

Abstract
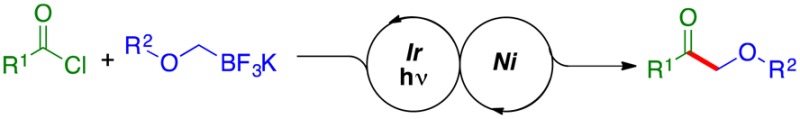
A visible-light, single-electron-transfer (SET), photoredox cross-coupling for the synthesis of α-alkoxyketones has been developed. In this method, various aliphatic and aromatic acyl chlorides were successfully coupled with structurally diverse potassium alkoxymethyltrifluoroborates, producing the corresponding α-alkoxyketones with high yields. In this operationally simple and mild cross-coupling protocol, the desired ketones are obtained in one step from bench stable starting materials by a bond connection that is unique to both alkylboron chemistry and photoredox/Ni catalysis.
Among the many virtues of organoboron reagents is their tolerance of sensitive functional groups, which can be used to advantage for late-stage incorporation of functional groups within highly elaborated substructures. In counterbalance to this is the decidedly nonpolar nature of the carbon–boron bond, which attenuates the nucleophilic reactivity of alkylboron reagents toward nearly all electrophiles. As a consequence of this limited reactivity, an effective means to activate alkylboron reagents in a highly selective manner is of utmost importance and value in order to take full advantage of their desirable physical and chemical properties.
We, and others, have recently unveiled novel reactivity patterns of alkyltrifluoroborates based on single electron transfer (SET) oxidation, generating radicals that can take part in a variety of diverse transformations.1 As an important extension of this, we introduced a novel concept for dual catalytic cross-coupling in which the effective transmetalation occurs via an SET pathway, exploiting the production of radicals generated upon oxidation of the boron reagents.2 Visible light photoredox catalysis based on simultaneous activation and engagement of the reactive partners with two distinct catalysts through low-barrier, open-shell processes has emerged as a valuable paradigm for the design of single electron-transfer pathways.3 Under the dual catalysis paradigm, the synergistic roles of an Ir photoredox catalyst and a Ni catalyst enable broad application in the cross-coupling of diverse potassium alkyltrifluoroborates with aryl and heteroaryl bromides under remarkably mild conditions (i.e., weak base, room temperature).2a,4
In an effort to expand the scope of this paradigm for cross-coupling, attention was turned toward its application to halide electrophiles outside of the aryl/heteroaryl manifold. The goal was to provide access to useful substructures in an approach that was underrepresented in terms of previous alkylboron chemistry. Acyl chlorides were targeted as the first of such electrophiles.
Because of their enhanced reactivity, acyl halides represent virtually the only class of electrophiles that can react with organoboron “ate” complexes in intermolecular reactions.5 Despite their relatively high reactivity as electrophiles, acyl chlorides have been used relatively sparingly in reactions with alkylboron compounds. One of the inherent drawbacks of the methods applied to date is the necessity to handle air-sensitive trialkylboranes, from which the requisite “ate” complexes are generated. Additionally, only one of the three alkyl groups on boron is transferred. The latter issue can be resolved by employing alkyl-9-BBN reagents from which appropriate “ate” complexes can be generated, but air sensitivity remains an issue, and with the 9-BBN derivatives one is additionally restricted in terms of functional group incorporation and accessibility to diverse substructures.
Boronic acids and boronate esters resolve both of these particular issues associated with trialkylboron “ate” complexes, but not without some added burdens of their own. Knochel developed a transmetalative protocol that was quite effective as a route to ketones,6 but this protocol required the use of 6 equiv of flammable dialkylzinc reagents and 2 equiv of CuCN to effect the acylation. Beyond this, there appear to be only two reports of transition-metal-catalyzed cross-coupling between alkylboronic acids and carboxylic acid chlorides. In the first, a single example (phenethylboronic acid) was reported of a Pd-catalyzed process that was carried out at 70 °C with no yield or detailed procedure.7 The second was restricted to cyclopropylboronic acids (possessing significant Csp2-bond character), which also involved a Pd-catalyzed procedure requiring 2 equiv of acid chloride and 2 equiv of Ag2O in toluene at 80 °C.8
To broaden the range of electrophiles to which the photoredox/Ni dual catalytic cross-coupling could be applied, the reaction of acyl chlorides with potassium α-alkoxymethyltrifluoroborates was chosen as a test case (eq 1). The synthesis of α-alkoxyketones has attracted much attention. These structures serve as versatile and valuable building blocks in the construction of various natural products, as well as being of some importance in food chemistry, where they have been identified as flavor components in a wide variety of foods.9
Although there are many techniques available for the synthesis of α-alkoxyketones,10 often these methods are limited and demonstrate a lack of generality because of their harsh conditions. Also in currently available protocols, α-alkoxyketones are generally obtained by multistep syntheses in low overall yields. Thus, a complementary, operationally simple, mild, and high yield synthesis route would enjoy a privileged role in accessing these structures.
