

Abstract
Glycans are critical to protein biology and are useful as disease biomarkers. Many studies of glycans rely on clinical specimens, but the low amount of sample available for some specimens limits the experimental options. Here we present a method to obtain information about protein glycosylation using a minimal amount of protein. We treat proteins that were captured or directly spotted in small microarrays (2.2 × 2.2 mm) with exoglycosidases to successively expose underlying features, and then we probe the native or exposed features using a panel of lectins or glycan-binding reagents. We developed an algorithm to interpret the data and provide predictions about the glycan motifs that are present in the sample. We demonstrated the efficacy of the method to characterize differences between glycoproteins in their sialic acid linkages and N-linked glycan branching, and we validated the assignments by comparing results from mass spectrometry and chromatography. The amount of protein used on-chip was about 11 nanograms. The method also proved effective for analyzing the glycosylation of a cancer biomarker in human plasma, MUC5AC, using only 20 μL of the plasma. A glycan on MUC5AC that is associated with cancer had mostly 2,3 linked sialic acid, whereas other glycans on MUC5AC had a 2,6 linkage of sialic acid. The on-chip glycan modification and probing (on-chip GMAP) method provides a platform for analyzing protein glycosylation in clinical specimens and could complement the existing toolkit for studying glycosylation in disease.
Graphical abstract
INTRODUCTION
Deciphering glycan structures on glycoproteins is an important goal for better understanding the roles of protein glycosylation in normal and disease biology1. For disease research, studies using clinical specimens are particularly valuable, as they enable a direct look at the location and amount of expression of glycans in the biological context. But such studies are challenging using current methods of analyzing glycans, mainly because the methods require more sample than is typically available from clinical specimens. The use of mass spectrometry and chromatography to analyze protein glycosylation requires purification of a protein in microgram quantities2, which is not possible if the specimen is available only in small amounts or if the protein of interest has too low a concentration. Furthermore, the throughput and precision of methods that require multiple preparation and purification steps normally would not be high enough to determine differences among patient cohorts. Although specialized centers have automated systems with powerful capabilities3, new methods nevertheless are needed to provide complementary information and to broaden access to glycan analysis.
Here we developed a method that combines the low-volume and multiplexing capabilities of microarrays with the precision of affinity reagents and enzymes to probe and modify glycans. Researchers have used affinity reagents, including lectins and glycan-binding antibodies, for distinguishing closely related or rare glycan motifs in a variety of experimental formats4 such as histochemical staining5 and lectin microarrays6–7. A format that is valuable for probing glycosylation on specific proteins is a sandwich ELISA-type microtiter assay, in which a surface-bound antibody captures a protein out of solution, and a solution-phase lectin binds to glycans on the captured protein. Researchers used such an assay to analyze glycosylation variants of alpha-fetoprotein8 and prostate-specific antigen9–10, among others. We multiplexed and miniaturized the format using antibody microarrays11. The microarray was particularly valuable for biomarker studies because it required only microliters of sample and concentrations of the targeted proteins in the low ng/mL range11. Moreover, we obtained the throughput and precision necessary for examining large patient cohorts. Using antibody-lectin sandwich arrays, we identified glycan biomarkers in the plasma of pancreatic cancer patients12 and demonstrated that specific protein glycoforms can be better biomarkers than the core proteins13–15. Other groups also have effectively used this format for disease and biomarker studies16–18.
A limitation in the use of lectins is that they provide information on only one glycan motif that is accessible to the lectin. Additional information about the underlying glycan structures would be valuable in most studies and can be provided by LC-MS based approaches, but as noted above, the information can be difficult to obtain owing to sample limitations. An alternative approach is to combined the use of lectins with the use exoglycosidases to remove terminal saccharides and expose underlying features, analogous to the well-established method of sequential glycosidase digestions followed by chromatographic separations19. We postulated that such a method could apply to proteins captured on antibody arrays; instead of examining changes in glycan size, we could examine changes in lectin binding profile. The strategy is to measure binding across a panel of lectins incubated with and without prior enzymatic digestion, and then to predict which glycan motifs are present based on the integrated information.
The possibility of acquiring such data was clear from previous work using sialidase on proteins captured by antibody arrays11 and on glycans in glycan arrays20. But the experimental methods are only one part of the challenge; one also needs software to interpret the data. Manual or qualitative interpretation becomes ineffective when attempting to integrate information from multiple lectins and from changes in binding patterns upon enzymatic treatment, especially when accounting for the unique and complex binding specificity of each lectin. We approached this problem by building on our previous work in informatics for glycan analysis. Our group was the first to present an algorithm for the automated analysis of glycan array data—a method called Motif Segregation21—which was followed by additional, useful methods for glycan array analysis22–24. A large repository of glycan array data is available through the Consortium for Functional Glycomics (CFG), and we analyzed the set in its entirety to obtain the binding specificities of hundreds of lectins and glycan-binding antibodies25. The analysis was critical for developing a method to predict the glycan motifs in a biological sample based on measurements from a panel of lectins26.
Starting from the above foundations, in this research we developed the ability to treat a microarray of proteins with exoglycosidases and to probe the glycans both in their native form and in their enzyme-treated form. Furthermore, we developed an algorithm to interpret the data to provide evaluations of the glycan motifs present on the glycoproteins. In this report we present 1) a description of the experimental method and analysis algorithm; 2) the testing of the method on control proteins and comparing the results to data from orthogonal methods (mass spectrometry and chromatography); and 3) the application of the method to interrogating biomarker glycosylation using a small amount of a clinical specimen. The biomarker was MUC5AC obtained from 20 μL of human plasma, and we sought to answer a specific question about a glycan that is upregulated in pancreatic cancer.
