with active CD (A‐CD, n = 18), patients with active UC (A‐UC, n = 16), CD patients in remission (R‐CD, n = 14), UC patients in remission (R‐UC, n = 15) and healthy donors (n = 13) during colonoscopy. All patients were recruited at the Department of Gastroenterology, the Shanghai Tenth People's Hospital (Shanghai, China). The diagnosis of CD or UC was based on clinical, radiological, endoscopic examination and histological findings. The baseline characteristics, including disease categories, duration of the disease and medical history, are described in Table 1. The severity of disease was evaluated according to international standard criteria such as Crohn's Disease Activity Index (CDAI) for the diagnosis of CD patients and Mayo scores for UC patients. All studies were approved by the Institutional Review Board for Clinical Research of the Shanghai Tenth People's Hospital, Tongji University. Written informed consent was obtained from all subjects involved in this study.
Table 1.
Clinical characteristics of inflammatory bowel disease (IBD) patients
| Biopsy samples | |||||
|---|---|---|---|---|---|
| Con | CD (A) | UC (A) | CD (R) | UC (R) | |
| Number of patients | 18 | 38 | 29 | 14 | 15 |
| Age (years) | 37·1 ± 5·6 | 30·3 ± 9·2 | 35·4 ± 14·4 | 33·2 ± 5·1 | 35·6 ± 6·4 |
| Gender | |||||
| Male | 9 | 15 | 14 | 8 | 6 |
| Female | 9 | 23 | 15 | 6 | 9 |
| Disease duration (month) | 41·3 ± 23·6 | 42·5 ± 18·8 | 39 ± 20·4 | 40·2 ± 17·5 | |
| Current therapy | |||||
| 5‐aminosalicylates | 16 | 10 | 5 | 7 | |
| Azathioprine | 5 | 3 | 1 | 2 | |
| Corticosteroids | 7 | 6 | 3 | 6 | |
| IFX | 10 | 10 | 5 | 0 | |
| Disease extent (UC) | |||||
| E1 | 5 | 3 | |||
| E2 | 16 | 10 | |||
| E3 | 8 | 2 | |||
| Disease location | |||||
| L1 | 7 | 2 | |||
| L2 | 14 | 9 | |||
| L3 | 17 | 3 | |||
| L4 | |||||
| CRP (mg/l) | 33·8 ± 15·3 | 30·9 ± 11·7 | 28·7 ± 14·5 | 29·3 ± 12·2 | |
CD = Crohn's disease; UC = ulcerative colitis; IFX= infliximab; CRP = C‐reactive protein.
Anti‐TNF treatment for CD patients
Ten patients with active CD were treated with anti‐TNF monoclonal antibody (mAb) [infliximab (IFX), 5 mg/kg; Cilag AG, Schaffhausen, Swizerland] at weeks 0, 2 and 6 26, according to the manufacturer's instructions. All patients were monitored weekly during follow‐ups and intestinal biopsies were collected at weeks 0 and 12 after the first infusion. The efficacy of IFX therapy was assessed according to the CDAI and mucosal healing by endoscopy, as described previously 27.
Mice and dextran sulphate sodium (DSS)‐induced colitis model
Female C57BL/B6 mice were purchased from Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai, China) and maintained under specific pathogen‐free (SPF) conditions. DSS‐induced acute colitis in mice was performed as described previously 28. Briefly, 6 − 8‐week‐old female C57BL/B6 mice (10 mice per group) were given 2% DSS (molecular mass, 36000–50000; MP Biomedicals, LLC, Solon, OH, USA) in drinking water for 7 days, and switched to normal drinking water for an additional 3 days. The mice (10 mice per group) with only drinking water served as control. The severity of inflammation was evaluated by monitoring daily body weight, diarrhoea and bloody stools. Animal experiments in this study were approved by the Institutional Animal Care and Use Committee at Tongji University.
Antibodies and reagents
Dulbecco's modified Eagle's medium (DMEM), Roswell Park Memorial Institute (RPMI)−1640 medium, fetal bovine serum (FBS), penicillin and streptomycin were purchased from HyClone (Logan, UT, USA). Anti‐human HIF‐1α mAb was purchased from Novus Biologicals, Inc. (Littleton, CO, USA). Anti‐IL‐33 antibody was purchased from Abcam (Cambridge, MA, USA). Antibodies against P‐extracellular‐regulated kinase (ERK) and p‐p38 were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti‐Fas antibody was purchased from Merck Millipore (Billerica, MA, USA). The cytokines, including TNF, IL‐17A, interferon (IFN)‐γ, IL‐23, IL‐10 and transforming growth factor (TGF)‐β, were purchased from BioLegend (San Diego, CA, USA). LPS (Escherichia coli 055:B5), peptidoglycan (PGN), flagellin, polyI:C, SP600125, Bay 11‐7082, YC‐1 and dimethyloxalylglycine (DMOG) were purchased from Sigma‐Aldrich (St Louis, MO, USA).
Ex‐vivo mucosal tissue culture
Fresh colonic inflamed biopsies were obtained from patients with A‐CD (n = 10) and A‐UC (n = 10) during endoscopic examination. The samples (two biopsies/well) were cultured in 1 ml complete RPMI‐1640 medium containing 10% fetal bovine serum (FBS), penicillin (100 U/ml) and streptomycin (100 μg/ml) in the presence of 50 μg/ml IFX or control human immunoglobulin (Ig)G (HIg) at 37ºC for 24 h. The biopsies were then harvested for further analysis with quantitative real‐time–polymerase chain reaction (qRT–PCR).
Isolation of IECs
Fresh IECs were isolated as described previously 29. Briefly, the colon and caecum were removed from the euthanized mice, cut into small pieces and rinsed thoroughly with cold phosphate‐buffered saline (PBS) to remove all debris and blood. The colonic biopsies from patients with IBD obtained during endoscopic examination were washed thoroughly with cold PBS. The colonic tissues were incubated at 37°C with shaking for 2 × 20 min in 20 ml Hanks's balanced salt solution without Ca+ and Mg+ (Cellgro Mediatech, Manassas, VA, USA), containing 5 mM ethylenediamine tetraacetic acid (EDTA) (Sigma‐Aldrich), 5% FBS and 10 mM HEPES buffer (Cellgro Mediatech). The epithelial layer was then pooled and passed through a 70‐μm strainer. The pellet of epithelial cells was resuspended in 4 ml of 20% Percoll‐RPMI solution (GE Healthcare, Uppsala, Sweden), layered over 2 ml of 40% Percoll‐RPMI solution and centrifuged at 720 g for 20 min at room temperature. IECs were collected from the interphase.
