

Summary
Refractory coeliac disease (RCD) is a form of coeliac disease (CD) resistant to gluten‐free diet and associated with elevated risk of complications. Many effector cytokines over‐produced in the gut of patients with RCD are supposed to amplify the tissue‐destructive immune response, but it remains unclear if the RCD‐associated mucosal inflammation is sustained by defects in counter‐regulatory mechanisms. The aim of the present study was to determine whether RCD‐related inflammation is marked by high Smad7, an intracellular inhibitor of transforming growth factor‐β 1 (TGF‐β 1) activity. Smad7 was evaluated in duodenal biopsy samples of patients with RCD, patients with active CD, patients with inactive CD and healthy controls by Western blotting, immunohistochemistry and real‐time PCR. In the same samples, TGF‐β 1 and phosphorylated (p)‐Smad2/3 were evaluated by ELISA and immunohistochemistry, respectively. Pro‐inflammatory cytokine expression was evaluated in RCD samples cultured with Smad7 sense or antisense oligonucleotide. Smad7 protein, but not RNA, expression was increased in RCD compared with active and inactive CD patients and healthy controls and this was associated with defective TGF‐β 1 signalling, as marked by diminished p‐Smad2/3 expression. TGF‐β 1 protein content did not differ among groups. Knockdown of Smad7 in RCD biopsy samples reduced interleukin‐6 and tumour necrosis factor‐α expression. In conclusion, in RCD, high Smad7 associates with defective TGF‐β 1 signalling and sustains inflammatory cytokine production. These results indicate a novel mechanism by which the mucosal cytokine response is amplified in RCD and suggest that targeting Smad7 can be therapeutically useful in RCD.
Keywords: gluten, inflammation, mucosal immune response, transforming growth factor‐β
Abbreviations
- ACD
active coeliac disease
- CD
coeliac disease
- ICD
inactive coeliac disease
- IELs
intraepithelial lymphocytes
- IL
interleukin
- RCD
refractory coeliac disease
- TCR
T‐cell receptor
- TGF‐β1
transforming growth factor‐β 1
Introduction
Refractory coeliac disease (RCD) is a form of coeliac disease (CD) characterized by the persistence or recurrence of symptoms/signs of malabsorption and villous atrophy despite a gluten‐free diet for > 12 months in the absence of other disorders. This condition does not necessarily require the presence of a positive CD‐specific serology.1, 2, 3, 4, 5 RCD is usually divided into two types taking into account the immunophenotyping of intraepithelial lymphocytes (IELs): type I (RCDI) characterized by normal and polyclonal IEL phenotype and type II (RCDII) with aberrant IELs deficient in CD3/T‐cell receptor complexes, but containing intracellular CD3ε and a clonal rearrangement of the γ‐chain of the T‐cell receptor.6, 7, 8 Unlike RCDI, RCDII is associated with a poor prognosis because it may more frequently evolve into enteropathy‐associated T‐cell lymphoma.9
The pathogenesis of RCD is not yet fully understood, but activation of cytotoxic IELs and enhanced production of effector cytokines by immune cells are supposed to contribute to the tissue damage in this disorder.10, 11, 12 This hypothesis is in line with the clinical improvement of most patients with RCDI following treatment with immunosuppressive drugs.8, 13 The fact that RCD‐associated mucosal inflammation proceeds uncontrolled despite gluten withdrawal from the diet suggests that the immune response leading to epithelial injury differs from that seen in active CD (ACD).14 Indeed, production of interleukin‐13 (IL‐13), a T helper type 2‐related cytokine, is higher in patients with RCD than in patients with ACD.15 Similarly, other canonical inflammatory cytokines, such as tumour necrosis factor‐α (TNF‐α) and IL‐6, are preferentially produced in RCD and not in ACD.14
Transforming growth factor‐β 1 (TGF‐β 1) is a multifunctional cytokine that plays a critical role in controlling inflammatory responses and oral tolerance.16, 17 Studies in transgenic mice expressing both human CD4 and HLA‐DQ8 molecules have documented the role of TGF‐β 1 in preventing intestinal inflammation in response to oral challenge by gliadin peptides.18 The ability of TGF‐β 1 to negatively control inflammatory responses is mainly mediated by binding of the cytokine to a heterodimeric receptor, which leads to the phosphorylation and activation of the intra‐cellular proteins Smad2 and Smad3.19 Once activated, Smad2 and Smad3 form a complex with Smad4, which translocates to the nucleus where it inhibits transcription of many inflammatory genes.20 Expression of phosphorylated Smad3 (p‐Smad3) is markedly reduced in the gut of patients with inflammatory bowel diseases, and this defect has been associated with high Smad7, an inhibitor of TGF‐β‐induced Smad3 phosphorylation. Indeed, knockdown of Smad7 with a specific Smad7 antisense oligonucleotide restores TGF‐β 1 signalling thus suppressing inflammatory pathways.21, 22 In contrast, in the duodenal mucosa of patients with ACD, TGF‐β 1 signalling is reduced, but this phenomenon seems to rely on IL‐15‐mediated c‐jun N‐terminal kinase activation rather than induction of Smad7.23
Based on these observations and on the fact that the RCD‐linked immune response shows similarities with that seen in inflammatory bowel disease, we hypothesized that RCD‐related inflammation could be associated with elevated levels of Smad7. Therefore, this study was aimed at examining whether high Smad7 contributes to sustain the effector cytokine response in RCD.
