

Summary
Non‐coding RNAs (ncRNAs), including microRNAs (miRNAs) and long non‐coding RNAs (lncRNAs), are RNA molecules that do not translate into protein. Both miRNAs and lncRNAs are known to regulate gene expression and to play an essential role in T cell differentiation and function. Both systemic lupus erythematosus (SLE), a prototypic systemic autoimmune disease, and rheumatoid arthritis (RA), a representative disease of inflammatory arthritis, are characterized by a complex dysfunction in the innate and adaptive immunity. T cells play a central role in cell‐mediated immune response and multiple defects in T cells from patients with SLE and RA have been observed. Abnormality in T cell signalling, cytokine and chemokine production, T cell activation and apoptosis, T cell differentiation and DNA methylation that are associated closely with the aberrant expression of a number of miRNAs and lncRNAs have been implicated in the immunopathogenesis of SLE and RA. This review aims to provide an overview of the current state of research on the abnormal expression of miRNAs and lncRNAs in T cells and their roles in the immunopathogenesis of SLE and RA. In addition, by comparing the differences in aberrant expression of miRNAs and lncRNAs in T cells between patients with SLE and RA, controversial areas are highlighted that warrant further investigation.
Keywords: non‐coding RNAs, rheumatoid arthritis, systemic lupus erythematosus, T lymphocytes
Introduction
Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease and rheumatoid arthritis (RA) is a representative disease of inflammatory arthritis. Typically, SLE appears in women during the second to fourth decade of life, whereas RA occurs predominantly later in life, with a female to male ratio of 3 : 1. The immunopathogenesis of SLE and RA is extremely complex, involving both the innate and adaptive immune responses. These two diseases have distinct serological markers, organ involvement and specific environmental risk factors. Many important immunological dimensions, such as cytokine production, T cell function, balance of T helper type 17 cells (Th17), regulatory T cells (Treg) and epigenetic regulation are found to be altered in these diseases 1, 2.
In recent years, transcriptomic and bioinformatic studies have discovered the existence of thousands of non‐coding RNAs (ncRNAs). These are functional RNA molecules that are transcribed from DNA but not translated into protein. They are classified conventionally into long ncRNAs (lncRNAs) and short ncRNAs (sncRNAs), with more or fewer than 200 nucleotides of the RNA length, respectively. SncRNAs can be divided further into microRNAs (miRNAs) (21–25 nucleotides long), small nucleolar RNAs (snoRNAs) and piwi‐interacting RNAs (piRNAs). While miRNAs are well studied and known to induce mRNA degradation or translational inhibition via the RNA interference pathway, much less is known about lncRNAs. LncRNAs appear to play a role in a wide variety of biological processes, including the activation or repression of gene expression, and also function as part of the cell structure.
Recently, lncRNAs and miRNAs have been shown to be involved in the innate and adaptive immune responses. Aberrant expression of miRNAs and lncRNAs has also been found in patients with different autoimmune diseases, including SLE and RA. A number of comprehensive reviews have addressed the abnormal expression of miRNAs in different tissues and cells from patients with SLE 3, 4, 5 and RA 6 as well as the aberrant expression of lncRNAs 7. As T cells play a critical role in the pathogenesis of autoimmune diseases, this review aims to clarify the current knowledge of ncRNAs, including miRNAs and lncRNAs, in their contribution to T cell dysfunction in patients with SLE and RA. We reviewed papers on aberrant expression of ncRNAs, including miRNAs and lncRNAs, in T cells from patients with SLE and RA, and summarized their findings with our current understanding of the immunopathogenesis of SLE and RA. In addition, by comparing the differences in aberrant expression of miRNAs and lncRNAs in T cells between patients with SLE and RA, we highlight controversial areas that warrant further investigation.
Aberrant expression of ncRNAs in T cells from patients with systemic lupus erythematosus
Aberrant expression of miRNAs
The pathogenesis of SLE is extremely complex. Genetic, epigenetic, environmental, hormonal and immunoregulatory factors can all contribute to its development. Both the innate and adaptive immune systems are impaired in patients with SLE, which leads to a loss of self‐tolerance and the production of numerous autoantibodies. T cells and their signalling abnormalities are important contributors to the immunopathogenesis of SLE. Diverse instances of T cell dysfunction, such as cell signalling aberrations, altered gene transcription and T cell subset alteration, have been well characterized in patients with SLE 8.
Cell signalling abnormalities
One of the early studies investigating the roles of miRNAs in the pathogenesis of SLE demonstrated a decreased expression of miR‐146a in the peripheral blood mononuclear cells (PBMCs) from patients with SLE 9. Later, a decreased expression of miR‐146a was confirmed in CD4+ T cells from patients with SLE 10. The expression levels of miR‐146a correlated with the SLE disease activity index and the occurrence of lupus nephritis. Treating SLE patients with mycophenolic acid was shown to increase the expression levels of miR‐146a 10. In addition, miR‐146a could repress the production of type I interferons (IFN‐α and IFN‐β) and its downstream signalling pathway, possibly through a down‐regulation of interferon regulatory factor‐5 (IRF‐5), signal transducer and activator of transcription 1 (STAT‐1), interleukin‐1 receptor‐associated kinase 1 (IRAK1) and tumour necrosis factor receptor‐associated factor 6 (TRAF6) levels 9, 11. Although type I IFNs are responsible for modulating the innate immune response, they can also affect T cell function 12. SLE T cells exhibit distinct type I IFN signatures compared with normal T cells 13. It should be noted that miR‐146a also targets STAT‐1, which could transduce signals from other cytokines, such as IFN‐γ, to affected T cell function. Using microarray analysis, Stagakis et al. showed that the expression levels of miR‐21, miR‐25, miR‐106b, miR‐148a, miR‐148b and miR‐324‐3p were up‐regulated, whereas those of lethal‐7a (let‐7a), let‐7d, let‐7g, miR‐296 and miR‐196a were down‐regulated in CD4+ T cells from patients with SLE. As the expression level of miR‐21was correlated strongly with SLE disease activity, the authors further investigated the detailed functions of miR‐21. They demonstrated that silencing of miR‐21 could reverse the activated phenotype of T cells from patients with SLE, including enhanced CD4 T cell proliferation, interleukin (IL)‐10 production, CD40L expression and their capacity to promote B cell maturation into plasma cells 14. Moreover, silencing of miR‐21 could ameliorate autoimmune splenomegaly in an animal model of lupus 15. Our previous report also showed under‐expressed miR‐145 and over‐expressed miR‐224 in T cells from SLE patients with relatively low disease activity 16. In another study, increased expression of miR‐224 is also noted in CD4+ T cells from patients with SLE with low disease activity 17. Transfection of Jurkat cells with miR‐145 suppressed STAT‐1 expression, and STAT‐1 is an important transcription factor for cytokines, such as IFN‐γ signalling 18. Increased miR‐224 expression was found to suppress apoptosis inhibitory protein 5 (API5) expression 16. API5, also called anti‐apoptosis clone 11 (AAC11), is a nuclear protein that has been shown to prevent cell apoptosis after growth factor withdrawal 19. An increase in T cell apoptosis is well known in patients with SLE, and previous studies have demonstrated that this phenomenon is mediated by a Fas‐related pathway 20, 21. In addition, we found that increased expression of miR‐224 in SLE T cells accelerated activation‐induced cell death through the inhibition of API5, thus providing another possible explanation for the increased apoptosis of SLE T cells.