 |
1 |
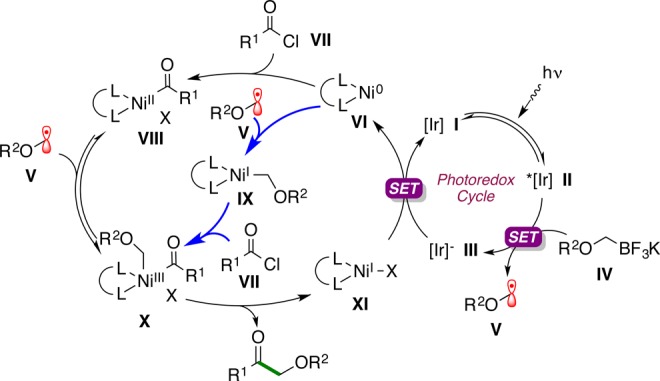
Scheme 1 shows the mechanism for Ir-photoredox/Ni dual catalysis that we proposed would lead to the desired transformation. The mechanistic scheme is adapted from our studies on cross-coupling of alkyltrifluoroborates with aryl and heteroaryl halides.11
Scheme 1. Proposed Mechanism for the Photoredox Cross-Coupling of Acyl Chlorides with Potassium Alkoxymethyltrifluoroborates.
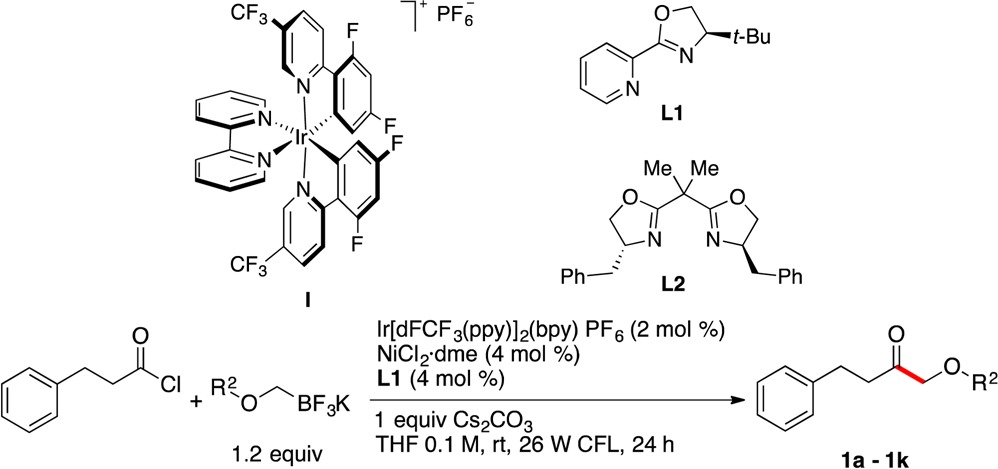
To validate the visible-light photoredox cross-coupling of acyl chlorides with potassium alkoxymethyltrifluoroborates, hydrocinnamoyl chloride and potassium benzyloxymethyltrifluoroborate were chosen as model coupling partners in the presence of Ir[dF(CF3)ppy]2(bpy)PF6, [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, bpy = 2,2′-bipyridine] I as a photocatalyst. An extensive screening of various reaction parameters (e.g., solvent, Ni catalyst, ligand, and additive) was carried out by means of microscale high-throughput experimentation (HTE) to determine suitable conditions for cross-coupling.12 As a result of the initial screens, the treatment of hydrocinnamoyl chloride with potassium benzyloxymethyltrifluoroborate in the presence of 3 mol % of I, 6 mol % of NiCl2·dme, 6 mol % of 4-tert-butyl-2-(2-pyridyl)oxazoline L1, and 1 equiv of lutidine in THF at room temperature produced the desired α-alkoxyketone in good yield (see Supporting Information, Table S1, entry 1, 74% yield). Further investigations revealed that changing the base from lutidine to Cs2CO3 provided a significant improvement in yield (Table S1, entry 2, 83%). Also, a decrease of catalyst loading to 2 mol % of I, 4 mol % of NiCl2·dme, and 4 mol % of L1 did not affect the yield of the reaction considerably and is economically and environmentally beneficial (entry 5). Lower concentrations (0.05 M) increase the yield of the reaction, while higher molarities (0.2 M) result in diminished yields (Table S1, entries 6 and 7, respectively). Finally, control experiments were carried out to confirm the essential roles of the photocatalyst, Ni catalyst, and additives (Table S1, entries 3–5).
