

Abstract
Transition metal oxides show great promise as Earth-abundant catalysts for the oxygen evolution reaction in electrochemical water splitting. However, progress in the development of highly active oxide nanostructures is hampered by a lack of knowledge of the location and nature of the active sites. Here we show, through atom-resolved scanning tunnelling microscopy, X-ray spectroscopy and computational modelling, how hydroxyls form from water dissociation at under coordinated cobalt edge sites of cobalt oxide nanoislands. Surprisingly, we find that an additional water molecule acts to promote all the elementary steps of the dissociation process and subsequent hydrogen migration, revealing the important assisting role of a water molecule in its own dissociation process on a metal oxide. Inspired by the experimental findings, we theoretically model the oxygen evolution reaction activity of cobalt oxide nanoislands and show that the nanoparticle metal edges also display favourable adsorption energetics for water oxidation under electrochemical conditions.
Earth abundant transition metal oxides show great promise as catalysts for the oxygen evolution reaction. Here, the authors reveal a self-assisted water dissociation mechanism and favourable theoretical adsorption energetics for water oxidation at the edge sites of cobalt oxide nano-islands.
Water is an almost unavoidable element in surface chemistry—in some cases essential, and in others undesirable. The nature of water interactions with metal oxides is a topic of special interest for geochemistry, corrosion, catalysis and electrochemistry1,2,3,4,5. In particular, the chemistry of water on oxide surfaces is fundamental to the operation of electrolyzers that drive the electrochemical splitting of water into hydrogen and oxygen. A particular bottleneck for the development of new renewable energy technologies based on this process is the oxygen evolution reaction (OER)6—one of the electrochemical half reactions for water splitting—where the current catalysts of choice are made from oxides of expensive noble metals, unfeasible for large-scale application. However, recent progress has shown that inexpensive oxides based on 3d transition metals are promising substitutes7,8,9,10,11,12,13, and detailed theoretical predictions are available for many active oxide phases14,15,16. Not all sites of an oxide nanoparticle are equally active, and hence current efforts are being directed towards optimizing activity empirically through nanostructuring17,18,19, or via enhancement by contact synergy with noble metals such as Au8,20. In particular, exfoliated layered double hydroxides have emerged as potential Earth-abundant catalysts for OER, especially cobalt oxy-hydroxide, β-CoOOH (refs 21, 22, 23), and analogous compounds from Ni and Fe. The β-CoOOH active phase adopts a two-dimensional sheet structure in which octahedral Co is sandwiched between O layers. This structure was recently suggested through indirect evidence to host the active sites for the OER on the edges of the β-CoOOH layers19. Other studies have considered basal plane O vacancies and under coordinated Co atoms as the active sites24. However, the precise nature and location of the active sites is still under debate.
In this article, we investigate the mechanism of hydroxylation resulting from water dissociation on cobalt oxide nanoislands synthesized on a single gold (111) crystal25, exposing both basal plane and edge sites on the island perimeter, which structurally resemble those in a β-CoOOH layer. We find that the only sites capable of dissociating H2O in our experiments are located on the particle edges. These results highlight a special nature and reactivity of edge sites and propose that such minority sites should be payed special attention and considered in the efforts to further understand the detailed OER pathway on oxides. Inspired by our experimental study that emphasize significant differences between edge and basal plane sites, we extend the studies by means of density functional theory (DFT) calculations on the adsorption energetics of OER intermediates to address the potential of layered cobalt oxide structures as OER catalysts. The calculations reveal very favourable properties and a low theoretical overpotential for cobalt terminated edges, that is, the same sites also responsible for the observed water dissociation. Our theoretical modelling of the process observed in the experiment furthermore indicates that all the steps in the dissociation process, as well as in the following hydroxyl migration, are assisted by an additional water molecule, acting to lower the energy barriers involved.
Results
Hydroxylation of CoO nanoislands
The majority (95%) of the cobalt oxide nanoislands synthesized on Au(111) are single Co–O bilayers that coexist with a minority (5%) of double Co–O bilayers, both oriented with a pristine close-packed O-terminated (111) basal plane parallel to the substrate with a cobalt layer in direct contact with the gold support (Fig. 1). These structures and their relative abundance remain essentially unchanged from low coverage (∼10%) to the monolayer limit (∼95%). The predominant hexagonal shape of the nanoislands dictates two different low-index edge structures terminating the islands, one exposing a row of cobalt atoms (Co edge), and the other a row of oxygen atoms (O edge) (Fig. 1b). The edge structure of the double bilayers, which adopts the wurtzite structure, is more complicated and expresses both of the cobalt and oxygen terminations.