Materials and methods
Patients and samples
Forty‐three patients undergoing upper endoscopy at the Gastrointestinal Unit of Tor Vergata University Hospital (Rome, Italy) or S. Matteo Hospital, University of Pavia, were enrolled. The study population included 12 patients on a gluten‐containing diet who were diagnosed as patients with ACD; all these patients were positive for both serum anti‐endomysium IgA antibody and anti‐tissue transglutaminase‐2 antibody and had villous atrophy on histological examination according to Marsh classification (grade 3 B–C).24, 25 Ten patients with a previously confirmed diagnosis of CD, who were asymptomatic, negative for both anti‐endomysium IgA antibody and anti‐tissue transglutaminase‐2 antibodies, and had normal duodenal histology while receiving a strict gluten‐free diet, were considered as patients with inactive CD (ICD). Eleven individuals who were negative for both antibodies while receiving a gluten‐containing diet and had no villous atrophy were considered as a control group. The remaining 10 patients had a previous diagnosis of CD, and persistent malabsorption symptoms and histological evidence of villous atrophy (Marsh, grade 3 A–C) despite a long‐term gluten‐free diet. In all those patients, gluten‐free diet was accompanied by negativity of both anti‐endomysium IgA antibody and anti‐tissue transglutaminase‐2 and a complete diagnostic work‐up for other disorders was negative. Therefore, a diagnosis of RCD was made in all these patients. Five out of 10 patients were subsequently classified as RCDI; four of them were receiving steroids but the other was taking no drugs. The other five patients were classified as RCDII: three patients had ulcerative jejunoileitis and were receiving steroids, while two were taking no drugs.
Duodenal biopsies were taken from the distal duodenum and oriented on tissue paper and immediately either fixed in formalin and then embedded in paraffin or taken for RNA and protein extraction.
Each patient who took part in the study gave written informed consent.
Immunohistochemistry
Paraffin‐embedded duodenal sections of four patients with ACD, four patients with ICD, seven patients with RCD (four RCDI, three RCDII) and four healthy controls were analysed for Smad7 and p‐Smad2/3 using anti‐human Smad7 mouse monoclonal antibody (R&D Systems, Minneapolis, MN: 1 : 50 final dilution) or anti‐human p‐Smad2/3 rabbit polyclonal antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA; 1 : 1000 final dilution). Isotype control IgG‐stained sections were prepared under identical immunohistochemical conditions as described above, replacing the primary antibody with a purified mouse normal IgG control antibody (R&D Systems, Minneapolis, MN).
Total protein extraction and Western blotting
All reagents were from Sigma‐Aldrich (Milan, Italy), unless specified. Duodenal biopsies of 10 patients with ACD, 10 patients with ICD, 10 patients with RCD (five RCDI, five RCDII) and 10 healthy controls were lysed on ice with a buffer containing 10 mm HEPES (pH 7·9), 10 mm KCl, 0·1 mm EDTA and 0·5% Nonidet P‐40, supplemented with 1 mm dithiothreitol, 10 mg/ml aprotinin, 10 mg/ml leupeptin, 1 mm PMSF, 1 mm Na3VO4 and 1 mm NaF. Lysates were clarified by centrifugation at 12 000 g for 30 min at 4° and separated by electrophoresis on 10% SDS–polyacrylamide gels. Smad7 was detected using a monoclonal mouse anti‐human antibody (final concentration 1 µg/ml, R&D Systems) followed by a horseradish peroxidase‐conjugated rabbit anti‐mouse IgG monoclonal antibody (Dako, Milan, Italy). The reactions were detected with a sensitive enhanced chemiluminescence kit (Pierce, Rockford, IL). After the analysis, blots were stripped and incubated with a mouse anti‐human β‐actin antibody followed by a rabbit anti‐mouse antibody conjugated to horseradish peroxidase, to ascertain equivalent loading of lanes.
