

Summary
This study aimed to examine whether acute intermittent porphyria (AIP) is associated with systemic inflammation and whether the inflammation correlates with disease activity. A case–control study with 50 AIP cases and age‐, sex‐ and place of residence‐matched controls was performed. Plasma cytokines, insulin and C‐peptide were analysed after an overnight fast using multiplex assay. Long pentraxin‐3 (PTX3) and complement activation products (C3bc and TCC) were analysed using enzyme‐linked immunosorbent assay (ELISA). Urine porphobilinogen ratio (U‐PBG, µmol/mmol creatinine), haematological and biochemical tests were performed using routine methods. Questionnaires were used to register AIP symptoms, medication and other diseases. All 27 cytokines, chemokines and growth factors investigated were increased significantly in symptomatic AIP cases compared with controls (P < 0·0004). Hierarchical cluster analyses revealed a cluster with high visfatin levels and several highly expressed cytokines including interleukin (IL)‐17, suggesting a T helper type 17 (Th17) inflammatory response in a group of AIP cases. C3bc (P = 0·002) and serum immunoglobulin (Ig)G levels (P = 0·03) were increased significantly in cases with AIP. The U‐PBG ratio correlated positively with PTX3 (r = 0·38, P = 0·006), and with terminal complement complex (TCC) levels (r = 0·33, P = 0·02). PTX3 was a significant predictor of the biochemical disease activity marker U‐PBG in AIP cases after adjustment for potential confounders in multiple linear regression analyses (P = 0·032). Prealbumin, C‐peptide, insulin and kidney function were all decreased in the symptomatic AIP cases, but not in the asymptomatic cases. These results indicate that AIP is associated with systemic inflammation. Decreased C‐peptide levels in symptomatic AIP cases indicate that reduced insulin release is associated with enhanced disease activity and reduced kidney function.
Keywords: chemokines, complement, cytokines, human, inflammation
Introduction
Acute intermittent porphyria (AIP) is an autosomal dominant metabolic disorder caused by a mutation in the hydroxymethylbilane synthase gene, which encodes an enzyme involved in haem synthesis 1. A rare condition worldwide, with a prevalence in European populations of only 1/75 000 to 10–20/100 000 2, 3, AIP is prevalent in Nordland county in Norway, with a prevalence as high as 600/100 000 in one municipality 4. Many patients have only one porphyria attack over a lifetime, and few suffer chronic AIP symptoms. Individuals carrying the AIP gene mutation are classified as symptomatic (15–20%) or asymptomatic (80–85%) 1. Symptomatic AIP presents typically with acute, severe abdominal pain, nausea, vomiting, constipation, dark red urine and muscle weakness 1. Most attacks last from a few days up to 2 weeks. Bulbar or phrenic nerve paresis may ensue occasionally and lead to life‐threatening respiratory failure 1, 5. A high prevalence of hypertension and kidney impairment has been documented 3. Some mutations, including the W198X mutation, carry an increased risk of hepatocellular carcinoma (HCC), which is found in approximately one‐third of patients with this mutation 3, 6.
Established triggers for AIP attack include infections, alcohol, smoking, hormonal factors, physical or mental stress and medications affecting haem synthesis 1, 2. In terms of the mechanisms whereby these triggers cause illness, fasting, fever and stress are known to induce haem oxygenase‐1 activity, leading to a reduction in the free haem pool and induction of 5‐aminolevulinic synthase‐1 (ALAS1) 1. Acute phase proteins and interleukin (IL)‐6 may also increase ALAS1 synthetase activity in liver cells 7. ALAS1 is increased in the liver in AIP patients, due probably to intracellular haem deficiency 8. Some patients with AIP, however, may suffer severe forms of the disease without any known triggers. Treatment for this group is demanding, and requires the avoidance or treatment of triggering factors, a high oral sugar intake, glucose and insulin infusion or infusion of haem. In these patients, ALAS1 mRNA in the liver is decreased by insulin through the peroxisome proliferator‐activated receptor co‐activator 1α 9. The fact that liver transplantation is an efficient cure for AIP strengthens the evidence that the role of liver metabolism in AIP pathogenesis is central 10. In addition, inflammation may possibly affect haem synthesis through hepcidin 11.
Low‐grade inflammation has been reported to play a role in several systemic diseases, such as atherosclerosis and type II diabetes 12, 13. AIP could therefore be associated with low‐grade inflammation. The accumulation of certain damage‐associated molecular patterns (DAMPs) has been shown to stimulate the immune cells and inflammation in various diseases 14. Haem and porphyrins have been reported to activate the complement system 15, 16. Sato et al. showed that dietary supplementation with 5‐aminolevulinic acid (ALA) induced T cell responses in chickens through oxidative stress 17. Delaby et al. reported lower prealbumin levels in patients with symptomatic AIP, probably as a result of malnutrition or hepatic inflammation 18.
Although there is evidence for a possible role of inflammation in AIP, it has not been determined whether patients with AIP have increased systemic inflammation. The aims of this study were to examine systemic inflammation in AIP by plasma biomarkers and biochemical disease activity in the form of urine porphyrin precursor levels, and to determine whether inflammatory markers are associated with biochemical disease activity, insulin release and kidney function.
Materials and methods
Study design and participants
A case–control study was conducted in 50 individuals with AIP, 15 asymptomatic and 35 symptomatic carriers of the AIP gene mutation and 50 controls matched for age, sex and place of residence (Table 1). Of the 50 AIP patients, 48 had the W198X mutation. The patients and matched controls were recruited from the Norwegian counties of Nordland, Troms, Trøndelag and Oslo. The 42 healthy controls consisted of 22 men and 20 women, with a mean age of 48.8 years, without inflammatory diseases. The single‐draw blood samples were obtained in a period outside AIP attacks. Only one patient had chronic AIP symptoms.
Table 1.