EXPERIMENTAL SECTION
Protein and Antibody Microarray Fabrication and Use
The antibodies, lectins, control proteins, and enzymes were purchased from various sources (Table S-1). We printed the capture antibodies or glycoproteins onto coated microscope slides (PATH, Grace Bio-Labs, Bend, OR) using a robotic arrayer (2470, Aushon Biosystems, Billerica, MA) (see the Supporting Information for additional details). Each slide contained 192 identical arrays arranged in an 8 × 24 grid with 2.25 mm spacing between arrays, and each array had the same antibodies or proteins printed in six-replicate. After printing, hydrophobic borders were imprinted onto the slides (SlideImprinter, The Gel Company, San Francisco, CA) to segregate the arrays and allow for multiple, separate sample incubations on each slide27.
The assays were modified from the protocol described previously11 (see the Supporting Information for additional details). For experiments involving the analysis of human plasma, we diluted each sample 2-fold into a buffer (1X PBS with 0.1% Tween-20, 0.1% Brij-35, species-specific blocking antibodies, and protease inhibitor) and incubated the sample on an antibody array overnight at 4 °C. We prepared α2-3 Neuraminidase (P0728L, New England Biolabs, Ipswich) or α2-3, 6, 8 Neuraminidase (P0720S, New England Biolabs, Ipswich) at a concentration of 250 U/mL in the supplied reaction buffer and incubated each separately on arrays containing the captured or spotted glycoproteins overnight at 37°C. The arrays not treated with enzymes were incubated with the enzyme reaction buffer in the same conditions. We incubated each array with a biotinylated lectin solution (3 μg/mL in 1X PBS with 0.1% Tween-20 and 0.1%BSA) and subsequently with Cy5-conjugated streptavidin (43-4316, Invitrogen, Carlsbad, CA) (2 μg/mL in the same buffer as the lectins). The slides were scanned for fluorescence at 633 nm using a microarray scanner (LS Reloaded, TECAN, Morrisville, NC).
Data Analysis and Software
The fluorescence images were quantified and analyzed using custom, in-house software28. (see the Supporting Information for additional details). For the calculation of motif scores we used custom software written in Java (Sun Microsystems), and we used Matlab (R2015b, Mathworks) for calculating the motif prediction scores. For final data processing and figure making, we used Microsoft Excel, GraphPad Pro, and Deneba Canvas.
Human Plasma Samples
All collections took place at the University of Pittsburgh Medical Center following informed consent of the participants and prior to any surgical or medical procedures. The donors were patients with pancreatic cancer (n = 206) and patients with pancreatitis or benign biliary obstruction (n = 49). All blood samples (EDTA plasma) were collected according to the standard operating procedure from the Early Detection Research Network and were frozen at −70 °C or colder within 4 hours of time of collection. Aliquots were shipped on dry ice and thawed no more than three times prior to analysis.
Glycan Analysis by Mass Spectrometry
N-glycans released by PNGaseF digestion of fetuin and transferrin were extracted from precipitated protein and dried by vacuum centrifugation in preparation for ethyl esterification of terminal sialic acid residues. The ethyl esterification protocol, including the modification and enrichment, was adapted from Reiding et al.29 as previously reported30. Glycans were spotted with CHCA matrix and analyzed by MALDI-FTICR as previously described30.
Glycan Analysis by Chromatography
N-glycans were released from transferrin and AGP by in-gel digestion and analyzed by sequencing chromatography according to the method of Royle et al.3 with modifications. (See the Supporting Information for details.)
Safety Considerations
Human blood plasma is a potential source of infectious agents. Researchers should handle specimens collected from human subjects with the appropriate precautions.
RESULTS
On-Chip Glycan Modification and Probing
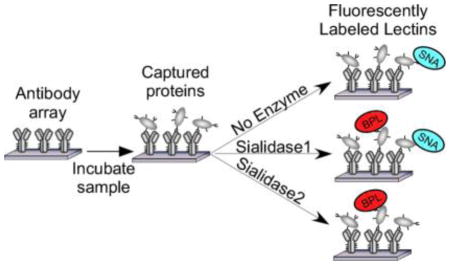
To acquire data on the glycosylation of a protein, we probe the protein with a panel of lectins that are incubated either with or without prior modification of the glycans by an exoglycosidase (Fig. 1). The signals are quantified and used in an algorithm to predict the glycan motifs that are present on the proteins. To test the method, we sought to distinguish between two related motifs: alpha 2,3-linked sialic acid and alpha 2,6-linked sialic acid. Enzymes are available that differentially cleave these features (Fig. 2A). One cleaves only 2,3-linked sialic acid (referred to as sialidase 1) and another is a pan-sialidase that cleaves 2,3, 2,6, 2,8 and 2,9 linkages (referred to as sialidase 2). We also selected a panel of lectins that bind either sialic acid in one of its linkages or that bind non-sialylated glycans that would be exposed upon removal of sialic acid (Fig. 2B). We then applied these reagents to the analysis of purified glycoproteins that we had printed in microarrays on microscope slides.