Cell culture
The intestinal epithelial cancer cell line HT‐29 [American Type Culture Collection (ATCC), Manassas, VA, USA) was cultured under stimulation with TNF, IL‐17A, IFN‐γ, IL‐23, IL‐10 and TGF‐β (10 ng/ml), respectively, or stimulated with LPS (100 ng/ml), PGN (1 μg/ml), flagellin (100 ng/ml) and polyI:C (10 μg/ml), respectively, for 24 h. The transcriptional levels of HIF‐1α were assessed by qRT–PCR. Moreover, HT‐29 cells were also cultured under stimulation with different concentrations of TNF (1, 5, 10, 20, 50 ng/ml) for 24 h or stimulated with TNF (10 ng/ml) for 0, 12, 24 and 48 h, respectively, and the expression of HIF‐1α mRNA was analysed by qRT–PCR. For HIF‐1α over‐expressing assay, 1 μM DMOG was added to HT‐29 cells as described previously 30, and the cells were then harvested after 24 h of culture to determine the efficiency of gene over‐expression.
HIF‐1α small interfering RNA (siRNA)
siRNA transfection was performed according to the manufacturer's instructions (Genechem, Shanghai, China). Briefly, HT‐29 cells were seeded at 5 × 104 cells per well of 24‐well plates. After 24 h culture, the cells were transfected with HIF‐1α‐specific siRNA or the scramble siRNA (50 nM) using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in 500 μl opti‐MEM medium. The efficiency of knock‐down was measured 48 h post‐transfection.
RNA extraction and qRT–PCR
Total RNA was isolated from HT‐29 cells and colonic tissues using TriZol reagent (Invitrogen), and 400 ng RNA was reverse‐transcribed with 5 × All‐In‐One RT MasterMix kit (Applied Biological Materials, Inc., Richmond, BC, Canada). qRT–PCR was performed using a SYBR Green PCR kit (TaKaRa, Dalian, China) to quantify the expression of HIF‐1α and IL‐33. The mRNA expression of HIF‐1α and IL‐33 was relative to human housekeeping gene glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). The primer sequences of HIF‐1α and IL‐33 are shown in Table 2. The relative amount of transcript was analysed using the 2–ΔΔCT method.
Table 2.
Primer sequence used for quantitative real‐time–polymerase chain reaction (qRT–PCR) analysis
| Human | 5′‐3′ | Mouse | 5′‐3′ | ||
|---|---|---|---|---|---|
| HIF‐1α | Forward | TCCAGTTACGTTCCTTCGATCA | HIF‐1α | Forward | GAAACGACCACTGCTAAGGCA |
| Reverse | TTTGAGGACTTGCGCTTTCA | Reverse | GGCAGACAGCTTAAGGCTCCT | ||
| IL‐33 | Forward | CACCCCTCAAATGAATCAGG | IL‐33 | Forward | TCCTTGCTTGGCAGTATCCA |
| Reverse | GGAGCTCCACAGAGTGTTCC | Reverse | TGCTCAATGTGTCAACAGACG | ||
| ST2 | Forward | GACGGCGACCAGGTCCTT | VEGF | Forward | GCTACTGCCGTCCGATTGAG |
| Reverse | GGGCTCCGATTACTGGAAACA | Reverse | ACTCCAGGGCTTCATCGTTACAG | ||
| GAPDH | Forward | CCATCACCATCTTCCAGGAG | TNF | Forward | CATCTTCTCAAAATTCGAGTGACAA |
| Reverse | CCTGCTTCACCACCTTCTTG | Reverse | TGGGAGTAGACAAGGTACAACCC | ||
| GAPDH | Forward | TGATGACATCAAGAAGGTGGTGAAG | |||
| Reverse | TCCTTGGA GGCCATGTAGGCCAT | ||||
ST2 = receptor suppression of tumorigenicity 2; GAPDH = glyceraldehyde‐3‐phosphate dehydrogenase; TNF = tumour necrosis factor; VEGF = vascular endothelial growth factor; IL = interleukin; HIF‐a = hypoxia inducible factor‐1α.
Immunohistochemistry
To localize HIF‐1α expression, colonic biopsy specimens were obtained from patients with active CD or UC or healthy donors, fixed in 10% formalin overnight, and embedded in paraffin. The sections were then stained with anti‐HIF‐1α mAb as a primary antibody, followed by staining with horseradish peroxidase‐conjugated anti‐IgG (dilution 1 : 400) at room temperature. The colour reaction was developed with 3,3′‐diaminobenzidine. The sections were counterstained with haematoxylin. As negative controls, sections were treated with isotype‐matched mouse IgG1 or PBS instead of primary antibody. The positive cells that were stained with anti‐HIF‐1α antibody were observed under light microscopy.
Western blot analysis
The total protein of human colonic tissues was isolated through radio‐immunoprecipitation assay (RIPA) and phenylmethanesulphonyl fluoride (PMSF, 1 mM). Samples were then resolved on sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) by standard procedures. Immunoblotting was performed with human primary antibodies to HIF‐1α, IL‐33, P‐P38, P‐ERK and β‐actin (Sigma‐Aldrich), respectively. Blots were washed and incubated with secondary goat anti‐rabbit IgG‐horseradish peroxidase antibody (1 : 2000) (Santa Cruz Biotecnology, Inc., Santa Cruz, CA, USA). Finally, labelled bands were detected by enhanced chemiluminescence.
Luciferase reporter assay
Briefly, a 2‐kb fragment of the human IL‐33 promoter was cloned by PCR to generate IL‐33‐pGL3 reporters in pGL3‐basic vector. HIF‐1α transcription factor plasmid (HIF‐1α TF) and control plasmid (HIF‐1α TF‐NC) were constructed for the over‐expression of HIF‐1α. HIF‐1‐binding site mutants (Mut.A, Mut.B, Mut.C) were generated by the GeneArt® Site‐Directed Mutagenesis System from Life Technologies (Shanghai, China). HT‐29 cells were co‐transfected with IL‐33‐pro (0·1 μg) and HIF‐1α TF (0·3 μg), pGL3‐basic vector (0·1 μg) and HIF‐1α TF (0·3 μg), IL‐33‐pro (0·1 μg) and HIF‐1α TF‐NC (0·3 μg), pGL3‐basic vector (0·1 μg) and HIF‐1α TF‐NC (0·3 μg), respectively. After 24 h of culture, the cells were lysed and analysed for luciferase activity using the Dual‐Luciferase® Reporter Assay System (Promega, Madison, WI, USA). HT‐29 cells were co‐transfected with HIF‐1‐binding site mutants and HIF‐1α TF for 24 h. The luciferase activity was then analysed with the Dual‐Luciferase® Reporter Assay System.
