

Abstract
The ability of FNR to sense and respond to cellular O2 levels depends on its [4Fe-4S]2+ cluster. In the presence of O2, the [4Fe-4S]2+ cluster is converted to a [2Fe-2S]2+ cluster, which inactivates FNR as a transcriptional regulator. In this study, we demonstrate that ∼2 Fe2+ ions are released from the reaction of O2 with the [4Fe-4S]2+ cluster. Fe2+ release was then used as an assay of reaction progress to investigate the rate of [4Fe-4S]2+ to [2Fe-2S]2+ cluster conversion in vitro. We also found that there was no detectable difference in the rate of O2-induced cluster conversion for FNR free in solution compared to its DNA-bound form. In addition, the rate of FNR inactivation was monitored in vivo by measuring the rate at which transcriptional regulation by FNR is lost upon the exposure of cells to O2; a comparison of the in vitro and in vivo rates of conversion suggests that O2-induced cluster conversion is sufficient to explain FNR inactivation in cells. FNR protein levels were also compared for cells grown under aerobic and anaerobic conditions.
By sensing and responding to environmental O2, facultative anaerobes are able to adopt the most energy-efficient metabolic processes for promoting cell growth under a variety of conditions. In Escherichia coli, the shift between aerobic and anaerobic metabolism, which is controlled primarily by the global transcriptional regulator FNR (13, 23, 32), is known to involve the repression and activation of hundreds of genes (25). However, the reactions that control the steps from O2 sensing to changes in gene expression have not been fully described. The present study focuses on the O2-sensing mechanism of FNR and the rapidity with which FNR reacts with O2 in order to adapt to shifting environmental conditions through appropriate changes in gene expression.
Our current model for cellular O2 sensing by FNR is based upon the critical discovery that FNR is directly inactivated by O2 in vitro (12, 15, 16, 19). FNR is a homodimeric DNA-binding protein containing approximately one [4Fe-4S]2+ cluster per subunit (called 4Fe-FNR) (3, 16, 19). The ability of FNR to function as a transcription factor depends on the integrity of a [4Fe-4S]2+ cluster, which promotes a conformation that is necessary for FNR dimerization, site-specific DNA binding, and transcriptional regulation (12, 16, 19, 21). The O2 sensitivity of FNR is also mediated by the [4Fe-4S]2+ cluster; in the presence of O2, the [4Fe-4S]2+ cluster is converted to a [2Fe-2S]2+ cluster both in vitro and in vivo (15, 16, 24). The [2Fe-2S]2+ form of FNR (called 2Fe-FNR) is monomeric in solution and is inactive for DNA binding and transcriptional regulation (19, 28). Finally, the observation that there is no significant difference in FNR protein levels (34) or in FNR Fe-S cluster biogenesis under aerobic and anaerobic growth conditions (3, 25) suggests that the O2-dependent conversion of the [4Fe-4S]2+ cluster of FNR is the key step that senses changes in O2 availability.
Nevertheless, while the conversion of 4Fe-FNR to 2Fe-FNR is sufficient to inactivate FNR in vitro, additional pathways may also play a regulatory role in vivo by determining the steady-state level of 4Fe-FNR that is available to be inactivated by O2. For example, there is evidence to suggest that 2Fe-FNR can be reconverted to active 4Fe-FNR when O2 is removed from cultures (24). Yet recent studies indicate that under aerobic conditions, 2Fe-FNR is converted to apo-FNR possibly through destruction of the [2Fe-2S]2+ cluster by superoxide, a by-product of aerobic metabolism (28). In addition, it is unknown whether the apo-FNR generated by the superoxide pathway is a substrate for the resynthesis of 4Fe-FNR in vivo. A previous study indicated that FNR present in aerobic cells can be activated upon a shift to anaerobic conditions in the presence of a protein synthesis inhibitor (8). However, it is unknown whether the FNR that was activated following the shift arose from apo-FNR, which was generated by cluster degradation, or from newly synthesized FNR, which had yet to acquire a Fe-S cluster. While it is still unresolved whether newly synthesized FNR protein is biochemically distinct from apo-FNR, a previous study has shown that the [4Fe-4S]2+ cluster can be assembled in vitro into FNR that has been purified (either aerobically or anaerobically) under conditions that produce clusterless FNR, suggesting that perhaps the apo-FNR form can be recycled to 4Fe-FNR in cells (32). An understanding of the rates by which all of these processes occur in cells is critical for developing a comprehensive model of O2 sensing by FNR.
As a first step toward addressing this question, we monitored the rate of conversion of 4Fe-FNR to 2Fe-FNR in vitro by measuring the release of iron ions in the reaction. The rate of inactivation of FNR by O2 in cells has also been measured in order to analyze the relevance of the in vitro reaction to the in vivo process. Finally, we also examined the effect of the interaction of FNR with DNA on the rate of 4Fe-FNR to 2Fe-FNR conversion by O2.
MATERIALS AND METHODS
Isolation of 4Fe-FNR.
4Fe-FNR was isolated from anaerobically prepared cells in a Coy anaerobic chamber as previously described, by using a PolyCAT A column in a Beckman high-performance liquid chromatography system, followed by concentration over a Biorex 70 column (27). Protein, iron, and sulfide content of the purified 4Fe-FNR was measured as previously described (5, 16, 27). The 4Fe-FNR preparations used in these studies were >95% pure (as judged by sodium dodecyl sulfate [SDS]-polyacrylamide gel electrophoresis). On the basis of the sulfide determination, which has a standard error of 3 to 4% (5), 64% of the FNR molecules contained a [4Fe-4S] cluster and an average Fe/S ratio of 1.23 ± 0.02. The reproducibility of the protein determinations was within 5%. Throughout the paper, reported 4Fe-FNR concentrations refer to the concentration of FNR molecules containing [4Fe-4S] clusters.
