

Summary
Type B coxsackievirus (CV‐B) infections are involved frequently in the triggering of several autoimmune diseases such as myocarditis, dilated cardiomyopathy, pericarditis, pancreatitis, type 1 diabetes, encephalitis, thyroiditis or Sjögren's syndrome. Serological and virological evidence suggests that maternal infections during pregnancy can play a role in the appearance of these diseases in offspring. The current study aims to explore the effect of an in‐utero CV‐B infection on the fetal thymus, the central site for programming immunological self‐tolerance. In this perspective, female Swiss albino mice were inoculated intraperitoneally or orally with the diabetogenic CV‐B4 E2 strain at gestational days 10 or 17. Offspring were killed at different post‐inoculation times, and their thymuses were analysed for evidence of infection and alterations in thymic T cell subsets. In‐utero CV‐B infection of the thymus was demonstrated during the course of vertical transmission, as attested by viral RNA and infectious virus detection in most analysed samples. No histopathological changes were evident. Thymic T cells were not depleted, despite being positive for viral RNA. As evidenced by flow cytometry analysis, CV‐B infection of the fetal thymus induced significant changes of thymic T cell populations, particularly with maternal inoculation at gestational day 10. Altogether, these findings suggest that CV‐B infection of the fetal thymus may play an important role in the genesis of autoimmune diseases.
Keywords: fetal thymus, mouse model, T cell differentiation, type B Coxsackieviruses (CV‐B), vertical transmission
Introduction
Type B Coxsackieviruses (CV‐B) are small, non‐enveloped, positive sense single‐stranded RNA viruses, belonging to the species Enterovirus B, genus Enterovirus, family Picornaviridae 1. Infections with CV‐B are involved in a variety of acute and chronic diseases that have an autoimmune component, including myocarditis, dilated cardiomyopathy, pericarditis 2, 3, 4, pancreatitis 5, type 1 diabetes (T1D) 6, 7, 8, encephalitis 9, thyroiditis 10 or Sjögren's syndrome 11.
Like all Enteroviruses, CV‐B are essentially transmitted through the faecal–oral route, and their vertical transmission has also been described, especially through the transplacental route 12, 13, 14. This materno–fetal transfer of CV‐B infection seems to be widely involved in the most severe diseases affecting fetuses, newborns and young infants 15, 16, 17, 18, 19.
Several mechanisms have been suspected to be involved in triggering or accelerating the autoimmune process by CV‐B, essentially molecular mimicry between viral and host antigens, bystander activation of autoreactive clones, persistent infection of the target tissues and induced thymus dysfunction (for a review see 7, 20). As the thymus is the central site of self‐tolerance establishment during fetal life 21, 22, 23, its infection with CV‐B, especially at that time, could disturb its tolerogenic functions and thus contribute to the genesis of CV‐B‐associated autoimmune diseases (for a review see 24, 25).
In this context, we know that the diabetogenic CV‐B4 E2 strain is able to infect in‐vitro‐cultured total thymic cells 26, thymic epithelial cells 27, 28 and fetal thymic organ cultures (FTOC) 29, 30. CV‐B4 E2 reaches the thymus during the course of a systemic infection of Swiss albino mice inoculated through the oral route, with viral RNA detection for at least 70 days post‐inoculation (p.i.) 31. CV‐B4 infection of cultured fetal human thymic fragments, as well as mouse FTOC, also disturbs thymocyte populations in both models 29, 30. Such abnormalities in thymocyte populations have also been reported in vivo in a mouse model of CV‐B4 E2‐induced T1D 32. A drastic repression of type 2 insulin‐like growth factor (Igf2, a major β cell autoantigen involved in negative selection of thymocytes autoreactive towards antigens of the insulin family) expression, following CV‐B4 E2 infection of a murine thymic epithelial cell line, has been documented equally 28.
Altogether, these data argue for a role of thymic CV‐B4 infection as a potential mechanism in the genesis of associated autoimmune diseases. However, an essential point remains to be assessed. Indeed, despite that it is well established that thymus is active mainly during fetal life, the issue of thymus infection during the course of vertical transmission has not been documented thoroughly 33, 34.
Thus, the current study aims to investigate whether CV‐B4 E2 can reach the fetal thymus during the course of an in‐utero infection and whether such an infection could disturb the major thymic function, namely, the T cell differentiation process.
Materials and methods
Virus
The diabetogenic strain CV‐B4 E2 (kindly provided by J. W. Yoon, Julia McFarlane Diabetes Research Centre, Calgary, Alberta, Canada) was propagated in human epithelial type 2 (HEp‐2) cells (BioWhittaker, Walkersville, MD, USA) in Eagle's minimal essential medium (MEM; Gibco BRL, Invitrogen, Gaithersburg, MD, USA) supplemented with 10% heat‐inactivated fetal calf serum (FCS; Gibco BRL), 1% (2 mM) L‐glutamine (BioWhittaker), 50 µg/ml streptomycin, 50 IU/ml penicillin (Gibco BRL), 1% non‐essential amino acids (Gibco BRL) and 0·05% (2·5 µg/ml) fungizone (Amphotericin B; Apothecon). Supernatants were collected 3 days p.i., clarified by centrifugation at 2000 g for 10 min, divided into aliquots and stored at −80°C. Virus titres in stocks were determined on HEp‐2 cells by the plaque assays method and expressed as plaque‐forming units (p.f.u.)/ml.
Mice
All experiments were conducted by following the standards of general ethics guidelines and approved by the Faculty of Pharmacy, University of Monastir, Tunisia. Animals used in this investigation were maintained in specific pathogen‐free conditions with unlimited access to food and water. Adult outbred Swiss albino mice (Pasteur Institute, Tunis, Tunisia) were mated (three females per male were caged together) until successful fertilization was noted. The day the vaginal plug was observed was considered as the first day of gestation (day 1G).