These conditions were used to explore the scope of the alkoxymethyltrifluoroborate component in this Csp2–Csp3 cross-coupling protocol in reactions with hydrocinnamoyl chloride (Table 1). All types of alkoxymethyltrifluoroborates utilized, containing primary, secondary, and tertiary alkoxy groups (entries 1–8, 9, and 10, respectively), were successfully coupled to give the corresponding products in moderate to very good yields. To demonstrate the scalable nature of this coupling, a reaction on 5.9 mmol scale to generate 1a was performed. In this reaction, using reduced loadings of the catalysts (1 mol % of I and 2 mol % of Ni/L1), the desired product was obtained in 75% yield (entry 1, Table 1). In contrast to traditional cross-couplings, in this proposed SET mechanism radical intermediates are produced from alkoxymethyltrifluoroborate precursors. Therefore, the stereochemical outcome of the reaction is presumably determined by the rate of reductive elimination from diastereomeric complexes X (Scheme 1), where the ligands on Ni are enantioenriched.10 Consequently, use of an appropriate chiral ligand would enable the stereoconvergent synthesis of enantioenriched α-alkoxyketones from racemic organotrifluoroborate starting materials via prochiral radical intermediates. Using hydrocinnamoyl chloride and racemic potassium 3-benzyloxy-3-trifluoroboratopropylbenzene as a model reaction, a screen of various chiral ligands under the developed conditions was conducted. Use of 2,2′-isopropylidenebis[(4R)-4-benzyl-2-oxazoline, L2] as a ligand afforded the desired ketone 1k in 91% yield with an enantiomeric ratio (er) of 81:19 (entry 11, Table 1).
Table 1. Scope of Potassium Alkoxymethyltrifluoroborates in Cross-Coupling with Hydrocinnamoyl Chloride.
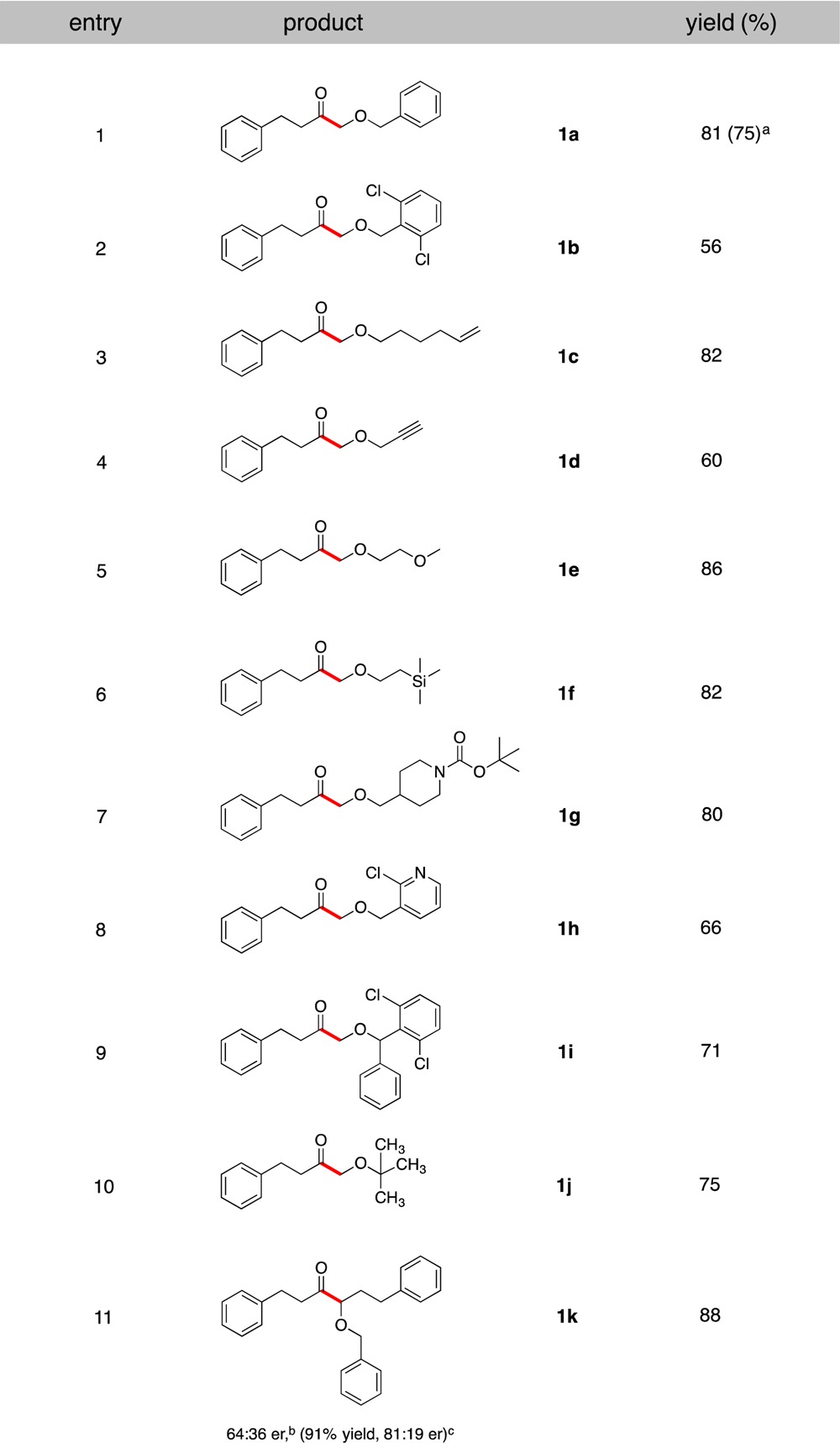
Reaction completed on 5.9 mmol scale with 1 mol % Ir photocatalyst and 2 mol % NiCl2·dme/L1.
Determined by SFC using L1 under the optimized conditions.
Determined by SFC using 4 mol % L2 as a ligand at −25 °C. er = enantiomeric ratio.