Kinetic analysis of the O2-dependent loss of iron from 4Fe-FNR.
The loss of iron from 4Fe-FNR was measured through formation of a ferene-Fe2+ complex, which absorbs at 593 nm with an extinction coefficient of 39.6 mM−1 cm−1. Reaction mixtures (final volume, 2 ml) were prepared in a Coy anaerobic chamber in sealed screw-cap cuvettes (total capacity, 2.6 ml), containing a final concentration of 2 μM 4Fe-FNR and 100 μM ferene in the presence of 10 mM KPO4 buffer (pH 6.8, 10% glycerol, 0.4 M KCl). A range of [O2] were achieved by addition of known volumes of O2-saturated buffer (880 μM) that was prepared by bubbling a solution of 10 mM KPO4 buffer (pH 6.8, 10% glycerol, 0.4 M KCl) with 100% O2 for 60 to 90 min at room temperature. Formation of the ferene-Fe2+ complex was monitored over time as changes in A593 with a Perkin Elmer λ2 spectrophotometer, in which the water thermostat-regulated cell holder was maintained at 25°C by a circulating water bath.
The rate of cluster loss was compared with the rate of Fe2+ release through the simultaneous analysis of reactions containing 5 μM 4Fe-FNR in anaerobic 10 mM KPO4 buffer (pH 6.8, 10% glycerol, 0.4 M KCl) with or without 100 μM ferene. At time zero, the samples were pipetted under aerobic conditions to introduce air and spectra were collected from 350 to 700 nm at 2.5-min intervals. The release of Fe2+ was monitored as an increase in absorption at 593 nm in the ferene-containing sample, while the loss of the [4Fe-4S]2+ cluster was monitored as a decrease in absorption at 420 nm in a parallel reaction mixture without ferene.
The reaction of the [4Fe-4S]2+ cluster of FNR with O2 was modeled as reaction 1.
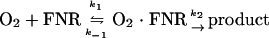 |
The progress curves for the O2-dependent release of Fe2+ from 4Fe-FNR (monitored as A593) under pseudo-first order conditions were fitted to a exponential reaction model (31), using the equation y = y0 + (ymax − y0)(1 − e[−kobs · x]), by computing fits for the parameters y0, kobs, and ymax. The kobs values determined were plotted against the corresponding [O2], and the resulting plot was fitted as a hyperbolic curve by using the equation kobs = a[O2]/(b + [O2]), by computing fits for the parameters a (max kobs) and b (Kd). At high O2 concentrations, kobs approaches k2 (31), the rate constant for the second step in reaction 1.
The effect of FNR-DNA binding on the loss of Fe2+ from 4Fe-FNR.
Fe2+ release from 4Fe-FNR was monitored in the presence or absence of DNA containing a consensus FNR-binding site (5′-CGCAATAAATTTGATGTACATCAAATTTTAGGCA-3′; underlined bases represent the consensus site) (18), as the increase in A593 characteristic of the ferene-Fe2+ complex (as described above). Double-stranded DNA (dsDNA) was prepared by annealing 200 μM complementary 34-bp single-stranded DNA oligonucleotides at 70°C and allowing the mixture to cool slowly to room temperature. Reaction mixtures were prepared in a Coy anaerobic chamber and contained 2 μM 4Fe-FNR in 50 mM KPO4 buffer (pH 6.8, 10% glycerol, 0.2 M KCl) in the presence or absence of 3 μM dsDNA. Alternatively, the reaction of 3 μM FNR in the presence or absence of 3 μM dsDNA was analyzed in 37.25 mM KPO4 buffer (pH 6.8, 25 mM potassium glutamate, 6.25% glycerol, 0.2 M KCl, 5.8 mM dithiothreitol) (18). Following ∼45-min incubation under anaerobic conditions at room temperature to allow for the formation of 4Fe-FNR/DNA complexes, ferene (100 μM) was added and O2 was subsequently introduced by drawing the mixture up and down with a pipette under an aerobic atmosphere. The change in A593 of the reactions was monitored over time in a Perkin Elmer λ2 spectrophotometer at 25°C.
Assay of FNR activity in cells.
Strains containing lacZ transcriptional fusions to the FNR-repressed promoter Pndh (PK3286, fnr+; PK3289, Δfnr) (18) or the FNR-activated promoter PdmsA (PK3292, fnr+; PK3293, Δfnr) (17) were grown in 500 ml of glucose minimal medium under anaerobic conditions by sparging with a 95% N2-5% CO2 gas mixture as previously described (27). At an optical density at 600 nm (OD600) of ∼0.2, O2 was added to the gas mixture to produce a final mixture of 25% O2-70% N2-5% CO2. Aliquots (in triplicate) were collected at various times before and after the O2 shift for the measurement of β-galactosidase activity by adding cells directly to an ice-cold tube containing chloramphenicol. The β-galactosidase activity per milliliter of culture [ONPG (o-nitrophenyl-β-d-galactopyranoside) hydrolyzed/(min · ml)] was plotted as a function of time to indicate changes in the rate of lacZ expression following the shift, as a measure of the corresponding changes in FNR activity.
Determination of cellular FNR levels by quantitative Western blot analysis.
To calculate the number of FNR molecules per cell, MG1655 was grown in M9 minimal glucose medium at 37 or 25°C to an OD600 of ∼0.4 (Perkin Elmer λ2 spectrophotometer). Aerobic or anaerobic culture conditions were achieved by sparging cells as described in “Assay of FNR activity in cells” above. Aliquots (250 μl) of each culture (in triplicate) were centrifuged to pellet the cells, the supernatant was removed, and the pellets were frozen at −20°C. The cell pellets were thawed, resuspended in 10 μl SDS-loading buffer, heated for 10 min at 90°C, and loaded onto a 12% SDS-polyacrylamide gel for electrophoresis along with aliquots of known amounts of purified FNR protein. The proteins were then transferred to a nitrocellulose membrane by Western transfer, and FNR levels were detected using α-FNR primary antibodies and fluorescein isothiocyanate-labeled anti-rabbit secondary antibodies (BD Pharmingen). The fluorescence of the resulting blots was then quantified using a Hitachi FM-BioII fluorescent scanner and Molecular Dynamics ImageQuant software. The number of cells in each aliquot was determined by plating dilutions from the same cultures on Tryptone-yeast extract medium and growing at 37°C overnight for viable cell counts. All samples were analyzed in triplicate.