Figure 1. On-chip glycan modification and probing.

Glycoproteins were immobilized onto a planar surface, and the glycans on the proteins were probed by a panel of lectins, either with or without prior modification of the glycans using enzymes. We quantified the binding of each lectin under each condition and then used an algorithm to predict the motifs that are present on the glycoprotein.
Figure 2. Test case: distinguishing α2,3 from α2,6 sialic acid.

A) We used two enzymes. Sialidase 1 cleaves only α2,3 sialic acid, and sialidase 2 cleaves all linkages of sialic acid. B) We selected lectins based on their primary specificities against the motifs targeted by the enzymes and the motifs expected to be exposed after enzyme treatment. The fine specificities of the lectins are more complex than depicted here. C) The images show the fluorescence signals from microarrays produced by direct spotting of purified glycoproteins. We present representative examples from each treatment condition. D) The heatmaps show the relative signals under each condition as well as the differences between signals obtained with and without enzyme treatment, indicated by ΔSialidase1 and ΔSialidase2. To normalize the signals, all the values obtained with a given lectin were divided by the highest value for that lectin, making the range 0 to 1 for the original values and the range −1 to 1 for the differences.
The amount of protein in each microarray is about 170 picograms (170 × 10−12 grams), based on a protein concentration of 250 μg/mL, a spot volume of 170 pL, and 4 replicate spots per array. The entire analysis used 63 microarrays, based on 7 lectins, 3 conditions (no enzyme, sialidase 1, and sialidase 2), and 3 replicate arrays per condition. Thus the protein consumption was around 170 × 63 = 11 nanograms, equivalent to 140 femtomoles for an 80 kD/mole protein such as transferrin. Here the value of low-volume microarrays is apparent. The 2.2 × 2.2 mm arrays use 1.5 μL per incubation, so the lectin and enzyme consumption is 63 × 1.5 μL = 95 μL for the entire analysis.
The fluorescence images of the lectin binding showed major differences between the lectins and conditions (Fig. 2C). The quantified data (Fig. 2D) showed that the binding of lectins with specificity for sialic acid decreased upon enzymatic treatment, whereas the binding of lectins with specificity for underlying glycans increased. The differences between signals obtained with enzymatic treatment and without enzymatic treatment more clearly showed the changes and the differences between the proteins (Fig. 2D). SNA, which binds primarily 2,6-linked sialic acid, showed loss of binding after application of sialidase 2 but not sialidase 1. In contrast, MAL-1, which binds primarily 2,3-linked sialic acid, showed loss of binding after application of either sialidase. Thus the changes in binding qualitatively matched our expectations.
We evaluated the reproducibility of the measurements using 3 independent experiments (Fig. S-3). The coefficients of variation between the replicates averaged 0.42 for transferrin and 0.37 for fetuin, which is acceptable given the developmental stage of the assays. The average Pearson correlations across the replicate sets were 0.94 for transferrin and 0.95 fetuin. These numbers indicate stability in the measurements and the ability to provide consistent motif predictions.
Automated Interpretation of Lectin Binding and Glycan Modification
An important requirement for the usability of the method is an algorithm to interpret the measurements. Such a process requires quantification both of the specificities of the lectins and of the expected changes in binding following enzymatic modification. For this step we used glycan array data and a bioinformatics method called motif segregation21. First we defined a set of substructures of glycans, referred to as motifs, that represent potential binding determinants of lectins. We defined 158 motifs (Table S-2) covering sialylated and non-sialylated features commonly found in mammalian glycans. Next, using glycan array data obtained from the Consortium for Functional Glycomics, we determined the presence or absence of each motif in each glycan on the array. (For example, the glycan ‘Galβ1-4(Fucα1-3)GlcNAcβ1’ contains the motif ‘terminal Galβ1-4’, but the glycan ‘Galβ1-3(Fucα1-3)GlcNAcβ’ does not.) Finally, using the binding intensities to each glycan for a particular lectin, we calculate a score for each motif based on the difference in intensities between the glycans that contain a motif and those that do not. In this way we calculated the 158 “motif scores” for each lectin.
To obtain the expected changes in lectin binding upon enzymatic modification, we defined modified motifs corresponding to the original motifs. For example, the motif Siaα2-3Galβ1-4GlcNAcβ becomes Galβ1-4GlcNAcβ after treatment with sialidase 1 (the sialidase with specificity for only 2,3-linked sialic acid), but the motif Siaα2-6Galβ1-4GlcNAcβ is unchanged after treatment with sialidase 1 (Fig. 3A). We calculated motif scores using the modified motifs, and we then determined the differences between the modified and the original motif scores for each lectin and enzyme (Fig. 3B). The latter we refer to as “delta motif scores.”
Figure 3. Motif scores for the original and modified motifs.