Organ cultures
Duodenal biopsies of five healthy patients were placed on Transwell (transwell permeable support, Costar; Corning Inc., Corning, NY, USA) in 24‐well plates containing X‐VIVO medium supplemented with 1% penicillin/streptomycin (P/S) and 50 μg/ml gentamycin in the presence of IL‐6 (10 μg/ml) (Peprotech, London, UK), TNF‐α (10 μg/ml) (R&D Systems), IL‐15 (25 μg/ml) (R&D Systems) or no stimulation. The culture was performed in an organ culture chamber at 37° in a 5% CO2/95% O2 atmosphere. After 24 hr, total proteins were extracted and Smad7 was detected by Western blotting as previously described.
Duodenal biopsies of three patients with RCD were placed on Transwell (transwell permeable support, Costar; Corning Inc.) in 24‐well plates containing X‐VIVO medium supplemented with 1% P/S and 50 μg/ml gentamycin in the presence of either Smad7 sense or Smad7 antisense oligonucleotide (both used at 10 μg/ml).21 The culture was performed in an organ culture chamber at 37° in a 5% CO2/95% O2 atmosphere for 24 hr and at the end the samples were used for RNA extraction. The supernatants were collected and analysed for IL‐6 and TNF‐α content by ELISA.
Total protein extraction and ELISA
Total proteins extracted from duodenal biopsies of 12 patients with ACD, eight patients with ICD, eight patients with RCD (five RCDI, three RCDII) and 11 healthy controls were analysed for active TGF‐β 1 using a sensitive commercial ELISA kit (R&D Systems). Interleukin‐6 and TNF‐α were analysed in the organ culture supernatants of RCD biopsies incubated with Smad7 sense or Smad7 antisense oligonucleotide using sensitive commercial ELISA kits (R&D Systems).
RNA extraction, cDNA preparation and real‐time PCR
Total RNA of 11 patients with ACD, nine patients with ICD, seven patients with RCD (four RCDI, three RCDII) and 11 healthy controls was extracted using a Pure Link mRNA mini kit according to the manufacturer's instructions (Life Technologies, Milan, Italy). A constant amount of RNA (1 μg/sample) was retro‐transcribed into cDNA and then 1 μl of cDNA/sample was amplified using a Sybergreen mastermix (BioRad, Milan, Italy). PCR was performed using the following conditions: denaturation 1 min at 95°; annealing 30 seconds at 62° for Smad7 and at 60° for β‐actin; primers sequence was as follows: Smad7 forward 5′‐CGGATCTCAGGCATTCCTCG‐3′, reverse 5′‐GGCACAGCATCTGGACAGTC‐3′. RNA was also extracted from duodenal biopsies cultured as indicated above and used for assessing cytokine transcripts by PCR using the following conditions: denaturation 1 min at 95°; annealing 30 seconds at 61° for IL‐6, at 62° for TNF‐α and at 60° for β‐actin. Primer sequence were as follows: IL‐6 forward 5′‐CCACTCACCTCTTCAGAACG‐3′, reverse 5′‐GCCTCTTTGCTGCTTTCACAC‐3′; TNF‐α forward 5′‐AGGCGGTGCTTGTTCCTCAG‐3′, reverse 5′‐GGCTACAGGCTTGTCACTCG‐3′; β‐actin forward 5′‐AAGATGACCCAGATCATGTTTGAGACC‐3′, reverse 5′‐AGCCAGTCCAGACGCAGGAT‐3′ was used as internal control.
RNA expression was calculated relative to the housekeeping β‐actin gene on the base of the ∆∆Ct algorithm.
Data analysis
Differences between groups were compared using the Mann–Whitney U‐test or the unpaired t‐test. Statistical differences were assessed with the graphpad prism statistical PC program (GraphPad Software, San Diego, CA). A P‐value of < 0·05 was considered statistically significant.
Results
Smad7 protein, but not RNA, is increased in patients with RCD
To determine whether Smad7 expression is increased in patients with RCD, total proteins were extracted from duodenal biopsy samples of patients with RCD, patients with ACD, patients with ICD and healthy controls and analysed for Smad7 by Western blotting. Smad7 was detectable in all the samples with no apparent difference between patients with ACD and those with ICD or controls. In contrast, immunoreactivity corresponding to Smad7 was more pronounced in patients with RCD in comparison to controls (Fig. 1a). Densitometric analysis of immunoblots revealed that Smad7 expression was significantly higher in patients with RCD than in ACD, ICD and controls (Fig. 1a, right panel).