Baseline patient demographic characteristics of the study population
| Controls | a AIP cases | OR (CI) | P | |
|---|---|---|---|---|
| Demographic features | ||||
| Age, years (s.d.) | 50·4 (18·6) | 50·6 (18·4) | 0·38 | |
| Height, cm (s.d.) | 171·6 (9·5) | 172·1 (9·7) | 0·84 | |
| Weight, kg (s.d.) | 80·2 (14·0) | 81·0 (15·7) | 0·60 | |
| Body mass index, kg/m2 (s.d.) | 27·2 (3·9) | 27·2 (3·9) | 0·92 | |
| Woman, n (%) | 21 (42%) | 21 (42%) | ||
| Men, n (%) | 29 (58%) | 29 (58%) | ||
| Symptomatic a AIP, n (%) | 35 (70%) | |||
| Asymptomatic a AIP, n (%) | 15 (30%) | |||
| Inflammatory diseases | ||||
| Gout, n (%) | 2 (4%) | 8 (16%) | 4·57 | 0·09 |
| Inflammation in the musculoskeletal system, n (%) | 6 (12%) | 3 (6%) | 0·47 | 0·49 |
| Rheumatoid arthritis, n (%) | 1 (2%) | 1 (2%) | 1·00 | 1·00 |
| Ankylosing spondylitis, n (%) | 2 (4%) | 1 (2%) | 0·49 | 1·00 |
| Gastrointestinal inflammatory disease, n (%) | 3 (6%) | 3 (6%) | 1·00 | 1·00 |
| Bacterial infection recently, n (%) | 1 (2%) | 1 (2%) | 1·00 | 1·00 |
| Viral infection recently, n (%) | 1 (2%) | 0 (0%) | 0·33 | 1·00 |
| Diabetes mellitus, n (%) | 3 (6%) | 4 (8%) | 1·36 | 1·00 |
| Anti‐inflammatory drugs and allopurinol, current use | ||||
| Allopurinol, n (%) | 1 (2%) | 4 (8%) | 4·26 | 0·36 |
| NSAIDs, n (%) | 5 (10%) | 3 (6%) | 0·57 | 0·72 |
| Immunosuppressants, n (%) | 3 (6%) | 1 (2%) | 0·32 | 0·62 |
| Prednisolone, n (%) | 2 (4%) | 1 (2%) | 0·49 | 1·00 |
| Penicillin V, n (%) | 1 (2%) | 0 (0%) | 0·33 | 1·00 |
| Autoantibodies and biomarkers | ||||
| S c ‐Rheumatoid factor ≥ 6 U/l, n (%) | 7 (14%) | 4 (8%) | 0·53 | 0·53 |
| S c ‐Anti‐nuclear ab b > 20 U/l, n (%) | 8 (16%) | 10 (20%) | 1·31 (0·47–3·66) | 0·80 |
| S c ‐Anti‐thyroid peroxidase ab b > 60 kU/l, n (%) | 2 (4%) | 6 (12%) | 3·27 | 0·27 |
| S c ‐Anti‐citrullinated peptide ab b > 20 U/l, n (%) | 2 (4%) | 0 (0%) | 0·19 | 0·50 |
| S c ‐Anti‐cardiolipin, U/l, n (%) | 1 (2%) | 0 (0%) | 0·33 | 1·00 |
| S c ‐Urate > 400 μmol/l, n (%) | 12 (24%) | 15 (30%) | 1·35 (0·56–3·30) | 0·65 |
| S c ‐Cystatin C, mg/l (IQR) | 0·97 (0·85–1·03) | 0·98 (0·86–1·11) | 0·43 | |
| Relative eGFR d CKD‐EPI, ml/min/1·73 m2 (IQR) | 87 (69–99) | 83 (63–100) | 0·54 | |
| Ethanol intake and smoking | ||||
| Ethanol intake, g per day (IQR) | 6·3 (0–14·7) | 3·2 (0–11·9) | 0·28 | |
| Never smokers, n (%) | 19 (38%) | 19 (38%) | ||
| Former smokers, n (%) | 25 (50%) | 21 (42%) | 0·84 (0·35–1·99) | 0·83 |
| Current smokers, n (%) | 6 (12%) | 10 (20%) | 1·67 (0·50–5·51) | 0·55 |
The data represent the mean values (s.d.), n (%) or median values and interquartile range (IQR). Wilcoxon's matched‐pairs signed‐rank test was used for the demographic features; Fisher's exact test was used for all other variables. OR = odds ratio; CI = confidence interval; n = 50 matched pairs, except for daily alcohol intake n = 46 matched pairs. OR for smoking was calculated by comparing former smokers against never smokers in cases versus controls and current smokers against never smokers in cases versus controls. aAIP = acute intermittent porphyria; bab = antibodies; cS = serum; drelative estimated glomerular filtration rate (eGFR) Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) based on cystatin C levels; NSAIDs = non‐steroidal anti‐inflammatory drugs; s.d. = standard deviation.
Ethics, consent and permission
The Regional Committee for Medical and Health Research Ethics approved the study. Written informed consent was obtained from all participants. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration. This trial is registered with ClinicalTrials.gov number NCT01617642.
Medical evaluation
A physician completed a questionnaire for each participant during the interview on the same day that the blood samples were taken. Individuals with the AIP gene mutation were questioned about the presence or absence of AIP symptoms, time of diagnosis, number and durations of attacks and suspected triggering and relieving factors. They were asked specifically about abdominal pain, vomiting, constipation, muscle ache, muscle weakness, decreased sensitivity, paresis, headache, tiredness, epilepsy, palpitations, dark or red urine and psychiatric symptoms. Participants were also asked about their smoking and alcohol habits, physical activity and emotional stress. All participants gave a past medical history, history of current illness and drug history before the interview. Dietary intake was registered using a 7‐day food diary. Trained nurses measured height and weight and calculated body mass index (BMI).
Blood sampling and laboratory methods
Blood samples were obtained by venepuncture between 8 a.m. and 9 a.m. after an overnight fast using Vacuette citrate and ethylenediamine tetraacetic acid (EDTA) serum tubes (Greiner Bio‐one GmbH, Frickenhausen, Germany). The EDTA tubes for plasma cytokine and complement analysis were placed immediately onto crushed ice and centrifuged at 1500 g for 15 min at +4°C, and the plasma was stored at −80°C until analysis.
Multiplex technology
The cytokines were analysed in EDTA plasma using a Bio‐Plex Human Cytokine 27‐plex kit (Bio‐Rad Laboratories Inc., Hercules, CA, USA). The following cytokines, chemokines and growth factors were analysed: interleukin (IL)‐1β, IL‐1RA (IL‐1 receptor antagonist), IL‐2, IL‐4, IL‐5, IL‐6, IL‐7, chemokine (C‐X‐C) motif 8 (CXCL8), IL‐9, IL‐10, IL‐12 (p70), IL‐13, IL‐15, IL‐17, chemokine (C‐C motif) ligand 5 (CCL5), chemokine ligand 11 (CCL11), basic fibroblast growth factor (FGF), granulocyte colony‐stimulating factor (G‐CSF), granulocyte–macrophage colony‐stimulating factor (GM‐CSF), interferon (IFN)‐γ, CXCL10, CCL2, CCL3, CCL4, platelet‐derived growth factor‐BB (PDGF‐BB), tumour necrosis factor (TNF) and vascular endothelial growth factor (VEGF). Insulin, visfatin and C‐peptide were analysed using the human Bio‐plex Pro human diabetes immunoassay kit from Bio‐Rad. The analysis was performed using the Bio‐Plex 200 system (Bio‐Rad).
Enzyme immunoassay
Long‐pentraxin 3 (PTX3) was analysed in EDTA plasma using an enzyme‐linked immunosorbent assay (ELISA) kit (R&D Systems Inc., Minneapolis, MN, USA). The complement activation products C3bc and soluble C5b‐9 (sC5b‐9) were analysed in EDTA plasma using enzyme‐linked immunosorbent assays (ELISAs), as described previously 19. Both assays are based on monoclonal antibodies to neoepitopes exposed in the activation products and not present in the native components. Optical density was measured using an MRX microplate reader (Dynex Technologies, Denkendorf, Germany) 20. Anti‐transglutaminase and anti‐endomysium were analysed using Quanta LiteTM h‐tTG immunoglobulin (Ig)A ELISA kits (Inova Diagnostics, Inc., San Diego, CA, USA). Anti‐nuclear antibody (ANA) screen, rheumatoid factor and anti‐CCP were analysed in serum using Quanta LiteTM ANA, rheumatoid factor (RF) IgM and CCP3 IgG ELISA kits, respectively (Inova Diagnostics, Inc.). Anti‐cardiolipin was analysed using the indirect enzyme immunoassay (EIA) test RELISA® cardiolipin IgG and IgM (ImmunoConcepts N.A. Ltd., Sacramento, CA, USA).
Routine biochemistry tests
Routine haematology parameters were analysed using a Siemens ADVIA 2120 Hematology System (Siemens Healthcare Diagnostics Ltd, Camberley, UK). HbA1c was analysed using a Tosoh G8 high‐performance liquid chromatography (HPLC) system. Serum levels of urate, IgA, IgG and IgM, urine creatinine and other clinical chemistry parameters in urine and serum were measured using an Advia®1800 system (Siemens Medical Solutions Diagnostics, Tokyo, Japan) and reagents from Siemens Healthcare Diagnostics Ltd. Anti‐thyroid peroxidase antibodies (anti‐TPO) levels were analysed using an Advia Centaur® system (Siemens). Levels of C3, C4 and high‐sensitivity C‐reactive protein (CRP) in serum were analysed using a BN ProSpec® nephelometer (Siemens Healthcare Diagnostics Ltd). Urine porphobilinogen (U‐PBG) levels were analysed using a kit from BioRad Laboratories (München, Germany) and Ehrlich reagent, and urine porphyrins were analysed as described previously 21. The U‐PBG ratio (µmol porphobilinogen/mmol creatinine) was subsequently calculated.