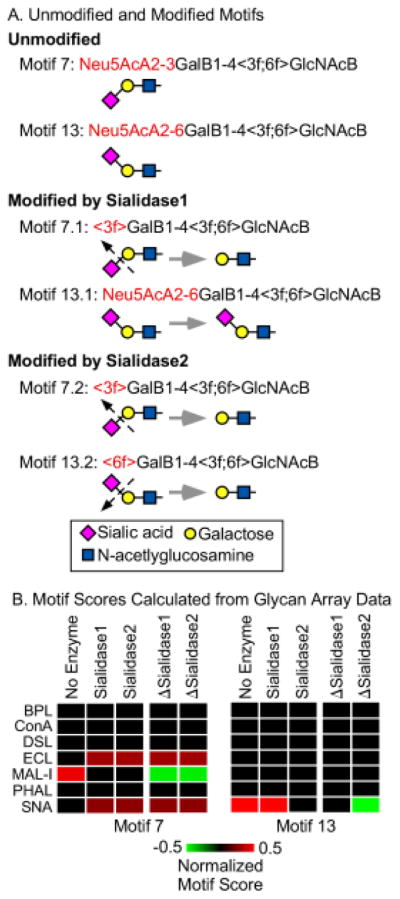
A) We defined 158 motifs covering the variants we were probing. For each motif, we defined modified versions representing treatment by either sialidase 1 or sialidase 2. Two representative motifs are illustrated here. The angled brackets refer to attributes of the immediately following monosaccharide. (See supplementary information for details on the motif language and the motifs.) B) For each of the 158 motifs we calculated a motif score from glycan array data (obtained from the Consortium for Functional Glycomics) for each lectin, using both the original and modified motifs. We then calculated the differences between the modified and original motif scores, indicated by ΔSialidase1 andΔSialidase2.
Next, we arrived at a “motif prediction score.” The motif score for each lectin is multiplied by the normalized binding intensity for the corresponding lectin, and the resulting products are summed over all lectins (Fig. 4A). In addition, the delta motif scores are multiplied by the delta intensities and summed over the lectins (Figs. 4B and 4C). Finally, the motif prediction scores are summed over all conditions. When applied to transferrin, several motifs showed high scores only after the use of enzymatic modifications (Fig. 4D).
Figure 4. Calculation of the motif prediction scores for transferrin.

A) Each motif score for a particular lectin was multiplied by the fluorescence signal for that lectin. The resulting products were summed over all lectins for each motif, giving the motif prediction scores for the unmodified glycans. The calculation was the same for the glycans after modification by sialidase 1 (panel B) and sialidase 2 (panel C), using the Δmotif scores and the Δintensities. The final step was to sum the motif prediction scores over all conditions. D) The top motif was 6-sialyl N-acetyl-lactosamine connected to unbranched mannose. Some of the top 5 motifs had high scores only after summing over the enzyme conditions.
Comparison to Orthogonal Methods
We next assessed the accuracy of the method by comparing results on the control glycoproteins to data from orthogonal glycan analysis methods. A cluster of the top motifs calculated for 5 glycoproteins showed clear groupings among the proteins (Fig. 5A). The groupings mainly were defined by the linkage of sialic acid, branching, and terminal mannose or galactose. According to motif prediction, fetuin displays more 2,3-linked than 2,6-linked sialic acid on simple N-glycan extensions (motifs 37 and 55) and extensions with branching (motifs 44 and 62), but transferrin has more 2,6-linked sialic acid. AGP is predicted to have more branched than unbranched glycans, in contrast to transferrin and fetuin.
Figure 5. Validation of predicted motifs.

A) The heatmap of top prediction motifs shows both similarities and differences among the 5 proteins. The top motifs showed a relative dominance of motifs with α2,3 sialic acid in fetuin and motifs with α2,6 sialic acid in transferrin. AGP showed higher amounts of branched motifs (e.g. motifs 62 and 44) relative to transferrin, which showed higher unbranched motifs (e.g. motif 55). B) A quantification by mass spectrometry showed that the top glycans in transferrin primarily displayed α2,6 sialic acid, but the top glycans in fetuin primarily displayed α2,3 sialic acid. The glycans shown are probable structures based on biosynthetic rules. C) Quantitative glycan analysis by chromatography confirmed differences between transferrin and AGP in the branching of N-linked glycans. D) The top 15 motifs in either fetuin or transferrin had probable assignments in the top glycans found by MS, whereas related motifs with low MP scores had no probably assignments.
We obtained mass spectrometry analysis of fetuin and transferrin (Fig. S-1) and chromatographic analysis of transferrin and AGP (Fig. S-2). The mass spectrometry analysis used ethyl esterification of sialic acids29 to distinguish 2,3 from 2,6 sialic acid linkages, and the chromatography enable quantification of the percentage of branched glycans. The MS data revealed that the top N-linked glycans on fetuin primarily have 2,3-linked sialic acid, but the top glycans on transferrin primarily have 2,6-linked sialic acid (Fig. 5B). The chromatographic analysis showed that transferrin mainly has unbranched N-linked glycans, unlike AGP with mainly branched glycans (Fig. 5C).
Thus the motif predictions of 2,3 relative to 2,6 sialic acid, as well as differences in the amount of branching on N-linked glycans, agreed with the results from independent methods. The prediction of terminal mannose on transferrin but not fetuin agreed with the mass spectrometry data, which showed glycans with terminal mannose only on transferrin (Fig. S-1). Terminal mannose is not commonly observed on transferrin, so its presence could be due to contamination, but the agreement between the methods suggests the identification is not false.
We further explored the validity of the findings by asking whether the top predicted motifs could be reasonably assigned to the top glycan compositions found by MS. Among the top 15 motifs found in either fetuin or transferrin, all but one could be assigned to the probable structures found by MS (Fig. 5D). In contrast, among motifs that are closely related but had low motif prediction scores, none could not be assigned to the probable structures. The lack of assignment of one top motif could be due to a false interpretation of the MS results, given that other structures are possible besides the ones shown. In any case the comparison supports the validity of the predicted motifs.