An increased and prolonged calcium influx due to T cell receptor (TCR) ζ chain defect 22 leading to a decreased IL‐2 production 23 is a well‐documented finding in SLE T cells. Our previous study showed that calcium influx could regulate the expression of several miRNAs, and among them, miR‐524‐5p and miR‐449b were over‐expressed in SLE T cells 24. Increased expression of miR‐524‐5p directly paralleled the disease activity in patients with SLE. Transfection of miR‐524‐5p also enhanced IFN‐γ production in activated Jurkat cells. Regarding the downstream calcium signalling pathway, decreased expression of miR‐31 in SLE T cells indicated another mechanism. Under‐expression of miR‐31 contributed to a decreased production of IL‐2 in lupus T cells via its targeting of Ras homologue gene family, member A (RhoA), which inhibited the expression of nuclear factor of activated T cells (NFAT) 25.
Ding et al. showed that the expression of miR‐142‐3p and miR‐142‐5p is down‐regulated significantly in CD4+ T cells from patients with SLE compared with healthy controls. Increased expression of miR‐142‐3p and miR‐142‐5p was observed in CD4+ T cells from patients with SLE with treatment of mycophenolic acid 10. The expression of miR‐142 was controlled by histone modifications and DNA methylation. MiR‐142‐3p/5p levels were found to be correlated inversely with the putative SLE‐related targets signalling lymphocytic activation molecule‐associated protein (SAP), CD84 and IL‐10 26. SAP is an intracellular adaptor molecule in T cells that transmits a signal to control its activation. Conversely, there is also a report showing that SAP protein expression was decreased in SLE T cells 27. Further studies are required to elucidate this discrepancy.
Overall, the decreased expression of miR‐146a and miR‐142‐3p/5p as well as the increased expression of miR‐21 and miR‐224 could contribute to cell signalling abnormalities in SLE T cells, including the activation of type I IFN pathway, activation of T cells, stimulation of B cells and increased apoptosis (Fig. 1).
Figure 1.
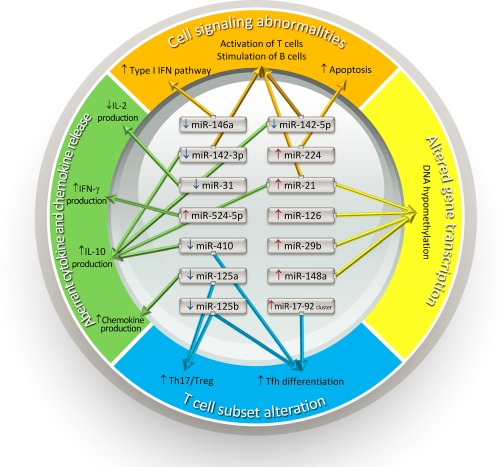
Various aberrantly expressed microRNAs involving in cell signalling abnormalities, altered gene transcription, aberrant cytokines and chemokines release, and T cell subset alteration in T cells of patients with systemic lupus erythematosus that are implicated in the immunopathogenesis of systemic lupus erythematosus. IFN = interferon; IL = interleukin; Tfh = T follicular helper cells; Th = T helper cells; Treg = regulatory T cells.
Altered gene transcription
The study of reversible and potentially heritable changes in gene expression without alterations in the DNA sequence, known as epigenetics, is an emerging area of investigation into the pathogenesis of SLE. DNA methylation and histone modifications are the two key epigenetic pathways that interplay to regulate gene expression and genome functions. Abnormalities of DNA methylation and histone modifications have been shown to lead to an aberrant increase of autoimmune‐associated gene expression in patients with lupus 28. Expression of miR‐148a, miR‐126, miR‐29b and miR‐21 is increased in CD4+ T cells from patients with SLE. MiR‐148a, miR‐126 and miR‐29b could target and inhibit DNA (cytosine‐5)‐methyltransferase 1 (DNMT1) translation directly, while miR‐21 could do so indirectly. As DNMT1 is one of the methyltransferases that regulates the process of DNA methylation, the inhibition of DNMT1 translation can lead to hypomethylation of DNA in CD4 T cells from patients with SLE, which can result in increased expression of many proinflammatory genes, such as CD11a, CD70 and epidermal growth factor‐like 7 (EGFL7) 29, 30, 31.
Using high‐throughput sequencing to systematically profile the DNA methylation and expression patterns of both miRNAs and mRNAs on a genome‐wide scale in SLE CD4+ T cells, Zhao et al. demonstrated that miR‐142‐3p, miR‐505 and miR‐324‐5p were down‐regulated, whereas miR‐126, miR‐451 and miR‐181b were up‐regulated in purified CD4+ T cells from patients with SLE compared with normal controls. In addition, 36 up‐regulated miRNAs were found near cytosine–phosphate–guanine (CpG) sites and were hypomethylated in SLE CD4+ T cells compared with normal controls, whereas only eight down‐regulated miRNAs were hypermethylated in SLE CD4+ T cells. Thus, DNA methylation appears to potentially influence miRNAs expression 32.
