

Abstract
Primary cilia are microtubule‐based structures present on most mammalian cells that are important for intercellular signaling. Cilia are present on a subset of endothelial cells where they project into the vessel lumen and are implicated as mechanical sensors of blood flow. To test the in vivo role of endothelial cilia, we conditionally deleted Ift88, a gene required for ciliogenesis, in endothelial cells of mice. We found that endothelial primary cilia were dispensable for mammalian vascular development. Cilia were not uniformly distributed in the mouse aorta, but were enriched at vascular branch points and sites of high curvature. These same sites are predisposed to the development of atherosclerotic plaques, prompting us to investigate whether cilia participate in atherosclerosis. Removing endothelial cilia increased atherosclerosis in Apoe −/− mice fed a high‐fat, high‐cholesterol diet, indicating that cilia protect against atherosclerosis. Removing endothelial cilia increased inflammatory gene expression and decreased eNOS activity, indicating that endothelial cilia inhibit pro‐atherosclerotic signaling in the aorta.
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Molecular Biology of Disease
Introduction
Primary cilia are signaling organelles that protrude from the surface of many types of cells. They play important roles in chemosensation in a wide variety of systems 1. In development, cilia are best known for their obligate role in vertebrate Hedgehog (Hh) signaling, but also function in other intercellular signaling pathways, including some forms of GPCR signaling 1. In addition to chemosensation, cilia are thought to function in some contexts as mechanosensors 2, 3, 4, 5, 6, 7. For example, kidney epithelial cells may use cilia to sense urine flow 3, 8, 9. Cultured mouse aortic ECs flux calcium and synthesize nitric oxide in response to the onset of flow 7. These responses depend on both Pkd proteins and primary cilia, suggesting that EC cilia are mechanosensors that sense blood flow 6. In zebrafish, cilia and Pkd2 cooperate to regulate calcium flux and angiogenesis 5. Mouse endothelial cells (ECs) line the lumen of blood vessels and can possess cilia, especially in areas of disturbed flow 10. The function of these cilia has been unclear.
Results and Discussion
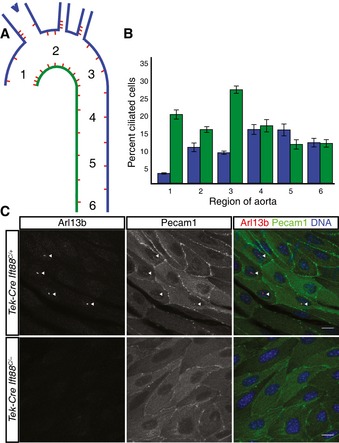
To examine the distribution of cilia within the aorta, we stained wild‐type mouse aortas for the ciliary marker Arl13b. ECs were identified by costaining for Pecam1 (CD31). Cilia projected from the apical surface of a subset of ECs into the lumen of the vessel. The distribution of ciliated cells within the vasculature was not uniform: Cilia were more common on the lesser curvature (more ventral and caudal side) of the aortic arch and less common on the greater curvature (more dorsal and rostral side) (Fig 1A and B). The distribution of ciliated cells was more uniform in the descending thoracic aorta, where blood flow is less disturbed. Intriguingly, these are the same areas susceptible to atherosclerosis in humans and mouse models 11, 12. Our findings are concordant with a previous study that found that cilia are enriched in curved and branched regions of the aorta 10.
Figure 1. Endothelial primary cilia are non‐uniformly distributed in the adult mouse aorta and are dispensable for development.
-
A, BPrimary cilia are enriched at branch points and the lesser curvature of the aorta of wild‐type mice, schematized in (A) and quantified in (B). The color in (A) indicates which side of the aorta is quantified in (B). Numbers indicate the region of the aorta (A) whose proportion of ciliated cells is quantified in (B). Error bars are ± 1 SEM and n = 7 mice.
-
CAortic endothelial cilia are ablated in Tek‐Cre Ift88 C/− mice. Endothelial cells and cilia are labeled by staining for Pecam1 and Arl13b, respectively. Scale bars are 10 μm.
We developed mice lacking EC cilia by conditionally deleting intraflagellar transport 88 (Ift88) 13, a gene essential for ciliogenesis and cilia maintenance, using Tek‐Cre. Tek‐Cre is expressed by embryonic day (E) 7.5 in angioblasts (Fig EV1A) 14 and induces recombination in ECs and the hematopoietic lineage. Tek‐Cre Ift88 C/− mice were born in normal ratios, viable and fertile (Fig EV2A). Tek‐Cre activated the ROSA26 mTmG reporter by gestational day E7.5 specifically in angioblasts, indicating that Tek‐Cre activity is early and concomitant with the differentiation of angioblasts (Fig EV1A). Tek‐Cre maintained the activity of the ROSA26 mTmG reporter in ECs later in embryonic development and in postnatal animals, indicating that it activated recombination pervasively throughout ECs (Fig EV1B and D). We confirmed that EC cilia are specifically absent in Tek‐Cre Ift88 C/− mice by immunofluorescent staining juvenile and adult aortas for Arl13b (Figs EV1F and G, and 1C). To confirm that EC cilia are dispensable for vascular development, we deleted a different gene essential for ciliogenesis, Kif3a, in ECs. Like Tek‐Cre Ift88 C/− mice, Tek‐Cre Kif3a C/− mice were born in normal ratios and postnatally viable (Fig EV2A), and exhibited loss of EC cilia (Fig EV1E).
