

Abstract
Kindlins co-activate integrins alongside talin. They possess, like talin, a FERM domain (4.1-erythrin–radixin–moiesin domain) comprising F0–F3 subdomains, but with a pleckstrin homology (PH) domain inserted in the F2 subdomain that enables membrane association. We present the crystal structure of murine kindlin-3 PH domain determined at a resolution of 2.23 Å and characterise its lipid binding using biophysical and computational approaches. Molecular dynamics simulations suggest flexibility in the PH domain loops connecting β-strands forming the putative phosphatidylinositol phosphate (PtdInsP)-binding site. Simulations with PtdInsP-containing bilayers reveal that the PH domain associates with PtdInsP molecules mainly via the positively charged surface presented by the β1–β2 loop and that it binds with somewhat higher affinity to PtdIns(3,4,5)P3 compared with PtdIns(4,5)P2. Surface plasmon resonance (SPR) with lipid headgroups immobilised and the PH domain as an analyte indicate affinities of 300 µM for PtdIns(3,4,5)P3 and 1 mM for PtdIns(4,5)P2. In contrast, SPR studies with an immobilised PH domain and lipid nanodiscs as the analyte show affinities of 0.40 µM for PtdIns(3,4,5)P3 and no affinity for PtdIns(4,5)P2 when the inositol phosphate constitutes 5% of the total lipids (∼5 molecules per nanodisc). Reducing the PtdIns(3,4,5)P3 composition to 1% abolishes nanodisc binding to the PH domain, as does site-directed mutagenesis of two lysines within the β1–β2 loop. Binding of PtdIns(3,4,5)P3 by a canonical PH domain, Grp1, is not similarly influenced by SPR experimental design. These data suggest a role for PtdIns(3,4,5)P3 clustering in the binding of some PH domains and not others, highlighting the importance of lipid mobility and clustering for the biophysical assessment of protein–membrane interactions.
Keywords: lipid clustering, molecular dynamics, PH domain, surface plasmon resonance, X-ray crystallography
Introduction
Integrin-mediated adhesion between cells and the extracellular matrix is essential for the development of multicellular organisms. Integrins are heterodimeric (α and β) transmembrane receptors comprising a large extracellular domain for ligand binding (such as fibronectin, collagen, and vitronectin) and short cytoplasmic tails [1,2], and are responsible for bidirectional signal transduction across the cell membrane. The cytoplasmic tail of the β-subunit, despite its short length, is critical for integrin activation and has been shown to interact with several regulatory proteins. Talin was the first essential intracellular activator of integrins to be identified, and it contains an amino-terminal FERM domain (4.1-erythrin–radixin–moiesin domain) complemented with a long rod-like domain at its carboxy-terminal end [3,4]. The later identification of the kindlin family of proteins revealed them to be key co-activators of integrins, alongside talin. Talin and kindlin interact directly with the cytoplasmic tails of integrin β-subunits, with kindlin binding to a membrane-distal NPxY motif and talin binding to a membrane-proximal NPxY motif [5–8]. Biophysical data indicate that talins and kindlins are capable of binding simultaneously to their respective NPxY sites [5], while kindlins may be earlier recruited to integrins during activation than talin is [9].
In humans, three isoforms of kindlin proteins, kindlins-1, -2, and -3, have been identified and shown to adopt tissue-specific patterns of expression. Kindlin-1 is expressed in the epithelial cells of the skin and gut, kindlin-2 is widely expressed but most notably in striated and smooth muscle, and kindlin-3 is expressed particularly in haematopoietic tissues but also in endothelial cells [10,11]. Abnormal expression of, or mutations in, kindlins can result in severe disease. For example, kindlin-1 mutations are associated with Kindler syndrome, a rare genetic dermatitis [12], whereas aberrant expression of kindlin-2 is associated with oncogenesis, especially on relocation to the nucleus where it appears to have a role in the control of transcriptional activity leading to the loss of tumour-suppressor signals [13,14].
In contrast, a nuclear role has not been shown for kindlin-3, but it is overexpressed in chronic lymphocytic leukaemia (which led to its discovery) [15] and when overexpressed in breast cancer, it may play a key angiogenetic role [16]. The importance of kindlin-3 in blood clot formation is, however, clear — for example, mice carrying a QW > AA substitution in the integrin-binding F3 subdomain have compromised clotting ability [17] because kindlin-3 activity is essential for platelet activation [18]. As detailed above, nuclear localisation of kindlin-2 is oncogenic [13,14,19], and another recent intriguing insight is that kindlin-2 is the preferred binding partner of β1 integrins over kindlin-3 [20]. This suggests that one possible mechanism linking the two is that overexpression of kindlin-3 could dislodge kindlin-2 from an integrin-bound state, leading to its nuclear relocalisation.
In any case, membrane binding by kindlins is thus not only an important factor in integrin activation, but may also determine their cell signalling impact and even the cell's fate. The membrane-binding capacity of kindlins has been proved to be essential for integrin activation and is dependent on their PH domains [4,21–23] (see Supplementary Figure S1 for a sequence alignment) and on a long loop within the F1 FERM subdomain that is conserved and similar to an equivalent loop in talin [24]. Deletion of the kindlin-3 PH domain eliminates its ability to participate in the adhesion and migration of B cells mediated by the leucocyte integrin LFA-1 [25].
Our previous work on the kindlin PH domains focused on kindlin-1, where we solved its crystal structure to reveal an isoform-specific salt bridge occluding the canonical inositol phosphate-binding site [23]. Molecular dynamics indicated that the salt bridge is dynamic and led us to make a mutant lacking it which had altered inositol phosphate-binding properties. We showed, however, that in either case, the affinity for inositol phosphate ligands is relatively weak (10−4 M), although with a specific preference for PtdIns(3,4,5)P3. We also showed that the apparent affinity was influenced by the buffer choice for an experiment: phosphate-based buffers seem to interfere with the apparent affinity measured, suggesting a lack of specificity in the PtdIns(3,4,5)P3 interactions taking place [23].
Here, we report the crystal structure of the kindlin-3 PH domain together with molecular dynamics-based and biophysical characterisation of its inositol phosphate lipid-binding properties. We show that the kindlin-3 PH domain has a hydrogen bond-based occlusion to its (canonical) PtdInsP-binding cleft, and that, like the kindlin-1 PH domain, it binds surface-immobilised inositol phosphates with rather low (10−4 M) affinity. Using lipid nanodiscs as a model membrane system and molecular dynamics simulations to study the interaction of the kindlin-3 PH domain with different combinations of lipid species including inositol phosphate lipids, we show that lipid clustering is likely to be a significant factor in its binding of PtdIns(3,4,5)P3 in bilayer membranes. On this basis, we propose that a subset of PH domains is capable of binding to multiple inositol phosphates simultaneously and so via an avidity effect has its interaction with target membranes strengthened. This would explain how an apparently low affinity for inositol phosphates can be biologically relevant to the localisation of kindlins to the plasma membrane and their subsequent activation of integrins.
Results
Overall structure of kindlin-3 PH domain
The crystal structure of the kindlin-3 PH domain exhibits an archetypal pleckstrin homology (PH) superfamily fold at its core [26], with a partly open seven-stranded β-barrel capped at one end by a C-terminal α-helix (Figure 1; see Table 1 for data collection and refinement statistics). Similar to that of kindlin-1 and -2 [22,23], the PH domain of kindlin-3 contains an additional C-terminal amphipathic helix extension, which is not present in other PH domains (Figure 1A). A salt bridge in the kindlin-1 PH domain (Arg380–Glu416) has been shown to occlude its canonical phosphoinositol-binding site [23], and in the kindlin-3 PH domain, this salt bridge is replaced by a hydrogen bond (Arg360–Gln396) in the equivalent position (Figure 1A). As shown in Figure 1B, the kindlin-3 PH domain has a mixed surface charge distribution with a pronounced concentration of positive charge at the canonical inositol phosphate-binding site.
Figure 1. Overall structure of kindlin-3 PH domain.

(A) Crystal structure of the kindlin-3 PH domain. The protein is coloured rainbow-wise from its amino (blue) to carboxy (red) terminals, and the canonical inositol phosphate-binding site is highlighted with an arrow (left). The hydrogen bonds within the open end of the PH domain are shown (right). (B) Surface electrostatic charge of the kindlin-3 PH domain. (C) Average root mean square fluctuation (RMSF) of the kindlin-3 PH domain during atomistic MD simulations in water (i.e. in the absence of a bilayer) for two different force fields (i.e. GROMOS and OPLS). The magnitude of the average RMSF of each residue was mapped on the kindlin-3 PH domain crystal structure.