The observation that SLE develops predominantly in women and that men with Klinefelter's syndrome (XXY) have a 14‐fold increased risk of developing lupus compared with men in the general population suggested that the second X chromosome could contribute to the increased risk of SLE. By comparing the mRNA and miRNA expression profiles in experimentally demethylated T cells from women and men as well as in T cells from women and men with lupus, Hewagama et al. identified that miR‐98, let‐7f‐2*, miR‐188‐3p, miR‐421 and miR‐503 were over‐expressed in women relative to men. They also reported that Casitas B‐lineage lymphoma (CBL) protein, an E3 ubiquitin‐protein ligase, was targeted by miR‐98 and miR‐188‐3p 33. Their findings supported that CBL down‐regulation in T cells could alter T cell receptor signalling pathways and thus potentially contribute to lupus pathogenesis.
While the participation of histone modifications in the pathogenesis of SLE is well documented 28, currently there are still no reports concerning the possible roles of miRNAs in the abnormal histone modification in patients with SLE. Overall, regarding altered gene transcription, the increased expression of miR‐148a, miR‐126, miR‐29b and miR‐21 appears to contribute to the DNA hypomethylation in SLE T cells.
Aberrant cytokine and chemokine release
RANTES (regulated on activation, normal T cell‐expressed and secreted), a proinflammatory chemokine that induces migration of circulating leucocytes to sites of inflammation, has been found to be significantly higher in the serum of patients with SLE 34. Normally, miRNA‐125a regulates RANTES expression negatively by targeting Kruppel‐like factor 13 (KLF13) in activated T cells. However, miR‐125a was under‐expressed in patients with SLE, which could lead to an elevated expression of RANTES 35. Therefore, both RANTES and miR‐125a could be involved in the pathogenesis of SLE.
MiR‐410 has been shown to be another key regulatory factor in the pathogenesis of SLE. Liu et al. demonstrated that miR‐410 could suppress the transcription activity of STAT‐3, and silence of STAT‐3 down‐regulated IL‐10 expression in CD3+ T cells in turn 36. This pathway therefore provides an explanation to the decreased level of miR‐410 and the elevated serum IL‐10 observed in patients with SLE 37. Over‐expression of IL‐10 has been implicated in SLE development through inducing the production of autoantibodies by B cells 38.
Overall, regarding aberrant cytokine and chemokine releases, the increased expression of miR‐524‐5p and decreased expression of miR‐125a, miR‐142‐3p/5p, miR‐410, miR‐31 and miR‐21 could contribute to the increased IFN‐γ, IL‐10 and RANTES, as well as the decreased IL‐2 secretion in SLE T cells (Fig. 1).
T cell subset alteration
Treg, a subpopulation of T cells, suppress activation of the immune system actively and prevent pathological self‐reactivity. Pan et al. showed that miR‐125a could stabilize both the commitment and immunoregulatory capacity of Treg. MiR‐125a knock‐out mice have been shown to be susceptible to autoimmune encephalomyelitis, because decreased expression of miR‐125a disrupted Treg‐mediated immune homeostasis leading to inflammation 39. In addition, the genome‐wide target analysis revealed that miR‐125a could suppress expression of several genes, including STAT‐3, IFN‐γ and IL‐13, relative to effector T cell function. Among these targets, STAT‐3 is a key signalling transducer for Th17 and T follicular helper (Tfh) cell differentiation. An imbalance of Th17/Treg is known to contribute to the immunopathogenesis of SLE 1.
Another study exploring miRNA expression profiles in PBMCs from patients with SLE revealed eight up‐regulated and 26 down‐regulated miRNAs. Among these miRNAs, miR‐125b was confirmed to be down‐regulated in T cells from patients with SLE with STAT‐3 found to be its target gene 40. As increased expression levels of STAT‐3 41 can promote Th17 cell differentiation, miR‐125b can play a role in the regulation of Th17 cell differentiation.
Recently, overactivity of Tfh, a CD4+ helper T cell subset specialized for the provision of help to B cells, has been shown to be involved in the pathogenesis of SLE 42. Production of high levels of IL‐21 is a hallmark of Tfh, and elevated plasma levels of IL‐21 are also observed in SLE patients compared with healthy controls. Data from a murine model also suggested that IL‐21 signalling is fundamental to the pathogenesis of SLE. In addition, several studies have implicated that decreased expression of miR‐410, miR‐125a and miR‐125b could contribute to the increased expression of STAT‐3 36, 39, 40 and STAT‐3 is known to be important for Tfh cell differentiation. Finally, the miRNA cluster miR‐17‐92 appeared to be critical for Tfh cell differentiation and function 43, 44. Several members of the miR‐17‐92 cluster, including miR‐17, miR‐17a, miR‐18a, miR‐19a, miR‐19b and miR‐20a, have been shown to be over‐expressed in CD4+ T cells from patients with SLE 45.
Rasmussen et al. showed that miR‐155 levels are decreased significantly in CD4+ T cells from patients with SLE compared with healthy controls. They also found that over‐expression of miR‐155 could lead to a higher level of STAT‐3 phosphorylation, which would increase IL‐21 production differentially in CD4+ T cells from patients with SLE compared with controls 46. Moreover, the role of miR‐155 in the pathogenesis of SLE was investigated using a knock‐out mouse model. MiR‐155 deficiency Faslpr/lpr mice were found to be resistant to the development of SLE lesions by the regulation of a target gene S1pr1 of miR‐155 47. Therefore, the decreased miR‐155 in SLE T cells could play a negative feedback loop to control STAT‐3 phosphorylation and IL‐21 production.