RESULTS
Fe2+ is released in the reaction of O2 with 4Fe-FNR.
One goal of this study was to perform a kinetic analysis of the O2-dependent conversion of 4Fe-FNR to 2Fe-FNR. As a first step, we developed an assay to monitor the [4Fe-4S]2+ cluster to [2Fe-2S]2+ cluster conversion under conditions where O2 could be added in excess relative to 4Fe-FNR and where product formation could be easily monitored in real time. Since our previous Mössbauer analyses of the cluster conversion process suggested that Fe2+ is one product of the reaction of 4Fe-FNR with O2 (16, 24), we examined the effectiveness of using the Fe2+-specific chelator ferene to monitor reaction progress. Because the extinction coefficient for the Fe2+-ferene complex is much higher than that of either the [4Fe-4S]2+ or [2Fe-2S]2+ cluster, using Fe2+-ferene to monitor reaction progress offered the advantage that [O2] could be varied over a wide range while still maintaining O2 in excess of 4Fe-FNR, which was not possible when monitoring changes in Fe-S cluster absorption in the visible region. In addition, this reaction could be carried out at 25°C which, given our experimental setup, allowed us to vary the amount of O2 in solution.
Fe2+ release was monitored by measuring absorption by the ferene-Fe2+ complex at 593 nm. In the presence of a range of initial [O2] from ∼80 to 440 μM, an average of 4.33 ± 0.06 μM Fe2+ ions were reproducibly released from 2 μM [4Fe-4S]2+-FNR (8 μM with respect to cluster sulfide and 9.84 μM with respect to the initial cluster-bound Fe) within ∼2 min in the presence of 100 μM ferene (Fig. 1; also data not shown). These results demonstrated that 2.17 Fe2+ ions (44% of the initial cluster Fe) were released during the conversion of 4Fe-FNR to 2Fe-FNR. The ferene-Fe2+ complex and the 2Fe-FNR produced from the reaction were stable for hours at 25°C (data not shown; 28), suggesting that the reaction of O2 with 4Fe-FNR is irreversible under these solution conditions. No change in absorption at 593 nm was observed in a reaction of 4Fe-FNR with excess ferene under anaerobic conditions, demonstrating that the [4Fe-4S]2+ cluster was stable to ferene under these conditions and that the release of Fe2+ was O2 dependent, as expected. Finally, a comparison of the progress of the reaction of O2 with 4Fe-FNR as measured either by monitoring cluster conversion using visible spectroscopy or by Fe2+ release shows that the processes are concurrent (Fig. 2); in agreement with these results, the loss of sulfide from the reaction of O2 with the [4Fe-4S] cluster of FNR (16) was not influenced by the presence of ferene (data not shown). Therefore, we conclude that the observed release of Fe2+ can be used as an indicator of the progress of cluster conversion.
FIG. 1.

Release of Fe2+ from the [4Fe-4S]2+ cluster of FNR upon reaction with O2. 4Fe-FNR (2 μM) was mixed with 160 (open square), 240 (filled circle), 320 (open diamond), or 400 μM (filled triangle) O2 in the presence of 100 μM ferene at 25°C. Absorption at 593 nm indicates formation of the Fe2+-ferene complex. The single exponential curve fit for each condition is represented as a line; all fits produced an R2 of ≥0.997.
FIG. 2.

Comparison of the rates of [4Fe-4S]2+ cluster conversion with the release of Fe2+. 4Fe-FNR (5 μM) was incubated in anaerobic buffer in the presence (filled circles) or absence (filled triangles) of 100 μM ferene at 25°C. At time zero, O2 was introduced and the loss of the [4Fe-4S]2+ cluster was monitored as a decrease in absorption at 420 nm, and the release of Fe2+ was monitored as an increase in absorption at 593 nm.
While these data indicate that ∼2 Fe ions released during the [4Fe-4S]2+ to [2Fe-2S]2+ cluster conversion are Fe2+, our previous Mössbauer analysis of the cluster conversion process indicated that both Fe3+ and Fe2+ accumulated as reaction products (16, 24). To determine whether the Fe3+ arises from the subsequent oxidation of Fe2+ in the presence of O2, we compared the amount of chelatable Fe2+ from reactions in which ferene was either present during the entire reaction or was added after the reaction was complete. When ferene was added about 4 min after the reaction was initiated, the amount of Fe2+ chelated by the ferene was diminished by about 40% (data not shown). This is consistent with the assumption that Fe2+ is the form released in the reaction, which is then subject to oxidation to the Fe3+ form under aerobic conditions. If, instead, both Fe2+ and Fe3+ were reaction products then ferene would be expected to bind equivalent amounts of Fe2+ when introduced at any stage in the reaction. In addition, the incubation of ferrous ammonium sulfate in aerobic buffer prior to the addition of ferene greatly reduced the formation of the ferene-Fe2+ complex, indicating that Fe2+ is oxidized in the presence of O2 if ferene is not present to chelate it. Therefore, we conclude that Fe2+ and not Fe3+ is the product of the cluster conversion process and that cluster conversion can be represented by formation of the ferene-Fe2+ complex.
Kinetic analysis of the reaction of O2 with 4Fe-FNR.