Figure 1. Cobalt oxide nanoislands on a Au(111) surface.

(a) Representative STM image showing a 30% ML synthesis of CoO nanoislands. Single and double bilayers are labelled. (b) Ball structure model showing a pristine hexagonal single bilayer island expressing both cobalt and oxygen edges. Scale bar: (a) 100 Å.
To study water dissociation, we exposed the cobalt oxide nanoislands to water vapour (p(H2O)=2 × 10−7 mbar) at room temperature and investigated the surface with X-ray photoelectron spectroscopy (XPS). Figure 2a shows the O 1s XPS spectrum of the islands as a function of water exposure measured in Langmuir (denoted by the unit L, where 1 L=1 × 10−6 torr for 1second). The O 1s spectrum radically changes as a function of exposure. This can be monitored in more detail by deconvolution of the spectra into individual peaks, corresponding to distinct oxygen species. We find that three peaks are required to produce a satisfactory fit for the entire series. The low binding energy (BE) peak is predominant in the spectrum recorded for pristine nanoislands and represents lattice oxygen (Olat) within the nanoislands. The higher binding energy peak at 532.0 eV, on the other hand, increases significantly with water exposure and can be assigned to hydroxyl groups (OOH)2. This hydroxylation results from water dissociation on the nanoislands and causes a corresponding drop in Olat as a function of water exposure, indicating that lattice oxygen atoms are hydroxylated in the process (Fig. 2b). At high water exposures (>600 l), no further change is observed, thus implying that saturation hydroxylation has been reached with a ratio of Olat to OOH of 1:1. We ascribe the weak intermediate energy peak to buried lattice oxygen present in the double bilayer islands (Odbl). The peak's intensity does not show dramatic changes throughout the experiment and the Odbl do not appear accessible for hydroxylation under our experimental conditions.
Figure 2. Hydroxylation of CoO nanoislands monitored by XPS.

(a,b) XPS spectra (hν=625 eV) and individual peak areas of the O 1s core level during water exposure at 2 × 10−7 mbar. (c) Ball model showing the individual oxygen species OOH and Olat.
The progress of hydroxylation of the islands was investigated in parallel in atom-resolved scanning tunnelling microscopy (STM)26 movies by dosing water into the chamber in situ during continuous STM scanning at room temperature (Supplementary Movie 1 (single bilayer) and 2 (double bilayer)). Point-like features start to appear on the basal planes of both island types (Fig. 3a,b) with a density directly correlated with water exposure. On both structures they occupy Olat positions and are assigned to hydroxylated oxygen atoms, that is, H adsorbed on Olat sites. STM shows that hydroxyl groups are present in low numbers on even pristine islands due to residual water vapour in the rest gas, which is in agreement with the small tail seen in the XPS data at 0 l exposure (Fig. 2a). Hydroxyl groups on single bilayer islands usually appear as faint protrusions (Fig. 3a, inset i), while appearing as ‘donut'-shaped depressions on double bilayer islands (Fig. 3b, inset ii). A variety of other imaging modes are also possible (Supplementary Fig. 1) since the STM contrast on oxides is in general highly sensitive to the STM tip termination27.
Figure 3. In situ STM movies and saturation level of hydroxylation.

(a,b) Image sequences from atom-resolved STM movie (supplementary) recorded during water exposure showing the initial stage of hydroxylation on single- (a) and double- (b) bilayer CoO nanoislands. The insets show high magnification atom-resolved STM images of hydroxyl groups on a single- (i) and double-bilayer (ii). (c) STM image of a heavily hydroxylated single bilayer island with areas of ordered 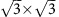 R30° superstructure. (d) DFT-calculated hydroxylation structure of the CoO basal plane as a function of H chemical potential and pressure at room temperature (upper scale). Scale bars: (a) 50 Å (inset 12 Å), (b) 50 Å (inset 6 Å), (c) 50 Å.
R30° superstructure. (d) DFT-calculated hydroxylation structure of the CoO basal plane as a function of H chemical potential and pressure at room temperature (upper scale). Scale bars: (a) 50 Å (inset 12 Å), (b) 50 Å (inset 6 Å), (c) 50 Å.