In summary, decreased expression of miR‐125a/b and miR‐410 as well as increased expression of miR‐17‐92 cluster appear to contribute to the increased differentiation of Tfh and Th17 in SLE.
Aberrant expression of lncRNAs
Studies have suggested that abnormal expression of lncRNAs might be involved with a number of diseases, including RA, autoimmune thyroid disease and psoriasis. However, less is known about the aberrant expression of lncRNAs in T cells from patients with SLE. Wu et al. showed two large intergenic non‐coding RNAs (lincRNAs), linc0949 and linc0597, were under‐expressed significantly in PBMCs in patients with SLE compared with healthy controls and patients with RA. LincRNAs, one of the several classes of lncRNAs, are so named because lincRNA genes do not overlap or locate in close proximity to protein‐coding genes. Expression of linc0949 was decreased in patients with lupus nephritis, one of the most common manifestations of organ damage in patients with SLE, and its level was increased significantly with treatment 48. Compared with the studies regarding miRNAs, lncRNAs are still a relatively new field of research and the significance of aberrant expression in SLE T cells is still waiting to be revealed.
Aberrant expression of ncRNAs in T cells from patients with rheumatoid arthritis
Early theories of the pathogenesis of RA have been focused mainly on autoantibodies and immune complexes, T cell‐mediated antigen‐specific responses, T cell‐independent cytokines network and aggressive tumour‐like behaviour of rheumatoid synovium 49. More recently, research on citrullinated proteins produced by enzymatic deimination of arginine residues in proteins by peptidylarginine deiminases (PADIs) and the formation of anti‐citrullinated protein antibodies (ACPAs) have advanced our understanding of the immunopathogenesis of RA. An important genetic risk factor for RA is the human leucocyte antigen (HLA) class II locus and it is implicated in the progression from protein citrullination to ACPA‐positive RA. A proposed mechanism is that antigen‐specific T cells can provide help to ACPA‐producing B cells, which allow for maturation of the ACPA response 50.
In addition, multiple T cell dysfunction, including abnormal T cell selection, abnormal T cell activation, premature T cell senescence, potent reactivity to the formation of neo‐autoantigens such as aggregated immunoglobulins (Ig)Gs or citrullinated peptides, imbalance of Th17/Treg has been characterized in RA patients or animal models of RA 51, 52. Even in the very early stages of RA, an abnormal CD4+ T cell genetic signature could be found in RA patients 53. Furthermore, inhibition of T cells co‐stimulation by soluble cytotoxic T lymphocyte antigen 4 recombinant proteins effectively suppresses joint inflammation in RA patients clinically 54. It is clear that T cells play a crucial role in the pathogenesis of RA.
Aberrant expression of miRNAs
Cytokines. The imbalance of proinflammatory and anti‐inflammatory cytokines are well characterized in the pathogenesis of RA 55, 56. Tumour necrosis factor (TNF)‐α, IL‐6 and IL‐1β are also critical proinflammatory cytokines in the pathogenesis of RA. These cytokines are involved in the regulation of cellular immune responses in many ways, such as controlling the development and function of Treg, generating T cell memory and modulating the secretion of cytokines 57, 58, 59. Inhibition of these cytokines can lead to rapid improvement of all clinical measurements of RA and, most importantly, suppress joint destruction and potentially improve survival 60, 61.
Li et al. showed that miR‐146a expression was up‐regulated significantly in CD4+ T cells from synovial fluid and peripheral blood of patients with RA. The expression level of miR‐146a was correlated positively with levels of TNF‐α. Moreover, in‐vitro studies showed that TNF‐α up‐regulated miR‐146a expression in T cells and over‐expressed miR‐146a could suppress T cell apoptosis 62. However, Pauley et al. showed that miR‐146a could repress the production of TNF‐α in PBMCs from patients with RA 63, thus providing a negative feedback loop for the repression of inflammatory response 64. Furthermore, it is known that activation of IL‐17 signalling is central in the pathogenesis of psoriasis. A study using an imiquimod‐induced mouse model of psoriasis showed that genetic deficiency in miR‐146a could lead to an earlier onset and exacerbated skin lesions, with increased expression of IL‐17‐induced keratinocyte‐derived inflammatory mediators 65, 66. In patients with RA, miR‐146a has also been shown to be up‐regulated in the IL‐17‐producing T cells 65, 66. Therefore, miR‐146a could play a negative regulatory role in the inflammatory response by influencing the expression of IL‐17 and TNF‐α.
One of the early studies for the aberrant expression of miRNAs in RA T cells indicated that miR‐223 is over‐expressed in T cells and CD4+ naive T lymphocytes, but not in Th17 cells from patients with RA 67. Elevated expression of miR‐223 was also found in T cells from early RA patients before treatment 68. A study from our group showed that miR‐223 and miR‐34b were over‐expressed in RA T cells. The expression levels of miR‐223 were correlated positively with the levels of rheumatoid factor (RF) in RA patients. Increased miR‐223 expression could impair IGF‐1‐mediated IL‐10 production in activated RA T cells in vivo, which might contribute to an imbalance between proinflammatory and anti‐inflammatory cytokines 69. It should be noted that there are still debates on the role of miR‐223 in the immunopathogenesis of RA. Several studies showed that over‐expression of miR‐223 could suppress osteoclastogenesis by blocking the differentiation of osteoclasts 70, 71, which might prevent joint damage in RA patients. In contrast, Li et al. demonstrated that the inhibition of miR‐233 expression was associated with reduced disease severity using a mouse model of collagen‐induced arthritis 72.
Positive correlations between increased expression of miR‐451 in peripheral blood T cells and RA disease activity score (DAS28), erythrocyte sedimentation rate levels and serum levels of IL‐6 have been reported in studies of patients with RA 73. A study using influenza‐infected murine dendritic cells showed that IL‐6 could stimulate the expression of miR‐451 and that the increased expression of miR‐451 could suppress the expression of IL‐6 74. This negative regulatory role of miR‐451 in the expression of IL‐6 could provide a possible explanation between miR‐451 and the inflammatory response in RA patients.