Application to Clinical Specimens
We next asked whether we could apply the method to glycoproteins captured out of biological fluid. We studied the nature of a glycoform of MUC5AC that we previously showed is elevated in pancreatic cancer12. The glycan is sialylated version of a stem cell marker detected by an antibody called TRA-1-6031. TRA-1-60 detects the non-sialylated glycan, so we removed the sialic acid using sialidase prior to detection (Fig. 6A). The sialylated glycan, called LSTa, is attached to the protein MUC5AC and is strongly elevated in the plasma of cancer patients relative to patients with benign pancreatic disease (Fig. 6B)12. The linkage of the sialylation, whether 2,3-linked or 2,6-linked, was not evident from our previous assay because we used the broad-specificity sialidase. Therefore we sought to determine the linkage of sialic acid on LSTa attached to MUC5AC.
Figure 6. Probing glycans on MUC5AC captured from plasma.

A) The TRA-1-60 antibody detects a non-sialylated glycan. To detect the glycan on MUC5AC, we captured MUC5AC from plasma using antibody arrays, treated the captured protein with sialidase, and probed with the antibody. B) The application of the method in panel A to samples from pancreatic cancer patients and subjects with benign pancreatic conditions revealed elevated levels in a significant number of cancer patients. C) Antibody arrays incubated with a cancer patient sample were probed with either BPL, ECL, or SNA using either no enzyme modification or treatment with one of the sialidases. D) We applied the motif prediction algorithm using either just the no enzyme condition or the sum over all conditions. The cluster shows the top 20 motifs from either method. Motif 50 was high using only in the summed value. E) A comparison of motifs 50, 68, and 88 showed that without summing over enzymatic modifications, no values were achieved for motifs 50 and 68. F) We probed a cancer patient sample and a control sample with the TRA-1-60 antibody using the 3 conditions. The pattern of increased binding after the sialidases agrees with the motif prediction result.
We tested whether on-chip GMAP, used with a small set of lectins, could provide accurate information for MUC5AC captured out of a plasma sample from a cancer patient and another from a control subject (Fig. 6C). The motif prediction scores calculated without the use of enzymatic modifications showed mainly 2,6-linked sialic acid motifs and type-2 N-acetyl-lactosamine (LacNAc) (Fig. 6D), which is more common than type-1 LacNAc. and the motifs containing the LSTa antigen did not have positive motif prediction scores (Fig. 6D). When we incorporated enzymatic modifications into the calculations, a motif representing the LSTa antigen (type-1 and type-2 LacNAc in sequence) with 2,3-linked sialic acid (motif 50) showed up as one of the top motifs. Comparison motifs with 2,6-linked sialic acid (motif 68) or no sialic acid (motif 88) had low scores (Fig. 6D), and the control plasma sample showed low scores for all motifs (not shown). Information about the sialylation of the LSTa antigen was derived only with the use of enzymated modifications (Fig. 6E).
The amount of plasma used in the complete analysis was 20.3 μL, based on 3 lectins, 3 conditions, 3 replicates, 1.5 μL/array, and a 2-fold dilution of the plasma. The concentration of MUC5AC in the plasma was not quantified because we did not have a calibration standard. Such a standard would be difficult to produce because the capture antibody (clone 45M1) poorly recognizes synthesized portions of MUC5AC, and because we are detecting specific glycoforms rather than the core protein. (The ability of the 45M1 clone to capture MUC5AC was not affected by varying glycosylation, according to a previous analysis.) Previous estimates of the detection limit for the antibody-lectin sandwich assay for an analyte in blood plasma are around 10 ng/mL, or 130 pM for an 80 kD/mole protein. Given a concentration in the plasma of 1 μg/mL for MUC5AC, total protein required was 0.2 μg, or around 1 femtomole for a 250kD/mole protein (the molecular weight of MUC5AC is variable depending on fragmentation and glycosylation). In addition, the reproducibility of the measurements was good; the average coefficient of variation across 3 replicates was 0.17, and the average Pearson correlation between sets was 0.87 (Fig. S-3).
We could assess the accuracy of the prediction by examining the binding of the TRA-1-60 antibody with and without treatment by each of the sialidases. Binding increased greatly upon application of the sialidase specific for 2,3-linked sialic acid (sialidase 1) and increased slightly more with the broader sialidase (sialidase 2) (Fig. 6F). Thus the pattern of increased binding of TRA-1-60 agrees with the motif prediction of primarily 2,3-linked sialic acid on the LSTa antigen.
DISCUSSION
Because of the importance of characterizing protein glycoforms in clinical samples, researchers will need methods that are compatible with limited sample volumes or low concentrations of the targeted proteins. Here we demonstrate a framework for achieving that goal using microscale capture, enzymatic modification, and lectin probing of the glycans, combined with an algorithm for the automated interpretation of the data. We validated the calculations of particular motif predictions by comparing results from control proteins to data obtained from MS and chromatography. The amount of protein consumed on-chip was only ~11 ng. We then demonstrated that without purification from large amounts of sample, we could learn about a specific feature of MUC5AC glycosylation that is associated with cancer, namely the sialic acid linkage on the LSTa glycan. The amount of protein required was about 0.2 μg of a 1 μg/mL protein in 20 μL of plasma. In contrast, most of the previous studies of mucin glycosylation used cell lines or several milliliters of a human specimen in order to purify at least 5–10 μg of protein, as in investigations of MUC132 and MUC1633 glycosylation.