Statistical analysis
Data were expressed as mean ± standard error of the mean (s.e.m.) and statistical analysis was performed using Prism version 5.0 software (Graphpad Software, San Diego, CA, USA). Differences between groups were evaluated by using the unpaired two‐tailed Student's t‐test. Statistical significance was set at P < 0·05. Pearson's correlation was used to analyse the correlation of HIF‐1α and IL‐33 mRNA expression in colonic tissues from IBD patients.
Results
Highly elevated HIF‐1α and IL‐33 in inflamed mucosa from patients with IBD
It has been shown that HIF‐1α is increased in inflamed mucosa of IBD patients using immunohistochemistry 23. We first investigated HIF‐1α expression in inflamed intestinal mucosa of IBD at the transcriptional and translational levels. As shown in Fig. 1a, the levels of HIF‐1α mRNA were up‐regulated significantly in inflamed mucosa of both active CD and UC patients compared with normal controls, whereas the expression of HIF‐1α was decreased in the intestinal mucosa of CD patients in remission as well as UC patients in remission compared with that in active IBD patients. Western blot analysis confirmed further the increase of HIF‐1α in inflamed intestinal mucosa from IBD patients compared with healthy controls (Fig. 1b,c). To localize HIF‐1α‐expressing cells in intestinal mucosa, immunohistochemical staining was performed in intestinal biopsies from active IBD patients and healthy donors. There were more HIF‐1α‐positive cells in inflamed mucosa of patients with IBD compared with normal controls (Fig. 1d–f).
Figure 1.
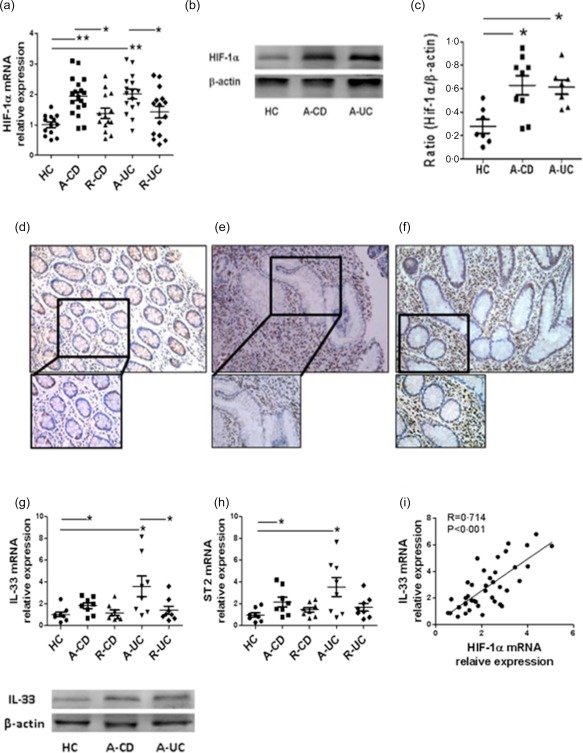
Hypoxia inducible factor‐1α (HIF‐1α) and interleukin (IL)−33 are expressed highly in inflamed mucosa of inflammatory bowel disease (IBD) patients. (a) Relative expression of HIF‐1α mRNA in colonic biopsies from healthy controls (HC, n = 13), inflamed mucosa of patients with active Crohn's disease (CD) (A‐CD, n = 18) and patients with active ulcerative colitis (UC) (A‐UC, n = 16), colonic tissues from CD patients in remission (R‐CD, n = 14) and UC patients in remission (R‐UC, n = 15) was determined by quantitative real‐time–polymerase chain reaction (qRT–PCR) and normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). * P < 0·05; ** P < 0·01. (b,c) Western blot analysis of HIF‐1α in healthy controls (HC), A‐CD and A‐UC. The levels of HIF‐1α were normalized to the β‐actin protein levels. * P < 0·05 versus HC. (d−f) Immunohistochemical staining for HIF‐1α‐positive cells in colonic mucosa from HC, A‐CD and A‐UC. Representative photographs from three independent experiments are shown. (g,h) Relative expression of IL‐33 and receptor suppression of tumorigenicity 2 (ST2) mRNA in colonic biopsies from HC (n = 8), R‐CD (n = 8) and R‐UC (n = 8), inflamed mucosa of A‐CD (n = 8) and A‐UC (n = 8) was determined by qRT–PCR analysis and normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). * P < 0·05. Western blot analysis of IL‐33 in HC, A‐CD and A‐UC. The levels of IL‐33 were normalized to the β‐actin protein levels. (i) Correlation of HIF‐1α with IL‐33 mRNA in intestinal epithelial cell (IECs) from patients with active IBD (n = 41) (Pearson's correlation coefficient, R = 0.714, P < 0·001).
We also analysed the expression of IL‐33 and ST2 in inflamed mucosa from patients with active CD and UC. As shown in Fig. 1g,h, the mRNA and protein levels of IL‐33 and ST2 were increased in inflamed mucosa of patients with active CD and UC compared with healthy controls. However, the mRNA levels of IL‐33 and ST2 were decreased in IBD patients in remission. Interestingly, HIF‐1α expression was correlated positively with IL‐33 expression in freshly isolated IECs from IBD patients (R = 0·714, P < 0·001) (Fig. 1i).
HIF‐1α and IL‐33 are increased in inflamed mucosa of mice with colitis induced by DSS
After confirming the correlation between HIF‐1α and IL‐33 in inflamed mucosa of patients with IBD, we used an in‐vivo study to determine the levels of HIF‐1α and IL‐33 in intestinal mucosa from an experimental colitis model. The colitis was induced in mice by administration of 2% DSS in drinking water for 7 days and switched to water for an additional 3 days 28. The severity of colitis was then assessed according to the changes of weight, colon length and histological scores. As shown in Fig. 2a, significant tissue architecture damage and inflammatory cell infiltration were observed in colonic mucosa from DSS‐treated mice. The weight and colon length were decreased while colonic tissue expression of TNF was increased in DSS‐treated mice compared with controls (Supporting information, Fig. S1a,b and Fig. S2b). Consistent with data from IBD patients described above, the expression of HIF‐1α and IL‐33 was also elevated in inflamed colonic tissues from colitic mice compared with controls (Fig. 2c,d). The increased activity of HIF‐1α was detected in inflamed colon from DSS‐treated mice by examining the expression of vascular endothelial growth factor (VEGF), which possesses the target HRE of HIF‐1α (Fig. 2e). In addition, the levels of IL‐33 were correlated positively with that of HIF‐1α in freshly isolated IECs from DSS‐treated mice (R = 0.8108, P < 0·01) (Fig. 2f). These data confirm further that HIF‐1α and IL‐33 are increased in inflamed mucosa during inflammatory responses, and may play a role in the pathogenesis of intestinal inflammation.