T cell subset alternation
The imbalance of Th17/Treg cell populations has been implicated in the pathogenesis of RA. Decreased expression of miR‐21 was noted in PBMCs and CD4+ T cells of patients with RA. A decreased miR‐21 expression was found to be associated closely with decreased mRNA levels of STAT‐5, which is an important transcription factor for Treg differentiation 75. In addition, miR‐21 can target the STAT‐3 gene, which plays a pivotal role in determining Th17 differentiation during RA 76, 77. Therefore, under‐expression of miR‐21 in RA patients could increase STAT‐3 and decrease STAT‐5 expression and, in turn, facilitate the differentiation of Th17 and inhibit the differentiation of Treg cells.
The role of miR‐146a and miR‐155 in Treg function was investigated in patients with RA. Diminished up‐regulation of miR‐146a in response to T cell stimulation was observed in Tregs of RA patients. In patients with active RA, the increased expression of inflammatory cytokine could be caused by an augmented expression and activation of STAT‐1, which is a direct target of miR‐146a. Thus, miR‐146a appears to contribute to the pathogenesis of RA by facilitating a proinflammatory phenotype of Tregs through increased activation of STAT‐1 78.
LncRNA in T cells from patients with rheumatoid arthritis
Several studies have investigated the role of aberrant expression of lncRNAs in PBMCs or synovium in the immunopathogenesis of RA 79, 80. Recently, Spurlock et al. demonstrated under‐expression of lincRNA‐p21 in whole blood samples from RA patients, which contributed to an increased nuclear factor (NF)‐κB activity. Treatment with methotrexate suppresses NF‐κB activity via an increase in lincRNA‐p21 81. In addition, a previous study from our group demonstrated that the expression levels of LOC100652951 and LOC100506036 were increased in T cells from patients with RA. A decreased expression level of LOC100652951 is found in patients with RA using biological agents. The expression level of LOC100506036, but not LOC100652951, was increased in Jurkat cells after activation. Transfection of small interfering RNA (siRNA) targeting LOC100506036 suppressed NFAT1 and sphingomyelin phosphodiesterase 1 (SMPD1) gene expression as well as inhibiting IFN‐γ production in activated Jurkat cells. NFAT1 plays a role in the inducible expression of cytokine genes in T cells and SMPD1 is known to regulate IL‐2 secretion and is induced by TNF‐α in T cells. Therefore, LOC100506036 could potentially involve in the inflammatory response of RA via regulating the expression of SMPD1 and NFAT1 82.
Overall, increased expression of miR‐223 and LOC100506036 as well as decreased expression of miR‐21 could contribute to the disrupted the proinflammatory and anti‐inflammatory cytokines balance, activation of T cells and increased Th17 differentiation, which could play a positive role in the immunopathogenesis of RA (Fig. 2).
Figure 2.
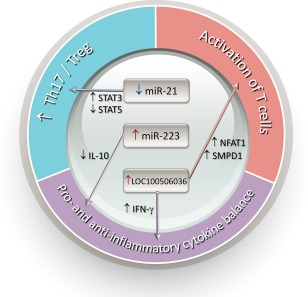
Various aberrantly expressed microRNAs involving in the activation of T cells, balance of proinflammatory and anti‐inflammatory cytokines, and balance of T helper type 17 (Th17) and regulatory T cells (Treg) in T cells of patients with rheumatoid arthritis that are implicated in the immunopathogenesis of systemic lupus erythematosus. IFN = interferon; IL = interleukin; NFAT = nuclear factor of activated T cells; SMPD = sphingomyelin phosphodiesterase; STAT = signal transducer and activator of transcription; Th = T helper cells.
Discussion
Although both SLE and RA are autoimmune diseases characterized by profound inflammation, two miRNAs show opposite expression profiles between patients with SLE and RA. First, the expression of miR‐146a is decreased in T cells from patients with SLE, but increased in T cells from patients with RA. As miR‐146a plays a negative feedback role in the inflammatory responses, the increased expression of miR‐146a in RA T cells can probably be considered as a failure to overcome the active inflammation.
Secondly, the expression of miR‐21 is increased in patients with SLE, but decreased in T cells from patients with RA. Nevertheless, most studies showed that an increase of miR‐21 in serum and immune cells from patients with various autoimmune diseases 83. Moreover, Dong et al. suggested that the decreased level of miR‐21 might facilitate Th17 differentiation by increasing STAT‐3 expression. However, another study showed that miR‐21 promoted Th17 differentiation by targeting SMAD‐7, a negative regulator of transforming growth factor (TGF)‐β signalling in animal models of experimental autoimmune encephalomyelitis 84. As a single miRNA is able to target hundreds of genes, the actual and overall effect of a single miRNA in a specific disease needs to be evaluated more carefully with a combination of observational human studies, cell manipulation studies and animal model studies. Clearly, much work needs to be conducted to clarify these issues.
Although many studies have addressed the aberrant expression of miRNAs in various tissues and cells from patients with RA, studies focusing on T cells are still relatively few compared to those with SLE. Studies of aberrant expression of miRNAs in T cells from SLE that cover abnormal T cell function, DNA hypomethylation, dysregulated cytokines production and T cell subset changes are all critical issues in the pathogenesis of SLE. Nevertheless, studies of miRNAs in RA T cells have focused mainly upon cytokines and Treg/Th17 imbalance. As global hypomethylation of DNA is also noted in RA T cells, how these miRNAs affect the expression of DNMT1 in T cells from patients with RA may be worth investigating 85. Furthermore, there are also numerous autoantibodies present in the serum of RA patients, yet no studies have addressed the role of miRNAs in T cells on the maturation of B cells. Some critical issues, such as smoking and ACPAs, are also important in the immunopathogenesis of RA. Although our previous work showed that ACPAs could suppress let‐7a expression in monocytes from patients with RA and facilitate the inflammatory responses 86, the effects of ACPAs on T cells remain unclear. Furthermore, it is of interest to note that PADI inhibitor could affect the expression of miRNAs 87. As PADI also plays a critical role in the development of RA, the relationship between miRNAs and PADI warrants further investigation.