Table 1. Data collection and structure refinement.
| Data collection | |
| Space group | P21212 |
| Cell dimensions | |
| a, b, c (Å) | 132.04, 36.19, 52.69 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 31.73–2.23 (2.31–2.23) |
| Rmerge | 0.049 (0.432) |
| I/σI | 23.7 (2.3) |
| Completeness (%) | 96 (93) |
| Redundancy | 3.1 (2.6) |
| CC_half | 0.998 (0.771) |
| Refinement | |
| Resolution (Å) | 31.73–2.23 (2.31–2.23) |
| No. of reflections | 38 032 (3014) |
| No. of unique reflections | 12 441 (1155) |
| Rwork/Rfree | 21.94 (27.07) |
| No. of atoms | |
| Protein | 2123 |
| Ligand/ion | 1 |
| Water | 13 |
| B-factors (Å2) | |
| Protein | 84.92 |
| Ligand/ion | 13.06 |
| Water | 61.66 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.11 |
| Ramachandran | |
| Favoured (outliers) | 98% (0%) |
The overall fold of the kindlin PH domain appears remarkably conserved. The kindlin-3 PH domain is closest in structure to the PH domain of kindlin-2 (RMSD = 0.69 Å), with the major conformational difference being in the β1–β2 loop. The close relationship between the kindlin PH domain structures is reflected in their position within a structural phylogenetic tree we constructed for a set of PH domains of different kinds of function and (where relevant) with different lipid-binding properties (Supplementary Figure S2). Of note, a previously constructed structural phylogeny places the previously determined kindlin-3 PH domain NMR structure in a different relationship to that of kindlins-1 and -2 to the crystal structure reported here [23]. This highlights the sensitivity of structural phylogenetic comparisons to the quality of the models used.
PH domains with canonical inositol phosphate-binding sites typically exhibit a highly positively charged core at the open end of the PH domain, whereby they interact with their ligand, as seen in PH domains of Grp1 [27] and PLCδ1 [28]. Non-canonical PH domains, such as Slm1 [29], Tiam1, and ArhGAP9 [30], lack the same kind of positive charge distribution and instead have their inositol phosphate-binding site on the side of the barrel mainly between the β1–β2 loop and the β5–β6 loop (Supplementary Figure S3). The PH domains of kindlins-3 and -2 have similar surface charge distribution to the PH domains with canonical inositol phosphate-binding sites (Figure 1B), indicating that inositol phosphate might interact in a canonical way through the β1–β2 loop. The previously reported crystal structure of the kindlin-1 PH domain is not as complete as either of the available crystallographic kindlin-2 or -3 models, lacking seven residues within the β1–β2 loop; its surface charge characteristics, however, seem to be similar to those of the other kindlin PH domains.
Atomistic molecular dynamics (AT-MD) simulation of the kindlin-3 PH crystal structure in solution (i.e. in the absence of any lipid bilayer) with two different force fields showed that the overall fold of the PH domain is retained during the simulations. The seven-stranded β-barrel in the core of the PH domain is very stable, whereas the unstructured regions connecting the β-strands are somewhat more flexible (Figure 1C). In particular, the β1–β2 loop is flexible with both simulation force fields, in good agreement with the crystal structure (Figure 1A). At the end of the simulation, the side chains of the positive residues on the β1–β2 loop remain exposed towards the open end of the barrel, presumably providing a site for inositol phosphate binding. With the exception of one simulation in which the Arg360–Gln396 hydrogen bond identified in the crystal structure breaks for an extended period of time, the Arg360–Gln396 hydrogen bond was generally preserved during the simulations although it breaks transiently before re-forming (Supplementary Figure S4).
Molecular dynamics simulations reveal a membrane-bound state of kindlin-3 PH domain and its interaction with PIP lipids
Coarse-grained molecular dynamics (CG-MD) simulations can be used to identify the molecular mechanism of peripheral membrane proteins binding to model membranes incorporated with anionic, for example, PIP lipids [31,32]. In particular, MD simulations have been used to study the interaction of many PH domains, e.g. DAPP1 PH [32] and Grp1 PH [33], with PIP lipids at the molecular level. A recent study has also demonstrated that MD simulations can be used to study the free energy of the interaction of the Grp1 PH domain with PIP2 and PIP3 molecules [34].
To examine the association of the new crystal structure of the kindlin-3 PH domain with model membranes, the PH domain was displaced away from a preformed bilayer containing 4 PIP3 molecules in each leaflet (concentration of ∼1.5% that mimics the in vivo concentration of PIP3 lipids in the plasma membrane), and an ensemble of 20 CG-MD simulations of 1.5 µs each was performed (Figure 2A). Calculation of the orientation of the PH domain relative to the bilayer (by calculating the Rzz component of its rotational matrix) suggests that the PH domain has a preferred orientation relative to the bilayer (Figure 2B). In this orientation, residues 360–372 (i.e. the positively charged loop between strands β1/β2) made the largest number of contacts with the PIP3 molecules. Regions 385–386, 410–415, and 434–436 also face towards the bilayer (Figure 2) and interact with PIP3 molecules. This orientation is rather different from, for example, the canonical orientation of the Grp1 PH domain relative to a bilayer [27,35]. Indeed, the helix at the end of the Grp1 PH domain was shown to be in a parallel orientation relative to the bilayer. We note, however, that in our ensemble we also observe secondary binding modes in which the kindlin-3 PH domain adopts an orientation that is similar to that of the Grp1 PH domain (Supplementary Figure S5E). Atomistic simulations starting from the preferred orientation of the PH domain on the PIP3-containing bilayer reveal that the PH domain retains this orientation relative to the bilayer.
Figure 2. Coarse-grained MD simulations of the kindlin-3 PH domain interacting with a PIP-containing lipid bilayer.

(A) Snapshots at the beginning and at the end of one of the simulations with 4 PIP3 molecules in each leaflet (Kin3PH-3 in Table 2). PIP3 molecules are shown in green, POPC and POPS phosphate atoms are shown in grey, and the kindlin-3 PH domain is shown in ice blue. (B) Rotation of the kindlin-3 PH domain (shown as the Rzz component of its rotational matrix) relative to the distance between the centre of mass of the protein and the centre of mass of the bilayer. (C) Final snapshot from one of the atomistic simulations that started using snapshots from the simulations shown in (A). (D) Number of PIP molecules interacting with the kindlin-3 PH domain. The number of PIP lipids is shown for Kin3PH-1, Kin3PH-2, and Kin3PH-3 simulations (Table 2) in orange, green, and light green, respectively. For this calculation, a PIP molecule is considered to be in contact with the protein if its phosphate is within 1 nm of the protein. The number of PIP molecules is shown only for the frames in which the protein is bound to the bilayer (i.e. the z component of the distance between the protein centre of mass and the bilayer centre of mass is smaller than 4.75 nm).
To examine whether the concentration of PIP3 in a membrane bilayer affects the association of the kindlin-3 PH domain to the membrane, MD simulations were then carried out with various concentrations of PIP3 lipids in the bilayer (7% PIP3 or 2% PIP3 in each leaflet, or 1% PIP3 in each leaflet; see Table 2). Despite the fact that when we reduce the number of PIP3 molecules in the bilayer, the kindlin-3 PH domain associates with the bilayer less frequently, in all cases it adopts a preferred orientation which is similar to the preferred orientation described above (Supplementary Figure S6). The PH domain adopted the same orientation when we ran simulations with 7% PIP2 lipids or with both PIP3 and PIP2 lipids in the bilayer (Kin3PH-1 and Kin3PH-6 in Table 2, respectively). Interestingly, atomistic simulations starting from four different snapshots of the kindlin-3 PH domain bound to PIP2-containing bilayers at the preferred orientation (six simulations overall; see Materials and Methods) resulted in three simulations in which the kindlin-3 PH domain retained its orientation relative to the bilayer and in three simulations in which the PH domain adopted the secondary binding mode at the end of the atomistic simulations (Supplementary Figure S5) as in subsequent potential of mean force (potential of mean force, PMF) calculations (see below).