Figure 2.
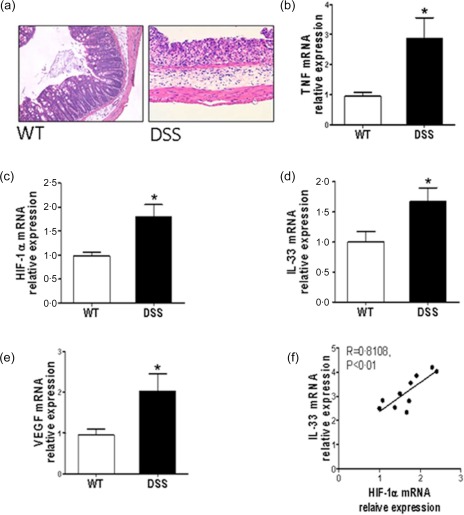
Hypoxia inducible factor‐1α (HIF‐1α) promotes interleukin (IL)−33 expression in inflamed colon of mice with dextran sulphate sodium (DSS)‐induced colitis. (a) Six to 8‐week‐old C57BL/B6 mice (10 mice per group) were given 2% DSS in drinking water for 7 days, and switched to drinking water for an additional 3 days. Haematoxylin and eosin (H&E) staining in colonic tissues from wild‐type (WT) and DSS‐treated mice. (b−e) Quantitative real‐time–polymerase chain reaction (qRT–PCR) analysis for tumour necrosis factor (TNF), HIF‐1α, IL‐33 and vascular endothelial growth factor (VEGF) in colonic tissues of WT and DSS‐treated mice. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a housekeeping gene. * P < 0·05 versus wild‐type (WT) mice. (f) A positive correlation between HIF‐1α and IL‐33 in isolated intestinal epithelial cells (IECs) from colonic tissues of DSS‐treated mice (Pearson's correlation coefficient, R = 0·8108, P < 0·01). Data are plotted as relative expression of HIF‐1α and IL‐33 mRNA (n = 10 mice per group). [Colour figure can be viewed at wileyonlinelibrary.com]
Effects of HIF‐1α on IL‐33 expression in IECs
Given that the levels of HIF‐1α and IL‐33 were increased simultaneously in inflamed mucosa from IBD patients and colitic mice, we then sought to investigate whether HIF‐1α regulates IL‐33 expression directly in IECs. To this end, HT‐29 cells were utilized in vitro to examine the effect of HIF‐1α on IL‐33 expression. As shown in Fig. 3a, HIF‐1α enhanced IL‐33 expression at the mRNA levels in HT‐29 cells, which was up‐regulated further by DMOG, a prolyl hydroxylase (PHD) inhibitor. The expression of HIF‐1α at protein levels was confirmed by Western blot (Supporting information, Fig. S2b). We then used si‐HIF‐1α to silence the expression of HIF‐1α. The efficiency of si‐HIF‐1α transfection was confirmed by its mRNA expression (Supporting information, Fig. S2a). Silencing of HIF‐1α expression resulted in a significant decrease of IL‐33 expression in HT‐29 cells (Fig. 3b).
Figure 3.
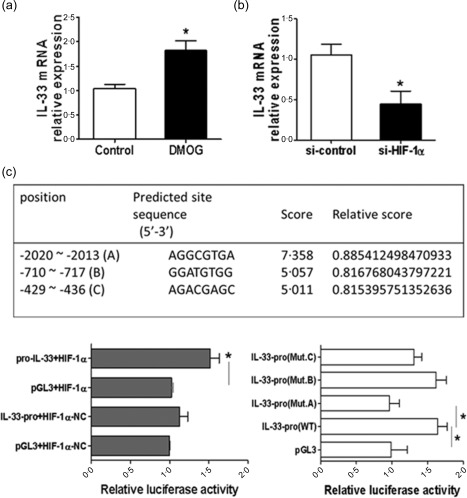
Hypoxia inducible factor‐1α (HIF‐1α) promotes intestinal epithelial interleukin (IL)−33 expression. (a) HT‐29 cells were treated with dimethyloxalylglycine (DMOG) (1 μM) or vehicle for 24 h, and the mRNA levels of IL‐33 were determined by quantitative real‐time–polymerase chain reaction (qRT–PCR). * P < 0·05 versus controls. (b) HT‐29 cells were transfected with HIF‐1α specific siRNA or the scramble siRNA, and the levels of IL‐33 mRNA were determined by qRT–PCR. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a housekeeping gene. * P < 0·05 versus controls. (c) HT‐29 cells were co‐transfected with IL‐33‐pro and HIF‐1α transcription factor (TF), pGL3‐basic vector and HIF‐1α TF, IL‐33‐pro and HIF‐1α TF‐NC, pGL3‐basic vector and HIF‐1α TF‐NC, and the luciferase activity was measured and normalized to Renilla activity (left panel). * P < 0·05. HT‐29 cells were co‐transfected with HIF‐1 TF together with IL‐33‐pro wild‐type (WT), IL‐33‐pro (Mut.A), IL‐33‐pro (Mut.B) or IL‐33‐pro (Mut.C) (right panel). * P < 0·05. Relative luciferase activity of each construct was measured and normalized to Renilla activity. Data represent mean ± standard error of the mean (s.e.m.) from three independent experiments. [Colour figure can be viewed at wileyonlinelibrary.com]
To investigate further whether there are specific HIF‐1α DNA binding sites in the promoter region of IL‐33, we co‐transfected HT‐29 cells with pGL3‐IL‐33 plasmid which contains the IL‐33 promoter sequence and the plasmid containing HIF‐1α transcription factor. Under HIF‐1α over‐expression conditions, the luciferase activity was enhanced significantly in HT‐29 cells after transfection with pGL3‐IL‐33 promoter construct compared with controls, suggesting that HIF‐1α binds the promoter region of IL‐33 and promotes the expression of IL‐33. Three putative binding sites were present through the JASPAR CORE database. To investigate whether these sites are essential for IL‐33 promoter activation, three mutants (Mut.A, Mut.B, Mut.C) in the IL‐33 promoter region were then constructed (Fig. 3c). The luciferase activity was reduced when transfected with Mut.A but not Mut.B or Mut.C containing pGL3‐IL‐33 plasmid as compared with the positive control (i.e. pGL3‐IL‐33 transfection) (Fig. 3c), indicating that HIF‐1α binds effectively to the binding site at −2020 to −2013 in the IL‐33 promoter region and regulates its expression.