STM images recorded on syntheses exposed to high water dosages further than that shown in the STM movies (>1,000 l) display the emergence of an ordered 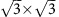 R30° superstructure (with respect to the Olat positions) of hydroxyls (Fig. 3c). In agreement, our density functional theory calculations within the Hubbard U approximation (DFT+U) show that the expected optimal OH coverage (θOH, defined as the ratio of hydroxyls and number of basal plane O atoms) under the experimental conditions for the defect-free basal plane is θOH=0.33, reflected by such a
R30° superstructure (with respect to the Olat positions) of hydroxyls (Fig. 3c). In agreement, our density functional theory calculations within the Hubbard U approximation (DFT+U) show that the expected optimal OH coverage (θOH, defined as the ratio of hydroxyls and number of basal plane O atoms) under the experimental conditions for the defect-free basal plane is θOH=0.33, reflected by such a 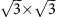 R30° overlayer structure of the adsorbed H (Fig. 3d). However, coexisting with the
R30° overlayer structure of the adsorbed H (Fig. 3d). However, coexisting with the 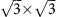 R30° superstructure are linear chains of hydroxyls which interconnect between domains, meaning the overall hydroxylation measured is closer to θOH≈0.5, as observed by XPS measurements (Fig. 2b). This is in contrast to the similar system of supported thin films of FeO, for which our calculations find an optimal θOH of 6% when supported on Au (Supplementary Fig. 2), consistent with previous experimental observations28. The tendency to form the
R30° superstructure are linear chains of hydroxyls which interconnect between domains, meaning the overall hydroxylation measured is closer to θOH≈0.5, as observed by XPS measurements (Fig. 2b). This is in contrast to the similar system of supported thin films of FeO, for which our calculations find an optimal θOH of 6% when supported on Au (Supplementary Fig. 2), consistent with previous experimental observations28. The tendency to form the 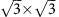 R30° superstructure is indicative of hydroxyl–hydroxyl repulsion, a phenomenon that has been also observed on other oxide surfaces1,29.
R30° superstructure is indicative of hydroxyl–hydroxyl repulsion, a phenomenon that has been also observed on other oxide surfaces1,29.
Reaction mechanism of water dissociation on CoO edge sites
The atom-resolved STM movies also reveal that the new hydroxyls originate from the edges of the islands before they diffuse to the basal plane (compare for example, panels 1 and 2 of Fig. 3b). This clearly suggests that island edges are the active sites for water dissociation and that hydroxyl groups diffuse from these sites to the centre of the island. To prove this, three different syntheses were made with different edge/basal plane ratios by varying the cobalt oxide island coverage from ∼10% (yielding small islands) to an almost complete monolayer CoO film (Fig. 4). These syntheses were exposed to the same total dosage of 200 l water, which is high enough to cause measureable hydroxylation in atom-resolved STM, but far from saturation. The surface hydroxyl coverage θOH was measured and compared with the effective island radius (reff, proportional to the inverse of edge to basal plane ratio) for a number of islands in STM images in each synthesis. The hydroxyl density θOH follows exactly a trend inversely proportional to the radius, in agreement with edge activity, as dissociation on the basal plane sites would result in a constant θOH independent of r.
Figure 4. Relationship between edge length and activity.

(a) STM images from a coverage series varying the amount of deposited and oxidized cobalt. Insets (100 × 100 Å2) show sample areas after exposure to 200 l H2O. (b) Effective island radius versus degree of hydroxylation after 200 l H2O estimated from atom-resolved STM images for different sized nanoislands showing an inversely proportional relationship. Note that the degree of hydroxylation was kept far below saturation (50%) to facilitate the image acquisition and counting of individual hydroxyls. The error bars indicate the s.d. on individual measurements of the hydroxylation and effective radius. Scale bars: (a) 75 Å.
Our DFT+U simulations on a series of systems consisting of two-dimensional periodic cobalt oxide stripes on a Au(111) surface (see Methods section30,31) reveal a favourable reaction pathway for water dissociation on the edges. Although these modelled stripes expose both the O-terminated basal plane together with Co- and O-terminated edges on opposite sides (Fig. 1b), no favourable reactions were found on the O edge, nor on the O-terminated basal plane of island. The lowest energy diagram obtained for the dissociation of a single water molecule on the Co edge is presented in Fig. 5a. This process starts with the adsorption of the water molecule to a Co-site in an on-top configuration (1) followed by the first H-dissociation, in which one hydrogen atom is transferred to the nearest neighbour oxygen on the basal plane. For this process, we find a relatively high activation barrier (Ea=1.01 eV), which is at first inspection inconsistent with the reactivity observed in the experiments at room temperature. Therefore, taking into account the water vapour background during the hydroxylation process, this step was modelled again in the presence of another water molecule in the vicinity. The influence of the additional water molecule is surprisingly strong, finding a significantly lower barrier of 0.41 eV. This demonstrates that the additional water assists water dissociation on the exposed Co sites by acting as a proton shuttle in form of a H3O species in the transition state (Fig. 5b). Such process resembles the autocatalytic water dissociation reported for bare metal surfaces32,33 and is the direct evidence that a water assisted dissociation process may proceed at under coordinated sites on a metal oxide.