Figure 1.
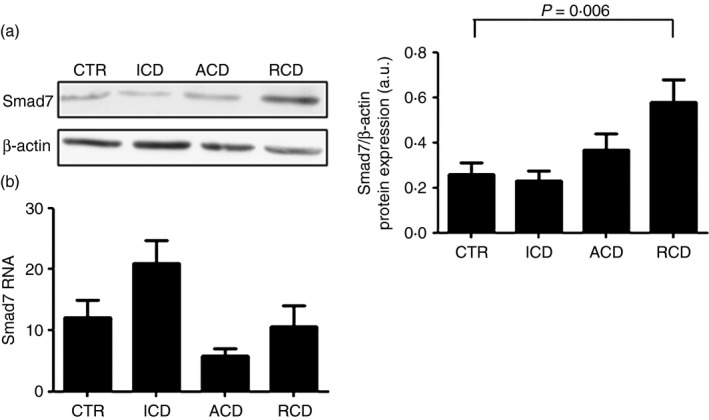
Smad7 protein expression is increased in patients with refractory coeliac disease (RCD). (a) Representative Western blots showing Smad7 and β‐actin in total proteins extracted from mucosal samples of one healthy control (CTR), one patient with active coeliac disease (ACD), one patient with inactive coeliac disease (ICD) and one patient with RCD. Right panel shows the quantitative analysis of Smad7/β‐actin ratio in mucosal samples taken from 10 healthy controls (CTR), 10 patients with ACD, 10 patients with ICD and 10 patients with RCD as measured by densitometry scanning of Western blots. Values are expressed in arbitrary units (a.u.) and indicate mean ± SEM of all samples. (b) Smad7 RNA expression is not increased in RCD patients. Smad7 RNA transcripts were evaluated in duodenal biopsies of seven CTR, 10 patients with ACD, eight patients with ICD and six patients with RCD by real‐time PCR, and levels were normalized to β‐actin. Data indicate mean ± SEM of all samples.
Next, we evaluated Smad7 RNA transcripts in the same groups by real‐time PCR: no significant difference was seen among groups (Fig. 1b). Taken together, these data suggest that Smad7 is differently regulated in the duodenum of patients with CD and up‐regulation of Smad7 occurs only in RCD.
In RCD, Smad7 is over‐expressed in both epithelial and lamina propria cells
To assess the cell sources of Smad7 in patients with RCD, we performed immunohistochemical analysis of the protein using paraffin‐embedded sections of duodenal biopsies of patients and controls. Smad7 staining was very faint in both epithelial and lamina propria mononuclear cells of patients with ACD, patients with ICD and healthy controls with no difference among groups. In contrast, Smad7‐positive cells were abundant in both the epithelial and lamina propria compartments of RCD (Fig. 2a) and the number of such cells did not differ between RCDI and RCDII (Fig. 2b).
Figure 2.
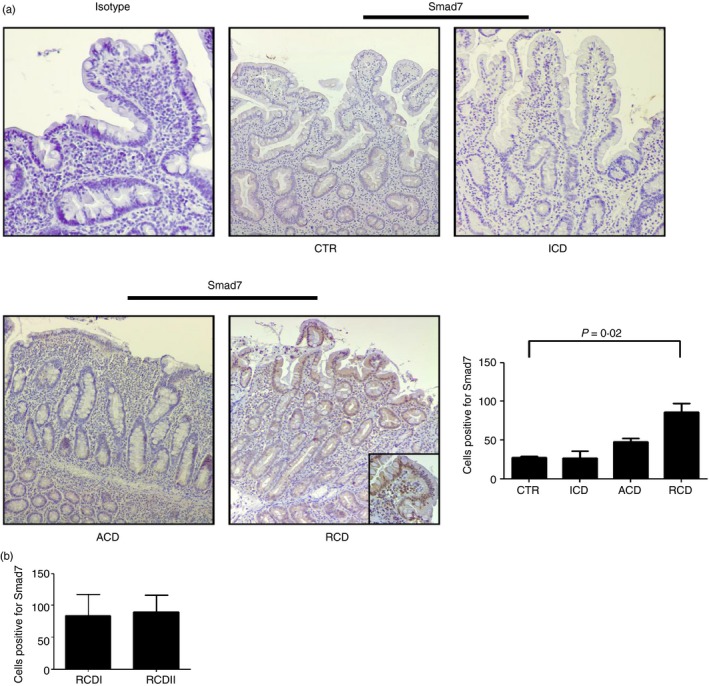
Smad7 expression is increased in both epithelial and lamina propria compartments in refractory coeliac disease (RCD). (a) Representative photomicrographs (×100 original magnification) of Smad7‐stained paraffin‐embedded sections of biopsy samples taken from one control (CTR), one patient with active coeliac disease (ACD), one patient with inactive coeliac disease (ICD) and one patient with RCD. In RCD, Smad7‐positive cells are