Statistical analyses
The Wilcoxon matched‐pairs signed‐rank test was used on the matched case–control data. The non‐detectable cytokine values were set to zero. The Mann–Whitney U‐test was used on the non‐matched case data. Spearman's rank correlation coefficient was used for the cases with AIP and in the calculations of the r‐ and two‐tailed P‐values because the data were not distributed normally. P < 0·05 was considered to be statistically significant. The statistical analysis was performed using Prism version 6.0 from GraphPad Software Inc. (San Diego, CA, USA). Multiple linear regression analyses were performed on data from the AIP cases using IBM SPSS Statistics for Macintosh version 23 (Armonk, NY, USA). In the hierarchical cluster analysis, the cytokine levels in each AIP case and matched controls were divided on the median levels of each cytokine in the 50 matched controls. Individuals with undetectable cytokine values were replaced with a random number below the lowest detection limit of each assay. The data were then imported into r version 3.1.3, Bug in Your Hair. The matrix results were then transformed using natural logarithm. The Bioconductor (version 3.3) library complex heatmap (version 1.10.2) was used to cluster and distance the cytoplex matrix for both row and columns with the following parameters: cluster = complete linkage, distance = Euclidian 22.
Results
Baseline characteristics and demographic features
The demographic features and baseline characteristics were similar in AIP patients and matched controls (Table 1). Of the 50 patients, 35 had symptomatic AIP and 48 had the W198X mutation. There was no difference in the prevalence of inflammatory diseases and the use of anti‐inflammatory medication between AIP cases and controls (Table 1). Finally, the levels of autoantibodies, biomarkers related to inflammatory diseases and the kidney function did not differ between the matched controls and AIP cases (Table 1).
Plasma levels of cytokines, chemokines, growth factors and other biomarkers in cases with acute intermittent porphyria compared with controls
The levels of all 27 cytokines, chemokines and growth factors were increased significantly (P < 0·0004) in the cases with AIP compared with the matched controls (Table 2). Median concentrations of proinflammatory cytokines, including TNF, IL‐1β, CXCL8, CCL2 and IL‐6, were increased significantly (P < 0·0001 for all) by 4·1‐, 1·8‐, 1·6‐, 1·5‐ and 2·5‐fold, respectively, in the cases with AIP compared with the matched controls (Table 2). Furthermore, median levels of IL‐7, IL‐12 (p70), IL‐17 and IFN‐γ (Table 2) were increased significantly (P < 0·0001 for all) by 2·3‐, 3·8‐, 24·6‐ and 7·7‐fold, respectively. The IL‐15 level (Table 2) was increased in cases with AIP, but with undefined fold‐change because the median concentration in the matched controls was undetectable.
Table 2.
Biomarker levels in acute intermittent porphyria cases and controls
| Controls (n = 50) | a AIP cases (n = 50) | § Fold increase | $ Healthy controls | |
|---|---|---|---|---|
| U b ‐PBG c (μmol/mmol creatinine) | 0·4 (0·3–0·5) | 2·6 (0·9–8·5)*** | 6·5 | |
| U b ‐total porphyrins (nmol/mmol creatinine) | 6·7 (4·4–10·6) | 25·7 (8·5–83)*** | 3·8 | |
| S d ‐ALT (IU/l) e | 24·5 (18·8–34) | 31·0 (23–45) # | 1·3 | |
| P f ‐IL‐1β g (pg/ml) | 0·9 (0·65–1·2) | 1·6 (0·9–3·3)*** | 1·8 | 0·9 (0·4–2·0) |
| P f ‐ IL‐1RA g (pg/ml) | 28·0 (13–50) | 64·0 (23–197)*** | 2·3 | 21 (0–117) |
| P f ‐IL‐2 g (pg/ml) | 1·0 (0·0–2·0) | 6·0 (1·0–16)*** | 6·0 | 0·03 (0·0–7·9) |
| P f ‐IL‐4 g (pg/ml) | 1·0 (0·2–1·0) | 2·0 (1·0–4·0)*** | 2·0 | 1·0 (0·0–2·0) |
| P f ‐IL‐5 g (pg/ml) | 1·1 (0·7–1·4) | 1·7 (1·3–4·0)*** | 1·6 | 1·0 (0·5–3·9) |
| P f ‐IL‐6 g (pg/ml) | 2·0 (0·6–3·0) | 5·0 (2·0–9·3)*** | 2·5 | 2·0 (0·02–6·9) |
| P f ‐IL‐7 g (pg/ml) | 3·0 (1·4–5·0) | 7·0 (5·0–14)*** | 2·3 | 3·0 (0·0–11) |
| P f ‐CXCL8 g (pg/ml) | 5·0 (3·0–7·0) | 8·0 (6·0–14)*** | 1·6 | 4·5 (0·8–19) |
| P f ‐IL‐9 g (pg/ml) | 5·5 (4·0–8·3) | 14·0 (8·8–22)*** | 2·6 | 5·5 (2·0–32) |
| P f ‐IL‐10 g (pg/ml) | 0·14 (0·0–0·53) | 1·4 (0·2–6·0)*** | 10·1 | 0·1 (0·0–9·5) |
| P f ‐IL‐12(p70) g (pg/ml) | 2·0 (0·2–5·3) | 13·5 (6·8–25)*** | 6·8 | 1·8 (0·0–32) |
| P f ‐IL‐13 g (pg/ml) | 2·2 (1·7–4·0) | 5·0 (2·2–8·3)*** | 2·3 | 2·1 (0·9–14) |
| P f ‐IL‐15 g (pg/ml) | 0·0 (0–0·12) | 1·25 (0·0–5·0)*** | j ND | 0·0 (0·0–2·0) |
| P f ‐IL‐17 g (pg/ml) | 0·65 (0·0–9·3) | 16·0 (3·5–55)*** | 24·6 | 0·5 (0·0–54) |
| P f ‐CCL2 h (pg/ml) | 11·0 (8·0–14·0) | 16·5 (13·0–22)*** | 1·5 | 10·0 (3·0–28) |
| P f ‐CCL3 h (pg/ml) | 1·0 (0·15–3·0) | 3·0 (2·0–6·0)*** | 3·0 | 1·0 (0·0–11) |
| P f ‐CCL4 h (pg/ml) | 35·0 (29–46) | 57·5 (45–69)*** | 1·6 | 34·0 (21–157) |
| P f ‐CCL5 h (ng/ml) | 0·22 (0·11–0·52) | 1·13 (0·17–10·7)*** | 5·1 | 0·21 (0·02–4·0) |
| P f ‐CCL11 h (pg/ml) | 43·0 (20–67) | 79·0 (59–105)*** | 1·8 | 35·5 (0·5–153) |
| P f ‐FGF basic (pg/ml) | 12·0 (4·0–23·5) | 31·0 (10–70)*** | 2·6 | 12·0 (0·0–71) |
| P f ‐G‐CSF (pg/ml) | 7·0 (1·5–15·0) | 29·5 (4·8–68)*** | 4·2 | 7·0 (0·0–60) |
| P f ‐GM‐CSF (pg/ml) | 4·0 (0·14–6·3) | 14·0 (5·0–31)*** | 3·5 | 3·0 (0·0–15) |
| P f ‐IFN‐γ (pg/ml) | 13·5 (0–53) | 104 (13·5–264)*** | 7·7 | 13·5 (0·0–117) |
| P f ‐CXCL10 (pg/ml) | 601 (402–914) | 963 (711–1162)** | 1·6 | 597 (205–2238) |
| P f ‐TNF (pg/ml) | 8·0 (4·6–21) | 33 (12–84)*** | 4·1 | 8·0 (0·02–54) |
| P f ‐VEGF (pg/ml) | 2·0 (0·42–5·0) | 16·5 (6·8–29)*** | 8·3 | 1·8 (0·0–17) |
| P f ‐PDGF‐BB (pg/ml) | 2·0 (0·27–11·3) | 19·0 (3·5 −155)*** | 9·5 | 1·5 (0–117) |
| P f ‐C3bc (CAU i /ml) | 5·7 (4·8–7·6) | 7·8 (6·4–9·1)* | 1·4 | |
| P f ‐C3bc/C3 ratio (CAU i /ml/g/l) | 5·4 (4·5–7·2) | 7·0 (5·7–8·0)* | 1·3 | |
| S‐Immunoglobulin G (g/l) | 10·0 (8·5–11) | 10·9 (10–12) # | 1·1 | |
| B‐HbA1c (%) | 5·3 (5·0–5·6) | 5·5 (5·3–5·7)* | 1·1 | |
| S d ‐Triglyceride | 1·0 (0·7–1·4) | 1·2 (1·0–1·9) # | 1·2 | |
| S d ‐Total cholesterol | 5·1 (4·6–5·7) | 5·7 (5·1–6·4)* | 1·1 |
The data represent the median values and interquartile range (IQR). The data were analysed using the Wilcoxon matched‐pairs signed‐rank test on AIP cases (n = 50) versus matched controls. The P‐values are exact, two‐tailed. # P < 0·05; *P <0·01; **P < 0·001; ***P < 0·0001 on AIP cases (n = 50) versus matched controls. All P‐values < 0·001 when comparing the cytokines in the AIP cases with the 42 healthy controls. §Fold increase, AIP cases median value (n = 50)/controls median values (n = 50), $cytokine values, median and 2·5–97·5 percentiles in the 42 healthy controls. aAIP = acute intermittent porphyria; bU = urine; cPBG = porphobilinogen; dS = serum; eIL = interleukin; fP = ethylenediamine tetraacetic acid (EDTA) plasma; gIU/l = international units per litre; hCCL = chemokine ligand; iCAU/ml = complement arbitrary units per millilitre; jn.d. = not determined; kB = EDTA blood.