In conclusion, among the various classes of ncRNA, the role of miRNA in T cells is the most studied, and has been implicated in the immunopathogenesis of SLE and RA. Previous studies have shown that miRNA could be involved in a variety of biological processes, including T cell differentiation, imbalance of proinflammatory/anti‐inflammatory cytokines, signalling pathway and hypomethylation. In contrast, studies of lncRNAs in T cells from patients with SLE and RA are still relatively scarce at present, but we anticipate that research in these areas will flourish in the near future.
Disclosures
None to declare.
References
- 1. Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2011; 365:2110–21. [DOI] [PubMed] [Google Scholar]
- 2. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010; 376:1094–108. [DOI] [PubMed] [Google Scholar]
- 3. Qu B, Shen N. miRNAs in the pathogenesis of systemic lupus erythematosus. Int J Mol Sci 2015; 16:9557–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yan S, Yim LY, Lu L, Lau CS, Chan VS. MicroRNA regulation in systemic lupus erythematosus pathogenesis. Immune Netw 2014; 14:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zan H, Tat C, Casali P. MicroRNAs in lupus. Autoimmunity 2014; 47:272–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen XM, Huang QC, Yang SL et al Role of micro RNAs in the pathogenesis of rheumatoid arthritis: novel perspectives based on review of the literature. Medicine (Balt) 2015; 94:e1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sigdel KR, Cheng A, Wang Y, Duan L, Zhang Y. The emerging functions of long noncoding RNA in immune cells: autoimmune diseases. J Immunol Res 2015; 2015:848790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Rheumatol 2010; 6:317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang Y, Luo X, Cui H et al MicroRNA‐146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum 2009; 60:1065–75. [DOI] [PubMed] [Google Scholar]
- 10. Tang Q, Yang Y, Zhao M et al Mycophenolic acid upregulates miR‐142‐3P/5P and miR‐146a in lupus CD4+T cells. Lupus 2015; 24:935–42. [DOI] [PubMed] [Google Scholar]
- 11. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF‐kappaB‐dependent induction of microRNA miR‐146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA 2006; 103:12481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Obermoser G, Pascual V. The interferon‐alpha signature of systemic lupus erythematosus. Lupus 2010; 19:1012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flint SM, Jovanovic V, Teo BW et al Leucocyte subset‐specific type 1 interferon signatures in SLE and other immune‐mediated diseases. RMD Open 2016; 2:e000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stagakis E, Bertsias G, Verginis P et al Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR‐21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis 2011; 70:1496–506. [DOI] [PubMed] [Google Scholar]
- 15. Garchow BG, Bartulos EO, Leung YT et al Silencing of microRNA‐21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med 2011; 3:605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu MC, Lai NS, Chen HC et al Decreased microRNA(miR)−145 and increased miR‐224 expression in T cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin Exp Immunol 2013; 171:91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinez‐Ramos R, Garcia‐Lozano JR, Lucena JM et al Differential expression pattern of microRNAs in CD4+ and CD19+ cells from asymptomatic patients with systemic lupus erythematosus. Lupus 2014; 23:353–9. [DOI] [PubMed] [Google Scholar]
- 18. Goropevsek A, Holcar M, Avcin T. The role of STAT signaling pathways in the pathogenesis of systemic lupus erythematosus. Clin Rev Allergy Immunol 2016. doi:10.1007/s12016-016-8550-y. [DOI] [PubMed] [Google Scholar]
- 19. Tewari M, Yu M, Ross B, Dean C, Giordano A, Rubin R. AAC‐11, a novel cDNA that inhibits apoptosis after growth factor withdrawal. Cancer Res 1997; 57:4063–9. [PubMed] [Google Scholar]
- 20. Emlen W, Niebur J, Kadera R. Accelerated in vitro apoptosis of lymphocytes from patients with systemic lupus erythematosus. J Immunol 1994; 152:3685–92. [PubMed] [Google Scholar]
- 21. Mysler E, Bini P, Drappa J et al The apoptosis‐1/Fas protein in human systemic lupus erythematosus. J Clin Invest 1994; 93:1029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liossis SN, Ding XZ, Dennis GJ, Tsokos GC. Altered pattern of TCR/CD3‐mediated protein‐tyrosyl phosphorylation in T cells from patients with systemic lupus erythematosus. Deficient expression of the T cell receptor zeta chain. J Clin Invest 1998; 101:1448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Juang YT, Wang Y, Solomou EE et al Systemic lupus erythematosus serum IgG increases CREM binding to the IL‐2 promoter and suppresses IL‐2 production through CaMKIV. J Clin Invest 2005; 115:996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu MC, Yu CL, Chen HC, Yu HC, Huang HB, Lai NS. Aberrant T cell expression of Ca2+ influx‐regulated miRNAs in patients with systemic lupus erythematosus promotes lupus pathogenesis. Rheumatology (Oxf) 2015; 54:343–8. [DOI] [PubMed] [Google Scholar]
- 25. Fan W, Liang D, Tang Y et al Identification of microRNA‐31 as a novel regulator contributing to impaired interleukin‐2 production in T cells from patients with systemic lupus erythematosus. Arthritis Rheum 2012; 64:3715–25. [DOI] [PubMed] [Google Scholar]
- 26. Ding S, Liang Y, Zhao M et al Decreased microRNA‐142‐3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum 2012; 64:2953–63. [DOI] [PubMed] [Google Scholar]
- 27. Karampetsou MP, Comte D, Kis‐Toth K, Terhorst C, Kyttaris VC, Tsokos GC. Decreased SAP expression in T cells from patients with systemic lupus erythematosus contributes to early signaling abnormalities and reduced IL‐2 production. J Immunol 2016; 196:4915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu H, Zhao M, Chang C, Lu Q. The real culprit in systemic lupus erythematosus: abnormal epigenetic regulation. Int J Mol Sci 2015; 16:11013–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pan W, Zhu S, Yuan M et al MicroRNA‐21 and microRNA‐148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol 2010; 184:6773–81. [DOI] [PubMed] [Google Scholar]