evident in both the epithelial and lamina propria compartments. Higher magnification photomicrograph (× 200) is shown in the insert. Staining with control IgG is also shown. Right panel shows the number of Smad7‐positive cells per high‐power field (hpf) in duodenal sections taken from three CTR, four patients with ACD, four patients with ICD and seven patients with RCD (four RCDI and three RCDII). Data indicate mean ± SE. (b) Number of Smad7‐positive cells in duodenal sections of four patients with RCDI and three patients with RCDII. Data indicate mean ± SE. [Colour figure can be viewed at wileyonlinelibrary.com]
TGF‐β 1‐associated Smad signalling is impaired in RCD
Since Smad7 is an inhibitor of TGF‐β 1/Smad signalling,26 we next evaluated expression of p‐Smad2/3 in patients with RCD and controls by immunohistochemistry. In the normal duodenum, both epithelial cells and lamina propria mononuclear cells expressed p‐Smad2/3 and the number of such cells did not differ between patients with ACD or ICD and controls (Fig. 3). In contrast, p‐Smad2/3‐positive cells were reduced in the epithelial and lamina propria compartments of patients with RCD (Fig. 3).
Figure 3.
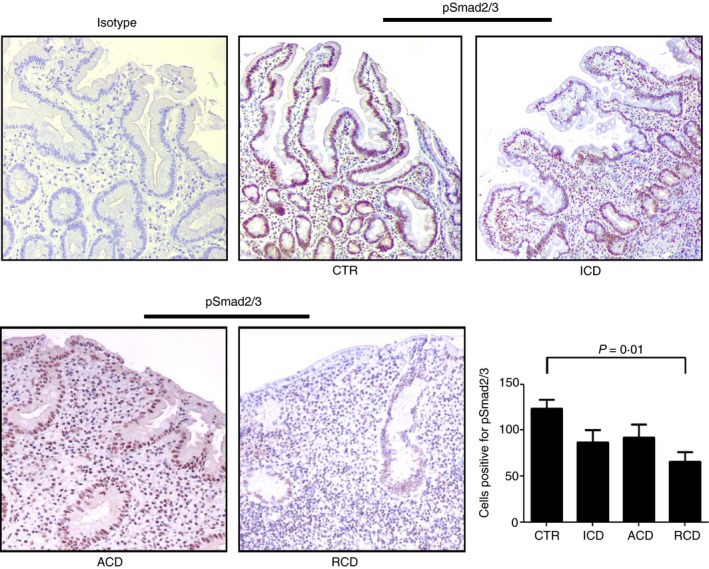
Transforming growth factor‐β 1 (TGF‐β 1)/Smad signalling is altered in refractory coeliac disease (RCD). Representative photomicrographs (× 100 original magnification) of p‐Smad2/3‐stained paraffin‐embedded sections of biopsy samples taken from one control (CTR), one patient with active coeliac disease (ACD), one patient with inactive coeliac disease (ICD) and one patient with RCD. In RCD, p‐Smad2/3 staining is reduced in both the epithelial and lamina propria compartments. The right panel shows the number of pSmad2/3‐positive cells per high‐power field (hpf) in duodenal sections taken from five CTR, three patients with ACD, three patients with ICD and six patients with RCD (three RCDI and three RCDII). Data indicate mean ± SE. [Colour figure can be viewed at wileyonlinelibrary.com]
RCD‐associated inflammation is associated with no change in TGF‐β 1 production
To examine whether the diminished p‐Smad2/3 expression in RCD relies on either high Smad7 or reduced synthesis of TGF‐β 1, we measured the levels of active TGF‐β 1 in total proteins extracted from duodenal biopsy samples of patients and controls by ELISA. As shown in Fig. 4, no significant difference in terms of TGF‐β 1 production was seen among groups.
Figure 4.