Mice inoculation
Pregnant mice were inoculated randomly, either at gestational days 10 (10G) or 17 (17G), intraperitoneally (i.p.) or orally (by gavage) with, respectively, 1·4 × 105 p.f.u. or 1·4 × 106 p.f.u. of CV‐B4 E2 contained in 200 µl culture medium. Naive mice served as negative controls. Offspring born to dams inoculated at day 10G were killed at day 17G, as well as at days 0 and 5 from birth, using isoflurane. Those born to dams inoculated at day 17G were killed at days 0 and 5 from birth. Thymuses were collected, washed with cold phosphate‐buffered saline (PBS), weighed and treated for viral RNA and infectious virus detection, histological examination and thymocyte analysis. The total body weight of each mouse was recorded at day 5 from birth, and the thymic index was calculated as: [thymus weight (g)/body weight (g)] × 100. More interest was given to the effect of oral inoculation, mimicking the natural way of contamination in humans, which is why some experiments were performed only with samples of offspring born to orally inoculated dams.
Viral genome detection
For each experimental condition (i.p. or oral inoculation at days 10G or 17G, or in the absence of inoculation), the presence of CV‐B4 E2 RNA was checked at different p.i. times (day 17G and days 0 and 5 from birth) in the thymuses of six mice (three offspring born to each of two dams), according to the procedure described below.
RNA extraction
Total RNA was extracted from washed thymuses by the acid guanidium thiocyanate–phenol–chloroform extraction procedure using Tri‐Reagent (Sigma, St Louis, MO, USA), as described by Chomczynski and Sacchi 35. Sterile nuclease‐free water and supernatant of CV‐B4 E2‐infected HEp‐2 cells, submitted to the same extraction procedure, served as negative and positive controls, respectively. Extracted RNA was then dissolved in 50 µl of nuclease‐free water (Promega, Madison, WI, USA), quantified using the Nanodrop 2000 (UV‐Vis Spectrophotometer; ThermoScientific, Waltham, MA, USA) and stored at −80°C until use in reverse transcription (RT)–polymerase chain reaction (PCR) assays.
Two‐step RT–PCR for CV‐B4 E2 RNA detection
For CV‐B4 E2 RNA amplification, we used primer sense 008: 5'‐GAGTATCAATAAGCTGCTTG‐3' and anti‐sense 007: 5'‐ATTGTCACCATAAGCAGCCA‐3' specific of the highly conserved 5' NC region of enterovirus genome and generating a 414 base pairs (bp) fragment 31. cDNA synthesis was first performed with approximately 100 ng of RNA in a final volume of 20 µl containing 200 U of M‐MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA), 0·1 µM of anti‐sense 007 primer, 0·5 mM each deoxynucleoside triphosphate (dNTP), 40 U of RNase inhibitor, 50 mM Tris‐HCl (pH 8·3), 75 mM KCl, 3 mM MgCl2 and 10 mM dithiothreitol (DTT). According to the manufacturer's instructions, secondary structures were first denatured by heating the samples for 5 min at 65°C in the presence of dNTPs and the corresponding primer. Tubes were then put on ice immediately, supplemented with buffer, DTT and RNase inhibitor and incubated for 2 min at 37°C. The RT reaction was finally performed at 37°C for 50 min, after adding the enzyme. The reaction was stopped by heating at 70°C for 15 min.
The PCR was carried out with 3 µl of cDNA samples and 0·4 µM each primer in a total volume of 50 µl containing 2·5 U of Taq Paq5000 DNA polymerase (Agilent Technologies, Edinburgh, UK), 0·2 mM each dNTP and 2 mM MgCl2. The PCR mixture was subjected to a first denaturation step for 3 min at 94°C, followed by 30 cycles of amplification, consisting of denaturation for 20 s at 94°C, annealing for 20 s at 55°C and extension for 30 s at 72°C, followed by a final extension step for 5 min at 72°C. All reactions were performed using a preheated Eppendorf thermal cycler.
RNA extracted from supernatant of CV‐B4 E2‐infected HEp‐2 cells was reverse‐transcribed and amplified according to the procedure described above, and served as a positive control. A negative control (no RNA) was also included in each reaction. Samples showing negative results were subjected to beta‐actin mRNA amplification as an internal control to ensure the integrity of extracted RNA and the absence of RT–PCR inhibitors.
Detection and analysis of amplification products
The amplified RT–PCR products were analysed by electrophoresis on a 2% agarose gel containing 0·5 mg/ml ethidium bromide (Sigma) and visualized using a Gel Doc 2000 system (Bio‐Rad, Hercules, CA, USA). A 100‐bp DNA ladder (Invitrogen) was used as a molecular mass marker.
Semi‐nested (sn) PCR
RT–PCR products showing negative results were subjected to a subsequent sn‐PCR with internal primer sense 006: 5'‐TCCTCCGGCCCCTGAATGCG‐3' and anti‐sense primer 007, generating a 155 bp fragment 36. A similar reaction mix and the same cycling programme were used, except that the hybridization temperature was 60°C. A positive control (DNA amplified from the RNA extract of supernatant of CV‐B4 E2‐infected HEp‐2 cells) and a negative control (no DNA) were included in each reaction.