Median levels of the anti‐inflammatory cytokines, including IL‐1RA and IL‐10, were also increased significantly (P < 0·0001 for both) by 2·3‐ and 10·1‐fold, respectively, in AIP cases compared with matched controls (Table 2). The levels of several chemokines, including CCL3 and CCL4, CCL5, CXCL10 and CCL11 (Table 2), were all increased significantly (P < 0·001 for all) in AIP cases compared with matched controls. Median levels of the growth factors FGF basic, VEGF, G‐CSF, GM‐CSF and PDGF‐BB were also increased significantly (P < 0·0001 for all) by 2·6‐, 8·3‐, 4·2‐, 3·5‐ and 9·5‐fold, respectively, in AIP cases compared with matched controls (Table 2). IL‐2, IL‐4, IL‐5, IL‐9 and IL‐13 were increased significantly (P < 0·0001 for all) in cases with AIP (Table 2). The levels of most cytokines were also increased in the AIP cases compared to the median level and the reference ranges in the 42 healthy controls (Table 2). The P‐values remained on the same level when we compared the cytokine levels with the 42 healthy controls instead of the 50 matched controls, except that the P‐values for CCL5 and FGF basic against the 42 healthy controls were P < 0·001 instead of P < 0·0001.
Serum ALT, IgG level and blood monocyte count were slightly, but significantly higher (P < 0·05) in AIP cases than matched controls (Table 2). Complement activation markers, including plasma C3bc and C3bc/C3 ratio, were higher (P < 0·01) in cases than matched controls (Table 2). Serum cholesterol and triglyceride levels were also significantly higher in cases than matched controls (Table 2). The biomarkers of AIP disease activity U‐PBG and total porphyrins were, as expected, significantly higher (P < 0·0001 for both) in cases than controls (Table 2). High‐sensitivity CRP, PTX3, serum IgA and IgM levels, total leucocyte count, platelet count and haemoglobin levels were not significantly different between the AIP cases and the matched controls (data not shown).
Cytokine levels in asymptomatic and symptomatic acute intermittent porphyria cases
We next examined whether cytokine levels were increased both in asymptomatic and symptomatic AIP cases. The levels of all 27 cytokines, chemokines and growth factors were increased significantly in cases with symptomatic AIP compared with matched controls, and eight of these are shown (Fig. 1). Furthermore, the levels of 23 of these 27 analytes were increased significantly in cases with asymptomatic AIP compared with matched controls, and seven of these are shown (Fig. 1). However, G‐CSF, CCL3, FGF‐basic and CXCL8 were not increased in asymptomatic AIP cases. The levels of cytokines and growth factors were also increased in many AIP cases compared to their respective reference ranges in 42 healthy controls (Fig. 1). The C3bc/C3 ratio was enhanced significantly by 1·3‐fold (P < 0·005) in the symptomatic AIP cases versus their matched controls. However, the levels of the complement activation markers terminal complement complex (TCC), C3bc/C3 ratio, C3 and C4 levels were not significantly different between the symptomatic and asymptomatic AIP cases (data not shown).
Figure 1.
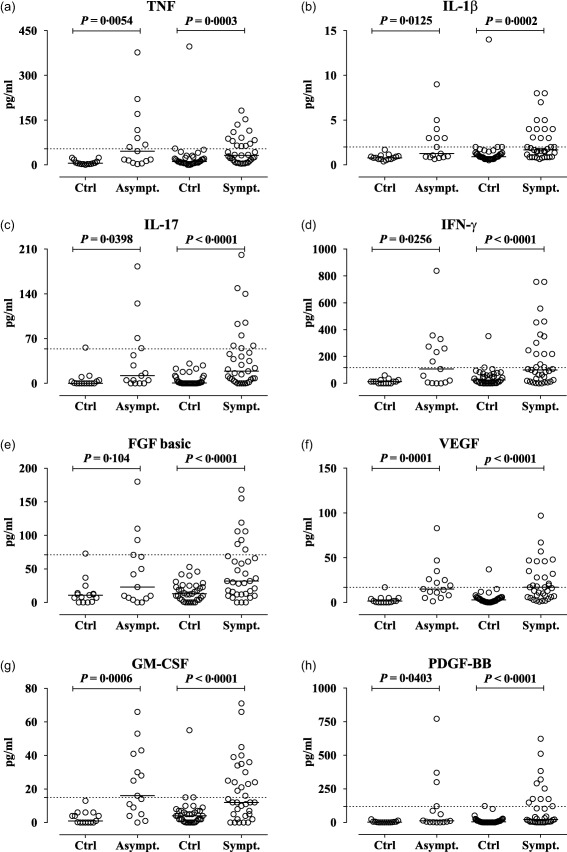
Increased levels of cytokines and growth factors in asymptomatic (Asympt.) and symptomatic (Sympt.) acute intermittent porphyria (AIP) cases compared with matched controls (Ctrl). (a) Increased levels of tumour necrosis factor (TNF), (b) interleukin (IL)‐1β and (c) IL‐17, (d) interferon (IFN)‐γ and the growth factors (e) basic fibroblast growth factor (FGF basic), and (f) vascular endothelial growth factor (VEGF), (g) granulocyte–macrophage colony‐stimulating factor (GM‐CSF) and (h) platelet‐derived growth factor (PDGF)‐BB in asymptomatic and symptomatic AIP cases compared with their respective matched controls (Ctrl). The concentration of cytokines and chemokines were measured using a multiplex cytokine assay. The results are expressed as pg/ml. The results from the age‐ and sex‐matched controls (n = 50, including 35 controls for the symptomatic cases) and the cases with AIP (n = 50, 35 symptomatic) are shown as scatter‐plots with the median. The horizontal dotted gridline indicate each cytokine's upper reference value (97·5 percentile) in 42 healthy controls. The data were analysed using the Wilcoxon matched‐pairs signed‐rank test.