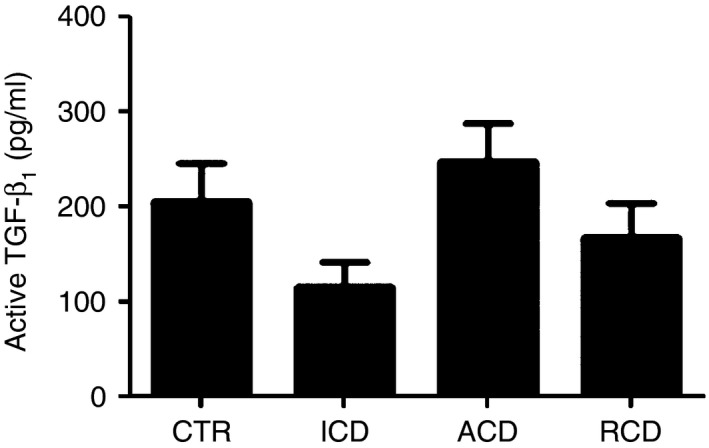
Active transforming growth factor‐β 1 (TGF‐β 1) content is not different between patients with refractory coeliac disease (RCD) and controls. Active TGF‐β 1 expression was evaluated in total proteins extracted from duodenal biopsy samples of 11 controls (CTR), 12 patients with active coeliac disease (ACD), eight patients with inactive coeliac disease (ICD) and eight patients with RCD by ELISA. Data indicate mean ± SEM.
Knockdown of Smad7 with an antisense oligonucleotide associates with diminished expression of inflammatory cytokines in RCD mucosa
To examine the role of Smad7 in RCD, we inhibited its expression using a specific antisense oligonucleotide, which was previously used to knockdown Smad7 in inflammatory bowel disease.21 Duodenal biopsies of patients with RCD were incubated with Smad7 antisense or sense oligonucleotide for 24 hr and then IL‐6 and TNF‐α transcripts were evaluated by RT‐PCR. Additionally, supernatants of such cultures were evaluated for the content of both cytokines by ELISA. Smad7 antisense reduced RNA and protein expression of IL‐6 and RNA transcripts of TNF‐α in all the samples (Tables 1 and 2) whereas TNF‐α protein was undetectable.
Table 1.
Knockdown of Smad7 decreases the RNA expression of interleukin‐6 (IL‐6) and tumour necrosis factor‐α (TNF‐α)
| Smad7 sense | Smad7 antisense | |
|---|---|---|
| IL‐6 RNA (relative expression) | 371.6 | 64.6 |
| 155.7 | 101.4 | |
| 29.6 | 10 | |
| TNF‐α RNA (relative expression) | 223.2 | 144.6 |
| 3.1 | 1.6 | |
| 5.4 | 1.2 |
IL‐6 and TNF‐α RNA transcripts in mucosal duodenal explants of three patients with refractory coeliac disease after treatment with Smad7 sense or antisense oligonucleotide for 24 hr. At the end, biopsy samples were used to extract RNA and real‐time PCR for both cytokines was performed as indicated in the Materials and methods section. Data indicate cytokine RNA relative expression as measured in mucosal samples cultured with Smad7 sense or antisense in each of the three experiments.
Table 2.
Knockdown of Smad7 decreases the protein expression of interleukin‐6 (IL‐6)
| Smad7 sense | Smad7 antisense | |
|---|---|---|
| IL‐6 (pg/100 μg total proteins) | 372.4 | 53.2 |
| 392.4 | 186.2 | |
| 685 | 242.7 | |
| TNF‐α (pg/100 μg total proteins) | und. | und. |
| und. | und. | |
| und. | und. |
IL‐6 and tumour necrosis factor‐α (TNF‐α) protein content was evaluated in supernatants of mucosal duodenal explants of three patients with refractory coeliac disease incubated with either Smad7 sense or antisense oligonucleotide for 24 hr by ELISA. TNF‐α protein was undetectable (und.).
Discussion
This study was undertaken to examine whether RCD‐associated inflammation is marked by defects in counter‐regulatory mechanisms, which could contribute to sustain the tissue‐damaging immune response in this disorder. To this end, we focused the work on Smad7, an inhibitor of the immunosuppressive cytokine TGF‐β 1.26 Our decision to explore the role of Smad7 in RCD was also because we have previously shown that high Smad7 sustains pathogenic signals in the gut of patients with inflammatory bowel disease and in mice with experimental colitis.21, 27 Moreover, our studies in patients with ACD showed that inhibition of Smad7 with a Smad7 antisense oligonucleotide‐containing drug was accompanied by clinical benefit in more than two‐thirds of the patients.28 By Western blotting, we initially showed that Smad7 protein expression was increased in patients with RCD compared with controls. In contrast, Smad7 RNA transcripts did not differ between patients with RCD and controls, supporting the notion that, in the human gut, Smad7 can be post‐transcriptionally regulated.22, 29 Indeed, our previous work showed that in inflammatory bowel disease, high Smad7 is dependent on mechanisms that enhance its protein stability without affecting Smad7 gene transcription.22 The elevated levels of Smad7 in RCD were confirmed by immunohistochemical analysis of duodenal sections, which showed a marked expression of the protein in both epithelial and lamina propria mononuclear cells. Consistent with these observations is the demonstration that RCD‐associated inflammation is characterized by defective TGF‐β 1 activity, as indicated by the reduced expression of p‐Smad2/3. Interestingly, expression of active TGF‐β 1 did not differ between patients with RCD and controls, supporting the hypothesis that high Smad7 impairs TGF‐β 1 signalling in RCD.