Figure 5. Comparison of water assisted and non-assisted water dissociation mechanisms.

(a) Reaction intermediates are: (1) water adsorption, (2) first H dissociation onto a basal plane oxygen, (3) hopping of the first proton to the basal plane. Note the H ends up on the O-edge due to its close vicinity. (4) Second hydrogen dissociation onto a basal plane oxygen, and (5) hopping of the second proton. (b) Assisted- and non-assisted reaction transition states in 1st H dissociation and H hopping. Colour code Co: light red, O: dark red, Au: yellow.
The hydrogen originating from the dissociated water in configuration (2) subsequently diffuses away from the Co-metal edge to energetically preferable configurations on the basal plane O lattice of the cobalt oxide stripe as in experiment (and eventually onto the nearby O edge in the calculation (3)). Again, we find that the associated hydrogen hopping is much more efficient when it is assisted by water (Ea=0.23 eV), as opposed to the non-assisted hopping (Ea=1.06 eV), similar to a previous study on H diffusion a FeO thin film34. The fundamental effect of the reduction in the reaction barrier relates to the formation of H3O species in the transition state formed from the water and the hydrogen on the surface. Upon dissociation of the H3O species, the excess H adsorbs again on the surface with equal probability on all three neighbouring O sites leading to overall diffusion of H across the surface. In the STM movies presented in Fig. 3a,b, the hydrogen mobility is clearly evident due to the background water pressure during the acquisition of both movies. Contrary, at the base UHV pressure (10−10 mbar), hydroxyls are almost completely static, as expected from the calculations. Moreover, the mobility was directly shown to strongly correlate with water vapour pressure by recording an STM movie similar to Fig. 3a,b, but varying the water pressure in situ (Supplementary Fig. 3).
The second dehydrogenation of the OH intermediate on the Co edge (steps 3–4 in Fig. 5) is slightly higher in energy (Ea=0.57 eV) compared with the first H dissociation step, but again strongly water assisted, following the same mechanism. The water dissociation process is −0.54 eV downhill in total energy at 0 K, while energy neutral at typical conditions of the experiment. Overall, the calculations indicate that water plays a far greater role than previously thought, not only assisting hydrogen hopping, but also mediating all the elementary steps of its own dissociation.
The final state of the water dissociation process results in O adsorbed on the Co edge with H atoms spatially separated from the edge onto positions on the basal plane, as observed in the STM experiments. Further DFT+U simulations reveal that dissociation of additional water is not likely at room temperature when all Co edge sites are O-covered (Supplementary Figs 4 and 5), resulting in a maximum theoretical hydroxyl density of θOH=2nedge/nbasal, where nedge and nbasal are the number of sites on the island edge and basal plane, respectively. In a typical 30% ML synthesis (Fig. 1a), the measured ratio of edge atoms to basal plane atoms (nedge/nbasal) is between 0.10 and 0.15 giving an expected maximum hydroxylation of θOH=0.20–0.30. Surprisingly, we always observe a significantly higher hydroxylation of θOH ≈ 0.5 in both XPS and STM at saturation (Figs 2b and 3c). This excess hydroxylation in violation of the limit set by the available edge sites may be due to a number of contributing factors. Firstly, the ratio of edge atoms to basal plane atoms for a given island is calculated assuming perfect edges containing no kinks or single atom vacancies. However, such features can be observed at many island edges (Fig. 1a). Secondly, STM movies recorded during water exposure reveal that edge structures are highly dynamic upon water exposure with constant restructuring occurring (Supplementary Fig. 6). This suggests that fresh Co sites might be exposed dynamically even at room temperature. We rule out that such processes could be induced by the vicinity of the tip since independent XPS data shows the same degree of saturation. Therefore, we propose that dynamic rearrangements of the CoO edge structure might be generally occurring at room temperature and important for the dissociative properties of the islands.
Our experimental results of the model system directly reveal that exposed Co atoms act to dissociate water on the cobalt oxide nanoparticle edges, resulting in a hydroxylation of the oxide. Inspired by these findings based on studies under vacuum conditions, we have further investigated the energetics for electrochemical water oxidation on the cobalt oxide particle edges versus basal plane sites by means of DFT, addressing the existing hypothesis that the activity of layered cobalt oxyhydroxide nanocatalysts (strongly resembling our model structure) is related to the edge length19.