Hierarchical cluster analyses of cytokines
The hierarchical cluster analyses of cytokines in each AIP case and 50 matched controls divided the AIP cases and matched controls into two main clusters, (a) and (b) (Fig. 2). Cluster (b) featured high levels (red) of many cytokines and included five controls and 30 AIP cases, 21 of which were symptomatic AIP cases and nine were asymptomatic (Fig. 2). All five controls in cluster (b) with high cytokine levels had inflammatory diseases, such as undiagnosed rheumatoid arthritis and ankylosing spondylitis. Cluster (b2‐2) contains one control with undiagnosed, untreated rheumatoid arthritis and AIP cases. Many AIP cases in cluster (b2‐2) with high cytokine levels also had elevated U‐PBG and U‐ALA levels. The 30 AIP cases in cluster (b) had significantly higher cytokine and visfatin levels than the 20 AIP cases in cluster (a) (Supporting information, Table S1). However, the PBG ratio and kidney function were not significantly different in the AIP cases in cluster (b). The cytokines clustered in three groups, and the cluster 1 cytokines most elevated in the AIP cases were IL‐17, PDGF‐BB, IL‐10 and IL‐15. Finally, the AIP cases with the R167W mutation had similar cytokine levels to those with the W198X mutation (Fig. 2).
Figure 2.
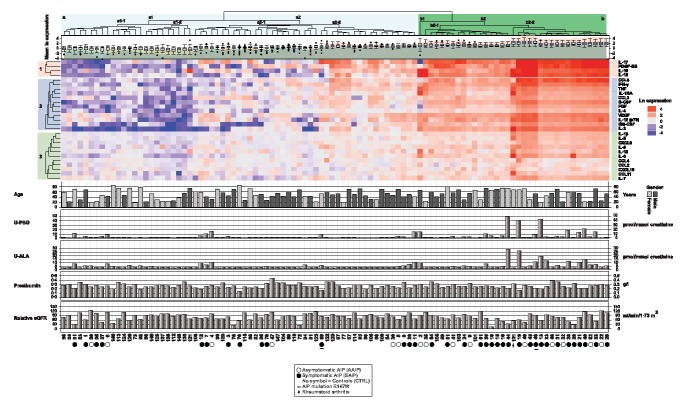
Hierarchical cluster analysis of cytokines in acute intermittent porphyria (AIP) cases and matched controls. The cytokine levels in each AIP case and matched controls were divided on the median levels of each cytokine in the 50 matched controls. The matrix results were then transformed using natural logarithm. The ln expression levels of individual cytokines are represented by a colour scale, with high values in red, intermediate levels in white and low cytokine levels in blue. The cluster analysis was performed using r with Euclidian distance and complete clustering. The main clusters of AIP cases and controls are named (a) and (b), and the subclusters are named (a1), (a2), (b1) and (b2) on the dendrogram. The cytokines clustered into three clusters named 1, 2 and 3. The mean ln expressions of all the cytokines levels are given as box‐plots below the dendrogram. The age in years, gender, U‐porphobilinogen (U‐PBG) and U‐ALA ratio expressed as µmol/mmol creatinine, pre‐albumin (g/l) and the relative estimated glomerular filtration rate (eGFR) Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) (ml/min/1·73 m2) based on cystatin C levels, are shown as a bar graphs below the heatmap for each AIP case and control. Asymptomatic (open circles) and symptomatic (filled circles) AIP cases are indicated below the heatmap, those with the R167W mutation are indicated with underscored symbols.
Correlations between disease activity, inflammatory markers and kidney function in cases with acute intermittent porphyria
Biochemical disease activity in AIP was measured by U‐PBG ratio. U‐PBG levels correlated positively with the inflammatory biomarker PTX3 (r = 0·38, P = 0·01) (Fig. 3). U‐PBG levels also correlated positively with IL‐9 (r = 0·31, P = 0·03) and IL‐7 levels (r = 0·30, P = 0·03) (Fig. 3). U‐PBG levels correlated positively with CCL4 levels (r = 0·30, P = 0·03, Fig. 3) and negatively with pre‐albumin levels (r = −0·31, P = 0·03) (data not shown). The U‐PBG level also correlated positively with the biomarkers of complement activation, including the C3bc/C3 ratio (r = 0·35, P = 0·01) and TCC levels (r = 0·33, P = 0·02) (Fig. 3). Examining the relationship between AIP disease activity and kidney function, we found that U‐PBG levels correlated positively with the levels of S‐cystatin C, a biomarker that is increased in the presence of impaired kidney function (r = 0·41, P = 0·003) (Fig. 3).
Figure 3.
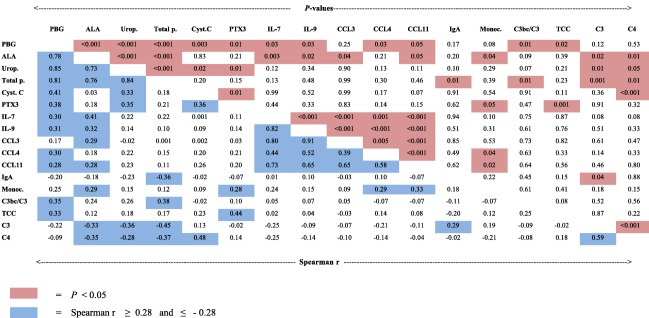
Correlation matrix of porphyrins and precursors, biomarkers of inflammation and kidney function. The data in the left lower part are Spearman's correlation coefficients, r, in the acute intermittent porphyria group (n = 50), r ≥ 0·28 and r ≤ −0·28 (blue colour). The pairwise correlation between the different variables is depicted. The corresponding significant P‐values (P < 0·05) are indicated by red colour in the upper right part. The following variables were included: urine porphobilinogen ratio (PBG); urine 5‐aminolevulinic acid ratio (ALA); uroporphyrin (Urop.); total porphyrins (Total p.); serum cystatin C (Cyst. C); plasma long pentraxin 3 (PTX3); interleukin (IL)‐7 and ‐9; chemokine ligand (CCL) 3, 4 and 11; serum immunoglobulin A (IgA); blood (B) monocyte count (Monoc.); serum C3bc/C3 ratio (C3bc/C3); the plasma levels of the terminal complement complex (TCC); complement C3 and C4.
Multiple regression analyses of inflammatory markers and potential confounding parameters that were associated independently with the biochemical disease activity
Multiple linear regression analyses were performed to describe further the relationship between the biochemical disease activity measured as the U‐PBG ratio and inflammatory markers in the AIP cases. PTX3 was associated independently with the disease activity in the AIP cases after adjusting for potential confounders and other inflammatory markers (Table 3). We tested several regression models, including various individual or pairs of cytokines, and the coefficient estimates of PTX3 remained unchanged. In addition, kidney function expressed as relative estimated glomerular filtration rate (eGFR) Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) based on cystatin C was a significant predictor in the multiple linear regression analysis (Table 3). The confounders tested including prealbumin, the ratio between energy consumption and energy requirements, catecholamines in urine, smoking pack‐years, percentage of alcohol of total energy consumption, fasting C‐peptide levels, glucose insulin ratio, age and gender were not significant themselves and altered neither the coefficient estimates nor the P‐values significantly (data not shown). We did not test drugs as a confounder, as only one AIP case received a porphyrinogenic drug.
Table 3.
Multiple linear regression of *U‐PBG (μmol/mmol creatinine), model R 2 = 0·308
| ‡B | 95% CI | P‐value | |
|---|---|---|---|
| *P‐long pentraxin‐3 | 0·591 | 0·054 to 1·129 | 0·032 |
| †Relative eGFR CKD‐EPI | −0·014 | −0·028 to −0·001 | 0·036 |
| S‐Prealbumin | −5·085 | −11·357 to 1·188 | 0·110 |
The data are regression coefficients, B, in the acute intermittent porphyria cases (n = 50). *Ln‐transformed. †Relative estimated glomerular filtration rate (eGFR) calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) cystatin C equation. ‡B = regression coefficients; CI = confidence interval.