To monitor the rate of cluster conversion, a representative set of progress curves generated at different [O2] (Fig. 1) levels were fitted to an equation describing a single exponential decay model for the pseudo-first order reaction of O2 with 4Fe-FNR (reaction 1). The resultant kobs values were plotted against the O2 concentration for each reaction (Fig. 3). The hyperbolic shape of the resulting curve suggests that k−1 is much greater than k2 (31), and therefore that the dissociation of 4Fe-FNR from O2 is rapid compared with the rate at which the 4Fe-FNR · O2 intermediate is resolved to product. Since k2 is the rate-limiting step for the reaction of 4Fe-FNR with O2, the asymptote of the plot of kobs versus [O2] approximates k2. Thus calculated, k2 is 0.0682 s−1, which suggests a minimal half-time of 10.2 s for the reaction of 4Fe-FNR at saturating [O2], at 25°C. It was not possible to determine whether O2 was lost through diffusion from the buffer into the headspace of the sealed cuvette (0.6 ml of the 2.6-ml total volume). However, such loss should be proportional for the various initial [O2] and would therefore not affect the calculation of k2. On the other hand, the value of Kd is dependent on the [O2]; based on the assumption that the loss of O2 is negligible under these conditions, the dissociation constant Kd was estimated to be ∼408 μM.
FIG. 3.

Plot of kobs versus [O2] for the release of Fe2+ from the [4Fe-4S]2+ cluster of FNR upon reaction with O2. The values of k2 and Kd were calculated for the reaction of O2 with 4Fe-FNR from the plot of kobs versus [O2] in Fig. 1.
Effect of DNA on the O2-induced conversion of 4Fe-FNR to 2Fe-FNR.
While it is known that the [4Fe-4S]2+ cluster of FNR is required for DNA binding, we are not aware of any experiments that have addressed whether DNA binding influences the rate of O2-induced [4Fe-4S]2+ cluster conversion. For example, DNA binding could protect the [4Fe-4S]2+ cluster if accessibility of the cluster to solvent (or O2) is hindered in the DNA-bound conformation. The effect of DNA binding on the rate of cluster conversion was determined by measuring O2-induced Fe2+ release from 4Fe-FNR in the presence of 34-bp dsDNA containing the consensus FNR-binding site. The reaction was performed under solution conditions similar to those used throughout this study and at a DNA concentration (3 μM) that was sufficient to bind all of the FNR protein (2 μM) (the apparent Kd is ∼1 nM; data not shown) (18). There was no appreciable difference in the rate of O2-induced Fe2+ release from 4Fe-FNR in the presence and absence of DNA (Fig. 4). The same result was obtained using slightly different solution conditions (see Materials and Methods and reference 18) previously employed for measuring DNA binding (data not shown). Therefore, we conclude that the interaction of DNA with 4Fe-FNR does not substantially influence the susceptibility of the [4Fe-4S]2+ cluster to O2. This suggests that in vivo, the rate of FNR inactivation by O2 is the same for both the DNA-bound and -unbound forms of FNR.
FIG. 4.

The effect of DNA on the release of Fe2+ from 4Fe-FNR. 4Fe-FNR (2 μM) was incubated at 25°C in the presence (filled diamonds) or absence (open diamonds) of 3 μM dsDNA to permit the formation of FNR · DNA complexes and mixed with anaerobic ferene (100 μM) prior to the introduction of air.
In vivo response of an FNR-repressed promoter to O2-induced inactivation of FNR.
To determine whether the cluster conversion rate measured in vitro is relevant for the in vivo O2-sensing mechanism, the rate of 4Fe-FNR inactivation (i.e., the consequence of O2-induced cluster conversion) was measured for FNR-regulated promoters at both 25 and 37°C. Growth at 25°C allowed us to directly compare the rates obtained in vitro, whereas growth at 37°C would allow comparison to other well-established physiological parameters. To establish the response time needed for O2 to induce changes in FNR-regulated gene expression, we monitored the increase in expression from the well-studied FNR-repressed promoter, Pndh (20). Attempts to detect the effects of FNR inactivation by measuring the levels of RNA produced from Pndh were unsuccessful because of low expression levels (data not shown); therefore, we investigated the effect of FNR inactivation on the production of β-galactosidase from constructs in which Pndh is fused to lacZ. In strains containing lacZ fused to Pndh, inactivation of FNR should lead to a rapid loss of Pndh repression and a corresponding increase in β-galactosidase activity per milliliter of culture, reflecting a new rate of synthesis; since β-galactosidase is a stable protein, the rate of its accumulation should reflect the rate of β-galactosidase synthesis.
To determine the length of time it took for Pndh to establish a new rate of synthesis following O2-mediated inactivation of FNR, β-galactosidase activity per milliliter of culture was measured from cells following a shift from anaerobic to aerobic growth conditions. Prior to the shift to aerobic conditions, β-galactosidase activity per milliliter of culture was low and changed little over time in cells grown at either 25 or 37°C (Fig. 5A and Fig. 6), reflecting the repressed state of the Pndh promoter under anaerobic conditions in fnr+ cells. Following the shift to aerobic growth conditions, β-galactosidase activity per milliliter of culture increased, achieving a new rate of synthesis ∼4 to 5 min postshift for cells grown at 37°C and 10.7 ± 1.9 min (average of three experiments) for cells grown at 25°C. No change in the rate of growth was observed immediately after the shift (Fig. 5B and 6). At 37°C, a plot of β-galactosidase activity per milliliter per OD600 versus time indicated that the fnr+ O2-treated cells achieve a similar specific activity of β-galactosidase as do cells lacking FNR (data not shown), indicating that these shifted cells eventually reach the same steady-state level of ndh expression as cells lacking FNR.
FIG. 5.