Virus titration by plaque assays
The thymuses of six mice (three offspring born to each of two dams) of each experimental condition (oral virus inoculation at days 10G or 17G, and in the absence of inoculation) were subjected to virus titration at different p.i. times (day 17G and days 0 and 5 from birth), according to the procedure described below. Snap‐frozen thymuses were weighed and crushed in 1% penicillin/streptomycin PBS, and then centrifuged at 12 000 g for 10 min at 4°C. Supernatants were diluted 10‐fold in MEM with 2% FCS, inoculated to confluent HEp‐2 cells (106 cells/well) in 12‐well culture plates. After 4 h of incubation, medium containing virus dilutions was removed, cells were washed with PBS and 1 ml of medium (9 vol) supplemented with 4% agarose solution (1 vol) were added to each well. Cultures were incubated at 37°C in a humidified atmosphere with 5% CO2 and examined daily for CV‐B4 cytopathic effect until 7 days p.i. Cells then were fixed with 1 ml of Carnoy's fixative (three parts EtOH + one part acetic acid) per well and stained with crystal violet for 30 min. Finally, wells were rinsed with water and plaques were counted for the highest dilution showing cytopathic effect. Results were expressed as mean titres (p.f.u/mg of tissue) ± standard deviation (s.d.).
Histological analysis
The thymuses of six mice (three offspring born to each of two dams) of each experimental condition (oral virus inoculation at days 10G or 17G and in the absence of inoculation) were subjected to histological examination at different p.i. times (day 17G and days 0 and 5 from birth), according to the procedure described below. Formalin‐fixed, paraffin‐embedded thymic lobes were cut into 4‐µm‐thick sections. The slices were deparaffinized twice in xylene for 10 min, then rehydrated through sequential concentrations of alcohol (100, 95, 70 and 50°) and finally with double‐distilled water. Slices then were stained with haematoxylin for 15 min, washed in running water, differentiated with 1% HCl/ethanol and incubated in water for 5 min. For cytoplasm staining, slices were immersed in 0·5% eosin solution for 3 min and washed in water for 10 min. Sections were dehydrated again through sequential concentrations of alcohol (50, 70, 95 and 100°). Finally, slides were mounted and observed under a light microscope.
Flow cytometry analysis of thymic T cell subsets
For each experimental condition (i.p. virus inoculation at days 10G and 17G, oral inoculation at days 10G and 17G, and in the absence of virus inoculation), whole thymuses of 12 mice (three offspring born to each of four dams) were tested at each of day 17G and days 0 and 5 after birth. Thymuses of the three offspring born to the same dam were pooled together.
Preparation of thymic T cell suspensions
Freshly harvested thymuses were put on ice immediately into a small Petri dish with fluorescence activated cell sorter (FACS) buffer [PBS ×1, 2% FCS, 2 mM ethylenediamine tetraacetic acid (EDTA)]. Cells were dissociated by crushing on a 70‐µm cell strainer filter. The obtained cell suspension was filtered and cells were washed twice by centrifugation. Pelleted cells were resuspended in FACS buffer, enumerated by trypan blue viability dye exclusion test and adjusted to a final concentration of 2–3 × 105 cells/100 µl for immunostaining and FACS analysis. Aliquots from some suspensions were submitted to extraction and RT–PCR analysis, as described above, for CV‐B4 RNA detection.
T cell staining and FACS analysis
Three‐colour cell staining was performed with CD90/Thy‐1 rat anti‐mouse monoclonal antibody (mAb) (clone G7) phycoerythrin (PE) conjugate, CD4 rat anti‐mouse mAb (clone GK1·5) phycoerythrin‐cyanin (PE‐Cy)®5 conjugate and CD8a rat anti‐mouse mAb (clone 53–6·7) fluorescein (FITC) conjugate (all from Life Technologies, Paisley, UK) antibody mixture, and the preparation was incubated for 30 min at 4°C in the dark. Cells were then washed twice by centrifugation in FACS buffer, resuspended in 500 µl of FACS buffer and analysed on an EPICS XL® (Beckman‐Coulter, Brea, CA, USA) flow cytometer.
Statistical analysis
Data are summarized as means ± s.d. Thymic index, total count of viable thymic T cells and proportions of T cell populations in infected animals were compared with those in controls using the Welch two‐sample t‐test. Statistical significance was defined by P‐values less than 0·05.
Results
Viral RNA was detected in the thymus of offspring born to CV‐B4 E2‐inoculated dams
Vertical transmission of CV‐B4 E2 infection from dams to their offspring's thymuses was checked by searching for the viral genome in that tissue, as described in the Materials and methods section. As shown in Table 1, CV‐B4 E2 RNA was detected in all sampled thymuses of offspring born to dams inoculated i.p. with CV‐B4 E2 at either days 10G or 17G, and to dams inoculated orally at day 10G, and seven of 12 analysed samples of offspring born to dams inoculated orally at day 17G.
Table 1.
Reverse transcription–polymerase chain reaction (RT–PCR) results for viral RNA detection in the thymus of offspring born to CV‐B4 E2‐inoculated dams
| Time of sampling | |||||
|---|---|---|---|---|---|
| Inoculation route | Inoculation time | Litters | Day 17G | Day 0 | Day 5 |
| Intraperitoneal | Day 10G | Dam 1 | 3/3 | 3/3 | 3/3 |
| Dam 2 | 3/3 | 3/3 | 3/3 | ||
| Day 17G | Dam 1 | – | 3/3* | 3/3* | |
| Dam 2 | – | 3/3* | 3/3* | ||
| Oral | Day 10G | Dam 1 | 3/3 | 3/3 | 3/3 |
| Dam 2 | 3/3 | 3/3 | 3/3 | ||
| Day 17G | Dam 1 | – | 0/3* | 3/3* | |
| Dam 2 | – | 2/3* | 2/3* | ||
*Results obtained by semi‐nested RT–PCR. Thymuses from six offspring (each three born to one different dam) were analysed at each time‐point for each experimental condition. Results are summarized as number of positive results/number of tested thymuses.
CV‐B4 E2 RNA was easily detectable by RT–PCR in thymuses of offspring born to dams inoculated at day 10G either by the i.p. or oral routes. However, thymuses of offspring born to dams inoculated at day 17G required a subsequent sn‐PCR round to reveal a positive result (Table 1).