Prealbumin, kidney function, C‐peptide, insulin and porphyrin precursor levels in symptomatic and asymptomatic cases with acute intermittent porphyria
Serum prealbumin levels (Fig. 4a) and kidney function measured as eGFR (Fig. 4b) were both significantly lower in symptomatic compared to asymptomatic AIP cases. C‐peptide (Fig. 4c) and insulin levels (Fig. 4d) after an overnight fast were significantly lower in the symptomatic AIP cases compared to their matched controls. The glucose/insulin ratio (Fig. 4e) and plasma visfatin levels (Fig. 4f) were increased significantly in symptomatic AIP cases compared to matched controls. In comparison, plasma glucagon levels were decreased in both asymptomatic and symptomatic AIP cases compared to their controls (data not shown). Levels of U‐ALA and U‐PBG were, as expected, increased significantly in both symptomatic and asymptomatic AIP cases (Fig. 4g,h). The number of symptoms in AIP correlated negatively with kidney function and pre‐albumin levels, but were not correlated significantly with the inflammatory markers (data not shown).
Figure 4.
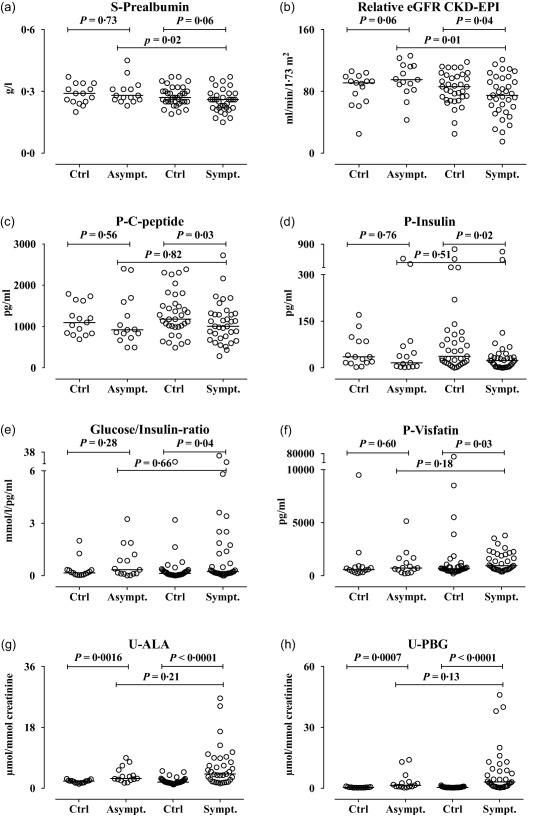
Prealbumin levels, kidney function, C‐peptide, insulin and glucose/insulin ratio, visfatin and porphyrin precursor levels in asymptomatic (Asympt.) and symptomatic (Sympt.) acute intermittent porphyria (AIP) cases compared with controls (Ctrl). (a) Serum prealbumin levels (g/l), (b) relative eGFR (estimated glomerular filtration rate) Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) (ml/min/1·73 m2), (c) plasma C‐peptide (pg/ml), (d) plasma insulin (pg/ml), (e) glucose/insulin ratio (mmol/l/pg/ml), (f) plasma visfatin levels (pg/ml), (g) urine delta‐amino levulinic acid (ALA, µmol/mmol creatinine), (h) urine porphobilinogen (PBG, µmol/mmol creatinine), in asymptomatic (Asympt.) and symptomatic (Sympt.) acute intermittent porphyria cases compared with their respective matched controls (Ctrl). Prealbumin, cystatin C, C‐peptide, insulin and visfatin were analysed using immunoassays. Glucose, ALA and PBG were analysed using standard biochemical assays. The results from the age‐ and sex‐matched controls (n = 50; of these 35 were controls for the symptomatic cases) and the cases with AIP (n = 50, 35 symptomatic) are shown as scatter‐plots with the median. The paired data were analysed using the Wilcoxon's matched‐pairs signed‐rank test. The asymptomatic and symptomatic AIP cases were compared using the Mann–Whitney U‐test.
Discussion
This study found evidence of higher levels of systemic inflammation in AIP cases than in controls matched for age, sex and place of residence. Virtually all inflammatory markers, including cytokines, chemokines and growth factors, were elevated significantly in individuals with AIP compared with the matched controls. Many AIP cases had cytokine levels above the reference ranges in the 42 healthy controls. Furthermore, levels of several inflammatory biomarkers were correlated with the U‐PBG ratio, a biomarker of AIP disease activity. AIP disease activity was associated with decreased insulin levels.
The association we found between AIP and inflammation may have several explanations. One probable explanation for the increased levels of the proinflammatory cytokines such as TNF and IL‐1β, the latter reflecting inflammasome activation 23, may be the accumulation of DAMPs 14, 23. Possible DAMPs candidates in AIP might be ALA, PBG and/or various porphyrins, in addition to tissue injury (Fig. 5). As uric acid, and especially uric acid crystals, are well‐known inflammatory stimuli, we cannot rule out totally that they may play a role as DAMPs in AIP cases. DAMPs may stimulate immunocompetent cells either directly by binding to receptors on the immune‐competent cells 12, 14, 23, or secondarily through proinflammatory, organ damage‐related complement or immune cell activation 15, 16. By analogy, diabetes mellitus is another metabolic disease in which DAMPs including glucose, cholesterol and fatty acids are known to be involved in insulin resistance and kidney failure 13. The reduced insulin levels and slightly enhanced HbA1c and triglyceride levels we found in AIP cases may be due to tissue damage, and indicate that DAMPs play a similar role in AIP to the role they are known to play in diabetes. Given the reduced insulin levels we found in symptomatic AIP cases – accompanied presumably by reduced cellular glucose uptake, reduced negative feedback on ALAS1 and enhanced haem synthesis and release of ALA and PBG – it seems reasonable to argue that symptomatic AIP seems to be associated with disturbed glucose metabolism.
Figure 5.
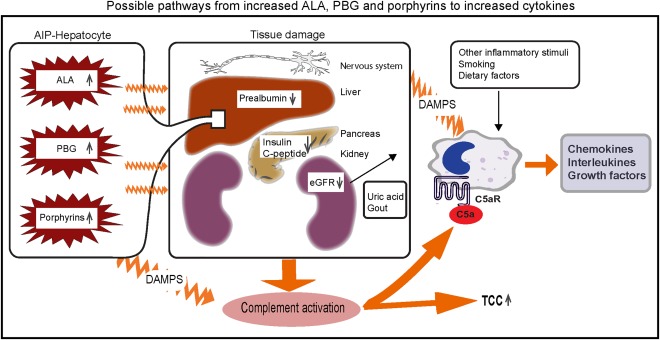
Possible pathways from increased ALA, PBG and porphyrins to increased cytokines. Increased levels of delta‐aminolevulinic acid (ALA), porphobilinogen (PBG) and porphyrins from the liver of acute intermittent porphyria (AIP) patients may cause tissue damage in several tissues, including the nervous system, the liver itself, pancreas and kidneys. Reduced prealbumin indicates inflammation in the liver, and reduced eGFR (estimated glomerular filtration rate) is due to kidney damage. Reduced insulin and C‐peptide secretion from the pancreas may be due to reduced hormone secretion or organ damage. Organ damage may activate complement measured as increased levels of the terminal complement complex (TCC) and C5a, which activates cells to release cytokines, interleukins and growth factors. Another pathway of cytokine release may possibly be the release of damage‐associated molecular patterns (DAMPS) from damaged cells including uric acid crystals in gout due to reduced kidney function which activate directly or indirectly immune cells to release cytokines, interleukins and growth factors. Whether enhanced levels of ALA, PBG and porphyrins may act as DAMPS themselves remains to be elucidated. Other inflammatory stimuli due to smoking and dietary factors, etc. may also activate cells to release cytokines.