Table 2. Summary of simulations.
| Simulation | Bilayer composition | Duration |
|---|---|---|
| Coarse-grained | ||
| Kin3PH-1 | POPC:POPS:PIP2 (∼73:20:7) | 20 × 1.5 µs |
| Kin3PH-2 | POPC:POPS:PIP3 (∼73:20:7) | 20 × 1.5 µs |
| Kin3PH-3 | POPC:POPS (∼78:20) + 4 PIP3 molecule per leaflet | 20 × 1.5 µs |
| Kin3PH-4 | POPC:POPS (∼79:20) + 2 PIP3 molecule per leaflet | 20 × 1.5 µs |
| Kin3PH-5 | POPC:POPS (∼79:20) + 1 PIP3 molecule per leaflet | 20 × 1.5 µs |
| Kin3PH-6 | POPC:POPS (∼73:20) + 5% PIP2 and 4 PIP3 molecule per leaflet | 20 × 1.5 µs |
| Kit3mutPH-1 | POPC:POPS (∼78:25) + 4 PIP3 molecule per leaflet — K363A, K367A mutation | 20 × 1.5 µs |
| Atomistic | ||
| Kin3PH_AT-1 | No bilayer — GROMOS force field | 3 × 100 ns |
| Kin3PH_AT-1 | No bilayer — OPLS force field | 3 × 100 ns |
| Kin3PH_AT-3 | POPC:POPS:PIP2 (∼73:20:7) | 6 × 100 ns |
| Kin3PH_AT-4 | POPC:POPS (78:20) + 4 PIP3 molecule per leaflet | 4 × 100 ns |
Analysis of the contacts between the PIP lipids and the PH domain during the simulations suggested that residues R362, K363, and K367 and residues K363, K367, and R370 made the largest number of contacts with PIP2 and PIP3 lipids, respectively. Our analysis also revealed that the PH domain induces clustering of the PIP lipids when it binds to the bilayer (Figure 2D). The clustering of PIP2 lipids is somewhat higher compared with PIP3 lipids. Note that the clustering of the PIP lipids occurs only in the leaflet in which the protein is bound. In the simulations with 7% of PIP2 or 7% PIP3 lipids in the bilayer, most of the time we observe four PIP3 and five PIP2 lipids, respectively, in an annulus of 1 nm from the protein surface. Given that recent studies have suggested an important role for anionic lipids and PIPs in integrin activation [36,37], the change in the local lipid environment by the binding of the PH domain may have some functional role.
Free energy of binding of the kindlin-3 PH domain to PIP(4,5)P2 and PIP(3,4,5)P3
Our analysis above revealed the molecular mechanism of the association of the kindlin-3 PH domain with the membrane, suggesting the occurrence of two different modes of interaction of the PH domain with the PIP-containing bilayer. To examine the strength of interaction of the PH domain with PIP2 and PIP3 molecules, we performed PMF calculations using a protocol we developed recently (see Materials and Methods and ref. [34]). PMF profiles for PIP3 and PIP2 lipids have a global minimum with a well depth of approximately −3 kcal/mol for PIP3 and a well depth of approximately −2.5 kcal/mol for PIP2 lipids. This suggests that the protein associates somewhat more strongly with PIP3 lipids (Figure 3). The first well (corresponding to the preferred orientation of the PH domain) in the profile of the PIP3 molecules was followed by a second shallower well (corresponding to a secondary PH orientation) at a distance of ∼2.5 nm. Examination of the interactions of the kindlin-3 PH domain with the lipids showed that, in the windows covering ∼1.5 to ∼2.3 nm protein–lipid separations, the protein is in a similar orientation to the preferred orientation (mode 1) described above in which the protein interacts with the bilayer mainly via the β1/β2 unstructured loop. In these windows, the PIP3 molecule is located between the β1/β2 and β5/β6 loops (i.e. in a non-canonical PIP-binding site) for most of the time (Supplementary Figure S7A). We note that we have also seen interactions of PIP molecules with this site in our encounter simulations as described above. Interestingly, in the second well the protein adopts an orientation that is similar to the secondary binding mode also observed in the simulations above, with the PIP3 molecule now located between the β1/β2 and β3/β4 loops (i.e. in the canonical PIP-binding site). In contrast, the profiles for PIP2 lipids and for the mutated form of kindlin-3 PH domain did not have a second well. In the simulations with a PIP2 molecule, the PIP2 is found both in the canonical (i.e. contacting the β1/β2 and β3/β4 loops) and in a non-canonical site (i.e. contacting the β1/β2 and β5/β6 loops; Supplementary Figure S7B). We note that the depth of the well for the binding of the Grp1 PH domain to PIP lipids using the same method was −5.3 kcal/mol for PIP3 and −3.8 kcal/mol for PIP2, suggesting that the binding of the kindlin-3 PH domain is weaker compared with the Grp1 PH domain binding a single lipid [34]. Disruption of the PH/PIP interactions by mutating the K363 and K367 residues which were identified to form a large number of contacts with the PIP lipids resulted in a reduced binding to PIP3 molecules by −1 kcal/mol.
Figure 3. PMF calculations of kindlin-3 PH domain with PIPs.
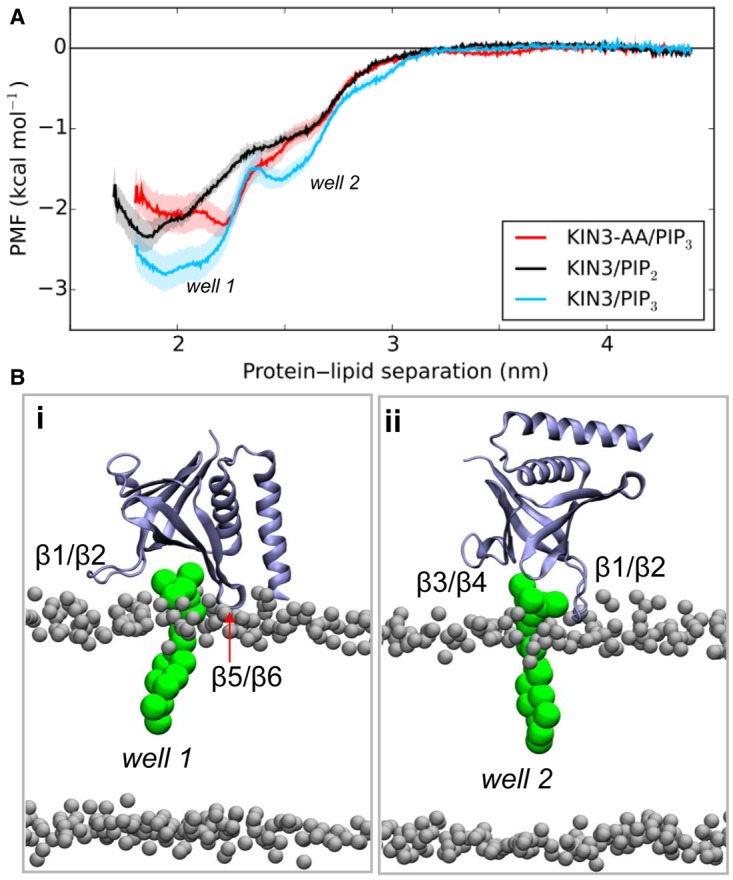
(A) PMF calculations for the binding of the WT and the mutated form of the kindlin-3 PH domain to PIP2 (black) and PIP3 (cyan and red) molecules. (B) Kindlin-3 PH/bilayer complexes corresponding to wells 1 and 2 in (A). The PIP3 molecules are shown in green and the PH domain in ice blue. For this calculation, the initial structures of WT or mutant kindlin-3 PH domains bound to PIP3 or PIP2 in a 80:20 POPC:POPS bilayer were generated by alignment with an existing Grp1–PH/PIP3 complex. The PH domain was pulled from the bound PIP lipid and bilayer along the membrane normal (z-axis). See Materials and Methods for more details.
An SPR study confirms that the kindlin-3 PH domain binds to multiple phosphatidylinositol phosphates
To verify the potential interaction between clustered phosphatidylinositol phosphates and the kindlin-3 PH domain observed in our MD simulations, we conducted surface plasmon resonance (SPR) experiments. Firstly, a biotinylated inositol phosphate was immobilised onto a streptavidin-derivatised sensor chip, as in previous studies [23]. The use of streptavidin-mediated immobilisation meant that the distance separating individual lipids would be larger than the size of the tetrameric streptavidin, and therefore than the PH domain itself, meaning that one PH domain would be able to bind no more than one lipid headgroup simultaneously, imposing one-to-one binding in the interaction (Figure 4A). In agreement with the MD simulation results, the kindlin-3 PH domain displays a preference for PIP3, though with a dissociation constant (KD) of ∼300 µM, in agreement with previous measurements [23]. The apparent binding capacity for PIP2 was much lower (KD = 1100 µM). Introduction of two point mutations (K363A and K367A) effectively abolished the binding capacity of both inositol phosphate moieties, indicating that the interaction is specific even if it is low affinity (Figure 4B). We note that, in the MD simulation with the mutated form of the kindlin-3 PH domain, the protein is capable of binding to the bilayer in a similar orientation to the wild type (WT); however, in many simulations, it quickly dissociates from the bilayer (Supplementary Figure S8). A naturally occurred IPRR (isoleucine–proline–arginine–arginine) insertion in the β1–β2 loop of kindlin-3 has been reported to disable integrin activation and thus lymphocyte adhesion and migration [21]. In agreement with results obtained by others using a pull-down assay with IP3 and IP4-coated beads, our SPR study in vitro revealed that the IPRR insertion directly disrupted the interaction between the PH domain and phosphoinositol moieties (Figure 4B,C), presumably because of the steric effect of an elongated β1–β2 loop.