Viral RNA was not detected in the thymuses of offspring born to negative control dams.
Infectious virus was isolated from the thymus of offspring born to CV‐B4 E2‐inoculated dams
Using the plaque assay method, infectious particles could be evidenced and titrated in all infected (positive for viral RNA) thymus samples, following oral inoculation at either days 10G or 17G (Fig. 1). Viral titres were markedly (50–2000‐fold) higher in thymuses of offspring born to dams inoculated at day 10G than in those born to dams inoculated at day 17G. Viral load decreased from days 17G and 0 to day 5 from birth, following an inoculation at day 10G. When inoculation was performed on day 17G, viral titres increased between days 0 and 5 from birth. No infectious virus could be evidenced, either in thymuses revealed negative for viral RNA or in those of offspring born to negative control dams.
Figure 1.
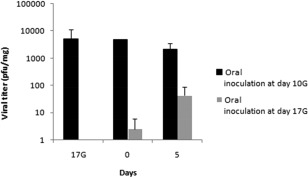
Viral progeny titration in CV‐B4‐infected thymuses. The thymuses of six mice (three offspring born to each of two dams) of each experimental condition (oral virus inoculation at days 10G or 17G, and in the absence of inoculation) were subjected to viral progeny titration, by the plaque assay method, at different post‐inoculation (p.i.) times (day 17 gestation and days 0 and 5 from birth). Data are representative of two independent experiments with n = 3 thymuses per group and per time‐point, in each. Results are plotted as mean titres plaque‐forming units (p.f.u.)/mg of tissue) ± standard deviations.
Thymus integrity was preserved after in‐utero CV‐B4 E2 infection
In order to assess the effect of vertically transmitted CV‐B4 E2 on thymus integrity, we first checked for eventual modifications on thymus size by calculating the thymic index. The latest, measured at day 5 from birth, was not decreased significantly in offspring born to dams inoculated orally at day 10G compared to those born to negative controls (0·47 versus 0·7%, respectively, P > 0·05) (Fig. 2). The thymic index following oral inoculation at day 17G was similar to that in negative controls (0·68%) (Fig. 2). The effect of the virus on thymus architecture was then evaluated by histological analysis of thymic lobes sections, as described in the Materials and methods section. Virus‐infected thymuses seemed similar to mock‐infected ones, presenting a normal architecture without obvious cell lysis, necrosis or any histopathological change, even when examined at higher magnification (Fig. 3).
Figure 2.
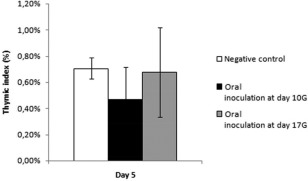
Effects of CV‐B4 on thymus weight. The thymic index [(thymus weight/whole body weight) × 100] was calculated at day 5 from birth for six mice (three offspring born to each of two dams) of each experimental condition [oral virus inoculation at days 10 gestation (G) or 17G, and in the absence of inoculation]. Data are representative of two independent experiments with n = 3 thymuses per group in each. Results are expressed as mean percentages ± standard deviations.
Figure 3.
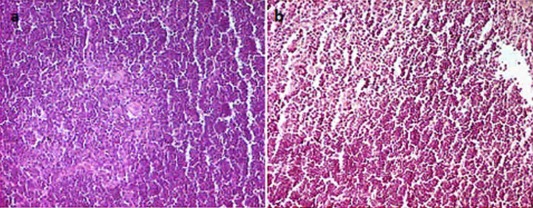
Effects of CV‐B4 on thymus integrity. The thymuses of six mice (three offspring born to each of two dams) of each experimental condition [oral virus inoculation at day 10 gestation (G) or 17G, and in the absence of inoculation] were subjected to histological examination at different post‐inoculation (p.i.) times (day 17G and days 0 and 5 from birth). No histopathological change was observed in all examined sections. Representative sections from a negative control thymus (a) and a thymus stemming from an intraperitoneal inoculation of CV‐B4 E2 at day 10G. (b) Both sampled at day 5 from birth, and examined after haematoxylin and eosin staining. Gr × 400. [Colour figure can be viewed at wileyonlinelibrary.com]
Viral RNA was detected in thymic T cells from infected offspring
As described in the Materials and methods section, RT–PCR for viral RNA was used to explore T cell infection in CV‐B4 E2‐infected thymuses. Thus, intracellular CV‐B4 E2 RNA could be found by RT–PCR in all analysed samples of thymic T cell suspensions generated from infected thymuses (Fig. 4). No trace of viral RNA was detectable in T cells from uninfected negative control samples.
Figure 4.
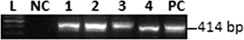
CV‐B4 E2 infects thymic T cells. Representative agarose gel electrophoresis of amplicons specific to CV‐B4 E2 RNA in thymic T cells of offspring, sampled the day of birth (day 0), from negative control dams (lane NC) and dams inoculated intraperitoneally at day 10G (lanes 1 and 2) or orally (lanes 3 and 4). L = 100 base pairs (bp) DNA ladder; PC = positive control.
CV‐B4 E2‐infected thymuses displayed abnormalities of T cell subsets
The effect of vertically transmitted CV‐B4 E2 on thymic T cells was assessed first by their enumeration by trypan blue viability dye exclusion test. As shown in Table 2, no signs of depletion were evident following CV‐B4 E2 infection, as no significant difference in total viable cell count was observed between i.p. or orally inoculated and control thymuses, either at days 0 or 5 from birth (P‐values > 0·05 versus mock).
Table 2.