The protein requirements of certain methods could be on par with on-chip GMAP. For example, a process of 2D gel separation, in-gel glycan release from individual protein spots, and analysis of intact glycans by chromatography was demonstrated for identifying a glycan released from possibly as low as 50 ng of acute phase proteins34. Also, in-situ glycan release from tissue and analysis by MALDI mass spectrometry could be compatible with similarly low protein quantities35. Such methods could provide complementary capabilities and could be used in combination with on-chip GMAP for added value. MS and chromatography would provide information about complete compositions, and used in a glycoproteomics mode can give site mapping and density, whereas on-chip GMAP would provide motif information with minimal sample usage and software to aid interpretation. The ambiguities from each method potentially could be cleared up by the use of all the methods in conjunction36. The features of on-chip GMAP that could be particularly useful for disease research include the ability to process many samples; low cost; and reproducible measurements allowing comparisons across populations. In addition, automated interpretations can improve accuracy and throughput while opening up the methods to researchers with little expertise in glycan analysis methods.
The on-chip GMAP strategy has several limitations. It does not give information about the complete composition of a glycan; it does not readily sort out information between glycans in samples with multiple glycans; and it does not provide information about site occupancy on a glycoprotein. In addition, the method currently is not quantitative for determining whether one motif is present at a higher occupancy than another within a given protein, because the computation of the motif prediction score is based on many factors that are variable between motifs. One could mitigate these limitations in various ways. For quantitation, one could use standards for the lectins and enzymes in order to calibrate the experimental measurements. To sort out the analysis of complex samples or proteins, one could simplify the sample by cleaving off components that are not relevant to the analysis, for example by removing N-glycans using the PNGase F enzyme prior to analyzing O-glycans. An enzyme to remove O-glycans is not available, so the reverse experiment is not possible, but the software potentially could distinguish between N-glycan and O-glycan motifs based on PNGaseF treatment after each round of lectin probing. The main goal in the current work was to test whether the results are valid, which was confirmed in the comparisons with MS and chromatography. Thus the method presented here does not represent a fully-developed system but rather a platform onto which we may add capabilities.
Since the method gives information only according to the lectins and enzymes used in the assay, an important area for further development is the validation of additional lectins and enzymes that could probe a broader range of glycans. The repertoire of reagents currently available covers many structures, but undoubtedly more lectins and enzymes are needed to probe uncommon features or non-mammalian glycans. Given the continual discoveries of lectins and enzymes with novel specificities, such resources will be forthcoming. In some cases, the required specificities may already be available but simply need to be identified. A database of analyzed and searchable glycan array data would be useful for finding reagents, as demonstrated earlier25.
Another area for development is improved information about the specificities of the lectins and enzymes. In the current state of development, we have incomplete information about most reagents. The progress in glycan array technology in its breadth of coverage and availability gives a good opportunity for filling in some of the information. For example, developers of glycan arrays have created arrays to cover mammalian glycans37–38, microbial glycans39–40, various types of sialylated structures41–43, N-linked glycans with asymmetric branching44, and others (reviewed in ref. 45). Ideally we will be able to merge information from multiple, diverse types of glycan arrays—as demonstrated earlier24—in order to overcome the limitations from any particular array. The structural modeling of lectin-glycan interactions also could provide insights into lectin specificities beyond what is possible from glycan array data46–47. Glycan arrays also would be useful for learning more about the specificities of exoglycosidases; one could probe changes in the glycans subsequent to treatment with an exoglycosidases. A study of influenza neuraminidase20 demonstrated the feasibility of such an approach.
CONCLUSIONS
The method presented here promises to be valuable in applications where sample amounts or analyte concentrations are low, or where precision comparisons over multiple samples are needed. Moreover, it should be valuable as a complement to orthogonal methods such as mass spectrometry and sequencing chromatography. The latter methods reveal glycan compositions and some structural information, but additional data about particular motifs could help to resolve ambiguities48. With further work to make the protocols routine and the reagents and software readily available, the method could improve accessibility of glycan analysis to researchers involved in a broad range of studies.
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute (Alliance of Glycobiologists for Cancer Detection, 1U01CA168896; R21CA186799); the National Institute of General Medical Sciences (1R41GM112750); the National Institute for Allergy and Infectious Disease (R21AI129872); the state of South Carolina SmartState Endowed Research program; the Strategic Positioning Fund (SPF2013/001) (“GlycoSing”) from the Biomedical Research Council (BMRC) of Agency for Science, Technology and Research (A*STAR), Singapore; and A*STAR’s Joint Council (JCO) Visiting Investigator Programme (“HighGlycoART”).
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflict of Interest Disclosure
The authors declare no competing financial interest.