- 30. Zhao S, Wang Y, Liang Y et al MicroRNA‐126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum 2011; 63:1376–86. [DOI] [PubMed] [Google Scholar]
- 31. Qin H, Zhu X, Liang J et al MicroRNA‐29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci 2013; 69:61–7. [DOI] [PubMed] [Google Scholar]
- 32. Zhao M, Liu S, Luo S et al DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J Autoimmun 2014; 54:127–36. [DOI] [PubMed] [Google Scholar]
- 33. Hewagama A, Gorelik G, Patel D et al Overexpression of X‐linked genes in T cells from women with lupus. J Autoimmun 2013; 41:60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu MM, Wang J, Pan HF et al Increased serum RANTES in patients with systemic lupus erythematosus. Rheumatol Int 2012; 32:1231–3. [DOI] [PubMed] [Google Scholar]
- 35. Zhao X, Tang Y, Qu B et al MicroRNA‐125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum 2010; 62:3425–35. [DOI] [PubMed] [Google Scholar]
- 36. Liu D, Zhang N, Zhang X, Qin M, Dong Y, Jin L. MiR‐410 Down‐regulates the expression of interleukin‐10 by targeting STAT3 in the pathogenesis of systemic lupus erythematosus. Cell Physiol Biochem 2016; 39:303–15. [DOI] [PubMed] [Google Scholar]
- 37. Yang X, Sun B, Wang H, Yin C, Wang X, Ji X. Increased serum IL‐10 in lupus patients promotes apoptosis of T cell subsets via the caspase 8 pathway initiated by Fas signaling. J Biomed Res 2015; 29:232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trifunovic J, Miller L, Debeljak Z, Horvat V. Pathologic patterns of interleukin 10 expression – a review. Biochem Med (Zagreb) 2015; 25:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pan W, Zhu S, Dai D et al MiR‐125a targets effector programs to stabilize Treg‐mediated immune homeostasis. Nat Commun 2015; 6:7096. [DOI] [PubMed] [Google Scholar]
- 40. Luo X, Zhang L, Li M et al The role of miR‐125b in T lymphocytes in the pathogenesis of systemic lupus erythematosus. Clin Exp Rheumatol 2013; 31:263–71. [PubMed] [Google Scholar]
- 41. Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O'Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev 2010; 21:425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gensous N, Schmitt N, Richez C, Ueno H, Blanco P. T follicular helper cells, interleukin‐21 and systemic lupus erythematosus. Rheumatology (Oxf) 2016. doi:10.1093/rheumatology/kew297. [DOI] [PubMed] [Google Scholar]
- 43. Baumjohann D, Kageyama R, Clingan JM et al The microRNA cluster miR‐17 approximately 92 promotes TFH cell differentiation and represses subset‐inappropriate gene expression. Nat Immunol 2013; 14:840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu T, Wieland A, Lee J et al Cutting edge: miR‐17‐92 is required for both CD4 Th1 and T follicular helper cell responses during viral infection. J Immunol 2015; 195:2515–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qin HH, Zhu XH, Liang J, Wu JF, Yang YS, Xu JH. The expression and significance of miR‐17‐92 cluster miRs in CD4+ T cells from patients with systemic lupus erythematosus. Clin Exp Rheumatol 2013; 31:472–3. [PubMed] [Google Scholar]
- 46. Rasmussen TK, Andersen T, Bak RO et al Overexpression of microRNA‐155 increases IL‐21 mediated STAT3 signaling and IL‐21 production in systemic lupus erythematosus. Arthritis Res Ther 2015; 17:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xin Q, Li J, Dang J et al miR‐155 Deficiency ameliorates autoimmune inflammation of systemic lupus erythematosus by targeting S1pr1 in Faslpr/lpr mice. J Immunol 2015; 194:5437–45. [DOI] [PubMed] [Google Scholar]
- 48. Wu Y, Zhang F, Ma J et al Association of large intergenic noncoding RNA expression with disease activity and organ damage in systemic lupus erythematosus. Arthritis Res Ther 2015; 17:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature 2003; 423:356–61. [DOI] [PubMed] [Google Scholar]
- 50. van Heemst J, van der Woude D, Huizinga TW, Toes RE. HLA and rheumatoid arthritis: how do they connect? Ann Med 2014; 46:304–10. [DOI] [PubMed] [Google Scholar]
- 51. Goronzy JJ, Weyand CM. Developments in the scientific understanding of rheumatoid arthritis. Arthritis Res Ther 2009; 11:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weyand CM, Yang Z, Goronzy JJ. T‐cell aging in rheumatoid arthritis. Curr Opin Rheumatol 2014; 26:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pratt AG, Swan DC, Richardson S et al A CD4 T cell gene signature for early rheumatoid arthritis implicates interleukin 6‐mediated STAT3 signalling, particularly in anti‐citrullinated peptide antibody‐negative disease. Ann Rheum Dis 2012; 71:1374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kremer JM, Westhovens R, Leon M et al Treatment of rheumatoid arthritis by selective inhibition of T‐cell activation with fusion protein CTLA4Ig. N Engl J Med 2003; 349:1907–15. [DOI] [PubMed] [Google Scholar]
- 55. Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest 2008; 118:3537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. O'Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol 2002; 2:37–45. [DOI] [PubMed] [Google Scholar]
- 57. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL‐17 and Th17 cells. Annu Rev Immunol 2009; 27:485–517. [DOI] [PubMed] [Google Scholar]
- 58. Schenten D, Nish SA, Yu S et al Signaling through the adaptor molecule MyD88 in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity 2014; 40:78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Croft M. The TNF family in T cell differentiation and function – unanswered questions and future directions. Semin Immunol 2014; 26:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morgan CL, Emery P, Porter D et al Treatment of rheumatoid arthritis with etanercept with reference to disease‐modifying anti‐rheumatic drugs: long‐term safety and survival using prospective, observational data. Rheumatology (Oxf) 2014; 53:186–94. [DOI] [PubMed] [Google Scholar]