Viable thymocyte T cell counts in mock‐ and CV‐B4 E2‐infected thymuses
| Time of sampling | ||
|---|---|---|
| Inoculation | Day 0 | Day 5 |
| Mock | 12 × 106 ± 2·12 × 106 | 12 × 106 ± 4·24 × 106 |
| CV‐B4 E2 (i.p.) | 10·2 × 06 ± 3·12 × 106 | 9·15 × 106 ± 0·21 × 106 |
| CV‐B4 E2 (oral) | 13 × 06 ± 0·71 × 106 | 9 × 106 ± 0·71 × 106 |
Data were generated from three independent experiments, representative of different experimental conditions [intraperitoneal (i.p.) inoculation at day 10G and oral inoculation at day 10G], with n = 3 thymuses per group and per time‐point in each. Results are expressed as means of 106 living T cell/thymus ± standard deviation.
We then focused upon thymic T cell (CD90+ cells, which represent more than 95% of the cells in our suspensions) maturation from the most immature stage double‐negative (DN) CD4–CD8– to double‐positive (DP) CD4+CD8+, then differentiation to mature single‐positive either CD4+CD8– (SP4) or CD4–CD8+ (SP8). For this purpose, proportion of these four populations was determined by flow cytometry analysis at each of day 17G and days 0 and 5 from birth in the different groups of infected offspring and compared with those in negative controls (Fig. 5).
Figure 5.
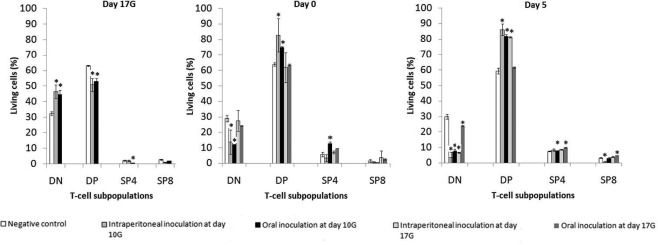
Effects of CV‐B4 E2 in‐utero infection on T cell differentiation. Thymic T cell suspensions were prepared from thymuses, collected at different post‐inoculation (p.i.) times, on offspring from negative control and CV‐B4 E2‐inoculated [either at day 10 gestation (G) or 17G, by the intraperitoneal or the oral route] dams, stained with anti‐CD90, anti‐CD4 and anti‐CD8 antibodies and analysed by fluorescence activated cell sorter (FACS). Histograms represent the DN, DP, SP4 and SP8 populations among total thymocytes (CD90+ cells). Data are representative of four independent experiments with n = 3 thymuses per group and per time‐point in each. Results are expressed as mean percentages ± standard deviations; *P < 0·05 versus mock. DN = double‐negative CD4–CD8–; DP = double‐positive CD4+CD8+; SP4 = single‐positive CD4+CD8–; SP8: single‐positive CD4–CD8+.
As illustrated in Fig. 5, several anomalies in thymic T cell subsets could be observed following in‐utero CV‐B4 E2 infection.
Virus inoculation of dams through the intraperitoneal route at day 10G disturbed the pattern of immature T cell populations significantly compared with negative controls. An increase in DN (46·4 ± 4·4% versus 32·4 ± 1·24%, P = 0·005) associated with a decrease in DP (50·8 ± 4·25% versus 63·03 ± 0·45%, P = 0·009) was evident at day 17G. Conversely, starting from day 0, we observed a decrease in DN (13·66 ± 7·86% versus 28·97 ± 2·16%, P = 0·025) together with an increase in DP (82·75 ± 10·8% versus 63·83 ± 1·04, P = 0·038) subset. At day 5, the decrease in DN (3·49 ± 3·45% versus 29·97 ± 1·67%, P = 0·0009) and increase in DP persisted (87·37 ± 2·31% versus 59·37 ± 1·79%, P = 0·0001), and the decrease in SP8 also became significant (0·37 ± 0·23% versus 3·15 ± 0·25%, P = 0·0003). No evident anomalies were observed in the SP4 population.
Inoculation through the oral route at day 10G induced a significant increase in DN (44·68 ± 1·93% versus 32·4 ± 1·25%, P = 0·0015) together with a significant decrease in DP (53 ± 2% versus 63·03 ± 0·45%, P = 0·009) and SP4 (0·44 ± 0·03% versus 2·07 ± 0·46%, P = 0.038), evident at day 17G. Starting from day 0 the profile inverted, as described above in mice inoculated i.p. at day 10G, showing a significant decrease in DN (11·8 ± 0·3% versus 28·97 ± 2·16%, P = 0·004) associated with an increase in DP (74·95 ± 0·45% versus 63·83 ± 1·04%, P = 0·0008) and SP4 (12·7 ± 0·7% versus 5·67 ± 1·51%, P = 0·007) populations. By day 5, the decrease in DN (7·8 ± 0·7% versus 29·97 ± 1·67%, P = 0·0004) and increase in DP persisted (81·7 ± 0·8% versus 59·37 ± 1·79%, P = 0·0005), but the proportion of SP4 became comparable to that in controls. No significant anomalies were observed in the SP8 population.
Virus inoculation of dams through the intraperitoneal route at day 17G did not induce any significant effect at day 0, but a significant decrease in DN (6·49 ± 0·28% versus 29·97 ± 1·67%, P = 0·001) as well as an increase in DP (81·1 ± 0·35% versus 59·37 ± 1·79%, P = 0·001) and SP4 (8·58 ± 0·25% versus 7·41 ± 0·21%, P = 0·004) subsets were evident at day 5. A non‐significant increase was observed in the SP8 population.
Finally, inoculation through the oral route at day 17G equally did not induce any significant effect at day 0, but a significant decrease in DN (23·7 ± 0·28% versus 29·97 ± 1·67%, P = 0·023) as well as an increase in SP4 (9·86 ± 0·2% versus 7·41 ± 0·21%, P = 0·0018) and SP8 (4·83 ± 0·21% versus 3·15 ± 0·25%, P = 0·006) subsets were evident at day 5, together with a non‐significant increase in DP population.