We also report no induction of Smad7 in duodenal biopsies of patients with ACD and patients with ICD in comparison with normal controls. Moreover, expression of p‐Smad2/3 did not differ between patients with ACD or ICD and normal controls, confirming previous studies showing that in CD, defective TGF‐β 1 activity is independent of Smad7‐mediated Smad signalling disruption.23, 29 Altogether, these data support the concept that RCD‐related inflammation is in part different from that seen in ACD.14
Further studies will be needed to evaluate which immune cells express Smad7, to examine if there is a cell‐specific regulation of Smad7 expression and to explore which role Smad7 plays in specific cell types. So far, these studies have been hampered by the limited availability of mucosal samples as no more than two to four biopsy samples could be collected from a single patient. Despite these limits, our ex vivo organ culture studies demonstrated that knockdown of Smad7 with a validated antisense oligonucleotide21 was associated with diminished expression of IL‐6 and TNF‐α, two cytokines over‐produced in RCD.14 Although the scarcity of biological samples did not allow us to examine whether inhibition of inflammatory cytokine expression following Smad7 knockdown is entirely dependent on activation of the TGF‐associated Smad pathway, the present findings highlight the positive effect of Smad7 on the ongoing mucosal inflammation and suggest that Smad7 could be a valid target for therapeutic intervention in patients with RCD.
In conclusion, our data show that elevated levels of Smad7 and defective TGF‐β 1 signalling characterize the detrimental inflammatory response in RCD and help to delineate a novel, gluten‐independent, pathogenic mechanism by which such a response could be amplified and/or perpetuated in RCD.
Authorship
Giovanni Monteleone is the guarantor of the article. Authors contributions: SS performed the experiments and statistical analysis and contributed to the drafting of the manuscript and interpretation of data. VDS, IM, GB and RI contributed to the experiments and the analysis of data. OAP and PG contributed to collection of the samples. AC and AO performed the immunohistochemistry. AV contributed to the histological evaluation of the biopsies. ADS contributed to collect the samples and to supervise parts of the project. FP and GRC contributed to supervise parts of the project. GM designed research, supervised the project, and wrote the manuscript.
Disclosures
Giovanni Monteleone has filed a patent related to the treatment of inflammatory bowel diseases with Smad7 antisense oligonucleotides. The remaining authors declare no conflict of interest.
References
- 1. Daum S, Cellier C, Mulder CJ. Refractory coeliac disease. Best Pract Res Clin Gastroenterol 2005; 19:413–24. [DOI] [PubMed] [Google Scholar]
- 2. Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of nonresponsive celiac disease: results of a systematic approach. Am J Gastroenterol 2002; 97:2016–21. [DOI] [PubMed] [Google Scholar]
- 3. Al‐Toma A, Verbeek WH, Mulder CJ. Update on the management of refractory coeliac disease. J Gastrointestin Liver Dis 2007; 16:57–63. [PubMed] [Google Scholar]
- 4. Ludvigsson JF, Leffler DA, Bai JC, Biagi F, Fasano A, Green PH et al The Oslo definitions for coeliac disease and related terms. Gut 2013; 62:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leffler DA, Dennis M, Hyett B, Kelly E, Schuppan D, Kelly CP et al Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol 2007; 5:445–50. [DOI] [PubMed] [Google Scholar]
- 6. Cellier C, Patey N, Mauvieux L, Jabri B, Delabesse E, Cervoni JP et al Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology 1998; 114:471–81. [DOI] [PubMed] [Google Scholar]
- 7. Olaussen RW, Lovik A, Tollefsen S, Andresen PA, Vatn MH, De Lange T et al Effect of elemental diet on mucosal immunopathology and clinical symptoms in type 1 refractory celiac disease. Clin Gastroenterol Hepatol 2005; 3:875–85. [DOI] [PubMed] [Google Scholar]
- 8. Malamut G, Afchain P, Verkarre V, Lecomte T, Amiot A, Damotte D et al Presentation and long‐term follow‐up of refractory celiac disease: comparison of type I with type II. Gastroenterology 2009; 136:81–90. [DOI] [PubMed] [Google Scholar]