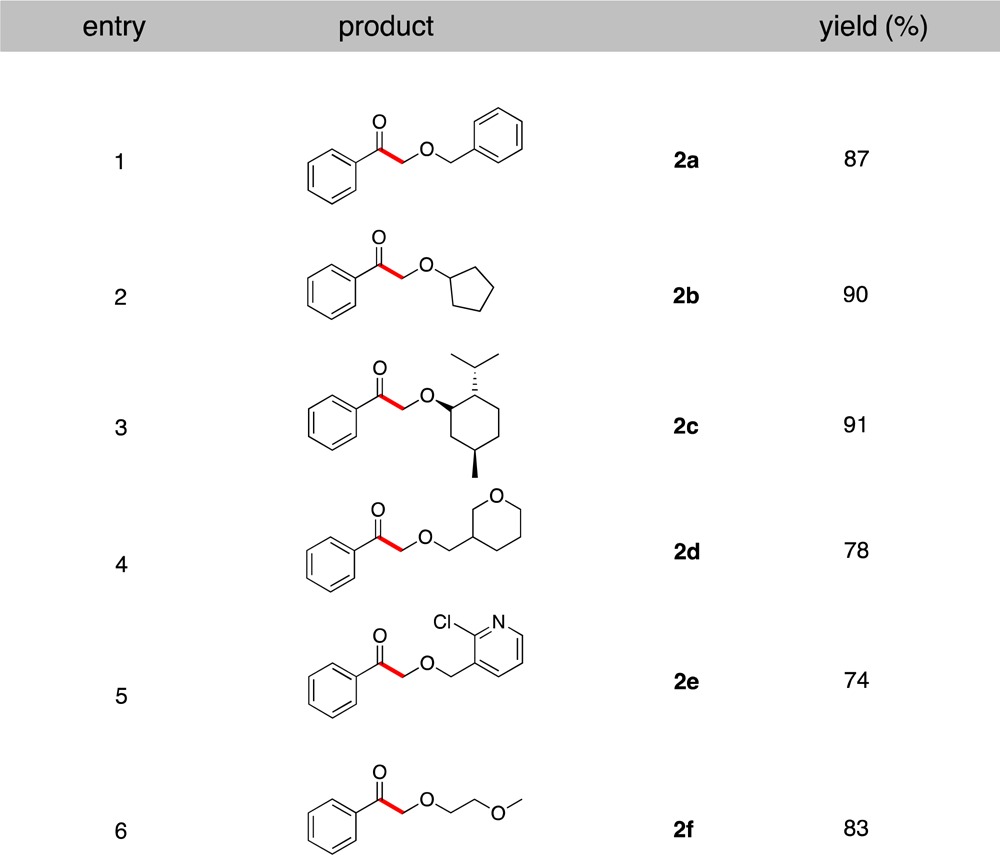
To demonstrate the efficiency of this protocol for the coupling of aromatic acyl chlorides, benzoyl chloride was reacted with different alkoxymethyltrifluoroborates under the optimal conditions (Table 2). All of the alkoxymethyltrifluoroborates utilized were successfully coupled, affording the desired products in yields up to 91%. These results serve to highlight the complementary nature of this novel method for the synthesis of both aliphatic and aromatic α-alkoxyketones.
Table 2. Scope of Potassium Alkoxymethyltrifluoroborates in Cross-Coupling with Benzoyl Chloride.
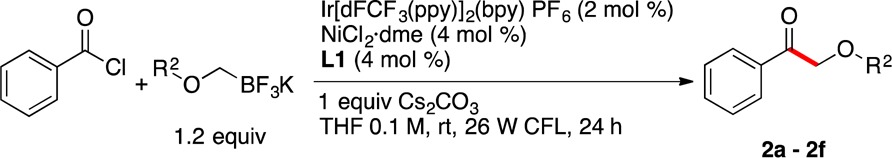
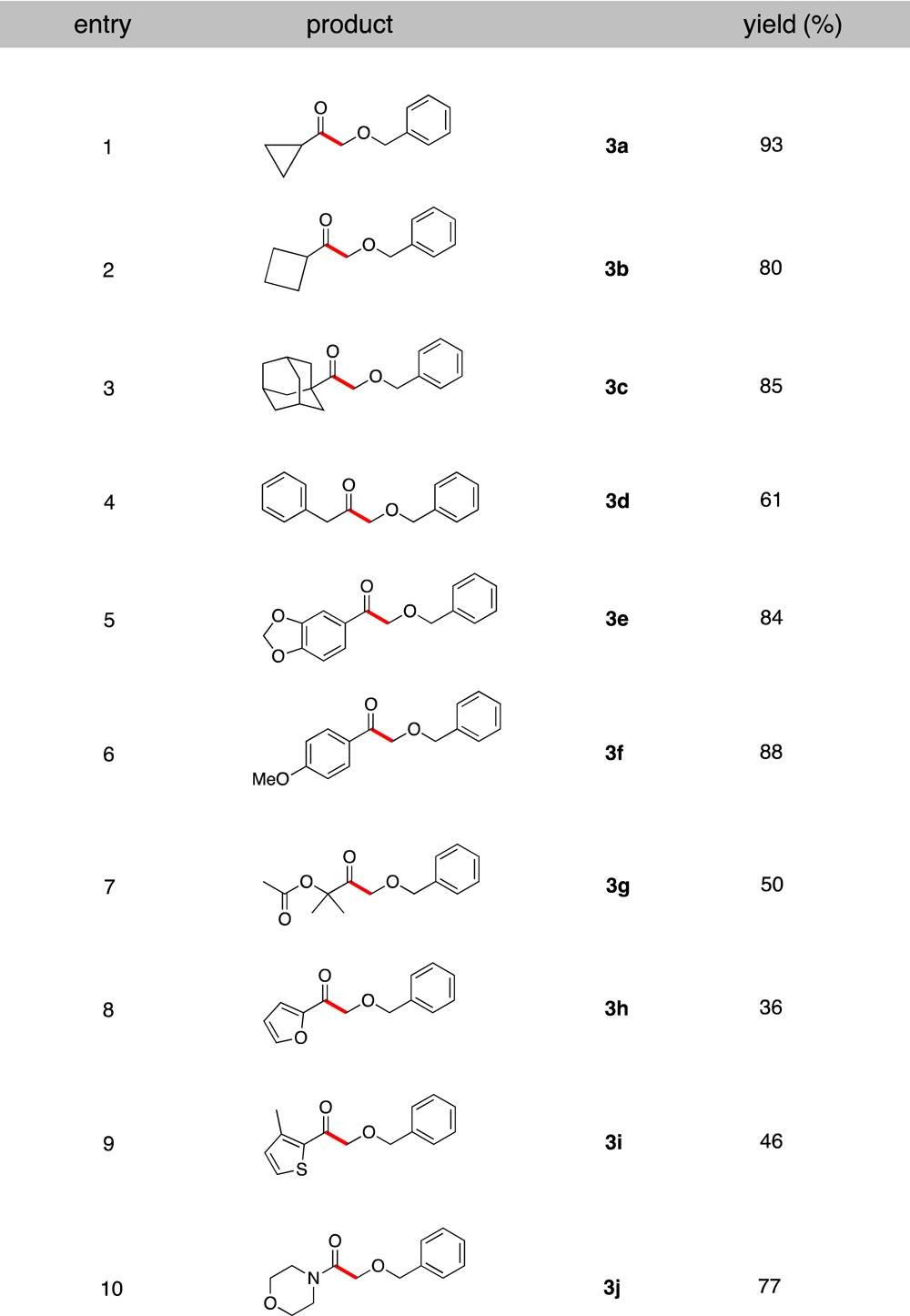
Next, attention was turned to exploring the scope of the acyl chloride component. Interestingly, as revealed in Table 3, we found that a broad range of acyl chlorides readily participate in this cross-coupling reaction. Cyclopropane- and cyclobutanecarbonyl chlorides were well tolerated, generating the corresponding α-alkoxyketones in 93% and 80% yields, respectively (entries 1 and 2). An acyl chloride bearing a bulky group, 1-adamantanecarbonyl chloride, provided the desired cross-coupled product with an excellent yield (entry 3, 85%). Additionally, the reaction with 4-morpholinecarbonyl chloride produced 2-(benzyloxy)-1-morpholinoethanone as an α-alkoxyamide in 77% yield (entry 10).
Table 3. Scope of Acyl Chlorides in the Cross-Coupling.

In conclusion, a visible light photoredox cross-coupling of acyl chlorides with potassium alkoxymethyltrifluoroborates has been developed to access a variety of α-alkoxyketones with high yields. This mild and operationally simple method is functional group tolerant and produces the desired ketones in one step from bench stable starting materials. The protocol described herein not only represents an expansion of the photoredox/Ni dual catalytic paradigm into new electrophile space but also constitutes one of the few methods to couple alkylboron reagents with acyl chlorides. Because of the scope of the reaction, this cross-coupling protocol provides an efficient means to synthesize complex α-alkoxyketones. Further development and application of this protocol is underway in our laboratory.
Acknowledgments
This research was supported by the NIGMS (R01 GM 113878). Frontier Scientific is thanked for the donation of boron compounds used in this investigation. Johnson-Matthey is acknowledged for the donation of IrCl3 used for the synthesis of the photocatalyst. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.5b03705.