The hierarchical cluster analysis of cytokines indicated that many of the AIP cases with the highest levels of several cytokines, especially IL‐17, PDGF‐BB, IL‐10 and IL‐15, also had high biochemical disease activity measured by U‐PBG and ALA. Gender, age, kidney function and prealbumin could not explain the distribution of AIP cases into clusters (a) and (b). The increased visfatin levels in AIP cases in cluster (b) with high cytokine levels indicate a role of the adipose tissue in the inflammation observed in these cases. The AIP cases with the most elevated cytokines clustered together with controls with inflammatory diseases, e.g. untreated rheumatoid arthritis and ankylosing spondylitis. IL‐17 was the cytokine with the highest relative fold increase in the AIP cases and was also identified in the most highly expressed cytokine cluster in the cluster analysis. IL‐17 is a proinflammatory cytokine derived from type 17 helper T cells and natural killer cells and is involved in the Th17 response in several chronic inflammatory conditions 24. IL‐1β, IL‐6 and TNF, which are involved in the development of Th17 cells, were also elevated significantly in the AIP cases 24. IL‐2 and IFN‐γ, which are involved in Th1 responses, were increased moderately in the AIP cases and were also identified in cytokine cluster 2. In comparison, IL‐5 and IL‐13, which are involved in Th2 responses were only slightly elevated in the AIP cases and were located in cytokine cluster 3. The inflammatory cytokine response in a cluster of AIP cases therefore appears to be dominated by a Th17 response similar to other chronic inflammatory conditions, such as rheumatoid arthritis and multiple sclerosis.
Complement plays an essential role in the innate immunological recognition of DAMPs. Frimat et al. showed that haem, which consists of protoporphyrin IX and iron, activates the alternative complement pathway in serum and on endothelial surfaces 15. It is well known that complement activation, in particular through the release of C5a, induces cytokine release in immune‐competent cells 25. We observed enhanced complement activation consistently in the AIP cases revealed by increased levels of C3bc and C3bc/C3 ratio in plasma. Thus, we provide new evidence that the complement system may also play a hitherto unrecognized role in the pathophysiology of AIP. The inflammatory response in AIP may be due partly to damage in the liver, kidney or other organs induced by ALA, PBG or porphyrins 12. Organ damage and cell death may activate complement, leading to immune cell activation and cytokine release similar to the complement activation during sterile inflammation, e.g. myocardial infarction 23, 25. Consistent with this, the AIP disease activity marker U‐PBG was correlated with the kidney function biomarker cystatin C, TCC levels and the C3bc/C3 ratio; the last two are indicators of ongoing complement activation. The negative correlation between C3, C4 and the levels of ALA, coproporphyrin and uroporphyrin in urine may be due to complement activation with consumption of C3 and C4 or liver damage with reduced synthesis of C3 and C4. However, the increases in serum ALT reflecting liver cell damage and plasma C3bc levels reflecting complement activation were modest in the cases with AIP compared with the controls, suggesting that other additional mechanisms are probably involved.
The multiple linear regression analyses showed that PTX3 was associated independently with AIP biochemical disease activity. This association of AIP with smouldering inflammation might provide pathophysiological insights into the long‐term consequences of AIP, including the enhanced risk of HCC and chronic kidney disease. PTX3 is produced by innate immune cells such as granulocytes, macrophages and dendritic cells and is involved in inflammation, arteriosclerosis and tissue repair 26. PTX3 is a pattern recognition molecule able to interact with several complement proteins, including binding to C1q, which leads to activation of the classical pathway. As PTX3 was correlated with enhanced disease activity, enhanced complement activation and decreased kidney function, we speculate that PTX3‐induced complement activation may be involved in the organ damage and/or tissue repair observed in the AIP cases 26. Therefore, our data indicate that PTX3 may play a key role in the low‐grade inflammation observed in the AIP cases. However, as the data set included 50 AIP patients, the results of the multiple linear regression analysis must be confirmed in larger studies. In this study, we held the number of variables in the multiple regression model low because of the limited number of cases and because it is highly probable that several of the cytokines expressions are inter‐related. Furthermore, we cannot exclude an unknown confounder that we have not taken into account in the multiple linear regression analyses.
Our data indicate that the basal level of inflammation might be a driving factor in disease development. First, many AIP cases had cytokine levels above the reference ranges in the healthy controls. Secondly, the PTX3 level was associated independently with disease activity in the multiple regression analysis. Thirdly, this study indicates that low‐grade inflammation is associated with enhanced biochemical disease activity in AIP. Furthermore, inflammation induced by bacterial and viral infections may provoke acute attacks in AIP. We speculate that this low‐grade inflammation may be involved in the development of organ damage, HCC, reduced kidney function and enhanced disease activity in some of the AIP cases. The decreased kidney function in AIP was shown previously to be associated with the development of tubulointerstitial nephropathy and focal cortical atrophy with progressive arteriosclerosis in the kidney 27. The prevalence of HCC is high among AIP patients 3, 6, but the mechanism(s) of carcinogenesis in AIP are unknown. HCC usually arise in a diseased liver with a dynamic inflammatory microenvironment. Several growth factors, including FGF, PDGF and VEGF, may act as possible pro‐angiogenetic factors secreted by stromal cells after chronic liver injury, and IL‐17 may be involved in tumour progression 28. In this study, we determined that several of these cytokines and growth factors were elevated outside attacks, also compared with the reference ranges, and we therefore speculate that they may be involved in HCC development in AIP patients. However, other mechanisms may also possibly explain the enhanced risk of HCC in AIP patients, including the direct or indirect carcinogenicity of ALA 3.
Immune‐competent cells in the liver may also contribute to inflammation in AIP. The innate hepatic immune system plays a role in defence against infection, liver damage and repair 29. Furthermore, prealbumin levels correlated negatively with the number of symptoms (data not shown) and with several inflammatory variables. In line with this, prealbumin levels were decreased in symptomatic AIP cases, due most probably to inflammation in the liver. The level of prealbumin correlated negatively with levels of the AIP disease activity biomarker U‐PBG, confirming that hepatic inflammation is important in the pathophysiology of AIP 18. It is unlikely that these lower prealbumin levels relate to impaired nutritional status in AIP cases, as BMI and nutritional intake (data not shown) were similar in both groups. Dowman et al.'s finding that two of three liver graft recipients from a patient with AIP developed AIP symptoms and increased U‐PBG levels suggests that AIP symptoms are derived only from metabolites from the liver 30. The finding that liver transplantation is a cure for AIP and normalizes the excretion of ALA and PBG confirms this hypothesis 10. Both porphyrins or porphyrin precursors from the liver may thus induce inflammation and organ damage and mediate the symptoms in AIP. However, most inflammatory biomarkers and cytokines were not correlated with ALA, U‐PBG or porphyrin levels, suggesting that the direct activation of immune‐competent cells by porphyrins and porphyrin precursors may only partially explain our results. It thus remains to be elucidated whether enhanced levels of ALA, PBG and porphyrins may act as DAMPs themselves in AIP.
The inflammation found in AIP patients may also be due to ALA, PBG and porphyrin‐induced free radical release, and consequent cytokine release 17, 31, 32. Free radicals may stimulate inflammation through direct activation of the transcription factor nuclear factor kappa B (NF‐κB), leading to the release of IL‐6, IL‐8 and other cytokines 23. The role of free radicals in the AIP‐associated inflammation should be examined in future experimental and clinical studies. Furthermore, the cytokine profile during AIP attacks and in other AIP mutations should be addressed in future studies.