Discussion
Because the thymus plays a specific role in self‐tolerance programming 21, 22, 23, thymus dysfunction could be a basic event in the pathogenesis of autoimmunity. This hypothesis remains relevant in an infectious context, because thymus infection is associated with thymus dysfunction (for a review see 24, 25). This line of thought becomes even more plausible when the infection occurs in fetal life, the period during which thymus ensures T cell differentiation and negative selection (for a review see 37).
Despite CV‐B association with several autoimmune diseases, the issue of CV‐B infection of the thymus has not been investigated extensively. Most of the available data arise from in‐vitro experiments 26, 27, 28, 29, 30. The few studies carried in vivo used adult or young mice, but never pregnant mice and fetuses 31, 32. Thymus infection by CV‐B has already been reported through antigen detection in dead newborns 33, 34 but, through our original experimental approach, the current study provides direct evidence for a thymus infection in the course of an in‐utero vertical transmission, thus strengthening the hypothesis of a role of thymic CV‐B infection in the pathogenesis of autoimmune diseases.
Indeed, the use of outbred mice (which reflect more accurately the natural variations inside a population) and their inoculation in the second or the third weeks of gestation (as the inoculation period during pregnancy may influence the outcome of the infection 38, 39) with CV‐B4 E2 (a strain known for its diabetogenicity), as performed in a previous investigation by Bopegamage et al. 39 are, together, likely to constitute an ideal system for addressing this issue. With regard to virus inoculation, we first began by the intraperitoneal route to ‘favour’ virus spreading to the thymus and observe the eventual occurrence of anomalies, such as described previously in vitro. We then tested and focused upon the oral route to verify if the offspring's thymus could be reached following inoculation through the natural route of contamination in humans, and especially if similar effects could be obtained. However, a 10‐fold higher virus dose (the same dose used by Bopegamage et al. 39) was used for oral inoculation, as intestinal defences are susceptible to attenuate the infection 40. In addition, offspring sampling from day 17G allowed us to evidence prenatal transmission through the transplacental route, even though perinatal transmission cannot be excluded.
Viral genome detection in sampled thymuses demonstrates clearly that CV‐B4 E2 can reach the offspring's thymus in the course of vertical transmission, regardless of the route and time of inoculation. The need for a subsequent sn‐PCR to detect the viral genome in thymuses of offspring born to dams inoculated at day 17G may be due to the fact that the virus did not replicate enough in that tissue to reach a threshold detectable by our RT–PCR. Indeed, infectious virus titration showed lower viral levels following inoculation at day 17G than at day 10G. These results are extremely unlikely to be due to contamination with blood, as viral RNA is detectable only transiently in blood during the course of systemic infection 31, as well as in the placenta and other sampled tissues in the current study (data not shown), and as the virus is replicating (as proved by progeny detection).
Virus infections of the thymus are associated frequently with structural and/or functional alterations of that gland (for a review see 24, 25). Such alterations would be more pronounced when infection occurs during fetal development. Indeed, Lozovskaia et al. 34 described preterm thymic involution, hypotrophy and immaturity in the majority of perinatal and neonatal death cases associated with CV‐A and CV‐B infections.
Intriguingly, fetal thymus infection in our model did not result in any evident atrophy (as attested by thymic index calculation), histopathological change or developmental delay. This is reminiscent of previous findings in CV‐B4 E2‐infected murine FTOC 30. Similarly to the situation in the murine FTOC model, no significant T cell depletion could be seen (which is in agreement with thymic index data), despite viral RNA being detectable in those cells in the current investigation. As discussed previously, CV‐B4 can infect the thymus without obvious cell lysis 30. Our observations do not accord with what has been described in other models of fetal thymus infection. Indeed, feline immunodeficiency virus inoculated in kittens during fetal development induces severe neonatal thymus atrophy 41. By the same method, thymus atrophy with cortical involution and severe cortical depletion of thymic T cells were observed following in‐utero infection of piglets with porcine reproductive and respiratory syndrome virus 42. Those discrepancies may be explained by a relatively lower lytic potential of CV‐B4 E2 compared to other viruses, especially in tissues such as thymus, a potential site of persistence for that virus, as documented previously 28, 29, 31. Of note, CV‐B4 E2 RNA was still detectable at day 21 postpartum in this study (data not shown), and the reduction of the cytocidal potential is a strategy used commonly by viruses to establish persistence 43.
In contrast, in‐utero CV‐B4 E2 infection of the mouse thymus seems to disturb the T cell differentiation process, as revealed by analysis of the proportion of T cell subsets.
This effect would depend upon the inoculation time, as inoculation at day 10G induced significant disturbances on immature T cells as soon as day 17G, whereas such anomalies following inoculation at day 17G manifested only at day 5 from birth. This gap would follow that of the infection, and is evocative of what we deduced from the results of viral genome detection by sn‐RT–PCR versus RT–PCR and from those of viral progeny titration. In other words, infection at day 10G would, at first, block DN to DP transition, then DP to SP differentiation, in particular to SP8. Infection at day 17G, arising at a later stage of gestation, would reach the thymus at a still insufficient threshold (a mean of 2·5 p.f.u./mg) to rapidly disturb T cell populations and/or at an advanced stage of T cell differentiation. Indeed, no significant evolution in the total number of viable T cells and in the proportions of the four T cell subpopulations could be seen during the time–course of our study, suggesting that T cell differentiation was already almost completed by day 17G.
With regard to the inoculation route, quite similar patterns of thymic T cell populations were observed following either i.p. or oral inoculation, with some differences among mature SP populations (that were less affected anyway).