- 9. Cellier C, Delabesse E, Helmer C, Patey N, Matuchansky C, Jabri B et al Refractory sprue, coeliac disease, and enteropathy‐associated T‐cell lymphoma. French Coeliac Disease Study Group. Lancet 2000; 356:203–8. [DOI] [PubMed] [Google Scholar]
- 10. Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G et al Reprogramming of CTLs into natural killer‐like cells in celiac disease. J Exp Med 2006; 203:1343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN et al Coordinated induction by IL15 of a TCR‐independent NKG2D signaling pathway converts CTL into lymphokine‐activated killer cells in celiac disease. Immunity 2004; 21:357–66. [DOI] [PubMed] [Google Scholar]
- 12. Hue S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J et al A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 2004; 21:367–77. [DOI] [PubMed] [Google Scholar]
- 13. Al‐Toma A, Verbeek WH, Hadithi M, von Blomberg BM, Mulder CJ. Survival in refractory coeliac disease and enteropathy‐associated T‐cell lymphoma: retrospective evaluation of single‐centre experience. Gut 2007; 56:1373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caruso R, Marafini I, Sedda S, Del Vecchio Blanco G, Giuffrida P, MacDonald TT et al Analysis of the cytokine profile in the duodenal mucosa of refractory coeliac disease patients. Clin Sci (Lond) 2014; 126:451–8. [DOI] [PubMed] [Google Scholar]
- 15. Gross S, van Wanrooij RL, Nijeboer P, Gelderman KA, Cillessen SA, Meijer GA et al Differential IL‐13 production by small intestinal leukocytes in active coeliac disease versus refractory coeliac disease. Mediators Inflamm 2013; 2013:939047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aoki CA, Borchers AT, Li M, Flavell RA, Bowlus CL, Ansari AA, Gershwin ME. Transforming growth factor β (TGF‐β) and autoimmunity. Autoimmun Rev 2005; 4:450–9. [DOI] [PubMed] [Google Scholar]
- 17. Weiner HL. Induction and mechanism of action of transforming growth factor‐β‐secreting Th3 regulatory cells. Immunol Rev 2001; 182:207–14. [DOI] [PubMed] [Google Scholar]
- 18. Black KE, Murray JA, David CS. HLA‐DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J Immunol 2002; 169:5595–600. [DOI] [PubMed] [Google Scholar]
- 19. Abdollah S, Macías‐Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TGFβRI phosphorylation of Smad2 on Ser465 and 467 is required for Smad2/Smad4 complex formation and signalling. J Biol Chem 1997; 272:27678–85. [DOI] [PubMed] [Google Scholar]
- 20. Shi Y, Massagué J. Mechanisms of TGF‐β signaling from cell membrane to the nucleus. Cell 2003; 113:685–700. [DOI] [PubMed] [Google Scholar]
- 21. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF‐β 1 signaling in chronic inflammatory bowel disease. J Clin Invest 2001; 108:601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Monteleone G, Del Vecchio Blanco G, Monteleone I, Fina D, Caruso R, Gioia V et al Post‐transcriptional regulation of Smad7 in the gut of patients with inflammatory bowel disease. Gastroenterology 2005; 129:1420–9. [DOI] [PubMed] [Google Scholar]
- 23. Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V et al Inhibition of TGF‐β signaling by IL‐15: a new role for IL‐15 in the loss of immune homeostasis in celiac disease. Gastroenterology 2007; 132:994–1008. [DOI] [PubMed] [Google Scholar]
- 24. Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’). Gastroenterology 1992; 102:330–54. [PubMed] [Google Scholar]
- 25. Rubio‐Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA. American College of Gastroenterology . ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol 2013; 108:656–76; quiz 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW et al The Mad‐related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 1997; 89:1165–73. [DOI] [PubMed] [Google Scholar]
- 27. Boirivant M, Pallone F, Di Giacinto C, Fina D, Monteleone I, Marinaro M et al Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF‐β1‐mediated suppression of colitis. Gastroenterology 2006; 131:1786–98. [DOI] [PubMed] [Google Scholar]
- 28. Monteleone G, Neurath MF, Ardizzone S, Di Sabatino A, Fantini MC, Castiglione F et al Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn's disease. N Engl J Med 2015; 372:1104–13. [DOI] [PubMed] [Google Scholar]
- 29. Monteleone G, Pallone F, MacDonald TT. Smad7 in TGF‐β‐mediated negative regulation of gut inflammation. Trends Immunol 2004; 25:513–7. [DOI] [PubMed] [Google Scholar]