Effect of O2 on expression of Pndh in cells grown at 37°C. Normalized β-galactosidase activity per milliliter of culture (A) and OD600 (B) from PK3286 (fnr+, Pndh-lacZ) grown under continuous anaerobic growth conditions (open diamonds) or exposed to O2 at time zero (filled diamonds) at 37°C. A normalized β-galactosidase value of 1 represents 7 U/ml for anaerobic cells and 2.8 U/ml for O2-exposed cells. The error bars represent the standard errors of triplicate samples.
FIG. 6.

Effect of O2 on expression of Pndh-lacZ in cells grown at 25°C. β-galactosidase activity per milliliter of culture (filled triangles) and OD600 (open triangles) from PK3286 (fnr+, Pndh-lacZ) grown under anaerobic growth conditions and then exposed to O2 at time zero (arrow) at 25°C. The error bars represent the standard errors of triplicate samples.
Response of an FNR-activated promoter to O2-induced inactivation of FNR.
To compare the O2 response of an FNR-activated promoter with that previously described for Pndh, analogous experiments were performed at 37°C using cells containing the PdmsA-lacZ construct. O2-induced FNR inactivation should rapidly shut off FNR-activated expression of lacZ, preventing any further increase in β-galactosidase activity per milliliter. The introduction of O2 halted β-galactosidase accumulation (Fig. 7); since β-galactosidase is a stable protein, the resulting change in slope, from anaerobic to aerobic expression levels, is less pronounced than the response at Pndh, making the response time less accurate. Nonetheless, the response time is similar to that observed at Pndh. Furthermore, a plot of β-galactosidase activity per milliliter per OD600 versus time (data not shown) closely matches the results of a similar study on the regulation of PdmsA by FNR (30). In addition, these data support the hypothesis that the O2 response observed at Pndh is due to FNR inactivation, since after a similar time at PdmsA, no further synthesis of lacZ could be observed.
FIG. 7.

Effect of O2 on expression from PdmsA-lacZ in cells grown at 37°C. β-Galactosidase activity per milliliter of culture was calculated before and after exposure to O2 (at time zero; arrow) from strains PK3293, Δfnr (open triangles), and PK3292 (fnr+; filled triangles) grown at 37°C. The error bars represent the standard errors of triplicate samples.
Determination of FNR protein levels in cells.
To compare the time of FNR inactivation obtained in vivo with the rate of FNR cluster conversion determined from the Fe2+-ferene experiments, the number of FNR molecules present per cell was established for both aerobic and anaerobic growth conditions. Although previous data showed that FNR is present in ∼2,400 copies per cell, the growth conditions and the genetic background of the strain used in this earlier study were different than those used here (34). Therefore we used quantitative Western blots to determine FNR levels in cells grown under the appropriate conditions by comparison to known amounts of FNR protein (data not shown). From this analysis, we found that cultures grown at 25°C under anaerobic conditions contain ∼2,200 ± 200 molecules of FNR protein per cell, while aerobically grown cells contain ∼2,900 ± 300 molecules of FNR per cell. Cultures grown at 37°C under anaerobic conditions contain ∼2,600 ± 200 molecules of FNR protein per cell, while aerobically grown cells contain ∼4,100 ± 200 molecules of FNR per cell. Assuming a cell volume of 1 μm3, this produces a cellular [FNRmonomer] of ∼3.7 μM at 25°C under anaerobic conditions.
DISCUSSION
The kinetic data presented in this study provide key support for the model that [4Fe-4S]2+ to [2Fe-2S]2+ cluster conversion is sufficient to explain how FNR is inactivated by O2 in vivo. In addition, these results provide the groundwork to develop a more sophisticated model for the mechanism of cellular O2 sensing by FNR. Lastly, the finding that both of the Fe ions released during the O2-induced conversion of the [4Fe-4S]2+ cluster to a [2Fe-2S]2+ cluster are in the reduced (Fe2+) state also raises new questions concerning the mechanism by which O2 oxidizes the [4Fe-4S]2+ cluster.
The reaction of 4Fe-FNR with O2.
While we were preparing this report, Crack et al. (7) reported that H2O2 was produced from the reaction of FNR with O2, which led to the proposal of a reaction mechanism where two cluster irons are oxidized to Fe3+ during O2-induced cluster conversion to provide the two electrons to produce H2O2 from O2. Given the overall 2+ oxidation state of the [4Fe-4S]2+ cluster, the release of ∼2 Fe2+, as found in this study, does not support the proposal that cluster iron atoms are directly oxidized by O2 to produce the [2Fe-2S]2+ cluster and H2O2. In support of Fe3+ not being the predominant Fe ion released from the reaction of O2 with 4Fe-FNR, previous electron paramagnetic resonance (EPR) analysis of the cluster conversion process (15) did not detect significant amounts of Fe3+ after treatment of 4Fe-FNR with [O2] (Fe2+ is not observable by EPR). Furthermore, Mössbauer analysis (16, 24) showed that both Fe2+ and Fe3+ were observed after an initial 2-min reaction of 4Fe-FNR with excess O2 and that over time Fe3+ accumulated, apparently at the expense of Fe2+; it is likely that in the EPR experiments Fe2+ was not subsequently oxidized to Fe3+ because O2 was limiting. In contrast, reaction of 4Fe-FNR with the oxidant ferricyanide readily produced Fe3+ (15), indicating some differences in the effects of these two oxidants. In agreement with these previous data, when we added ferricyanide to [4Fe-4S]-FNR, no iron was complexed by ferene (data not shown); however, if ferricyanide oxidizes Fe2+ more efficiently than does O2, then a transient pool of Fe2+ may not be detectable with our experimental design. Finally, we consistently find, through numerous experiments of variable design, that the rate of the conversion of 4Fe-FNR to 2Fe-FNR, and the consequent release of Fe2+, is dependent on the concentration of O2; this finding is inconsistent with the proposal (7) that the rate-determining step in cluster conversion does not involve O2.