TNF up‐regulates HIF‐1α expression through activating nuclear factor (NF)‐κB
To investigate which cytokines or bacterial stimuli induce HIF‐1α expression in intestinal mucosa of IBD patients, we stimulated HT‐29 cells with different cytokines (IFN‐γ, IL‐17A, IL‐23, TNF, IL‐10, TGF‐β) or microbiota stimuli (LPS, PGN, flagellin, polyI:C) in vitro. TNF and LPS, but not other factors tested, induced HIF‐1α mRNA expression (Fig. 4a,b). Furthermore, TNF promoted the HIF‐1α expression in a dose‐ and time‐dependent manner (Fig. 4c,d). To investigate the potential role of TNF in the induction of HIF‐1a in vivo, 10 patients with active CD were infused with anti‐TNF mAb (5 mg/kg, infliximab, IFX) at weeks 0, 2 and 6. HIF‐1α expression was then determined in intestinal mucosa at week 12 after the first infusion. As shown in Fig. 4e, HIF‐1α was decreased in intestinal mucosa 12 weeks after IFX therapy compared with that before IFX treatment. Consistently, when freshly isolated biopsies from inflamed mucosa of 10 CD and 10 UC patients were stimulated with IFX or control human IgG (HIg) in vitro, HIF‐1α was also decreased after IFX treatment compared with controls (Fig. 4f,g). These data thereby indicate that TNF is the potent inducer to facilitate HIF‐1α expression in IEC.
Figure 4.
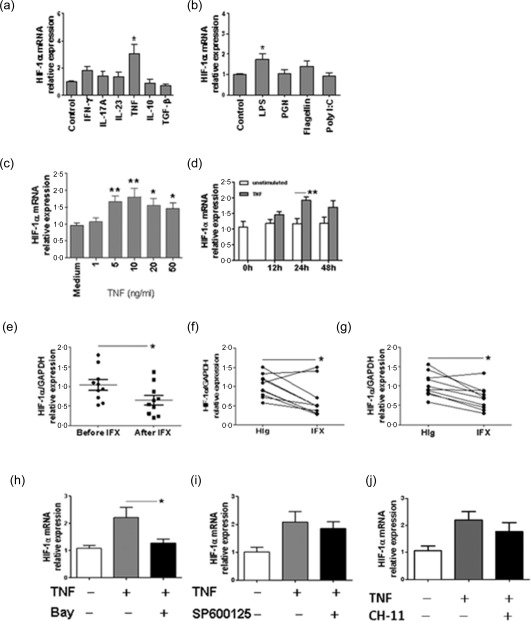
Tumour necrosis factor (TNF) induces hypoxia inducible factor‐1α (HIF‐1α) expression through activating nuclear factor (NF)‐кB. (a,b) HT‐29 cells (5 × 105/ml) were stimulated in vitro with interferon (IFN)‐γ (10 ng/ml), interleukin (IL)−17A (10 ng/ml), IL‐23 (10 ng/ml), TNF (10 ng/ml), IL‐10 (10 ng/ml), transforming growth factor (TGF)‐β (10 ng/ml), lipopolysaccharide (LPS) (100 ng/ml), peptidoglycan (PGN) (1 μg/ml), flagellin (100 ng/ml) and polyI:C (10 μg/ml) for 24 h, relative expression of HIF‐1α mRNA was determined by quantitative real‐time–polymerase chain reaction (qRT–PCR) analysis and normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). * P < 0·05 versus controls. (c) Relative expression of HIF‐1α mRNA in HT‐29 cells under stimulation with different concentrations of TNF (0, 1, 5, 10, 20, 50 ng/ml) for 24 h. Representative results from three independent experiments are shown. * P < 0·05; ** P < 0·01 versus data cultured in medium alone. (d) Relative expression of HIF‐1α mRNA in HT‐29 cells under stimulation with TNF (10 ng/ml) for 0, 12, 24 and 48 h. Representative results from three independent experiments are shown. ** P < 0·05. (e) Relative expression of HIF‐1α mRNA in colonic tissues from the same Crohn's disease (CD) patients (n = 10) before and after anti‐TNF monoclonal antibody (mAb) treatment [infliximab (IFX), 5 mg/kg]. GAPDH was used as a housekeeping gene. * P < 0·05. (f,g) Freshly isolated colonic biopsies from patients with CD (n = 10) (f) or ulcerative colitis (UC) (n = 10) (g) were cultured in the presence of IFX (50 μg/ml) or control human immunoglobulin (HIg, 50 μg/ml) for 24 h. The levels of HIF‐1α mRNA were determined by qRT–PCR and normalized to GAPDH. * P < 0·05. Data are compiled from three independent experiments. (h−j) HT‐29 cells were treated with TNF (10 ng/ml), TNF (10 ng/ml) and Bay 11‐7082 (1 μM) (h), TNF and SP600125 (10 μM) (i), or TNF and CH‐11 (200 ng/ml) (j) for 24 h, and the levels of HIF‐1α mRNA were determined by qRT–PCR. Representative results from three independent experiments are shown. * P < 0·05.
We then investigated the underlying mechanisms of HIF‐1α induction by TNF. HT‐29 cells were stimulated with TNF in vitro in the presence or absence of inhibitors specific for NF‐κB (Bay 11‐7082, 1 μM), JNK (SP600125, 10 μM) and anti‐Fas antibody (CH‐11, 200 ng/ml), respectively. After 24 h of culture, the expression of HIF‐1α mRNA was assessed by qRT–PCR. Interestingly, blockage of NF‐кB activity with Bay 11‐7082 but not SP600125 and CH‐11 reduced HIF‐1α mRNA expression significantly (Fig. 4h–j). Altogether, these results indicated that TNF induces HIF‐1α expression through induction of NF‐кB activation.