Figure 4. SPR study of the kindlin-3 PH domain with immobilised PIP2 and PIP3 headgroups.

(A) Schematic representation of SPR experiments with the immobilised lipid headgroup as the ligand and the PH domain as the analyte. (B) SPR measurements of the kindlin-3 PH domain WT and mutants with immobilised PtdIns(4,5)P2 and PtdIns(3,4,5)P3, respectively. On the left in each case, we show the background-subtracted raw sensogram data for the WT protein, while on the right we show binding saturation curves for WT, K363A K367A and IPRR insertion mutants. The highest concentration used is given on each set of sensogram curves; subsequent binding profiles derive from a series of twofold dilutions.
The observation in MD simulation that multiple phosphoinositol molecules may bind to more than one site on a single PH domain (Figures 2 and 3) led us to attempt further verification of this mode of interaction experimentally. Nanodiscs have been shown to be an effective model membrane system due to their stability and capacity to mimic the membrane bilayer [38,39], including the fluidity of lipids though within a limited bilayer area. To study the effect of phosphatidylinositol lipid concentrations on their binding by PH domains, we performed the SPR experiments in an inverted set-up. The biotinylated PH domain was immobilised onto the sensor chip and the nanodiscs were prepared as the analyte.
Nanodiscs incorporated with different components were stable in solution (Supplementary Figures S8D and S9A), with an estimated molecular mass of 120 kDa; they behaved as single species with a sedimentation coefficient of 4 S as assessed by analytical ultracentrifugation (Supplementary Figure S9B). It appears that the diameter of the nanodiscs correlates with the molar ratio of phosphatidylinositides incorporated in their membranes: nanodiscs with 5% PtdIns(3,4,5)P3 (diameter, d = 5.87 nm) were bigger than those with 3% PtdIns(3,4,5)P3 (d = 4.64 nm; Supplementary Figure S9C,E). Note that the diameters are small because they are average values; as shown in Supplementary Figure S9D, the nanodiscs are elliptical and of the expected size.
The SPR results showed that 5% PIP3-incoporated nanodiscs bind to the kindlin-3 PH domain strongly, with a much-enhanced affinity (KD = 0.4 µM; Figure 5) compared with that shown by the PH domain towards immobilised inositol phosphates. In contrast, nanodiscs with 5% PIP2 showed no binding to the kindlin-3 PH domain, demonstrating that PIP3 is its real ligand and suggesting that the apparent binding to immobilised PIP2 is due to a non-specific interaction. We also performed the same experiments with nanodiscs without incorporated phosphatidylinositol moieties and observed no binding in SPR experiments.
Figure 5. SPR study of the kindlin-3 PH domain with PtdIns(3,4,5)P3 incorporated nanodiscs.

(A) Schematic representation of SPR experiments with the immobilised PH domain as the ligand and lipid nanodiscs as the analyte in solution. (B) SPR measurement between the WT kindlin-3 PH domain and nanodiscs with 5% PtdIns(3,4,5)P3. The background-subtracted raw sensogram data for the WT protein are shown on the left and the resulting binding saturation curve on the right. The highest concentration used is given on the sensogram graph; subsequent binding profiles derive from a series of twofold dilutions. See Materials and Methods for experimental details.
There is evidence for PIP2 clustering within lipid bilayers [40], while other data suggest protein-driven PIP2 clustering in signalling complex formation [41]. Furthermore, while a study of yeast proteins has revealed that the interaction between one set of PH domains and membranes can be enhanced by co-operative interaction between PIPs and other lipids (such as POPS) [42], the interaction between the kindlin-3 PH domain and PIP3 seems to more rely on the concentration of PIP3 in the membrane, i.e. co-operativity among copies of the same lipid.
Grp1 PH domain binds inositol phosphate with 1:1 specificity
We further asked whether our observations are specific to the kindlin-3 PH domain (and similar domains) or could also be shown for PH domains which are already known to bind to inositol phosphate species in a classical, 1:1 interaction mode. If they could be shown for such well-characterised domains, then our results with the kindlin-3 PH domain would seem questionable. The crystal structure of the Grp1 PH domain in complex with Ins(1,3,4,5)P4 [equivalent to the PtdIns(3,4,5)P3 headgroup] reveals the ligand well embedded in a pocket formed by positively charged residues [43]. In line with this, we showed by SPR that the analyte/ligand design of our experiment does not significantly affect the apparent affinity of Grp1 for PIP3 compared with the effect it has on the kindlin-3 PIP3 binding observed. The Grp1 PH domain was shown to bind to immobilised PIP3 headgroups with a KD of 2.8 ± 0.4 µM (Figure 6A), and to immobilised PIP2 headgroups with a KD 10 times greater (Supplementary Figure S10), consistent with a previous study though with a lower absolute value due to methodological differences [27,44]. In contrast, the binding affinity to PIP3 within nanodiscs (5% of constituent lipids) has a KD of 0.58 µM. So, in the case of Grp1, there is only a five times difference in affinity between immobilised lipid headgroups and whole lipids in a lipid nanodisc (rather than the ∼100× enhancement observed with the kindlin-3 PH domain). This suggests that the Grp1–PH interaction with PIP3 at high affinity does not rely on the binding of multiple PIP3, though it does indicate an enhancement of lipid binding — most probably by a simple electrostatic attraction — in a membrane-like context [42].
Figure 6. SPR study of interaction between the Grp1PH domain and PtdIns(3,4,5)P3 in two experimental set-ups.

(A) Immobilised PtdIns(3,4,5)P3 headgroup as the ligand and the PH domain as the analyte. (B) Immobilised Grp1PH domain as the ligand and lipid nanodiscs as the analyte. The upper panel in each case gives the background-subtracted raw sensogram data and the lower panel gives the resulting binding saturation curve. The highest concentration used is given on the sensogram graphs (83 µM Grp1 PH domain in A; 3.4 µM nanodiscs in B); subsequent binding profiles derive from a series of twofold dilutions. See Materials and Methods for experimental details.
Discussion
The cytoplasmic surface of the plasma membrane serves as a platform for many critical biological processes. The recruitment of peripheral membrane proteins from the cytoplasm to the plasma membrane surface is spatiotemporally and accurately regulated. The PH domain is one of a selection of commonly used folds directing protein–membrane association and has been shown to confer specificity to different inositol phosphates on protein–membrane interactions.
PH domains have been shown to bind phosphatidylinositol phosphates (PtdInsPs) with different degrees of specificity [45] and the PH domains of the kindlins have a limited affinity for them [23], although the membrane interaction mediated by the PH domain is essential for integrin activation [23]. Phosphatidylinositol lipids are present in membranes at different concentrations, which may lead to variable activity and roles in membrane structure and membrane recognition. PtdIns(4,5)P2 (PIP2) has been reported to be the most abundant phosphatidylinositol lipid in cells [46] and can undergo phosphorylation to a PtdIns(3,4,5)P3 (PIP3) state; both PIP2 and PIP3 are found at the plasma membrane. Monophosphorylated inositide lipids are, in contrast, associated with compartments such as the Golgi and endoplasmic reticulum [46].
In the present study, we firstly solved the crystal structure of the kindlin-3 PH domain and then used the crystal structure as the basis for a multiscale molecular dynamics simulation study. MD simulation revealed that kindlin-3 PH domain interacts with the PIP lipids primarily via the loop connecting the β1 and β2 strands, and that it has a somewhat higher affinity for binding PtdIns(3,4,5)P3 than for PtdIns(4,5)P2 in a membrane-like system. Moreover, one kindlin-3 PH domain appears to bind multiple inositol phosphate lipids, at both canonical and non-canonical sites, by means of which the affinity of interaction between the PH domain and the membrane is greatly enhanced by an avidity effect. In contrast, with a PH domain (from Grp1) previously shown to have canonical and specific one-to-one binding to inositol phosphate lipid, this one-to-more binding mode was not observed. These observations from MD simulation were supported by our SPR experiments in two complementary set-ups. The first one involves immobilisation of biotinylated inositol phosphates on the sensor chip and use of the PH domain (WT and mutant) as the analyte, to mimic an isolated one-to-one binding mode. In this case, each PH domain could only bind to no more than one phospholipid due to the distance restraints imposed by streptavidin-mediated immobilisation. The other set-up attempted to mimic the bilayer plasma membrane, by immobilising the PH domain on the chip and using soluble membranes (nanodiscs) as the analyte. The affinity of interaction measured between the PH domain and inositol phosphate lipids was remarkably affected by experimental design.