The conventional OER mechanism in CoOx and NiOx catalysts in alkaline media is based on a single-site OH−/e− exchange mechanism16,35,36,37,38, although alternative mechanisms involving superoxo-type species have been also proposed7,35,39. Regardless of the type of mechanism, specific sites with favourable energetics for the O and OH intermediates need to exist for efficient OER to occur. We have modelled the reactivity under OER conditions of all the possible sites in gold supported Co–O and the related O–Co–O trilayer islands22 using the conventional OER mechanism (Table 1). For a better comparison with previous OER studies on Co-oxides16,36, these energies have been calculated at the PBE+U (U=3.32 eV) level. We find that all of these steps require the interaction of the OER intermediates (that is, OH*, O* and OOH*) at the under coordinated Co site at edges of the bilayer and trilayer nanoislands. Additionally, we have calculated the formation energies of vacancies on the fully O saturated basal plane, O edge and Co edge sites. Here, the O sites at the basal plane stand out as the sites with the highest oxygen vacancy formation energy (Supplementary Fig. 4). This finding is in agreement with the experiment, where we never observe O vacancies on the basal plane. Furthermore, the calculated energetics of the OH* and O* intermediates at the basal plane of both bilayers and trilayers are clearly outside the optimum range defined by a theoretical overpotential below 1 V (refs 14, 16, 37, 38). Therefore, calculations predict a very unfavourable OER activity for these sites (Table 1). This leaves the exposed Co edges as the only sites likely to dissociate water and act as OER active sites, with a calculated overpotential of only 330 mV for the O–Co–O trilayers.
Table 1. Calculated OER energetics at the available basal plane sites and edge sites of the bilayer (Co–O) and trilayer (O–Co–O) nanostripes.
| Specific site/OER energetics | ΔGOH*(eV) | ΔGO*(eV) | ΔGOOH*(eV) | Calculated overpotential ηcal (V) |
|---|---|---|---|---|
| OER active range (min, max) | (−0.5, 2.2) | (1.7, 4.4) | (2.7, 5.4) | <1 |
| Basal plane OER energetics | ||||
| Bilayers: OH-site on basal plane | −0.57 | 0.05 | NA | >1 (ads. too strong) |
| Bilayers: O-site on basal plane | −0.48 | −0.02 | NA | >1 (ads. too strong) |
| Trilayers: OH-site on basal plane | −1.09 | 0.06 | NA | >1 (ads. too strong) |
| Trilayers: O-site on basal plane | −1.39 | −0.77 | NA | >1 (ads. too strong) |
| Edge-site OER energetics | ||||
| Bilayers: top of the metal-edge | 1.37 | 3.03 | 4.53 | 0.43 |
| Trilayers: bottom of O-edge near Au | 0.17 | 1.99 | 3.31 | 0.60 |
| Trilayers: top of the metal-edge | 1.51 | 3.03 | 4.60 | 0.33 (optimal) |
The water dissociation, as studied in our experimental work, may not be the overall rate-limiting step in the OER mechanism, and in alkaline media the OER pathway does not necessarily involve the dissociation of water since OH is readily available from the solution. Furthermore, under biased electrochemical conditions the possibility exists that the dissociation at edge sites occurs more facile elsewhere on the cobalt oxide due to changes in the chemical state of the cobalt. However, our experimental study shows that the interactions between oxide and water on edges are remarkably different from basal plane sites for simple reactions such as the dissociation process. We propose that these sites might in general play a significant role in the water oxidation pathway, as well as other reactions. In addition, our calculations applicable to biased electrochemical conditions (using trilayer islands), again pinpoint the edges as the preferable active site for the OER, and thus support the recent indirect evidence that β-CoOOH layers host the active sites for the OER on the edges19. To confirm these findings, the well-defined number and controlled variation of edge sites in the model catalyst may be used to quantify edge reactivity in a working model electrode setup under electrochemical conditions. Lastly, the microscopic picture of water dissociation and water-assisted hydrogen mobility on cobalt oxide revealed in this work is also important for the general understanding and further modelling of the emerging class of Earth-abundant metal oxide catalysts for the OER, as well as other relevant catalytic reactions in aqueous media.