TNF induces IL‐33 expression in an HIF‐1α‐dependent manner
It has been shown that TNF induces IL‐33 and HIF‐1α expression in IEC 14, 25, and our data show further that ectopic expression of HIF‐1α enhanced IL‐33 expression. We then investigated whether TNF induces IL‐33 expression through HIF‐1α induction. As shown in Fig. 5a, treatment of HT‐29 cells with TNF resulted in an increase of IL‐33 mRNA, whereas the presence of YC‐1, a HIF‐1α inhibitor, decreased IL‐33 expression. We then used the specific si‐HIF‐1α to inhibit HIF‐1α expression and verify further the relationship among TNF, HIF‐1α and IL‐33. Consistently, specific interference of HIF‐1α with si‐HIF‐1α reduced the TNF‐induced IL‐33 expression in HT‐29 cells (Fig. 5b). As TNF has been shown to induce production of IL‐33 through activation of p38 and ERK pathways in rheumatoid arthritis synovial fibroblasts 31, we investigated whether HIF‐1α induces activation of ERK and p38 upon the stimulation of TNF in IEC. As shown in Fig. 5c, TNF activated ERK and p38 in HT‐29 cells. Interestingly, inhibition of HIF‐1α dampened TNF‐induced ERK and p38 activation. These data suggest that HIF‐1α regulates IL‐33 expression through activation of ERK and p38. To determine the effects of TNF on IL‐33 production in vivo, we assessed IL‐33 expression in inflamed colon from patients with active CD after anti‐TNF treatment. Consistent with in‐vitro data, the expression of IL‐33 in colonic tissue of active CD patients was decreased significantly after IFX treatment (Fig. 5d). The ex‐vivo inflamed colonic tissue cultures also demonstrated a decreased IL‐33 in anti‐TNF (IFX)‐treated colonic tissues from both CD and UC patients compared with controls (Fig. 5e,f). Collectively, these data indicate that TNF promotes the expression of IL‐33 in IECs in an HIF‐1α‐dependent manner.
Figure 5.
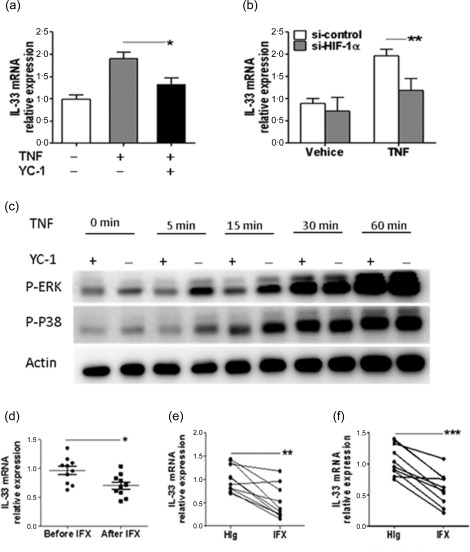
Tumour necrosis factor (TNF) induces interleukin (IL)−33 expression in a hypoxia inducible factor‐1α (HIF‐1α)‐dependent manner. (a) HT‐29 cells were treated with TNF (10 ng/ml), TNF (10 ng/ml) and YC‐1 (10 μM) or vehicle for 24 h, and the levels of IL‐33 mRNA were determined by quantitative real‐time–polymerase chain reaction (qRT–PCR). * P < 0·05. (b) HT‐29 cells transfected with si‐HIF‐1α or si‐control (50 nM) for 48 h were then stimulated with TNF (10 ng/ml) for 24 h, and IL‐33 mRNA was determined by qRT–PCR and normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) expression. ** P < 0·01. Representative results from three independent experiments are shown. (c) HT‐29 cells were first treated with or without YC‐1 (10 μM) for 24 h, and then stimulated with TNF (10 ng/ml) for 0, 5, 15, 30 and 60 min, respectively, the activation of extracellular‐regulated kinase (ERK) and p‐38 pathway was determined by Western blot. (d) Relative expression of IL‐33 mRNA in colonic tissues from the same Crohn's disease (CD) patients (n = 10) before and after anti‐TNF monoclonal antibody (mAb) treatment [infliximab (IFX), 5 mg/kg]. GAPDH was used as a housekeeping gene. * P < 0·05. (e,f) Freshly isolated colonic biopsies from patients with CD (n = 10) (e) or ulcerative colitis (UC) (n = 10) (f) were incubated in the presence of IFX (50 μg/ml) or control human IgG (HIg, 50 μg/ml) for 24 h. The levels of IL‐33 mRNA were analysed by qRT–PCR and normalized to GAPDH. ** P < 0·01; *** P < 0·001. Data are compiled from three independent experiments.
Discussion
Among multiple mechanisms involved in the pathogenesis of IBD, the inflammatory microenvironment of intestinal mucosa is considered to play a pivotal role in the immune regulation of intestinal inflammation. HIF‐1α, a key regulator in hypoxia, has been shown to play pleiotrophic roles in the development of intestinal inflammation through maintaining the integrity of IECs and the balance of immune response 32, 33, 34, and that the inflammatory hypoxia present in inflamed mucosa of IBD leads to the accumulation of HIF‐1α in intestinal mucosa 23. We reported in the current study that the expression of HIF‐1α and IL‐33 was increased simultaneously in inflamed mucosa of patients with IBD, and that there was a positive relationship between HIF‐1α and IL‐33. A functional binding site for HIF‐1α was identified in the IL‐33 promoter region. Moreover, TNF promoted expression of HIF‐1α through activating NF‐κB signalling and regulating IL‐33 expression in an HIF‐1α‐dependent manner. Therefore, we demonstrated for the first time that HIF‐1α induces the expression of IEC‐derived IL‐33 to regulate the intestinal homeostasis.
Several lines of evidence have implicated that IEC‐derived HIF‐1α maintains the integrity of intestinal barrier and that several HIF‐1α target genes (e.g. intestinal trefoil factor, CD73, multi‐drug resistance gene‐1) are involved in the protection of epithelial barrier 35, 36, 37. In this study, we confirmed first that HIF‐1α was over‐expressed in inflamed mucosa of patients with IBD and showed that IEC was an important source of HIF‐1α (Fig. 1a–f). Importantly, we also identified a novel target gene (IL‐33) of HIF‐1α. IL‐33 is considered as an alarmin, which can be released by epithelial cells on the condition of injury and functions as a dual‐faceted cytokine depending on the environment or the phase of intestinal inflammation. It mediates local inflammatory reactions, including epithelial damage, recruitment of neutrophils and macrophages. Previous studies have demonstrated that IL‐33 is increased in the sera 25 and inflamed mucosa of UC patients, and promotes intestinal inflammation by enhancing Th2‐associated immune responses 18. Recently, IL‐33 is also found to contribute to wound healing by inducing the restoration of epithelial barrier during the recovery phage of chronic DSS‐induced colitis in mice 38, and regulates intestinal homeostasis through induction of TGF‐β‐mediated Treg differentiation, function and adaptation to inflammatory responses in gut mucosa 19. A recent study indicates further that IL‐33 enhances the transepithelial resistance in T84 colon epithelial cells and decreases the epithelial permeability in wide‐type mice compared with ST2−/− mice 39. Therefore, we hypothesized that HIF‐1α regulates the production of IEC‐derived IL‐33 in response to low oxygen pressure during intestinal inflammation. Indeed, over‐expression of HIF‐1α in HT‐29 cells facilitated IL‐33 production (Fig. 3a). Consistently, silencing of HIF‐1α gene expression decreased IL‐33 expression (Fig. 3b). More importantly, we found a functional binding site for HIF‐1α in the IL‐33 promoter region (Fig. 3c), thereby confirming IL‐33 as a target of intestinal HIF‐1 activity through the promotion of the HIF‐1α expression.