Compared with liposomes, nanodiscs have obvious advantages for protein–lipid interaction studies in a membrane-like environment: they are more stable and have a limited number of lipids per disc. Nanodiscs made of MSP1E3D1 are estimated to accommodate ∼125 POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipids, with a diameter of ∼13 nm [47]; this is in agreement with our cryo-EM map of the nanodisc (Supplementary Figure S9D), which is fitted well (length ∼14 nm and width ∼9 nm) with the elongated and twisted conformation of the human apolipoprotein A1 [48]. In the SPR experiments here, the small and homogeneous size of the nanodisc membrane is critical for 1:1 model fitting and interpretation of the SPR results. Liposomes, with inherently larger diameters (e.g. 50–100 nm), would create multiple possible contact sites for PH domains, confounding binding affinity analysis. Nanodiscs, however, are capable of binding no more than one immobilised PH domain due to their limited size compared with the size and density of immobilised PH domains on the chip surface.
We find that PtsIns(3,4,5)P3 clustering enhances the affinity of kindlin PH domains for target membranes. This is a charge-based interaction as shown by its loss when positively charged lysine residues are mutated to alanine (Figure 4). Similarly, many other proteins have been shown to interact with clusters of PtdInsP2 lipids on the basis of opposite charge attraction. For example, a series of basic residues in the MARCKS peptide from a protein kinase C substrate are known to bind to PtdIns(4,5)P2 lipid clusters, as do syntaxin-1A, BAR domains, and N-WASP. N-WASP is an activator of Arp2/3, which in turn nucleates actin filament formation at membranes [49,50].
There is increasing evidence showing that PH domains can interact with multiple lipid species simultaneously. A recent crystal structure of the ASAP1 PH domain suggested that there were two anionic lipid-binding sites on the ASAP1 PH domain (a canonical site and an atypical site) [51], and recent comparative MD simulations have suggested that both sites are present for many PH domains [33]. Moreover, Vonkova et al. [42] studied 91 different PH domains and showed that lipid co-operativity is essential for their recruitment to the membrane. Clustering of lipids by the kindlin-3 PH domain is expected to change the local lipid environment. Given that kindlins act in synergy with talin, which was also shown to interact with anionic lipids [52], this may provide a mechanism for how talin and kindlins, in turn, associate with the integrin. Focal adhesion kinase has also been shown to bind to inositol phosphates, inducing clustering of a protein-mediated kind [2,23].
The orientation of the kindlin-3 PH domain on a target membrane indicated by this study, and the fact that kindlins have a rather long unstructured loop in the F1 domain allow us to form a hypothesis for how the kindlin head domain may interact with the membrane and with the integrin receptor. In particular, the orientation of the PH domain that we find, in combination with the short linker regions plugging it into the F2 subdomain, suggests that it acts as a buffer or spacer between the kindlin and the membrane surface, and thereby allows the F3 subdomain to interact with the membrane-distal NPxY integrin β-subunit tail motif rather than the membrane-proximal NPxY bound by talin [53] (Figure 7). Thus, in combination with the N-terminal F0 domain [54] and the long F1 loop [24], the PH domain provides kindlins with a specific location and binding mode for integrin activation.
Figure 7. A proposed model for integrin activation by kindlin-3 with recruitment by clustered inositol phosphates to a membrane.
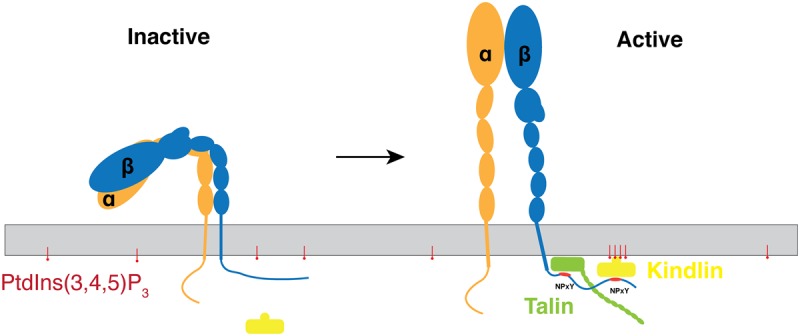
The activating conversion of integrins to an upright conformation is promoted by the binding of talin to the membrane-proximal NPxY and of kindlin to the membrane-distal NPxY on an integrin β-subunit cytoplasmic tail. Clustered PtdIns(3,4,5)P3 interacts with and recruits kindlin-3 from the cytosol to the membrane, thus facilitating the formation of an integrin–talin–kindlin ternary complex.
There are no high-resolution structural data for the rest of kindlin-3; however, it has been shown to have a similar structure to the talin head domain [5]. Therefore, further studies will focus on structure determination for kindlins and use structural and biophysical methods to test the hypothesis outlined above that binding of clustered phosphatidylinositol phosphate lipids is a mechanism employed by some proteins and specifically a subset of PH domains to enable the binding of membranes with an enhanced affinity.
Materials and methods
Constructs design, cloning, and mutagenesis
Mouse FERMT3 (UniProtKB: Q8K1B8) encoding kindlin-3 PH domain (residues 344–479) and Grp1 (general receptor of phosphoinositides 1, UniProtKB: O43739) PH domain (residues 264–392) were cloned into a pOPINJ vector, which contains a tandem His6 and GST (glutathione S-transferase) tag followed by a Human Rhinovirus-3C protease cleavage site in the N-terminus. The successful cloning was confirmed by DNA sequencing (Source Bioscience, U.K.). Mutagenesis and insertions were achieved by overlapping PCR following the standard protocol [55]. A DNA fragment encoding a 15-residue AviTag™ was inserted between the 3C protease cleavage site and the PH domain by PCR to enable in vivo protein biotinylation [56] using a BirA+ Escherichia coli strain for SPR experiments.
Expression of kindlin-3 PH domain and Grp1 PH domain
The construct encoding recombinant kindlin-3 PH domain was transformed into E. coli B834 strain. A single colony was picked from fresh transformants on an LB-agar–carbenicillin plate and cultured in 100 ml of LB supplemented with 50 µg/ml carbenicillin overnight. The pre-culture was diluted 50 times into LB media with antibiotics and incubated at 37°C with vigorous shaking at 200 rpm until OD600 reached ∼0.6. The expression of protein was induced by 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) at 18°C overnight. Cells were harvested by centrifugation at 6000 g for 20 min and pellets were frozen in a −80°C freezer until further use. Other proteins used in the present study were expressed using the same protocol.
Protein purification and crystallisation
The frozen cell pellet of the kindlin-3 PH domain (∼10 g of cell pellet from 2 l of overnight culture) was resuspended in lysis buffer [50 mM Tris (pH 7.5), 500 mM NaCl, and 0.1% Tween-20] and lysed with sonication, and the debris was separated by centrifugation at 40 000 g for 1 h. The resulting supernatant was incubated with glutathione sepharose equilibrated with lysis buffer for 2 h. The glutathione sepharose with protein bound was then washed with lysis buffer extensively. GST-3C protease was added to cleave the kindlin-3 PH domain from sepharose overnight at 4°C with gentle shaking. Supernatant containing a PH domain was concentrated and applied to size-exclusion chromatography (Superdex 75 16/60, GE Healthcare Life Sciences) in 10 mM Tris–HCl (pH 7.5), 150 mM NaCl, and 0.7 mM TCEP [tris(2-carboxyethyl)phosphine] buffer to obtain the purified proteins. The purification of kindlin-3 PH domain mutants and the Grp1 PH domain and biotinylated proteins was carried out in the same way. Successful in vivo biotinylation was verified by efficient binding to streptavidin magnetic beads (Sigma-49532).
Crystallisation screening was performed by means of sitting-drop vapour diffusion methods. Briefly, 100 nl of proteins at 20 mg/ml in 10 mM Tris–HCl (pH 7.5) and 150 mM NaCl were mixed with 100 nl of reservoirs and equilibrated against 95 µl of crystallisation reservoir (Hampton Research) at 293 K. Crystals appeared within 4 days and continued growing until 2 weeks in 4 M sodium nitrate and 0.1 M sodium acetate (pH 4.6). Crystals were flash-cooled with 25% (v/v) glycerol in the mother liquor as a cryoprotectant.
Data collection, processing, and structure determination
Diffraction data were collected using beamline I24 at Diamond Light Source (Didcot, U.K.) and then indexed, integrated, and scaled using the Xia2 pipeline [57]. The structure of the kindlin-3 PH domain was solved by molecular replacement (MR) using PHASER [58] with a polyalanine model of the kindlin-1 PH domain (PDB: 4BBK). The resulting model from MR was further built by Buccanneer [59] and corrected manually in Coot [60]. Refinement was accomplished using PHENIX.refine together with manual correction in Coot. Molecular graphics were prepared with PyMOL. Attempts at co-crystallising the kindlin-3 PH domain with Ins(1,3,4,5)P4 and at soaking the crystal with highly concentrated Ins(1,3,4,5)P4 resulted in a PH domain structure without ligand bound, presumably due to the low-affinity interaction between isolated Ins(1,3,4,5)P4 and the kindlin-3 PH domain.