ASSOCIATED CONTENT
- Figure S-1. Mass spectra of detach N-linked glycans from human transferrin and bovine fetuin. (Page S-9)
- Figure S-2. Chromatographic traces and peak assignments. (Page S-10)
- Figure S-3. Analysis of reproducibility. (Page S-11)
References
- 1.Thaysen-Andersen M, Packer NH, Schulz BL. Mol Cell Proteomics. 2016;6:1773–1790. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wada Y, Dell A, Haslam SM, Tissot B, Canis K, Azadi P, Backstrom M, Costello CE, Hansson GC, Hiki Y, Ishihara M, Ito H, Kakehi K, Karlsson N, Hayes CE, Kato K, Kawasaki N, Khoo KH, Kobayashi K, Kolarich D, Kondo A, Lebrilla C, Nakano M, Narimatsu H, Novak J, Novotny MV, Ohno E, Packer NH, Palaima E, Renfrow MB, Tajiri M, Thomsson KA, Yagi H, Yu SY, Taniguchi N. Mol Cell Proteomics. 2010;4:719–727. doi: 10.1074/mcp.M900450-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Royle L, Radcliffe CM, Dwek RA, Rudd PM. Methods Mol Biol. 2006:125–143. doi: 10.1385/1-59745-167-3:125. [DOI] [PubMed] [Google Scholar]
- 4.Sharon N. J Biol Chem. 2007;5:2753–2764. doi: 10.1074/jbc.X600004200. [DOI] [PubMed] [Google Scholar]
- 5.Ching CK, Black R, Helliwell T, Savage A, Barr H, Rhodes JM. J Clin Pathol. 1988;3:324–328. doi: 10.1136/jcp.41.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilobello KT, Krishnamoorthy L, Slawek D, Mahal LK. Chembiochem. 2005;6:985–989. doi: 10.1002/cbic.200400403. [DOI] [PubMed] [Google Scholar]
- 7.Kuno A, Uchiyama N, Koseki-Kuno S, Ebe Y, Takashima S, Yamada M, Hirabayashi J. Nat Methods. 2005;11:851–856. doi: 10.1038/nmeth803. [DOI] [PubMed] [Google Scholar]
- 8.Wu JT. Ann Clin Lab Sci. 1990;2:98–105. [PubMed] [Google Scholar]
- 9.Dwek MV, Jenks A, Leathem AJ. Clin Chim Acta. 2010;23–24:1935–1939. doi: 10.1016/j.cca.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Meany DL, Zhang Z, Sokoll LJ, Zhang H, Chan DW. J Proteome Res. 2009;2:613–619. doi: 10.1021/pr8007539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen S, LaRoche T, Hamelinck D, Bergsma D, Brenner D, Simeone D, Brand RE, Haab BB. Nat Methods. 2007;5:437–444. doi: 10.1038/nmeth1035. [DOI] [PubMed] [Google Scholar]
- 12.Tang H, Partyka K, Hsueh P, Sinha JY, Kletter D, Zeh H, Huang Y, Brand RE, Haab BB. Cell Mol Gastroenterology & Hepatology. 2016;2:201–221. doi: 10.1016/j.jcmgh.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao Z, Maupin K, Curnutte B, Fallon B, Feasley CL, Brouhard E, Kwon R, West CM, Cunningham J, Brand R, Castelli P, Crippa S, Feng Z, Allen P, Simeone DM, Haab BB. Mol Cell Proteomics. 2013;10:2724–2734. doi: 10.1074/mcp.M113.030700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yue T, Goldstein IJ, Hollingsworth MA, Kaul K, Brand RE, Haab BB. Mol Cell Proteomics. 2009;7:1697–1707. doi: 10.1074/mcp.M900135-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yue T, Maupin KA, Fallon B, Li L, Partyka K, Anderson MA, Brenner DE, Kaul K, Zeh H, Moser AJ, Simeone DM, Feng Z, Brand RE, Haab BB. PLoS ONE. 2011;12:e29180. doi: 10.1371/journal.pone.0029180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Tao SC, Bova GS, Liu AY, Chan DW, Zhu H, Zhang H. Anal Chem. 2011;22:8509–8516. doi: 10.1021/ac201452f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li D, Chiu H, Zhang H, Chan DW. Clin Proteomics. 2013;1:12. doi: 10.1186/1559-0275-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rho JH, Mead JR, Wright WS, Brenner DE, Stave JW, Gildersleeve JC, Lampe PD. J Proteomics. 2014:291–299. doi: 10.1016/j.jprot.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marino K, Bones J, Kattla JJ, Rudd PM. Nat Chem Biol. 2010;10:713–723. doi: 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- 20.Tappert MM, Smith DF, Air GM. J Virol. 2011;23:12146–12159. doi: 10.1128/JVI.05537-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter A, Yue T, Heeringa L, Day S, Suh E, Haab BB. Glycobiology. 2010;3:369–380. doi: 10.1093/glycob/cwp187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xuan P, Zhang Y, Tzeng TR, Wan XF, Luo F. Glycobiology. 2012;4:552–560. doi: 10.1093/glycob/cwr163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cholleti SR, Agravat S, Morris T, Saltz JH, Song X, Cummings RD, Smith DF. Omics: a journal of integrative biology. 2012;10:497–512. doi: 10.1089/omi.2012.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Cummings RD, Smith DF, Huflejt M, Campbell CT, Gildersleeve JC, Gerlach JQ, Kilcoyne M, Joshi L, Serna S, Reichardt NC, Parera Pera N, Pieters RJ, Eng W, Mahal LK. Glycobiology. 