Methods
Experimental methods
All syntheses and analysis was performed in UHV chambers with a base pressure of ∼1 × 10−10 mbar. Cobalt oxide nanoislands were synthesized on a clean and flat Au(111) surface using the synthesis described in (ref. 25). Briefly, this consists of evaporation of cobalt from an e-beam evaporator in an oxygen environment (1 × 10−6 mbar O2) and a post-annealing step at 523 K in the same environment and a final brief anneal at 523 K in UHV conditions. Milli-Q grade water was dosed from a glass vial into the UHV chamber via a leak valve. The water was first degassed by repeated freeze–pump–thaw cycles. The resultant islands were imaged using an ‘Aarhus'-type STM and XPS was done using the Matline endstation on the ASTRID II synchrotron, Aarhus (operating in ‘top up' mode). O 1s spectra were recorded at normal emission and a photon energy of 625 eV. The spectra were energy calibrated using the Au 4d 5/2 peak at a binding energy of 335.0 eV as a reference. There was no X-ray beam present on the sample during the water dosing.
Computational methods
DFT calculations were performed using the projector augmented wave (PAW) potentials40 within the Vienna ab initio simulation package41,42 VASP, version 5.3.5). As in the study of CoO on Ir(100)43, FeO on Pt(111)1,2, and also in our recent study of CoO nanoislands25, we adapted the PBE44 functional together with the Hubbard-U method45 with a value of Ueff =1 eV applied for the d-electrons of Co atoms. A standard energy cutoff of 400 eV and an approximately constant k-point mesh of 8 × 8 × 1 points per p(1 × 1) unit cell was kept in all slab calculations. In all cases, a metal-oxide bilayer and the upper most top layer of Au(111) were allowed to relax with the maximum force threshold of 0.02 eV Å−1, while at least 11 Å of vacuum was used to separate the layers. The magnetic structure was always taken to be the row-wise AFM structure, RW-AF-(2 × 1), known from FeO bilayers46.
The hydroxylation of basal planes was studied using infinite surfaces of periodicities ranging from p(1 × 1), p(✓3 × ✓3), p(2 × 2), p(3 × 3) to p(4 × 4) with the metal-oxide layer placed in a FCC stacking relative to a commensurate Au(111) lattice (Supplementary Fig. 2). One exception from this rule was the case of p(✓3 × ✓3) metal-oxide, where we also adopted incommensurate p(✓3 × ✓3)/Au(2 × 2) structures known from a previous work on FeO on Pt(111)47. To study the energetics of the edges of the nanoislands, we used stripes of periodicity p(2 × 4) CoO FCC stacked within the p(2 × 8) unit cell of Au(111), referred to as p(2 × 4/2 × 8). Size effects were also tested for some reaction intermediates using two times wider p(4 × 4/4 × 8) stripes leading to very similar results. The transition states for water dissociation and hydrogen hopping were located by means of the climbing image nudged elastic band (CI-NEB) method, using at least five images along the reaction coordinate45. The nature of the transition states was assessed by performing numerical frequency analysis to confirm the existence of only one imaginary vibrational mode.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Additional information
How to cite this article: Fester, J. et al. Edge reactivity and water-assisted dissociation on cobalt oxide nanoislands. Nat. Commun. 8, 14169 doi: 10.1038/ncomms14169 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Supplementary Figures
"STM movie (155Åx135Å, I=-0.28nA and V=-911.2mV) of a single bilayer CoO island during in-situ exposure to 2×10-7 mbar H2O at room temperature. Time difference between single images: 82.02s."
STM movie (150Åx120Å, I=-0.26nA and V=-806.0mV) of a double bilayer CoO island during in-situ exposure to 5×10-8 mbar H2O at room temperature. Time difference between single images: 36.7s."
Acknowledgments
The iNANO group gratefully acknowledges support from the Lundbeck Foundation and Villum Foundation. J.V.L. gratefully acknowledges financial support from the European Research Council (ERC starting grant no. 239834). We also acknowledge beamtime on the AU-Matline of the ASTRID2 synchrotron, Aarhus, Denmark. A.V., M.B. and M.G-M. gratefully acknowledge support from the U.S. Department of Energy Office of Basic Energy Science to the SUNCAT Center for Interface Science and Catalysis. A.V. acknowledges the support from the SLAC National Accelerator Lab LDRD program. M.G-M. acknowledges funding from the Agency for Administration of University and Research Grants of Catalonia (AGAUR, 2013 BP-A 00464). This research employed NERSC computational resources under DOE Contract No DE-AC02-05CH11231. We also gratefully acknowledge J. Knudsen for help with the XPS fitting.
Footnotes
The authors declare no competing financial interests.
Author contributions J.F. and A.S.W. performed the experiments and data analysis. M.G-M. and M.B. performed the calculations. Z.L. and L.L. helped with synchrotron and STM experiments. A.S.W. wrote the first draft and all authors contributed to the final version. A.V. and J.V.L. organized and designed the study.