Nanodisc preparation
POPC and POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) were purchased from Avanti Polar Lipids, and PtdIns(4,5)P2 (P-4516), PtdIns(3,4,5)P3 (P-3924), and Ins(1,3,4,5)P4 (Q-1345) were purchased from Echelon Biosciences. Nanodiscs were prepared following standard protocols [38]. Briefly, to prepare nandiscs containing 5% (mol/mol) PtdIns(3,4,5)P3, a mixture of 200 µl of 10 mg/ml POPC, 200 µl of 10 mg/ml POPE together with 670 µl of 0.5 mg/ml PtdIns(3,4,5)P3 in chloroform were dried on a clean Pyrex tube under argon. Chloroform residue was then removed by desiccation in a desiccator attached on a VARIO-SP diaphragm pump (Vacuubrand). The lipid film was subsequently hydrated by adding 1 ml of solubilisation buffer [20 mM HEPES (pH 7.5), 150 mM NaCl, and 17 mM Na-cholate] followed by vigorous votexing. The hydrated lipid film was then sonicated for 10 min in a water bath sonicator before 250 µl of MSP1E1 (7 mg/ml) proteins were added to the lipid mixture. Nanodiscs were self-assembled upon removal of Na-cholate by dialysing against 20 mM HEPES (pH 7.5) and 150 mM NaCl buffer overnight. The aggregate was removed by size-exclusion chromatography (Superdex 200 10/300, GE Healthcare Life Sciences), and the peak fractions corresponding to the size of monomeric nanodiscs species were collected (Supplementary Figure S9A) and used immediately for biophysical characterisation and SPR experiments.
Dynamic light scattering
Dynamic light scattering measurements were conducted on a Protein Solutions DynaPro instrument at 20°C. The nanodisc eluates from size-exclusion chromatography were diluted to ∼0.05 mg/ml in 10 mM HEPES (pH 7.5) and 150 mM NaCl before measurement in a cuvette.
Cryo-electron microscopy
After plunge-freezing in liquid ethane, nanodiscs on holey carbon grids were imaged using an FEI Tecnai F30 microscope operating at 200 kV and at a nominal magnification of 59 000×. Images were picked using Boxer [61] and reconstructed ab initio using IMAGIC [62] followed by the SPIDER [63] software. The resolution of the map was determined by Fourier shell correlation at FSC = 0.5 and was 23.5 Å.
Surface plasmon resonance
SPR experiments were performed using a Biacore T200 machine (GE Healthcare Life Sciences) at 20°C in 10 mM HEPES (pH 7.5) and 150 mM NaCl. PtdIns(4,5)P2 (C-45B6a) and PtdIns(3,4,5)P3 (C-39B6a) were purchased from Echelon Biosciences. The BIAcore CM5 chip (GE Healthcare Life Sciences) was firstly covered with streptavidin following the manufacturer's instructions before the biotinylated PIPs or biotinylated proteins were immobilised. The analyte with twofold serial dilutions was applied at a flow rate of 20 µl/min for 180 s followed by 600 s of dissociation time. The biosensor chip was regenerated after each sample injection cycle with a different buffer in the two experimental set-ups: in the case of immobilised PIPs on the sensor chip, 0.1% SDS was used; when the PH domain was immobilised, then the surface was regenerated by 2 M MgCl2. The data were fit with a 1:1 Langmuir adsorption model (B = BmaxC/(KD + C), where B is the response of bound analyte and C is the concentration of the analyte in the sample to calculate the dissociation constant (KD) using the BIAanalysis software. The molecular mass of the nanodiscs for SPR analysis was estimated to be 120 kDa.
Molecular dynamics simulations
CG-MD simulations
The CG-MD simulations were performed using GROMACS 4.5.5 [64] with the Martini 2.1 force field [65,66]. The simulation set-up is shown in Figure 2. A summary of all simulations performed is present in Table 2. Prior to the production simulations, all systems were energy minimised and then equilibrated for 500 ns with the protein backbone particles restrained. For each repeat simulation within an ensemble, the protein was rotated around the x, y, and z axes to form a different initial configuration. An ensemble of 20 simulations of 1.5 µs each was run with a time step of 20 fs. An elastic network model was applied to all backbone particles with a cut-off distance of 0.7 nm [67]. The LINCS algorithm was used to constrain to equilibrium bond lengths [68]. Lennard–Jones interactions were shifted to zero between 0.9 and 1.2 nm. Coulombic interactions were shifted to zero between 0 and 1.2 nm. The pressure was 1 bar and the temperature was 323 K. A Berendsen's algorithm [69] was used to control the pressure and the temperature with a coupling time of 1 ps.
AT-MD simulations
Conversion of CG to atomistic systems was made using a fragment-based approach [70]. The snapshot that was converted into an atomistic representation corresponded to the preferred orientation of the kindlin-3 PH domain in the density landscapes shown in Supplementary Figure S5. The GROMOS96 43a1 force field [71] was used with SPC water molecules. The temperature was 323 K. The velocity rescaling method [72] was used to control the temperature with a coupling time of 0.1 ps. The pressure was 1 bar, and it was controlled with semi-isotropic pressure coupling using the Parrinello–Rahman barostat [73] with a coupling time of 1 ps. Bond lengths were constrained to equilibrium lengths using the LINCS method. The time step was set at 2 fs. The particle mesh Ewald method was used to model the electrostatic interactions. For the atomistic simulation with the kindlin-3 PH domain in solution, both the GROMOS96 43a1 and the OPLS-AA force field within GROMACS were used. For these simulations, an isotropic pressure coupling was used. The temperature was 310 K.
PMF simulations
Initial structures of WT or mutant kindlin-3 PH domain bound to PIP3 or PIP2 in a 80:20 POPC:POPS bilayer were generated by alignment with an existing Grp1–PH/PIP3 complex (see ref. [34]). The system was converted into a coarse-grained representation. The PH domain was pulled from the bound PIP lipid along the membrane normal (z-axis) by fixing the centre of mass to a reference point, which was moved at a rate of 0.001 nm/ps for a total distance of 2.6 nm, while the PIP phosphate atom was restrained to its initial position. Snapshots with protein–lipid separations (measured from the PH domain centre of mass to the phosphate atom of the PIP) in 0.1 nm intervals for the first 2 and 0.2 nm thereafter were extracted and used as initial structures for umbrella sampling simulations. In each simulation, the phosphate atom was again restrained to its initial position and the z-distance of the PH centre of mass was restrained relative to the phosphate atom with a force constant of 1000 kJ/mol/nm2. To reduce the time required for adequate sampling, the PH domain centre of mass was also restrained in the x and y directions, by a force constant of 100 kJ/mol/nm. Each window was simulated for 1 µs (mutant), 1.4 µs (WT/PIP3), or 1.5 µs (WT/PIP2), with the distance between the PH centre of mass and the PIP atom bead along the z-axis recorded every 20 ps. PMF profiles were calculated using the GROMACS g_wham tool [74]. The time taken to reach convergence was judged from profiles generated for sequential time intervals; data before this time were discarded as equilibration before a final profile was calculated. Errors were obtained from bootstrap analysis. All profiles were shifted, so the value of the PMF in bulk was 0 kcal/mol.
Acknowledgements
T.N. was a Chinese Scholarships Council–University of Oxford scholar within the Nuffield Department of Medicine. We gratefully acknowledge the Diamond Light Source beamline staff and the support of Dr David Staunton with use of the Oxford Molecular Biophysics Suite within the University's Department of Biochemistry; the Division of Structural Biology is a part of the Wellcome Trust Centre for Human Genetics, Wellcome Trust Core Grant Number [090532/Z/09/Z]. Electron microscopy was performed using the Wellcome Trust-supported microscopy facility within the Division of Structural Biology established with a JIF award [060208/Z/00/Z] and further supported by a Trust equipment grant [093305/Z/10/Z]. A.C.K. and M.S.P.S. were also funded by the Wellcome Trust [WT092970MA].
Abbreviations
- AT-MD
atomistic molecular dynamics
- CG-MD
coarse-grained molecular dynamics
- EMT
epithelial-to-mesenchymal transition
- FERM domain
4.1-erythrin–radixin–moiesin domain
- FSC
Fourier shell correlation
- Grp1
general receptor of phosphoinositide 1
- GST
glutathione S-transferase
- IPRR
isoleucine–proline–arginine–arginine
- LAD-III
leucocyte adhesion deficiency type III
- MD
molecular dynamics
- MR
molecular replacement
- PH
pleckstrin homology
- PMF
potential of mean force
- POPS
palmitoyl-oleoyl glycerophosphoserine
- PtdInsP/PIP
phosphatidylinositol phosphate
- RMSD
root mean square deviation
- SPR
surface plasmon resonance
- WT
wild type.