Our data also demonstrate that TNF induced HIF‐1α expression through induction of NF‐κB activation (Fig. 4). To determine the role of HIF‐1α in TNF‐induced IL‐33, we stimulated HT‐29 cells with TNF and HIF‐1α inhibitor and found that inhibition of HIF‐1α suppressed the expression of IL‐33 in HT‐29 cells (Fig. 5a,b). These data thus supported the notion that local hypoxia or secretion of proinflammatory cytokines triggered by inflammation induces the adaptive response to the body injuries, which was consistent with the previous observation showing that TNF up‐regulated the expression of HIF‐1α 14 and IL‐33 in IECs 25. Together, we demonstrated that TNF regulates IL‐33 through HIF‐1α signalling, suggesting that increased mucosal HIF‐1α and IL‐33 in human IBD and murine colitis could be an important homeostatic response to limit inflammation in gut mucosa.
In summary, our studies demonstrate that HIF‐1α plays a critical role in the induction of IL‐33 expression and regulates the intestinal immune responses (Fig. 6). During intestinal inflammation, TNF produced by intestinal mucosal immune cells (e.g. CD4+ T cells, dendritic cells) induces expression of HIF‐1α in IEC via activating NF‐κB. HIF‐1α forms HIF heterodimer with β‐subunit and then binds to the promoter of IL‐33, leading to the over‐expression of IL‐33 in IECs and contributing to the maintenance of mucosal homeostasis. Future studies are needed to address the roles of HIF‐1α and IL‐33 in the regulation of the integrity of mucosal barrier and immune response in IBD and, importantly, they may serve as a novel therapeutic target in the management of IBD.
Figure 6.
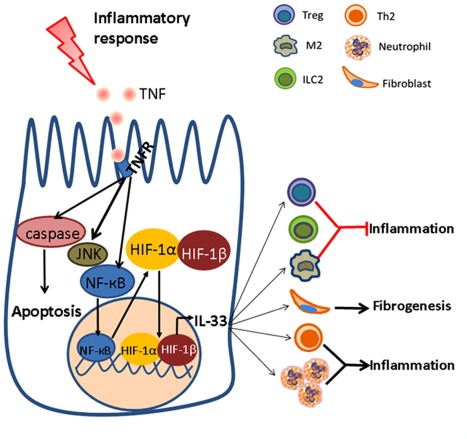
Potential role of the hypoxia inducible factor‐1α (HIF‐1α)–interleukin (IL)−33 axis in regulating intestinal mucosal inflammation in inflammatory bowel disease (IBD). Under inflammatory conditions, the intestinal mucosa encounters different micro‐environmental challenges, including increased expression of proinflammatory cytokines and low oxygen tension. An increase of tumour necrosis factor (TNF) could up‐regulate HIF‐1α expression through activating nuclear factor (NF)‐кB in a hypoxia‐independent manner, which further promotes the expression of IL‐33 in in intestinal epithelial cell (IECs) by binding hypoxic response elements (HRE) in the promoter region of IL‐33. Importantly, IL‐33 facilitates mucosal inflammation via enhancing T helper type 2 (Th2)‐associated immune response and neutrophil recruitment, and prevents inflammatory responses by inducing the differentiation of regulatory immune cells such as regulatory T cell (Treg) and alternatively activated (M2) macrophage. Additionally, IL‐33 may also activate fibroblast to accelerate wound healing. [Colour figure can be viewed at wileyonlinelibrary.com]
Disclosure
All authors have no financial disclosures to declare.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Animal model of dextran sulphate sodium (DSS)‐induced colitis in mice. C57BL/B6 mice (n = 10) were treated with 2% DSS in drinking water for 7 days, and switched to drinking water for additional 3 days. The control group (n = 10) was fed with fresh water for 10 days. The changes of the colon length (a) and body weight (b) were measured to assess the severity of colitis. ** P < 0·01 versus wild‐type (WT) mice; * P < 0·05 versus WT mice at the same time‐points.
Fig. S2. The efficiency of si‐hypoxia inducible factor‐1α (HIF‐1α) transfection and dimethyloxalylglycine (DMOG) treatment in HT‐29 cells. (a) HT‐29 cells were transfected with HIF‐1α specific siRNA (50 nM) or the scramble siRNA (50 nM) using lipofectamine 2000 in 500 μl opti‐modified Eagle's medium (MEM) medium. After 48 h transfection, the total RNA was extracted from HT‐29 cells for quantitative real‐time–polymerase chain reaction (qRT–PCR) analysis. * P < 0·05 versus data from cells transfected with scramble siRNA or cultured in medium alone. (b) Western blot was performed to determine the expression of HIF‐1α in HT‐29 cells after DMOG administration. Data are representative of three independent experiments.
Acknowledgements
This work is supported by grants from the National Natural Science Foundation of China (81270470, 81470822, 81630017) and the Science and Technology Commission of Shanghai Municipality (14401970100).