An alternative possibility to explain the mechanism of [4Fe-4S]2+ cluster oxidation is that cluster sulfide is oxidized by O2. In our previous analysis of the cluster conversion process, we found that only 70% of the sulfide originally in the cluster was detectable following the treatment of 4Fe-FNR with O2 (16). Given that 50% of the total sulfide would still be associated with the [2Fe-2S]2+ cluster, then 30% of the sulfide originally present in the [4Fe-4S]2+ cluster remains unaccounted for and must have been oxidized. Attempts to detect sulfide oxidation products such as polysulfide or sulfinic acid by chemical methods have thus far been unsuccessful (unpublished data). However, we are aware that the interpretation of chemical analyses of the reaction products might be complicated by slow reactions that could occur after the initial cluster conversion, such as the reaction of sulfur- and O2-derived products with each other or with the FNR protein. In addition, the further analysis of the fate of the sulfide is limited by the lack of an assay to measure protein-bound sulfide in real time as we did with Fe2+ release. Nevertheless, as neither ferric nor ferrous iron per se are soluble at the pH of the aerobic solution conditions utilized here, it is likely that the presence of a ferrous chelator makes the solubilization of iron as Fe2+ feasible at the expense of the missing sulfide (see above), which obviously is being oxidized. Furthermore, as mentioned above, our Mössbauer results (which were carried out in the absence of chelators) indicate that ferrous iron is formed upon oxidation of FNR by O2 and support the explanations given above.
Surprisingly, current evidence also does not support a simple one-electron reduction of O2 to form superoxide and Fe2+. This mechanism has been favored because there is precedent for Fe2+ release from a [4Fe-4S] cluster upon treatment of a dehydratase class of enzymes with superoxide (10). However, no superoxide was detected using the radical spin trap 5-5′-dimethyl-1-pyrroline-1-oxide for the reaction of 4Fe-FNR with O2, nor did we find any significant effect on either the amount or the kinetics of ferrous iron released when superoxide dismutase was present during the ferene assay (2). In addition, our finding that 2Fe-FNR is stable following the reaction of 4Fe-FNR with O2 also suggests that superoxide does not accumulate since 2Fe-FNR is destroyed by superoxide. Nevertheless, it is still formally possible that superoxide could be the O2-derived product of the reaction if it were removed faster than the rate at which superoxide reacts with 2Fe-FNR (t1/2 ≤ 4 min); for example, superoxide can spontaneously dismute to produce H2O2 with rate constants (k2) as high as 8 × 107 M−1 s−1 (11, 14). Thus, the observed production of H2O2 (7) might be explained by dismutation of superoxide. However, in this scenario, the initial release of superoxide and Fe2+ would also yield a [3Fe-4S]1+ cluster intermediate, rendering it difficult to explain the release of a second reduced Fe atom during the subsequent conversion to the [2Fe-2S]2+ cluster. Thus, more work is needed to find a model that satisfies all the observations reported to date.
One difference between the two studies that may be relevant in reconciling these data is that in vitro-reconstituted 4Fe-FNR was used by Crack et al. (7), whereas native 4Fe-FNR protein was used in the experiments described here. Although it is not obvious why these two proteins should have different properties, the reconstituted protein was reported to be unstable in the presence of ferrous and ferric chelators, in contrast to native 4Fe-FNR, which is stable under anaerobic conditions to either the Fe3+-specific chelator Tiron (data not shown) or the Fe2+-specific chelator ferene. In addition, the extinction coefficients reported for the reconstituted 4Fe-FNR (ɛ405 = 13,559 M−1 cm−1) and the 2Fe-FNR produced by exposing reconstituted 4Fe-FNR to O2 (ɛ420 = 11,040 M−1 cm−1) differ from those measured by us for purified native 4Fe-FNR (ɛ405 = 16,125 M−1 cm−1) and purified native 2Fe-FNR (ɛ420 = 8,382 M−1 cm−1). These distinctions suggest that there may be some important differences in the biochemical properties of FNR prepared by the different methods.
The rate of FNR inactivation is dependent on [O2] within a physiologically relevant range.
At the low cell densities and the sparging conditions used in these studies, the dissolved O2 in the culture media (25°C) was present in excess (∼220 μM) of the cellular [FNR] of 3.7 μM; accordingly, under these conditions, FNR inactivation rates should follow pseudo-first order kinetics and can be extrapolated from Fig. 3. These data also imply that at O2 levels in the range of microaerobic culture conditions, the rate of cluster conversion will be even slower, indicating that the amount of FNR active in cells at a particular dissolved [O2] should be largely determined by its rate of inactivation. This conclusion is supported by previous studies in which the in vivo activity of FNR was shown to be dependent on the dissolved [O2] in the media (4, 29).
O2-dependent cluster conversion of 4Fe-FNR can reasonably explain the loss of FNR activity by O2 in vivo.
Determining the in vitro rates of cluster conversion also allowed us to assess whether these values were compatible with in vivo measurements of FNR inactivation. If FNR was inactivated by O2 in vivo using this same mechanism (12, 16, 19), then we expect that the rate of inactivation in cells should be similar to the in vitro rate of cluster conversion taking into account the other kinetic steps involved in in vivo measurements of FNR activity, such as the time required for O2 diffusion into cells, FNR dissociation from ndh promoter DNA, transcription and translation of lacZ, and folding and assembly of β-galactosidase. Given the uncertainties associated with measuring in vivo kinetic constants for such processes, we recognize that such comparisons between in vitro and in vivo data are just approximations. Nevertheless, we found that using literature values for these steps, we could develop a kinetic model for the inactivation of FNR that was at least consistent with our in vitro data.