Author Contribution
T.N., A.K., M.S.P.S. and R.J.C.G. conceived and designed the work. All authors contributed to the experimental aspects of the work. T.N., A.K., M.S.P.S. and R.J.C.G. wrote the paper, which all authors commented on and approved.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Humphries J.D., Byron A. and Humphries M.J. (2006) Integrin ligands at a glance. J. Cell Sci. 119, 3901–3903 doi: 10.1242/jcs.03098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harburger D.S. and Calderwood D.A. (2009) Integrin signalling at a glance. J. Cell Sci. 122, 159–163 doi: 10.1242/jcs.018093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Critchley D.R. (2009) Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Ann. Rev. Biophys. 38, 235–254 doi: 10.1146/annurev.biophys.050708.133744 [DOI] [PubMed] [Google Scholar]
- 4.Calderwood D.A., Campbell I.D. and Critchley D.R. (2013) Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517 doi: 10.1038/nrm3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yates L.A., Füzéry A.K., Bonet R., Campbell I.D. and Gilbert R.J.C. (2012) Biophysical analysis of kindlin-3 reveals an elongated conformation and maps integrin binding to the membrane-distal β-subunit NPXY motif. J. Biol. Chem. 287, 37715–37731 doi: 10.1074/jbc.M112.415208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brahme N.N. and Calderwood D.A. (2012) Cell adhesion: a FERM grasp of the tail sorts out integrins. Curr. Biol. 22, R692–R694 doi: 10.1016/j.cub.2012.07.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moser M., Legate K.R., Zent R. and Fässler R. (2009) The tail of integrins, talin, and kindlins. Science 324, 895–899 doi: 10.1126/science.1163865 [DOI] [PubMed] [Google Scholar]
- 8.Shattil S.J., Kim C. and Ginsberg M.H. (2010) The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11, 288–300 doi: 10.1038/nrm2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachir A.I., Zareno J., Moissoglu K., Plow E.F., Gratton E. and Horwitz A.R. (2014) Integrin-associated complexes form hierarchically with variable stoichiometry in nascent adhesions. Curr. Biol. 24, 1845–1853 doi: 10.1016/j.cub.2014.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ussar S., Wang H.-V., Linder S., Fässler R. and Moser M. (2006) The kindlins: subcellular localization and expression during murine development. Exp. Cell Res. 312, 3142–3151 doi: 10.1016/j.yexcr.2006.06.030 [DOI] [PubMed] [Google Scholar]
- 11.Bialkowska K., Ma Y.-Q., Bledzka K., Sossey-Alaoui K., Izem L., Zhang X. et al. (2010) The integrin co-activator kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J. Biol. Chem. 285, 18640–18649 doi: 10.1074/jbc.M109.085746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siegel D.H., Ashton G.H.S., Penagos H.G., Lee J.V., Feiler H.S., Wilhelmsen K.C. et al. (2003) Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin–extracellular-matrix linker protein UNC-112, causes kindler syndrome. Am. J. Hum. Genet. 73, 174–187 doi: 10.1086/376609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao T., Guan L., Yu Y., Pei X., Zhan J., Han L. et al. (2013) Kindlin-2 promotes genome instability in breast cancer cells. Cancer Lett. 330, 208–216 doi: 10.1016/j.canlet.2012.11.043 [DOI] [PubMed] [Google Scholar]
- 14.Yu Y., Wu J., Wang Y., Zhao T., Ma B., Liu Y. et al. (2012) Kindlin 2 forms a transcriptional complex with β-catenin and TCF4 to enhance Wnt signalling. EMBO Rep. 13, 750–758 doi: 10.1038/embor.2012.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyd R.S., Adam P.J., Patel S., Loader J.A., Berry J., Redpath N.T. et al. (2003) Proteomic analysis of the cell-surface membrane in chronic lymphocytic leukemia: identification of two novel proteins, BCNP1 and MIG2B. Leukemia 17, 1605–1612 doi: 10.1038/sj.leu.2402993 [DOI] [PubMed] [Google Scholar]
- 16.Sossey-Alaoui K., Pluskota E., Davuluri G., Bialkowska K., Das M., Szpak D. et al. (2014) Kindlin-3 enhances breast cancer progression and metastasis by activating twist-mediated angiogenesis. FASEB J. 28, 2260–2271 doi: 10.1096/fj.13-244004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z., Chen X., Zhi H., Gao J., Bialkowska K., Byzova T.V. et al. (2014) Direct interaction of kindlin-3 with integrin αIIbβ3 in platelets is required for supporting arterial thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 34, 1961–1967 doi: 10.1161/ATVBAHA.114.303851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moser M., Nieswandt B., Ussar S., Pozgajova M. and Fässler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 doi: 10.1038/nm1722 [DOI] [PubMed] [Google Scholar]
- 19.Yu Y., Wu J., Guan L., Qi L., Tang Y., Ma B. et al. (2013) Kindlin 2 promotes breast cancer invasion via epigenetic silencing of the microRNA200 gene family. Int. J. Cancer 133, 1368–1379 doi: 10.1002/ijc.28151 [DOI] [PubMed] [Google Scholar]
- 20.Huet-Calderwood C., Brahme N.N., Kumar N., Stiegler A.L., Raghavan S., Boggon T.J. et al. (2014) Differences in binding to the ILK complex determines kindlin isoform adhesion localization and integrin activation. J. Cell Sci. 127, 4308–4321 doi: 10.1242/jcs.155879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hart R., Stanley P., Chakravarty P. and Hogg N. (2013) The kindlin 3 pleckstrin homology domain has an essential role in lymphocyte function-associated antigen 1 (LFA-1) integrin-mediated B cell adhesion and migration. J. Biol. Chem. 288, 14852–14862 doi: 10.1074/jbc.M112.434621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y., Zhu Y., Ye S. and Zhang R. (2012) Crystal structure of kindlin-2 PH domain reveals a conformational transition for its membrane anchoring and regulation of integrin activation. Protein Cell 3, 434–440 doi: 10.1007/s13238-012-2046-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yates L.A., Lumb C.N., Brahme N.N., Zalyte R., Bird L.E., De Colibus L. et al. (2012) Structural and functional characterization of the kindlin-1 pleckstrin homology domain. J. Biol. Chem. 287, 43246–43261 doi: 10.1074/jbc.M112.422089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouaouina M., Goult B.T., Huet-Calderwood C., Bate N., Brahme N.N., Barsukov I.L. et al. (2012) A conserved lipid-binding loop in the kindlin FERM F1 domain is required for kindlin-mediated αIIbβ3 integrin coactivation. J. Biol. Chem. 287, 6979–6990 doi: 10.1074/jbc.M111.330845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mory A., Feigelson S.W., Yarali N., Kilic S.S., Bayhan G.I., Gershoni-Baruch R. et al. (2008) Kindlin-3: a new gene involved in the pathogenesis of LAD-III, Blood 112, 2591 doi: 10.1182/blood-2008-06-163162 [DOI] [PubMed] [Google Scholar]
- 26.Lemmon M.A., Ferguson K.M. and Abrams C.S. (2002) Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 513, 71–76 doi: 10.1016/S0014-5793(01)03243-4 [DOI] [PubMed] [Google Scholar]
- 27.Lai C.-L., Srivastava A., Pilling C., Chase A.R., Falke J.J. and Voth G.A. (2013) Molecular mechanism of membrane binding of the GRP1 PH domain. J. Mol. Biol. 425, 3073–3090 doi: 10.1016/j.jmb.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson K.M., Lemmon M.A., Schlessinger J. and Sigler P.B. (1995) Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83, 1037–1046 doi: 10.1016/0092-8674(95)90219-8 [DOI] [PubMed] [Google Scholar]
- 29.Anand K., Maeda K. and Gavin A.-C. (2012) Structural analyses of the Slm1-PH domain demonstrate ligand binding in the non-canonical site. PLoS ONE 7, e36526 doi: 10.1371/journal.pone.0036526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ceccarelli D.F.J., Blasutig I.M., Goudreault M., Li Z., Ruston J., Pawson T. et al. (2007) Non-canonical interaction of phosphoinositides with pleckstrin homology domains of Tiam1 and ArhGAP9. J. Biol. Chem. 282, 13864–13874 doi: 10.1074/jbc.M700505200 [DOI] [PubMed] [Google Scholar]