Experimental procedures, HTE data, compound characterization data, and NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Sorin G.; Mallorquin R. M.; Contie Y.; Baralle A.; Malacria M.; Goddard J.-P.; Fensterbank L. Angew. Chem., Int. Ed. 2010, 49, 8721. 10.1002/anie.201004513. [DOI] [PubMed] [Google Scholar]; b Molander G. A.; Colombel V.; Braz V. A. Org. Lett. 2011, 13, 1852. 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lockner J. W.; Dixon D. D.; Risgaard R.; Baran P. S. Org. Lett. 2011, 13, 5628. 10.1021/ol2023505. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yasu Y.; Koike T.; Akita M. Adv. Synth. Catal. 2012, 354, 3414. 10.1002/adsc.201200588. [DOI] [Google Scholar]; e Chinzei T.; Miyazawa K.; Yasu Y.; Koike T.; Akita M. RSC Adv. 2015, 5, 21297. 10.1039/C5RA01826A. [DOI] [Google Scholar]; f Miyazawa K.; Koike T.; Akita M. Adv. Synth. Catal. 2014, 356, 2749. 10.1002/adsc.201400556. [DOI] [Google Scholar]; g Miyazawa K.; Yasu Y.; Koike T.; Akita M. Chem. Commun. 2013, 49, 7249. 10.1039/c3cc42695e. [DOI] [PubMed] [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also:; b Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Prier C. K.; Rankic D. A.; Macmillan D. W. C. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hopkinson M. N.; Sahoo B.; Li J.-L.; Glorius F. Chem. - Eur. J. 2014, 20, 3874. 10.1002/chem.201304823. [DOI] [PubMed] [Google Scholar]; c Schultz D. M.; Yoon T. P. Science 2014, 343, 985. 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lang S. B.; O'Nele K. M.; Tunge J. A. J. Am. Chem. Soc. 2014, 136, 13606. 10.1021/ja508317j. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lang S. B.; O’Nele K. M.; Douglas J. T.; Tunge J. A. Chem. - Eur. J. 2015, 21, 18589. 10.1002/chem.201503644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ryu D.; Primer D. N.; Tellis J. C.; Molander G. A. Chem. - Eur. J. 2016, 22, 120. 10.1002/chem.201504079. [DOI] [PubMed] [Google Scholar]; d El-Khatib M.; Serafim R. A. M.; Molander G. A. Angew. Chem., Int. Ed. 2016, 55, 254. 10.1002/anie.201506147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi E.-i.; Idacavage M. J. Org. React. 1985, 33, 1. 10.1002/0471264180.or033.01. [DOI] [Google Scholar]
- Langer F.; Waas J.; Knochel P. Tetrahedron Lett. 1993, 34, 5261. 10.1016/S0040-4039(00)73968-2. [DOI] [Google Scholar]
- Mallari J. P.; Shelat A.; Kosinski A.; Caffrey C. R.; Connelly M.; Zhu F.; McKerrow J. H.; Guy R. K. Bioorg. Med. Chem. Lett. 2008, 18, 2883. 10.1016/j.bmcl.2008.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Deng M.-Z. Org. Lett. 2000, 2, 1649. 10.1021/ol000013h. [DOI] [PubMed] [Google Scholar]