2014;6:507–517. doi: 10.1093/glycob/cwu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kletter D, Singh S, Bern M, Haab BB. Mol Cell Proteomics. 2013;4:1026–1035. doi: 10.1074/mcp.M112.026641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarter C, Kletter D, Tang H, Partyka K, Ma Y, Singh S, Yadav J, Bern M, Haab BB. Proteomics Clinical Applications. 2013:632–641. doi: 10.1002/prca.201300069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forrester S, Kuick R, Hung KE, Kucherlapati R, Haab BB. Molecular Oncology. 2007:216–225. doi: 10.1016/j.molonc.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ensink E, Sinha J, Sinha A, Tang H, Calderone HM, Hostetter G, Winter J, Cherba D, Brand RE, Allen PJ, Sempere LF, Haab BB. Anal Chem. 2015;19:9715–9721. doi: 10.1021/acs.analchem.5b03159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiding KR, Blank D, Kuijper DM, Deelder AM, Wuhrer M. Anal Chem. 2014;12:5784–5793. doi: 10.1021/ac500335t. [DOI] [PubMed] [Google Scholar]
- 30.Powers TW, Holst S, Wuhrer M, Mehta AS, Drake RR. Biomolecules. 2015;4:2554–2572. doi: 10.3390/biom5042554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews PW, Banting G, Damjanov I, Arnaud D, Avner P. Hybridoma. 1984;4:347–361. doi: 10.1089/hyb.1984.3.347. [DOI] [PubMed] [Google Scholar]
- 32.Parry S, Hanisch FG, Leir SH, Sutton-Smith M, Morris HR, Dell A, Harris A. Glycobiology. 2006;7:623–634. doi: 10.1093/glycob/cwj110. [DOI] [PubMed] [Google Scholar]
- 33.Kui Wong N, Easton RL, Panico M, Sutton-Smith M, Morrison JC, Lattanzio FA, Morris HR, Clark GF, Dell A, Patankar MS. J Biol Chem. 2003;31:28619–28634. doi: 10.1074/jbc.M302741200. [DOI] [PubMed] [Google Scholar]
- 34.Abd Hamid UM, Royle L, Saldova R, Radcliffe CM, Harvey DJ, Storr SJ, Pardo M, Antrobus R, Chapman CJ, Zitzmann N, Robertson JF, Dwek RA, Rudd PM. Glycobiology. 2008;12:1105–1118. doi: 10.1093/glycob/cwn095. [DOI] [PubMed] [Google Scholar]
- 35.Powers TW, Neely BA, Shao Y, Tang H, Troyer DA, Mehta AS, Haab BB, Drake RR. PLoS One. 2014;9:e106255. doi: 10.1371/journal.pone.0106255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Y, Lasanajak Y, Song X, Hu L, Ramani S, Mickum ML, Ashline DJ, Prasad BV, Estes MK, Reinhold VN, Cummings RD, Smith DF. Mol Cell Proteomics. 2014;11:2944–2960. doi: 10.1074/mcp.M114.039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC. Proc Natl Acad Sci U S A. 2004;49:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukui S, Feizi T, Galustian C, Lawson AM, Chai W. Nat Biotechnol. 2002;10:1011–1017. doi: 10.1038/nbt735. [DOI] [PubMed] [Google Scholar]
- 39.Stowell SR, Arthur CM, McBride R, Berger O, Razi N, Heimburg-Molinaro J, Rodrigues LC, Gourdine JP, Noll AJ, von Gunten S, Smith DF, Knirel YA, Paulson JC, Cummings RD. Nat Chem Biol. 2014;6:470–476. doi: 10.1038/nchembio.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D, Liu S, Trummer BJ, Deng C, Wang A. Nat Biotechnol. 2002;3:275–281. doi: 10.1038/nbt0302-275. [DOI] [PubMed] [Google Scholar]
- 41.Nycholat CM, McBride R, Ekiert DC, Xu R, Rangarajan J, Peng W, Razi N, Gilbert M, Wakarchuk W, Wilson IA, Paulson JC. Angew Chem Int Ed Engl. 2012:4860–4863. doi: 10.1002/anie.201200596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song X, Yu H, Chen X, Lasanajak Y, Tappert MM, Air GM, Tiwari VK, Cao H, Chokhawala HA, Zheng H, Cummings RD, Smith DF. J Biol Chem. 2011:31610–31622. doi: 10.1074/jbc.M111.274217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Padler-Karavani V, Song X, Yu H, Hurtado-Ziola N, Huang S, Muthana S, Chokhawala HA, Cheng J, Verhagen A, Langereis MA, Kleene R, Schachner M, de Groot RJ, Lasanajak Y, Matsuda H, Schwab R, Chen X, Smith DF, Cummings RD, Varki A. J Biol Chem. 2012;27:22593–22608. doi: 10.1074/jbc.M112.359323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, Boons GJ. Science. 2013;6144:379–383. doi: 10.1126/science.1236231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rillahan CD, Paulson JC. Annu Rev Biochem. 2011:797–823. doi: 10.1146/annurev-biochem-061809-152236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor ME, Drickamer K. Glycobiology. 2009;11:1155–1162. doi: 10.1093/glycob/cwp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grant OC, Tessier MB, Meche L, Mahal LK, Foley BL, Woods RJ. Glycobiology. 2016;7:772–783. doi: 10.1093/glycob/cww020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ashline DJ, Yu Y, Lasanajak Y, Song X, Hu L, Ramani S, Prasad V, Estes MK, Cummings RD, Smith DF, Reinhold VN. Mol Cell Proteomics. 2014;11:2961–2974. doi: 10.1074/mcp.M114.039925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.