- 31.Kalli A.C. and Sansom M.S. P. (2014) Interactions of peripheral proteins with model membranes as viewed by molecular dynamics simulations. Biochem. Soc. Trans. 42, 1418–1424 doi: 10.1042/BST20140144 [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto E., Kalli A.C., Akimoto T., Yasuoka K. and Sansom M.S. P. (2015) Anomalous dynamics of a lipid recognition protein on a membrane surface. Sci. Rep. 5, 18245 doi: 10.1038/srep18245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamoto E., Kalli A.C., Yasuoka K. and Sansom M.S.P. (2016) Interactions of pleckstrin homology domains with membranes: adding back the bilayer via high-throughput molecular dynamics. Structure 24, 1421–1431 doi: 10.1016/j.str.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naughton F.B., Kalli A.C. and Sansom M.S.P. (2016) Association of peripheral membrane proteins with membranes: free energy of binding of GRP1 PH domain with phosphatidylinositol phosphate-containing model bilayers. J. Phys. Chem. Lett. 7, 1219–1224 doi: 10.1021/acs.jpclett.6b00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lumb C.N., He J., Xue Y., Stansfeld P.J., Stahelin R.V., Kutateladze T.G. et al. (2011) Biophysical and computational studies of membrane penetration by the GRP1 pleckstrin homology domain. Structure 19, 1338–1346 doi: 10.1016/j.str.2011.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orłowski A., Kukkurainen S., Pöyry A., Rissanen S., Vattulainen I., Hytönen V.P. et al. (2015) PIP2 and talin join forces to activate integrin. J. Phys. Chem. B 119, 12381–12389 doi: 10.1021/acs.jpcb.5b06457 [DOI] [PubMed] [Google Scholar]
- 37.Schmidt T., Suk J.-E., Ye F., Situ A.J., Mazumder P., Ginsberg M.H. et al. (2015) Annular anionic lipids stabilize the integrin αIIbβ3 transmembrane complex. J. Biol. Chem. 290, 8283–8293 doi: 10.1074/jbc.M114.623504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borch J. and Hamann T. (2009) The nanodisc: a novel tool for membrane protein studies. Biol. Chem. 390, 805–814 doi: 10.1515/BC.2009.091 [DOI] [PubMed] [Google Scholar]
- 39.Wang X., Mu Z., Li Y., Bi Y. and Wang Y. (2015) Smaller nanodiscs are suitable for studying protein lipid interactions by solution NMR. Protein J. 34, 205–211 doi: 10.1007/s10930-015-9613-2 [DOI] [PubMed] [Google Scholar]
- 40.Ellenbroek W.G., Wang Y.-H., Christian D.A., Discher D.E., Janmey P.A. and Liu A.J. (2011) Divalent cation-dependent formation of electrostatic PIP2 clusters in lipid monolayers. Biophys. J. 101, 2178–2184 doi: 10.1016/j.bpj.2011.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Bogaart G., Meyenberg K., Risselada H.J., Amin H., Willig K.I., Hubrich B.E. et al. (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 doi: 10.1038/nature10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vonkova I., Saliba A.-E., Deghou S., Anand K., Ceschia S., Doerks T. et al. (2015) Lipid cooperativity as a general membrane-recruitment principle for PH domains. Cell Rep. 12, 1519–1530 doi: 10.1016/j.celrep.2015.07.054 [DOI] [PubMed] [Google Scholar]
- 43.Ferguson K.M., Kavran J.M., Sankaran V.G., Fournier E., Isakoff S.J., Skolnik E.Y. et al. (2000) Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol. Cell 6, 373–384 doi: 10.1016/S1097-2765(00)00037-X [DOI] [PubMed] [Google Scholar]
- 44.Pilling C., Landgraf K.E. and Falke J.J. (2011) The GRP1 PH domain, like the AKT1 PH domain, possesses a sentry glutamate residue essential for specific targeting to plasma membrane PI(3,4,5)P3. Biochemistry 50, 9845–9856 doi: 10.1021/bi2011306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moravcevic K., Oxley C.L. and Lemmon M.A. (2012) Conditional peripheral membrane proteins: facing up to limited specificity. Structure 20, 15–27 doi: 10.1016/j.str.2011.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Meer G., Voelker D.R. and Feigenson G.W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 doi: 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayburt T.H. and Sligar S.G. (2010) Membrane protein assembly into nanodiscs. FEBS Lett. 584, 1721–1727 doi: 10.1016/j.febslet.2009.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borhani D.W., Rogers D.P., Engler J.A. and Brouillette C.G. (1997) Crystal structure of truncated human apolipoprotein AI suggests a lipid-bound conformation. Proc. Natl Acad. Sci. U.S.A. 94, 12291–12296 doi: 10.1073/pnas.94.23.12291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lemmon M.A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 doi: 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- 50.Brown D.A. (2015) PIP2Clustering: from model membranes to cells. Chem. Phys. Lipids 192, 33–40 doi: 10.1016/j.chemphyslip.2015.07.021 [DOI] [PubMed] [Google Scholar]
- 51.Jian X., Tang W.-K., Zhai P., Roy N.S., Luo R., Gruschus J.M. et al. (2015) Molecular basis for cooperative binding of anionic phospholipids to the PH domain of the Arf GAP ASAP1. Structure 23, 1977–1988 doi: 10.1016/j.str.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goñi G.M., Epifano C., Boskovic J., Camacho-Artacho M., Zhou J., Bronowska A. et al. (2014) Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl Acad. Sci. U.S.A. 111, E3177–E3186 doi: 10.1073/pnas.1317022111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calderwood D.A., Zent R., Grant R., Rees D.J.G., Hynes R.O. and Ginsberg M.H. (1999) The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274, 28071–28074 doi: 10.1074/jbc.274.40.28071 [DOI] [PubMed] [Google Scholar]
- 54.Goult B.T., Bouaouina M., Harburger D.S., Bate N., Patel B., Anthis N.J. et al. (2009) The structure of the N-terminus of kindlin-1: a domain important for αIIbβ3 integrin activation. J. Mol. Biol. 394, 944–956 doi: 10.1016/j.jmb.2009.09.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heckman K.L. and Pease L.R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 doi: 10.1038/nprot.2007.132 [DOI] [PubMed] [Google Scholar]
- 56.Fairhead M. and Howarth M. (2014) Site-specific biotinylation of purified proteins using BirA. Meth. Mol. Biol. 1266, 171–184 doi: 10.1007/978-1-4939-2272-7_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winter G. (2010) Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 doi: 10.1107/S0021889809045701 [DOI] [Google Scholar]
- 58.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N. et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 doi: 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 doi: 10.1107/S0907444906022116 [DOI] [PubMed] [Google Scholar]
- 60.Emsley P. and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 doi: 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 61.Ludtke S.J., Baldwin P.R. and Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 doi: 10.1006/jsbi.1999.4174 [DOI] [PubMed] [Google Scholar]
- 62.van Heel M., Harauz G., Orlova E.V., Schmidt R. and Schatz M. (1996) A new generation of the IMAGIC image processing system. J. Struct. Biol. 116, 17–24 doi: 10.1006/jsbi.1996.0004 [DOI] [PubMed] [Google Scholar]
- 63.Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M. et al. (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 doi: 10.1006/jsbi.1996.0030 [DOI] [PubMed] [Google Scholar]
- 64.Hess B., Kutzner C., van der Spoel D. and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 doi: 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- 65.Marrink S.J., Risselada H.J., Yefimov S., Tieleman D.P. and de Vries A.H. (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 doi: 10.1021/jp071097f [DOI] [PubMed] [Google Scholar]
- 66.Monticelli L., Kandasamy S.K., Periole X., Larson R.G., Tieleman D.P. and Marrink S.-J. (2008) The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834 doi: 10.1021/ct700324x [DOI] [PubMed] [Google Scholar]
- 67.Atilgan A.R., Durell S.R., Jernigan R.L., Demirel M.C., Keskin O. and Bahar I. (2001) Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80, 505–515 doi: 10.1016/S0006-3495(01)76033-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hess B., Bekker H., Berendsen H. and Fraaije J.G.E.M. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472<missing element Fraaije Johannes G. E. M., Fraaije Johannes G. E. M. > [Google Scholar]
- 69.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A. and Haak J.R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 doi: 10.1063/1.448118 [DOI] [Google Scholar]
- 70.Stansfeld P.J. and Sansom M.S.P. (2011) From coarse grained to atomistic: a serial multiscale approach to membrane protein simulations. J. Chem. Theory Comput. 7, 1157–1166 doi: 10.1021/ct100569y [DOI] [PubMed] [Google Scholar]
- 71.Scott W.R.P., Hünenberger P.H., Tironi I.G., Mark A.E., Billeter S.R., Fennen J. et al. (1999) The GROMOS biomolecular simulation program package. J. Phys. Chem. A 103, 3596–3607 doi: 10.1021/jp984217f [DOI] [Google Scholar]
- 72.Bussi G., Donadio D. and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 doi: 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 73.Parrinello M. and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182 doi: 10.1063/1.328693 [DOI] [Google Scholar]
- 74.Hub J.S., de G: root B.L., and van der Spoel D. (2010). g_wham — a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 6, 3713–3720 doi: 10.1021/ct100494z [DOI] [Google Scholar]