FNR inactivation times were estimated from the cultures grown at 25°C since this is the temperature utilized in our in vitro studies. By subtracting the time estimated for the coupled transcription-translation of lacZ (∼2.5 min) (6, 9, 22) and the in vivo folding and assembly of β-galactosidase tetramers (∼4 min at 37°C) (1) from the time required to observe a change in the synthesis rate of β-galactosidase from the ndh-lacZ fusion strain following a shift from anaerobic to aerobic conditions (∼11 min, the average of three experiments), we obtained an inactivation time for FNR of ∼4.5 min at 25°C. We assumed that diffusion of O2 into the cell, which presumably occurs at diffusion-limited rates since previous studies suggest that the cell membrane does not serve as a barrier to O2 diffusion (26, 33), and the dissociation of inactivated FNR from DNA, which is <10 s (the Kd for nonspecific binding is 0.5 × 10−6 M) (19, 35), are rapid enough to make a negligible contribution to our calculation. By using the in vitro-determined rate constant for cluster conversion of 0.0240 s−1 for a well-aerated culture, i.e., [O2] at ∼220 μM, and assuming that at least 99% (seven half-lives of 28.9 s) of FNR must be inactivated in order to derepress Pndh, we would predict a minimal FNR inactivation time of 3.4 min in cells grown at 25°C. Thus, our in vivo estimate for the FNR inactivation time (4.5 min) from cultures grown at 25°C is similar to that predicted from the rate of Fe-S cluster conversion (3.4 min). Acknowledging the stated assumptions, we conclude that O2-dependent cluster conversion of 4Fe-FNR can reasonably explain the loss of FNR activity by O2 in vivo.
In summary, the data presented in this study support the hypothesis that the FNR cluster conversion pathway provides a facile system for responding to fluctuating levels of O2 in the environment. A future goal of our studies is to develop a comprehensive model for the cellular mechanism of O2 sensing by FNR. We recognize that such a model must also include steps that contribute to determining the steady-state levels of 4Fe-FNR in cells. In addition to the synthesis of the FNR protein, such steps include the acquisition of the [4Fe-4S]2+ cluster, superoxide-induced conversion of 2Fe-FNR to apo-FNR, reconversion of 2Fe-FNR to 4Fe-FNR, and the possible recycling of the apo-FNR form to regenerate active 4Fe-FNR. Determining the rates of these processes will require the development of new assays to measure Fe-S incorporation into FNR.
Acknowledgments
This work was supported by the NIH Molecular Biosciences training grant GM07215 (to V.R.S.) and NIH grant GM45844 (to P.J.K.).
We thank James Imlay (University of Illinois), Ruth Saecker (University of Wisconsin), John Yin (University of Wisconsin), and Judith Burstyn (University of Wisconsin) for helpful advice and discussions. We also gratefully acknowledge the data analysis assistance offered by Oleg Tsodikov (Massachusetts Institute of Technology) and the contributions made by Donna Bates (University of Wisconsin) toward method development. Finally, we thank the Kiley lab for helpful discussions and John Denu (University of Wisconsin) for critical reading of the manuscript.
REFERENCES
- 1.Agashe, V. R., S. Guha, H. C. Chang, P. Genevaux, M. Hayer-Hartl, M. Stemp, C. Georgopoulos, F. U. Hartl, and J. M. Barral. 2004. Function of trigger factor and DnaK in multidomain protein folding: increase in yield at the expense of folding speed. Cell 117:199-209. [DOI] [PubMed] [Google Scholar]
- 2.Bates, D. M. 1999. Role of iron-sulfur cluster conversion in the oxygen-sensing mechanism of the Escherichia coli transcription factor FNR. Ph.D. thesis. University of Wisconsin—Madison, Madison.
- 3.Bates, D. M., C. V. Popescu, N. Khoroshilova, K. Vogt, H. Beinert, E. Münck, and P. J. Kiley. 2000. Substitution of leucine 28 with histidine in the Escherichia coli transcription factor FNR results in increased stability of the [4Fe-4S]2+ cluster to oxygen. J. Biol. Chem. 275:6234-6240. [DOI] [PubMed] [Google Scholar]
- 4.Becker, S., G. Holighaus, T. Gabrielczyk, and G. Unden. 1996. O2 as the regulatory signal for FNR-dependent gene regulation in Escherichia coli. J. Bacteriol. 178:4515-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beinert, H. 1983. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem. 131:373-378. [DOI] [PubMed] [Google Scholar]
- 6.Bremer, H., and P. P. Dennis. 1987. Modulation of chemical composition and other parameters of the cell by growth rate, p. 1527-1540. In F. C. Neidhardt (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology, Washington, D.C.
- 7.Crack, J., J. Green, and A. J. Thomson. 2004. Mechanism of oxygen sensing by the bacterial transcription factor fumarate-nitrate reduction (FNR). J. Biol. Chem. 279:9278-9286. [DOI] [PubMed] [Google Scholar]
- 8.Engel, P., M. Trageser, and G. Unden. 1991. Reversible interconversion of the functional state of the gene regulator FNR from Escherichia coli in vivo by O2 and iron availability. Arch. Microbiol. 156:463-470. [DOI] [PubMed] [Google Scholar]
- 9.Farewell, A., and F. C. Neidhardt. 1998. Effect of temperature on in vivo protein synthetic capacity in Escherichia coli. J. Bacteriol. 180:4704-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flint, D. H., J. F. Tuminello, and M. H. Emptage. 1993. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 268:22369-22376. [PubMed] [Google Scholar]
- 11.Fridovich, I. 1978. The biology of oxygen radicals. Science 201:875-880. [DOI] [PubMed] [Google Scholar]
- 12.Green, J., B. Bennett, P. Jordan, E. T. Ralph, A. J. Thomson, and J. R. Guest. 1996. Reconstitution of the [4Fe-4S] cluster in FNR and demonstration of the aerobic-anaerobic transcription switch in vitro. Biochem. J. 316:887-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guest, J. R., J. Green, A. S. Irvine, and S. Spiro. 1996. The FNR modulon and FNR-regulated gene expression, p. 317-342. In E. C. C. Lin and A. S. Lynch (ed.), Regulation of gene expression in Escherichia coli. R. G. Landes Company, Austin, Texas.