References
- Merte L. R. et al. Water clustering on nanostructured iron oxide films. Nat. Commun. 5, 4193 (2014). [DOI] [PubMed] [Google Scholar]
- Henderson M. A. The interaction of water with solid surfaces: fundamental aspects revisited. Surf. Sci. Rep. 46, 1–308 (2002). [Google Scholar]
- Meyer B. et al. Partial dissociation of water leads to stable superstructures on the surface of zinc oxide. Angew. Chem. Int. Ed. Engl. 43, 6641–6645 (2004). [DOI] [PubMed] [Google Scholar]
- Bikondoa O. et al. Direct visualization of defect-mediated dissociation of water on TiO2 (110). Nat. Mater. 5, 189–192 (2006). [Google Scholar]
- Saavedra J., Doan H. A., Pursell C. J., Grabow L. C. & Chandler B. D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 345, 1599–1602 (2014). [DOI] [PubMed] [Google Scholar]
- Knudsen J. et al. Reduction of FeO/Pt (111) thin films by exposure to atomic hydrogen. Surf. Sci. 604, 11–20 (2010). [Google Scholar]
- Zhang M., de Respinis M. & Frei H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat. Chem. 6, 362–367 (2014). [DOI] [PubMed] [Google Scholar]
- Subbaraman R. et al. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 11, 550–557 (2012). [DOI] [PubMed] [Google Scholar]
- Liao L. et al. Efficient solar water-splitting using a nanocrystalline CoO photocatalyst. Nat. Nanotechnol. 9, 69–73 (2014). [DOI] [PubMed] [Google Scholar]
- Suntivich J., May K. J., Gasteiger H. A., Goodenough J. B. & Shao-Horn Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 334, 1383–1385 (2011). [DOI] [PubMed] [Google Scholar]
- Chou N. H., Ross P. N., Bell A. T. & Tilley T. D. Comparison of cobalt-based nanoparticles as electrocatalysts for water oxidation. ChemSusChem 4, 1566–1569 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang B. et al. Homogeneously dispersed multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016). [DOI] [PubMed] [Google Scholar]
- Ng J. W. D. et al. Gold-supported cerium-doped NiOx catalysts for water oxidation. Nat. Energy 1, 16053 (2016). [Google Scholar]
- Man I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011). [Google Scholar]
- Halck N. B., Petrykin V., Krtil P. & Rossmeisl J. Beyond the volcano limitations in electrocatalysis–oxygen evolution reaction. Phys. Chem. Chem. Phys. 16, 13682–13688 (2014). [DOI] [PubMed] [Google Scholar]
- Bajdich M., García-Mota M. n., Vojvodic A., Nørskov J. K. & Bell A. T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521–13530 (2013). [DOI] [PubMed] [Google Scholar]
- Cheng F. et al. Rapid room-temperature synthesis of nanocrystalline spinels as oxygen reduction and evolution electrocatalysts. Nat. Chem. 3, 79–84 (2011). [DOI] [PubMed] [Google Scholar]
- Song F. & Hu X. Ultrathin cobalt–manganese layered double hydroxide is an efficient oxygen evolution catalyst. J. Am. Chem. Soc. 136, 16481–16484 (2014). [DOI] [PubMed] [Google Scholar]
- Song F. & Hu X. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 5, 4477 (2014). [DOI] [PubMed] [Google Scholar]
- Seitz L. C., Hersbach T. J. P., Nordlund D. & Jaramillo T. F. Enhancement effect of noble metals on manganese oxide for the oxygen evolution reaction. J. Phys. Chem. Lett. 6, 4178–4183 (2015). [DOI] [PubMed] [Google Scholar]
- Yeo B. S. & Bell A. T. Enhanced activity of gold-supported cobalt oxide for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 133, 5587–5593 (2011). [DOI] [PubMed] [Google Scholar]
- Friebel D. et al. On the chemical state of Co oxide electrocatalysts during alkaline water splitting. Phys. Chem. Chem. Phys. 15, 17460–17467 (2013). [DOI] [PubMed] [Google Scholar]
- Trotochaud L., Ranney J. K., Williams K. N. & Boettcher S. W. Solution-cast metal oxide thin film electrocatalysts for oxygen evolution. J. Am. Chem. Soc. 134, 17253–17261 (2012). [DOI] [PubMed] [Google Scholar]
- Huang J. et al. CoOOH nanosheets with high mass activity for water oxidation. Angew. Chem. Int. Ed. Engl. 127, 8846–8851 (2015). [DOI] [PubMed] [Google Scholar]