Because the activation of adaptive immunity also stimulates cytokine release, we searched for autoimmune diseases and examined the levels of different autoantibodies in the AIP cases and controls. No significant differences were found in the frequency of inflammatory diseases and anti‐inflammatory medications between the controls and cases with AIP, indicating that autoimmunity and adaptive immunity responses do not account for the inflammatory response. However, the prevalence of gout was enhanced non‐significantly in AIP cases. Furthermore, the reduced baseline kidney function in the symptomatic AIP patients might have had an impact upon the levels of some cytokines. The accumulation of uric acid and other metabolites in kidney failure may induce inflammation. Reduced kidney function has been shown to increase haem synthesis and the excretion of ALA and PBG in porphyric mice 33. This is in line with our finding that reduced kidney function was the only confounding factor that predicted the biochemical disease activity in the AIP cases.
In conclusion, in this first study of its kind, to our knowledge, we found that numerous inflammatory markers, including all 27 cytokines measured, were increased in individuals with AIP compared with matched controls, and that several of these correlated with the biomarker of disease activity U‐PBG. Taken together, these two findings point towards a new pathophysiological mechanism in AIP. Our results suggest that probable porphyrin/precursor‐related hepatic damage may drive inflammation in AIP. The fact that symptomatic AIP cases also had evidence of reduced insulin release may indicate that decreased glucose uptake into cells may explain accelerated haem synthesis in at least some AIP cases. This metabolic similarity between diabetes mellitus and AIP, both of which show decreased insulin release and low‐grade inflammation, could usefully be studied further. In particular, the effect of treatment with a high carbohydrate intake and a small dosage of insulin in symptomatic AIP patients to lower the disease activity and prevent attacks should be examined in future studies.
Disclosure
The authors declare that they have no competing financial or other interest in relation to their work.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's website.
Table S1. Cytokine levels in the acute intermittent porphyria (AIP) cases in clusters (a) and (b).
Acknowledgements
This work was supported by grants from Northern, Southern and Eastern Norway Regional Health Authorities, The Research Council of Norway and the European Community's Seventh Framework Program under grant agreement no. 602699 (DIREKT).
References
- 1. Puy H, Gouya L, Deybach JC. Porphyrias. Lancet 2010; 375:924–37. [DOI] [PubMed] [Google Scholar]
- 2. Hift RJ, Thunell S, Brun A. Drugs in porphyria: from observation to a modern algorithm‐based system for the prediction of porphyrogenicity. Pharmacol Ther 2011; 132:158–69. [DOI] [PubMed] [Google Scholar]
- 3. Stewart MF. Review of hepatocellular cancer, hypertension and renal impairment as late complications of acute porphyria and recommendations for patient follow‐up. J Clin Pathol 2012; 65:976–80. [DOI] [PubMed] [Google Scholar]
- 4. Tollali G, Nielsen EW, Brekke OL. [Acute intermittent porphyria]. Tidsskr nor Laegeforen 2002; 122:1102–5. [PubMed] [Google Scholar]
- 5. Tracy JA, Dyck PJ. Porphyria and its neurologic manifestations In: Biller J, Ferro JM, eds. Handbook of clinical neurology. Amsterdam: Elsevier, 2014:839–49. [DOI] [PubMed] [Google Scholar]
- 6. Innala E, Andersson C. Screening for hepatocellular carcinoma in acute intermittent porphyria: a 15‐year follow‐up in northern Sweden. J Intern Med 2011; 269:538–45. [DOI] [PubMed] [Google Scholar]
- 7. Iwasa F, Sassa S, Kappas A. The effects of acute‐phase inducers and dimethyl sulphoxide on delta‐aminolaevulinate synthase activity in human HepG2 hepatoma cells. Biochem J 1989; 259:605–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yasuda M, Erwin AL, Liu LU et al Liver transplantation for acute intermittent porphyria: biochemical and pathologic studies of the explanted liver. Mol Med 2015; 21:487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Handschin C, Lin J, Rhee J et al Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC‐1alpha. Cell 2005; 122:505–15. [DOI] [PubMed] [Google Scholar]
- 10. Soonawalla ZF, Orug T, Badminton MN et al Liver transplantation as a cure for acute intermittent porphyria. Lancet 2004; 363:705–6. [DOI] [PubMed] [Google Scholar]
- 11. Weiss G. Iron metabolism in the anemia of chronic disease. Biochim Biophys Acta 2009; 1790:682–93. [DOI] [PubMed] [Google Scholar]
- 12. Medzhitov R. Origin and physiological roles of inflammation. Nature 2008; 454:428–35. [DOI] [PubMed] [Google Scholar]
- 13. Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol 2016; 12:13–26. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Q, Raoof M, Chen Y et al Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frimat M, Tabarin F, Dimitrov JD et al Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013; 122:282–92. [DOI] [PubMed] [Google Scholar]
- 16. Meurer M, Schulte C, Weiler A, Goerz G. Photodynamic action of uroporphyrin on the complement system in porphyria cutanea tarda. Arch Dermatol Res 1985; 277:293–8. [DOI] [PubMed] [Google Scholar]
- 17. Sato K, Matsushita K, Takahashi K et al Dietary supplementation with 5‐aminolevulinic acid modulates growth performance and inflammatory responses in broiler chickens. Poult Sci 2012; 91:1582–9. [DOI] [PubMed] [Google Scholar]
- 18. Delaby C, To‐Figueras J, Deybach JC, Casamitjana R, Puy H, Herrero C. Role of two nutritional hepatic markers (insulin‐like growth factor 1 and transthyretin) in the clinical assessment and follow‐up of acute intermittent porphyria patients. J Intern Med 2009; 266:277–85. [DOI] [PubMed] [Google Scholar]
- 19. Bergseth G, Ludviksen JK, Kirschfink M, Giclas PC, Nilsson B, Mollnes TE. An international serum standard for application in assays to detect human complement activation products. Mol Immunol 2013; 56:232–9. [DOI] [PubMed] [Google Scholar]
- 20. van Vuuren BJ, Bergseth G, Mollnes TE, Shaw AM. Electroluminescent TCC, C3dg and fB/Bb epitope assays for profiling complement cascade activation in vitro using an activated complement serum calibration standard. J Immunol Methods 2014; 402:50–6. [DOI] [PubMed] [Google Scholar]
- 21. Storjord E, Brekke OL, Nielsen EW. Safe usage of isotretinoin in a woman with latent acute intermittent porphyria. Acta Derm Venereol 2007; 87:267–8. [DOI] [PubMed] [Google Scholar]
- 22. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016; 32:2847–9. [DOI] [PubMed] [Google Scholar]
- 23. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010; 10:826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miossec P, Korn T, Kuchroo VK. Interleukin‐17 and type 17 helper T cells. N Engl J Med 2009; 361:888–98. [DOI] [PubMed] [Google Scholar]
- 25. Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol 2013; 190:3831–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garlanda C, Jaillon S, Doni A, Bottazzi B, Mantovani A. PTX3, a humoral pattern recognition molecule at the interface between microbe and matrix recognition. Curr Opin Immunol 2016; 38:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pallet N, Mami I, Schmitt C et al High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria. Kidney Int 2015; 88:386–95. [DOI] [PubMed] [Google Scholar]
- 28. Hernandez‐Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013; 144:512–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeong WI, Gao B. Innate immunity and alcoholic liver fibrosis. J Gastroenterol Hepatol 2008; 23(Suppl 1):S112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dowman JK, Gunson BK, Bramhall S, Badminton MN, Newsome PN. Liver transplantation from donors with acute intermittent porphyria. Ann Intern Med 2011; 154:571–2. [DOI] [PubMed] [Google Scholar]
- 31. Monteiro HP, Bechara EJ, Abdalla DS. Free radicals involvement in neurological porphyrias and lead poisoning. Mol Cell Biochem 1991; 103:73–83. [DOI] [PubMed] [Google Scholar]
- 32. Nathan C, Cunningham‐Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol 2013; 13:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Unzu C, Sampedro A, Sardh E et al Renal failure affects the enzymatic activities of the three first steps in hepatic heme biosynthesis in the acute intermittent porphyria mouse. PLOS ONE 2012; 7:e32978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's website.
Table S1. Cytokine levels in the acute intermittent porphyria (AIP) cases in clusters (a) and (b).