- a Mori M.; Inouye Y.; Kakisawa H. Chem. Lett. 1989, 1021. 10.1246/cl.1989.1021. [DOI] [Google Scholar]; b Edwards R. L.; Whalley A. J. S. J. Chem. Soc., Perkin Trans. 1 1979, 803. 10.1039/p19790000803. [DOI] [Google Scholar]; c Kuwahara Y.; Yen L. T. M.; Tominaga Y.; Matsumoto K.; Wada Y. Agric. Biol. Chem. 1982, 46, 2283. 10.1271/bbb1961.46.2283. [DOI] [Google Scholar]; d Nakatsu T.; Johns T.; Kubo I.; Milton K.; Sakai M.; Chatani K.; Saito K.; Yamagiwa Y.; Kamikawa T. J. Nat. Prod. 1990, 53, 1508. 10.1021/np50072a017. [DOI] [PubMed] [Google Scholar]; e Krafft M. E.; Dasse O. A.; Jarrett S.; Fievre A. J. Org. Chem. 1995, 60, 5093. 10.1021/jo00121a029. [DOI] [Google Scholar]; f Francis C. L.; Williamson N. M.; Ward A. D. Synthesis 2004, 2004, 2685. 10.1055/s-2004-831234. [DOI] [Google Scholar]; g Schreier P. J. Agric. Food Chem. 1980, 28, 926. 10.1021/jf60231a020. [DOI] [Google Scholar]; h Ramírez R.; Cava R. J. Agric. Food Chem. 2007, 55, 1923. 10.1021/jf062810l. [DOI] [PubMed] [Google Scholar]; i Zeppa G.; Giordano M.; Gerbi V.; Meglioli G. Ital. J. Food Sci. 2002, 14, 247. [Google Scholar]
- a Henze H. R.; Rigler N. E. J. Am. Chem. Soc. 1934, 56, 1350. 10.1021/ja01321a033. [DOI] [Google Scholar]; b Henze H. R.; Benz G. W.; Sutherland G. L. J. Am. Chem. Soc. 1949, 71, 2122. 10.1021/ja01174a060. [DOI] [Google Scholar]; c Wallace W. P.; Henze H. R. J. Am. Chem. Soc. 1942, 64, 2882. 10.1021/ja01264a046. [DOI] [Google Scholar]; d Barnes R. A.; Budde W. M. J. Am. Chem. Soc. 1946, 68, 2339. 10.1021/ja01215a059. [DOI] [Google Scholar]; e Castro B. Bull. Soc. Chim. Fr. 1967, 1540. [Google Scholar]; f Bartlett P. D.; Ross S. D. J. Am. Chem. Soc. 1948, 70, 926. 10.1021/ja01183a012. [DOI] [Google Scholar]; g Gulkova D.; Kraus M. J. Chem. Technol. Biotechnol. 1994, 61, 197. 10.1002/jctb.280610303. [DOI] [Google Scholar]; h Waters R. C.; Vander Werf C. A. J. Am. Chem. Soc. 1954, 76, 709. 10.1021/ja01632a021. [DOI] [Google Scholar]; i De Kimpe N.; De Cock W.; Stevens C. Tetrahedron 1992, 48, 2739. 10.1016/S0040-4020(01)88533-1. [DOI] [Google Scholar]; j De Kimpe N.; Sulmon P.; Moëns L.; Schamp N.; Declercq J.-P.; Van Meerssche M. J. Org. Chem. 1986, 51, 3839. 10.1021/jo00370a018. [DOI] [Google Scholar]; k Stevens C. L.; Dykstra S. J. J. Am. Chem. Soc. 1954, 76, 4402. 10.1021/ja01646a039. [DOI] [Google Scholar]; l Reich H. J.; Holtan R. C.; Bolm C. J. Am. Chem. Soc. 1990, 112, 5609. 10.1021/ja00170a026. [DOI] [Google Scholar]; m Colpaert F.; Mangelinckx S.; Rocchetti M. T.; De Kimpe N. Org. Biomol. Chem. 2011, 9, 549. 10.1039/C0OB00662A. [DOI] [PubMed] [Google Scholar]; n Foehlisch B.; Gehrlach E.; Stezowski J. J.; Kollat P.; Martin E.; Gottstein W. Chem. Ber. 1986, 119, 1661. 10.1002/cber.19861190518. [DOI] [Google Scholar]; o Zweifel G.; Horng A.; Plamondon J. E. J. Am. Chem. Soc. 1974, 96, 316. 10.1021/ja00808a084. [DOI] [Google Scholar]; p Koshino J.; Sugawara T.; Yogo T.; Suzuki A. Chem. Lett. 1983, 933. 10.1246/cl.1983.933. [DOI] [Google Scholar]; q Katritzky A. R.; Xie L.; Serdyuk L. J. Org. Chem. 1996, 61, 7564. 10.1021/jo960840p. [DOI] [PubMed] [Google Scholar]; r Rozen S.; Mishani E.; Kol M. J. Am. Chem. Soc. 1992, 114, 7643. 10.1021/ja00046a006. [DOI] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For more details about the screening through microscale High Throughput Experimentation (HTE), see Supporting Information; b Schmink J. R.; Bellomo A.; Berritt S. Aldrichimica Acta 2013, 46, 71. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.