- 14.Imlay, J. A., and I. Fridovich. 1991. Assay of metabolic superoxide production in Escherichia coli. J. Biol. Chem. 266:6957-6965. [PubMed] [Google Scholar]
- 15.Jordan, P. A., A. J. Thomson, E. T. Ralph, J. R. Guest, and J. Green. 1997. FNR is a direct oxygen sensor having a biphasic response curve. FEBS Lett. 416:349-352. [DOI] [PubMed] [Google Scholar]
- 16.Khoroshilova, N., C. Popescu, E. Münck, H. Beinert, and P. J. Kiley. 1997. Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc. Natl. Acad. Sci. USA 94:6087-6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamberg, K. E., and P. J. Kiley. 2000. FNR-dependent activation of the class II dmsA and narG promoters of Escherichia coli requires FNR-activating regions 1 and 3. Mol. Microbiol. 38:817-827. [DOI] [PubMed] [Google Scholar]
- 18.Lamberg, K. E., C. Luther, K. D. Weber, and P. J. Kiley. 2002. Characterization of activating region 3 from Escherichia coli FNR. J. Mol. Biol. 315:275-283. [DOI] [PubMed] [Google Scholar]
- 19.Lazazzera, B. A., H. Beinert, N. Khoroshilova, M. C. Kennedy, and P. J. Kiley. 1996. DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J. Biol. Chem. 271:2762-2768. [DOI] [PubMed] [Google Scholar]
- 20.Meng, W., J. Green, and J. R. Guest. 1997. FNR-dependent repression of ndh gene expression requires two upstream FNR-binding sites. Microbiology 143:1521-1532. [DOI] [PubMed] [Google Scholar]
- 21.Moore, L. J., and P. J. Kiley. 2001. Characterization of the dimerization domain in the FNR transcription factor. J. Biol. Chem. 276:45744-45750. [DOI] [PubMed] [Google Scholar]
- 22.Neidhardt, F. C. 1999. Bacterial growth: constant obsession with dN/dt. J. Bacteriol. 181:7405-7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patschkowski, T., D. M. Bates, and P. J. Kiley. 2000. Mechanisms for sensing and responding to oxygen deprivation, p. 61-78. In G. Storz and R. Hengge-Aronis (ed.), Bacterial stress responses. ASM Press, Washington, D.C.
- 24.Popescu, C. V., D. M. Bates, H. Beinert, E. Münck, and P. J. Kiley. 1998. Mössbauer spectroscopy as a tool for the study of activation/inactivation of the transcription regulator FNR in whole cells of Escherichia coli. Proc. Natl. Acad. Sci. USA 95:13431-13435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salmon, K., S. P. Hung, K. Mekjian, P. Baldi, G. W. Hatfield, and R. P. Gunsalus. 2003. Global gene expression profiling in Escherichia coli K12. The effects of oxygen availability and FNR. J. Biol. Chem. 278:29837-29855. [DOI] [PubMed] [Google Scholar]
- 26.Subczynski, W. K., J. S. Hyde, and A. Kusumi. 1989. Oxygen permeability of phosphatidylcholine—cholesterol membranes. Proc. Natl. Acad. Sci. USA 86:4474-4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutton, V. R., and P. J. Kiley. 2003. Techniques for studying the oxygen-sensitive transcription factor FNR from Escherichia coli. Methods Enzymol. 370:300-312. [DOI] [PubMed] [Google Scholar]
- 28.Sutton, V. R., A. Stubna, T. Patschkowski, E. Münck, H. Beinert, and P. J. Kiley. 2004. Superoxide destroys the [2Fe-2S]2+ cluster of FNR from Escherichia coli. Biochemistry 43:791-798. [DOI] [PubMed] [Google Scholar]
- 29.Tseng, C. P., J. Albrecht, and R. P. Gunsalus. 1996. Effect of microaerophilic cell growth conditions on expression of the aerobic (cyoABCDE and cydAB) and anaerobic (narGHJI, frdABCD, and dmsABC) respiratory pathway genes in Escherichia coli. J. Bacteriol. 178:1094-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tseng, C. P., A. K. Hansen, P. Cotter, and R. P. Gunsalus. 1994. Effect of cell growth rate on expression of the anaerobic respiratory pathway operons frdABCD, dmsABC, and narGHJI of Escherichia coli. J. Bacteriol. 176:6599-6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsodikov, O. V., and M. T. Record, Jr. 1999. General method of analysis of kinetic equations for multistep reversible mechanisms in the single-exponential regime: application to kinetics of open complex formation between Eσ70 RNA polymerase and λPR promoter DNA. Biophys. J. 76:1320-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Unden, G., S. Achebach, G. Holighaus, H. G. Tran, B. Wackwitz, and Y. Zeuner. 2002. Control of FNR function of Escherichia coli by O2 and reducing conditions. J. Mol. Microbiol. Biotechnol. 4:263-268. [PubMed] [Google Scholar]
- 33.Unden, G., S. Becker, J. Bongaerts, G. Holighaus, J. Schirawski, and S. Six. 1995. O2-sensing and O2-dependent gene regulation in facultatively anaerobic bacteria. Arch. Microbiol. 164:81-90. [PubMed] [Google Scholar]
- 34.Unden, G., and A. Duchene. 1987. On the role of cyclic AMP and the Fnr protein in Escherichia coli growing anaerobically. Arch. Microbiol. 147:195-200. [DOI] [PubMed] [Google Scholar]
- 35.Ziegelhoffer, E. C., and P. J. Kiley. 1995. In vitro analysis of a constitutively active mutant form of the Escherichia coli global transcription factor FNR. J. Mol. Biol. 245:351-361. [DOI] [PubMed] [Google Scholar]