References
- 1. Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut 2013; 62:1505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu Z, Cao AT, Cong Y. Microbiota regulation of inflammatory bowel disease and colorectal cancer. Semin Cancer Biol 2013; 23:543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peterson CT, Sharma V, Elmen L, Peterson SN. Immune homeostasis, dysbiosis and therapeutic modulation of the gut microbiota. Clin Exp Immunol 2015; 179:363–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cader MZ, Kaser A. Recent advances in inflammatory bowel disease: mucosal immune cells in intestinal inflammation. Gut 2013; 62:1653–64. [DOI] [PubMed] [Google Scholar]
- 5. Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol 2015; 8:969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 1997; 11:72–82. [DOI] [PubMed] [Google Scholar]
- 8. Makino Y, Cao R, Svensson K et al Inhibitory PAS domain protein is a negative regulator of hypoxia‐inducible gene expression. Nature 2001; 414:550–4. [DOI] [PubMed] [Google Scholar]
- 9. Semenza GL. HIF‐1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol 2001; 13:167–71. [DOI] [PubMed] [Google Scholar]
- 10. Hellwig‐Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin‐1beta and tumor necrosis factor‐alpha stimulate DNA binding of hypoxia‐inducible factor‐1. Blood 1999; 94:1561–7. [PubMed] [Google Scholar]
- 11. Liu H, Wang P, Cao M, Li M, Wang F. Protective role of oligomycin against intestinal epithelial barrier dysfunction caused by IFN‐gamma and TNF‐alpha. Cell Physiol Biochem 2012; 29:799–808. [DOI] [PubMed] [Google Scholar]
- 12. Peyssonnaux C, Cejudo‐Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: essential role of hypoxia inducible factor‐1alpha in development of lipopolysaccharide‐induced sepsis. J Immunol 2007; 178:7516–9. [DOI] [PubMed] [Google Scholar]
- 13. Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF‐1 activation in human monocytes via p44/42 MAPK and NF‐kappaB. Biochem J 2006; 396:517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia‐inducible factor induction by tumour necrosis factor in normoxic cells requires receptor‐interacting protein‐dependent nuclear factor kappa B activation. Biochem J 2003; 370:1011–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pastorelli L, De Salvo C, Vecchi M, Pizarro TT. The role of IL‐33 in gut mucosal inflammation. Mediators Inflamm 2013; 2013:608187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beltran CJ, Nunez LE, Diaz‐Jimenez D et al Characterization of the novel ST2/IL‐33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis 2010; 16:1097–107. [DOI] [PubMed] [Google Scholar]
- 17. Shadie AM, Herbert C, Kumar RK. Ambient particulate matter induces an exacerbation of airway inflammation in experimental asthma: role of interleukin‐33. Clin Exp Immunol 2014; 177:491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seidelin JB, Bjerrum JT, Coskun M, Widjaya B, Vainer B, Nielsen OH. IL‐33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett 2010; 128:80–5. [DOI] [PubMed] [Google Scholar]
- 19. Schiering C, Krausgruber T, Chomka A et al The alarmin IL‐33 promotes regulatory T‐cell function in the intestine. Nature 2014; 513:564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Besnard AG, Guabiraba R, Niedbala W et al IL‐33‐mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLOS Pathog 2015; 11:e1004607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang HR, Milovanovic M, Allan D et al IL‐33 attenuates EAE by suppressing IL‐17 and IFN‐gamma production and inducing alternatively activated macrophages. Eur J Immunol 2012; 42:1804–14. [DOI] [PubMed] [Google Scholar]
- 22. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL‐33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin‐EGFR interactions. Proc Natl Acad Sci USA 2015; 112:10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giatromanolaki A, Sivridis E, Maltezos E et al Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol 2003; 56:209–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007; 81:1–5. [DOI] [PubMed] [Google Scholar]
- 25. Pastorelli L, Garg RR, Hoang SB et al Epithelial‐derived IL‐33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci USA 2010; 107:8017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu C, Xia X, Wu W et al Anti‐tumour necrosis factor therapy enhances mucosal healing through down‐regulation of interleukin‐21 expression and T helper type 17 cell infiltration in Crohn's disease. Clin Exp Immunol 2013; 173:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu W, He C, Liu C et al miR‐10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut 2015; 64:1755–64. [DOI] [PubMed] [Google Scholar]
- 28. He C, Shi Y, Wu R et al miR‐301a promotes intestinal mucosal inflammation through induction of IL‐17A and TNF‐alpha in IBD. Gut 2016; 65:1938–50. [DOI] [PubMed] [Google Scholar]
- 29. Rimoldi M, Chieppa M, Salucci V et al Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol 2005; 6:507–14. [DOI] [PubMed] [Google Scholar]
- 30. Hindryckx P, De Vos M, Jacques P et al Hydroxylase inhibition abrogates TNF‐alpha‐induced intestinal epithelial damage by hypoxia‐inducible factor‐1‐dependent repression of FADD. J Immunol 2010; 185:6306–16. [DOI] [PubMed] [Google Scholar]
- 31. Hu F, Shi L, Mu R et al Hypoxia‐inducible factor‐1alpha and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PLOS ONE 2013; 8:e72650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia‐inducible factor‐1 is protective in murine experimental colitis. J Clin Invest 2004; 114:1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dang EV, Barbi J, Yang HY et al Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell 2011; 146:772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clambey ET, McNamee EN, Westrich JA et al Hypoxia‐inducible factor‐1 alpha‐dependent induction of FoxP3 drives regulatory T‐cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA 2012; 109:E2784–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furuta GT, Turner JR, Taylor CT et al Hypoxia‐inducible factor 1‐dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 2001; 193:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Synnestvedt K, Furuta GT, Comerford KM et al Ecto‐5′‐nucleotidase (CD73) regulation by hypoxia‐inducible factor‐1 mediates permeability changes in intestinal epithelia. J Clin Invest 2002; 110:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia‐inducible factor‐1‐dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 2002; 62:3387–94. [PubMed] [Google Scholar]
- 38. Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL‐33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm Bowel Dis 2012; 18:1900–9. [DOI] [PubMed] [Google Scholar]
- 39. Waddell A, Vallance JE, Moore PD et al IL‐33 signaling protects from murine oxazolone colitis by supporting intestinal epithelial function. Inflamm Bowel Dis 2015; 21:2737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Animal model of dextran sulphate sodium (DSS)‐induced colitis in mice. C57BL/B6 mice (n = 10) were treated with 2% DSS in drinking water for 7 days, and switched to drinking water for additional 3 days. The control group (n = 10) was fed with fresh water for 10 days. The changes of the colon length (a) and body weight (b) were measured to assess the severity of colitis. ** P < 0·01 versus wild‐type (WT) mice; * P < 0·05 versus WT mice at the same time‐points.
Fig. S2. The efficiency of si‐hypoxia inducible factor‐1α (HIF‐1α) transfection and dimethyloxalylglycine (DMOG) treatment in HT‐29 cells. (a) HT‐29 cells were transfected with HIF‐1α specific siRNA (50 nM) or the scramble siRNA (50 nM) using lipofectamine 2000 in 500 μl opti‐modified Eagle's medium (MEM) medium. After 48 h transfection, the total RNA was extracted from HT‐29 cells for quantitative real‐time–polymerase chain reaction (qRT–PCR) analysis. * P < 0·05 versus data from cells transfected with scramble siRNA or cultured in medium alone. (b) Western blot was performed to determine the expression of HIF‐1α in HT‐29 cells after DMOG administration. Data are representative of three independent experiments.