- Walton A. S. et al. Interface controlled oxidation states in layered cobalt oxide nanoislands on gold. ACS Nano 9, 2445–2453 (2015). [DOI] [PubMed] [Google Scholar]
- Heimann P., Van der Veen J. & Eastman D. Structure-dependent surface core level shifts for the Au (111), (100) and (110) surfaces. Solid State Commun. 38, 595–598 (1981). [Google Scholar]
- Mönig H. et al. Understanding scanning tunneling microscopy contrast mechanisms on metal oxides: a case study. ACS Nano 7, 10233–10244 (2013). [DOI] [PubMed] [Google Scholar]
- Wang W. et al. Observation of water dissociation on nanometer-sized FeO islands grown on Pt (111). Chem. Phys. Lett. 500, 76–81 (2010). [Google Scholar]
- Wu C., Castell M. R., Goniakowski J. & Noguera C. Stoichiometry engineering of ternary oxide ultrathin films: BaxTi2O3 on Au (111). Phys. Rev. B 91, 155424 (2015). [Google Scholar]
- Gu X. K., Ouyang R., Sun D., Su H. Y. & Li W. X. CO oxidation at the perimeters of an FeO/Pt (111) interface and how water promotes the activity: a first-principles study. ChemSusChem 5, 871–878 (2012). [DOI] [PubMed] [Google Scholar]
- Kudernatsch W. et al. Direct visualization of catalytically active sites at the FeO–Pt (111) interface. ACS Nano 9, 7804–7814 (2015). [DOI] [PubMed] [Google Scholar]
- Andersson K. et al. Autocatalytic water dissociation on Cu(110) at near ambient conditions. J. Am. Chem. Soc. 130, 2793–2797 (2008). [DOI] [PubMed] [Google Scholar]
- Michaelides A., Alavi A. & King D. A. Different surface chemistries of water on Ru{0001}: from monomer adsorption to partially dissociated bilayers. J. Am. Chem. Soc. 125, 2746–2755 (2003). [DOI] [PubMed] [Google Scholar]
- Merte L. R. et al. Water-mediated proton hopping on an iron oxide surface. Science 336, 889–893 (2012). [DOI] [PubMed] [Google Scholar]
- Diaz-Morales O., Ferrus-Suspedra D. & Koper M. T. The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation. Chem. Sci. 7, 2639–2645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Maxisch T. & Ceder G. Oxidation energies of transition metal oxides within the GGA+ U framework. Phys. Rev. B 73, 195107 (2006). [Google Scholar]
- Rossmeisl J., Qu Z.-W., Zhu H., Kroes G.-J. & Nørskov J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89 (2007). [Google Scholar]
- García-Mota M. N. et al. Importance of correlation in determining electrocatalytic oxygen evolution activity on cobalt oxides. J. Phys. Chem. C 116, 21077–21082 (2012). [Google Scholar]
- García-Melchor M., Vilella L. & Vojvodic A. Computationally probing the performance of hybrid, heterogeneous, and homogeneous iridium-based catalysts for water oxidation. ChemCatChem 8, 1792–1798 (2016). [Google Scholar]
- Kresse G. & Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999). [Google Scholar]
- Kresse G. & Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558 (1993). [DOI] [PubMed] [Google Scholar]
- Tröppner C. et al. Incommensurate moiré overlayer with strong local binding: CoO (111) bilayer on Ir (100). Phys. Rev. B 86, 235407 (2012). [Google Scholar]
- Perdew J. P., Burke K. & Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996). [DOI] [PubMed] [Google Scholar]
- Dudarev S., Botton G., Savrasov S., Humphreys C. & Sutton A. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505 (1998). [Google Scholar]
- Giordano L. et al. Interplay between structural, magnetic, and electronic properties in a FeO/Pt (111) ultrathin film. Phys. Rev. B 76, 075416 (2007). [Google Scholar]
- Giordano L. et al. Oxygen-induced transformations of an FeO (111) film on Pt (111): a combined DFT and STM study. J. Phys. Chem. C 114, 21504–21509 (2010). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
"STM movie (155Åx135Å, I=-0.28nA and V=-911.2mV) of a single bilayer CoO island during in-situ exposure to 2×10-7 mbar H2O at room temperature. Time difference between single images: 82.02s."
STM movie (150Åx120Å, I=-0.26nA and V=-806.0mV) of a double bilayer CoO island during in-situ exposure to 5×10-8 mbar H2O at room temperature. Time difference between single images: 36.7s."
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.