These current results do not accord fully with those we observed previously in murine FTOC infected at day 14G that, at 7 days p.i. (corresponding to day 0 in the current study), showed a significant increase in DN, SP4 and SP8, together with a significant decrease in DP 30. There is only a parallel in the proportions of immature T cells, which is evocative of that actually found at day 17G following inoculation at day 10G. A correct comparison between the results obtained with both systems cannot be made, as virus inoculation to pregnant dams was not performed at day 14G and because we do not know if the amount of virus that reaches offspring thymuses is comparable to the infectious dose used in the FTOC model. Even in comparable conditions, eventual discrepancies only remind us that, even though murine FTOC is a very useful experimental tool, this ex‐vivo model does not mimic perfectly the real situation in vivo.
Another key point of the current study is the follow‐up of thymic T cell subsets through sampling and measurement at different p.i. times until 5 days after birth, which allowed us to observe some dynamics in these populations following virus infection, and an effect of inoculation at day 17G that would not be detected if analysis was restricted to the day of delivery.
In conclusion, the current paper shows for the first time, to our knowledge, that CV‐B4 E2 can reach the murine fetal thymus during the course of an in‐utero transmission, thus opening the possibility to explore several ways by which the virus may disturb thymic functions. The infection promoted significant anomalies in thymic T cell subsets, regardless of the inoculation route and period. Although such anomalies have been reported already in T1D models 32, 44, their direct involvement in the development of autoimmunity is still unclear. The understanding of how CV‐B4 can lead to such disturbances also deserves to be investigated. Several studies are being conducted along this line in our laboratory.
Author contributions
H. J. designed the study, analysed the results and wrote the paper. A. H. performed all the experiments and analysed the results. H. J., H. J. and F. E. assisted A. H. in experiments with the murine model and the documentation of infection. S. A., G. B., H. M. and T. C. assisted A. H. in flow cytometry experiments. F. S. and D. H. provided some reagents. M. M. supervised the experiments of anatomohistopathology and analysed the slides. V. G., D. H. and M. A. supervised the study and corrected the paper.
Disclosure
The authors have no disclosures.
Acknowledgements
This work was supported by Ministère de l'Enseignement Supérieur et de la Recherche Scientifique, (LR99ES27), Tunisia.
References
- 1. Knowles NJ, Hovi T, Hyypiä T et al Picornaviridae In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, eds. Virus taxonomy: classification and nomenclature of viruses: ninth report of the international committee on taxonomy of viruses. San Diego, CA: Elsevier, 2012:855–80. [Google Scholar]
- 2. Huber SA. Autoimmunity in coxsackievirus B3 induced myocarditis. Autoimmunity 2006; 39:55–61. [DOI] [PubMed] [Google Scholar]
- 3. Knowlton KU. CVB infection and mechanisms of viral cardiomyopathy. Curr Top Microbiol Immunol 2008; 323:315–35. [DOI] [PubMed] [Google Scholar]
- 4. Rose NR. Autoimmunity in coxsackievirus infection. Curr Top Microbiol Immunol 2008; 323:293–314. [DOI] [PubMed] [Google Scholar]
- 5. Ramsingh AI. CVB‐induced pancreatitis and alterations in gene expression. Curr Top Microbiol Immunol 2008; 323:241–58. [DOI] [PubMed] [Google Scholar]
- 6. Jaïdane H, Hober D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab 2008; 34:537–48. [DOI] [PubMed] [Google Scholar]
- 7. Jaïdane H, Sauter P, Sane F, Goffard A, Gharbi J, Hober D. Enteroviruses and type 1 diabetes: towards a better understanding of the relationship. Rev Med Virol 2010; 20:265–80. [DOI] [PubMed] [Google Scholar]
- 8. Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta‐analysis of observational molecular studies. BMJ 2011; 342:d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bale JF Jr. Virus and immune‐mediated encephalitides: epidemiology, diagnosis, treatment, and prevention. Pediatr Neurol 2015; 53:3–12. [DOI] [PubMed] [Google Scholar]
- 10. Desailloud R, Hober D. Viruses and thyroiditis: an update. Virol J 2009; 6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Triantafyllopoulou A, Tapinos N, Moutsopoulos HM. Evidence for coxsackievirus infection in primary Sjögren's syndrome. Arthritis Rheum 2004; 50:2897–902. [DOI] [PubMed] [Google Scholar]
- 12. Basso NG, Fonseca ME, Garsia AG, Zuardi JA, Silva MA, Outani H. Enterovirus isolation from fœtal and placental tissues. Acta Virol 1990; 34:49–57. [PubMed] [Google Scholar]
- 13. Bendig JW, Franklin OM, Hebden AK et al Coxsackievirus B3 sequences in the blood of a neonate with congenital myocarditis, plus serological evidence of maternal infection. J Med Virol 2003; 70:606–9. [DOI] [PubMed] [Google Scholar]
- 14. Pereira L, Maidji E, McDonagh S, Tabata T. Insights into viral transmission at the uterine‐placental interface. Trends Microbiol 2005; 13:164–74. [DOI] [PubMed] [Google Scholar]
- 15. Kaplan MH, Klein SW, McPhee J, Harper RG. Group B coxsackievirus infections in infants younger than three months of age: a serious childhood illness. Rev Infect Dis 1983; 5:1019–32. [DOI] [PubMed] [Google Scholar]
- 16. Modlin JF, Rotbart HA. Group B coxsackie disease in children. The coxsackie B viruses. Curr Top Microbiol Immunol 1997; 223:53–80. [DOI] [PubMed] [Google Scholar]