- 61. Siebert S, Tsoukas A, Robertson J, McInnes I. Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacol Rev 2015; 67:280–309. [DOI] [PubMed] [Google Scholar]
- 62. Li J, Wan Y, Guo Q et al Altered microRNA expression profile with miR‐146a upregulation in CD4+ T cells from patients with rheumatoid arthritis. Arthritis Res Ther 2010; 12:R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK. Upregulated miR‐146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther 2008; 10:R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zeng Z, Gong H, Li Y et al Upregulation of miR‐146a contributes to the suppression of inflammatory responses in LPS‐induced acute lung injury. Exp Lung Res 2013; 39:275–82. [DOI] [PubMed] [Google Scholar]
- 65. Srivastava A, Nikamo P, Lohcharoenkal W et al MicroRNA‐146a suppresses IL‐17‐mediated skin inflammation and is genetically associated with psoriasis. J Allergy Clin Immunol 2016. doi:10.1016/j.jaci.2016.07.025. [DOI] [PubMed] [Google Scholar]
- 66. Niimoto T, Nakasa T, Ishikawa M et al MicroRNA‐146a expresses in interleukin‐17 producing T cells in rheumatoid arthritis patients. BMC Musculoskelet Disord 2010; 11:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fulci V, Scappucci G, Sebastiani GD et al miR‐223 is overexpressed in T‐lymphocytes of patients affected by rheumatoid arthritis. Hum Immunol 2010; 71:206–11. [DOI] [PubMed] [Google Scholar]
- 68. Sebastiani GD, Fulci V, Niccolini S et al Over‐expression of miR‐223 in T‐lymphocytes of early rheumatoid arthritis patients. Clin Exp Rheumatol 2011; 29:1058–9. [PubMed] [Google Scholar]
- 69. Lu MC, Yu CL, Chen HC, Yu HC, Huang HB, Lai NS. Increased miR‐223 expression in T cells from patients with rheumatoid arthritis leads to decreased insulin‐like growth factor‐1‐mediated interleukin‐10 production. Clin Exp Immunol 2014; 177:641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shibuya H, Nakasa T, Adachi N et al Overexpression of microRNA‐223 in rheumatoid arthritis synovium controls osteoclast differentiation. Mod Rheumatol 2013; 23:674–85. [DOI] [PubMed] [Google Scholar]
- 71. Sugatani T, Hruska KA. MicroRNA‐223 is a key factor in osteoclast differentiation. J Cell Biochem 2007; 101:996–9. [DOI] [PubMed] [Google Scholar]
- 72. Li YT, Chen SY, Wang CR et al Brief report: amelioration of collagen‐induced arthritis in mice by lentivirus‐mediated silencing of microRNA‐223. Arthritis Rheum 2012; 64:3240–5. [DOI] [PubMed] [Google Scholar]
- 73. Smigielska‐Czepiel K, van den Berg A, Jellema P et al Comprehensive analysis of miRNA expression in T‐cell subsets of rheumatoid arthritis patients reveals defined signatures of naive and memory Tregs. Genes Immun 2014; 15:115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rosenberger CM, Podyminogin RL, Navarro G et al miR‐451 regulates dendritic cell cytokine responses to influenza infection. J Immunol 2012; 189:5965–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dong L, Wang X, Tan J et al Decreased expression of microRNA‐21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J Cell Mol Med 2014; 18:2213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lee SY, Kwok SK, Son HJ et al IL‐17‐mediated Bcl‐2 expression regulates survival of fibroblast‐like synoviocytes in rheumatoid arthritis through STAT3 activation. Arthritis Res Ther 2013; 15:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ju JH, Heo YJ, Cho ML et al Modulation of STAT‐3 in rheumatoid synovial T cells suppresses Th17 differentiation and increases the proportion of Treg cells. Arthritis Rheum 2012; 64:3543–52. [DOI] [PubMed] [Google Scholar]
- 78. Zhou Q, Haupt S, Kreuzer JT et al Decreased expression of miR‐146a and miR‐155 contributes to an abnormal Treg phenotype in patients with rheumatoid arthritis. Ann Rheum Dis 2015; 74:1265–74. [DOI] [PubMed] [Google Scholar]
- 79. Stuhlmuller B, Kunisch E, Franz J et al Detection of oncofetal h19 RNA in rheumatoid arthritis synovial tissue. Am J Pathol 2003; 163:901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Song J, Kim D, Han J, Kim Y, Lee M, Jin EJ. PBMC and exosome‐derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin Exp Med 2015; 15:121–6. [DOI] [PubMed] [Google Scholar]
- 81. Spurlock CF III, Tossberg JT, Matlock BK, Olsen NJ, Aune TM. Methotrexate inhibits NF‐kappaB activity via long intergenic (noncoding) RNA‐p21 induction. Arthritis Rheumatol 2014; 66:2947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lu MC, Yu HC, Yu CL et al Increased expression of long noncoding RNAs LOC100652951 and LOC100506036 in T cells from patients with rheumatoid arthritis facilitates the inflammatory responses. Immunol Res 2016; 64:576–83. [DOI] [PubMed] [Google Scholar]
- 83. Wang S, Wan X, Ruan Q. The microRNA 21 in autoimmune diseases Int J Mol Sci 2016; 17:864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Murugaiyan G, da Cunha AP, Ajay AK et al MicroRNA‐21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest 2015; 125:1069–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. de Andres MC, Perez‐Pampin E, Calaza M et al Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res Ther 2015; 17:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lai NS, Yu HC, Yu CL, Koo M, Huang HB, Lu MC. Anti‐citrullinated protein antibodies suppress let‐7a expression in monocytes from patients with rheumatoid arthritis and facilitate the inflammatory responses in rheumatoid arthritis. Immunobiology 2015; 220:1351–8. [DOI] [PubMed] [Google Scholar]
- 87. Cui X, Witalison EE, Chumanevich AP et al The induction of microRNA‐16 in colon cancer cells by protein arginine deiminase inhibition causes a p53‐dependent cell cycle arrest. PLOS ONE 2013; 8:e53791. [DOI] [PMC free article] [PubMed] [Google Scholar]