- 17. Euscher E, Davis J, Holzman I, Nuovo GJ. Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet Gynecol 2001; 98:1019–26. [DOI] [PubMed] [Google Scholar]
- 18. Abzug MJ. Presentation, diagnosis, and management of enterovirus infections in neonates. Pediatr Drugs 2004; 6:1–10. [DOI] [PubMed] [Google Scholar]
- 19. Satosar A, Ramirez NC, Bartholomew D, Davis J, Nuovo GJ. Histological correlates of viral and bacterial infection of the placenta associated with severe morbidity and mortality in the newborn. Hum Pathol 2004; 35:536–45. [DOI] [PubMed] [Google Scholar]
- 20. Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol 2010; 6:279–89. [DOI] [PubMed] [Google Scholar]
- 21. Geenen V, Kroemer G. Multiple ways to cellular immune tolerance. Immunol Today 1993; 14:573–5. [DOI] [PubMed] [Google Scholar]
- 22. Martens H, Goxe B, Geenen V. The thymic repertoire of neuroendocrine self‐antigens: physiological implications in T‐cell life and death. Immunol Today 1996; 17:312–7. [DOI] [PubMed] [Google Scholar]
- 23. Geenen V, Bodart G, Henry S et al Programming of neuroendocrine self in the thymus and its defect in the development of neuroendocrine autoimmunity. Front Neurosci 2013; 7:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jaïdane H, Sané F, Hiar R et al Immunology in the clinic review series; focus on type 1 diabetes and viruses: enterovirus, thymus and type 1 diabetes pathogenesis. Clin Exp Immunol 2012; 168:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nunes‐Alves C, Nobrega C, Behar SM, Correia‐Neves M. Tolerance has its limits: how the thymus copes with infection. Trends Immunol 2013; 34:502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaïdane H, Gharbi J, Lobert PE et al Infection of primary cultures of murine splenic and thymic cells with coxsackievirus B4. Microbiol Immunol 2008; 52:40–6. [DOI] [PubMed] [Google Scholar]
- 27. Brilot F, Chehadeh W, Charlet‐Renard C, Martens H, Geenen V, Hober D. Persistent infection of human thymic epithelial cells by coxsackievirus B4. J Virol 2002; 76:5260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jaïdane H, Caloone D, Lobert PE et al Persistent infection of thymic epithelial cells with coxsackievirus B4 results in decreased expression of type 2 insulin‐like growth factor. J Virol 2012; 86:11151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brilot F, Geenen V, Hober D, Stoddart CA. Coxsackievirus B4 infection of human fetal thymus cells. J Virol 2004; 78:9854–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brilot F, Jaïdane H, Geenen V, Hober D. Coxsackievirus B4 infection of murine foetal thymus organ cultures. J Med Virol 2008; 80:659–66. [DOI] [PubMed] [Google Scholar]
- 31. Jaïdane H, Gharbi J, Lobert PE et al Prolonged viral RNA detection in blood and lymphoid tissues from Coxsackievirus B4 E2 orally inoculated Swiss mice. Microbiol Immunol 2006; 50:971–4. [DOI] [PubMed] [Google Scholar]
- 32. Chatterjee NK, Hou J, Dockstader P, Charbonneau T. Coxsackievirus B4 infection alters thymic, splenic, and peripheral lymphocyte repertoire preceding onset of hyperglycemia in mice. J Med Virol 1992; 38:124–31. [DOI] [PubMed] [Google Scholar]
- 33. Iwasaki T, Monma N, Satodate R, Kawana R, Kurata T. An immunofluorescent study of generalized Coxsackie virus B3 infection in a newborn infant. Acta Pathol Jpn 1985; 35:741–8. [DOI] [PubMed] [Google Scholar]
- 34. Lozovskaia LS, Osipov SM, Zubkova IV, Soboleva VD. Study of vertical transmission of coxsackie group enteroviruses in the etiology of congenital immunodeficiencies. Vopr Virusol 1997; 42:175–9. [PubMed] [Google Scholar]
- 35. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 1987; 162:156–9. [DOI] [PubMed] [Google Scholar]
- 36. Zoll GJ, Melchers WJ, Kopecka H, Jambroes G, Van Der Poel HJ, Lama JM. General primer‐mediated polymerase chain reaction for detection of enteroviruses: application for diagnostic routine and persistent infections. J Clin Microbiol 1992; 30:160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller JFA. Revisiting thymus function. Front Immunol 2014; 5:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lansdown AB. Influence of time of infection during pregnancy with coxsackievirus B3 on maternal pathology and foetal growth in mice. Br J Exp Pathol 1975; 56:119–23. [PMC free article] [PubMed] [Google Scholar]
- 39. Bopegamage S, Precechtelova J, Marosova L et al Outcome of challenge with coxsackievirus B4 in young mice after maternal infection with the same virus during gestation. FEMS Immunol Med Microbiol 2012; 64:184–90. [DOI] [PubMed] [Google Scholar]
- 40. Loria RM, Shadoff N, Kibrick S, Broitman S. Maturation of intestinal defenses against peroral infection with group B coxsackievirus in mice. Infect Immun 1976; 13:1397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnson CM, Papadi GP, Tompkins WA et al Biphasic thymus response by kitten inoculated with feline immunodeficiency virus during fetal development. Vet Pathol 1998; 35:191–201. [DOI] [PubMed] [Google Scholar]
- 42. Feng WH, Tompkins MB, Xu JS et al Thymocyte and peripheral blood T lymphocyte subpopulation changes in piglets following in utero infection with porcine reproductive and respiratory syndrome virus. Virology 2002; 302:363–72. [DOI] [PubMed] [Google Scholar]
- 43. Nathanson N, Gonzalez‐Scarano F. Viral persistence In: Nathanson N, ed. Viral pathogenesis and immunity, 2nd edn San‐Diego, CA: Elsevier, 2007:130–45. [Google Scholar]
- 44. Zipris D, Crow AR, Delovitch TL. Altered thymic and peripheral T‐lymphocyte repertoire preceding onset of diabetes in NOD mice. Diabetes 1991; 40:429–35. [DOI] [PubMed] [Google Scholar]