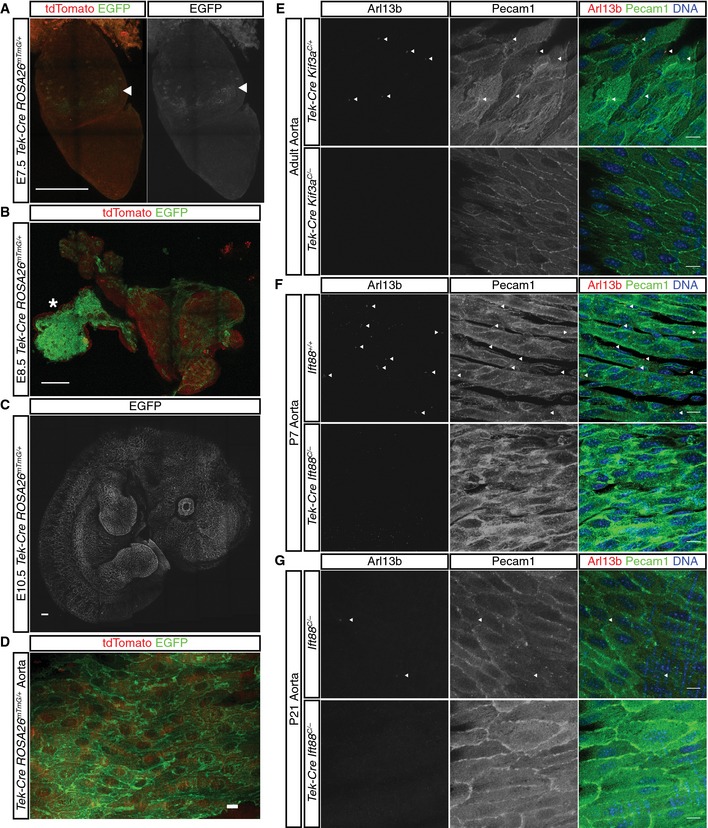
Figure EV1. Tek‐Cre mediates early, robust, and specific recombination in angioblasts.
-
A–DTek‐Cre is active in the mouse embryo in mTmG reporter mice by (A) E7.5 and labeling persists at (B) E8.5, (C) E10.5, and (D) in the aorta of adult mice. The arrowheads in (A) indicate the initial site of recombination in extraembryonic mesoderm. The asterisk in (B) indicates the yolk sac.
-
E, FPrimary cilia are efficiently removed from aortic endothelial cells by Tek‐Cre‐mediated removal of Ift88 at (E) P5 and (F) P14.
-
GPrimary cilia can also be ablated by Tek‐Cre‐mediated deletion of Kif3a. Endothelial cells and cilia are labeled by anti‐Pecam1 staining (green) and anti‐Arl13b staining (red).
Figure EV2. Tek‐Cre‐mediated depletion of Ift88, Kif3a, Smo, or Kif3a in a Pecam Gt/Gt background does not affect ratios at weaning, whereas Tek‐Cre Pkd2 mice are subviable.
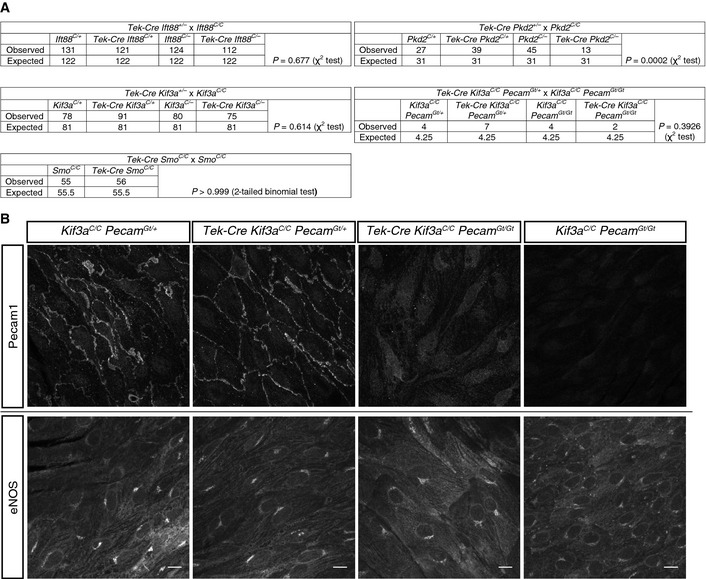
- Mice from the indicated crosses were weaned and genotyped at P21. Statistics (test indicated in table) suggest only Pkd2 endothelial cell deletion results in subviability.
- Littermate Tek‐Cre Kif3a Pecam mice of the indicated genotypes were sacrificed at P604 and stained for Pecam1 and eNOS (in different fields). Loss of Pecam1, EC cilia, or both did not affect aortic EC eNOS expression. Scale bars, 10 μm.
Most vertebrate Hh signaling depends on primary cilia, and the absence of a developmental phenotype in mice lacking EC cilia suggests that EC Hh signaling is not required for vascular development. To confirm EC Hh signaling is not required for vascular development, we generated Tek‐Cre Smo C/C mice which lack smoothened, a critical component of Hh signaling, in ECs. Tek‐Cre Smo C/C mice were also viable and did not display any discernable vascular phenotype (Fig EV2A). These data are consistent with previous results whereby Smo was deleted in the vasculature using Flk1‐Cre 15.
Angiogenesis is regulated by multiple pathways, including flow 16, 17, 18, 19. Vascular morphology was grossly normal in mice lacking EC cilia. To explore whether, as in zebrafish, EC cilia are required for development of small vessels, we quantified the outgrowth and degree of branching of retinal vasculature in day postnatal day (P) 5 mice. We found no significant differences in the growth of the retinal vasculature (Fig 2B) or the amount of branching (Fig 2C) between control and Tek‐Cre Ift88 C/− mice. Thus, unlike in the zebrafish 5, primary cilia and ciliary Hh signaling in ECs are dispensable for vascular development in the mouse.
Figure 2. Retinal angiogenesis and aortic endothelial polarity are unaffected in mice lacking EC cilia.
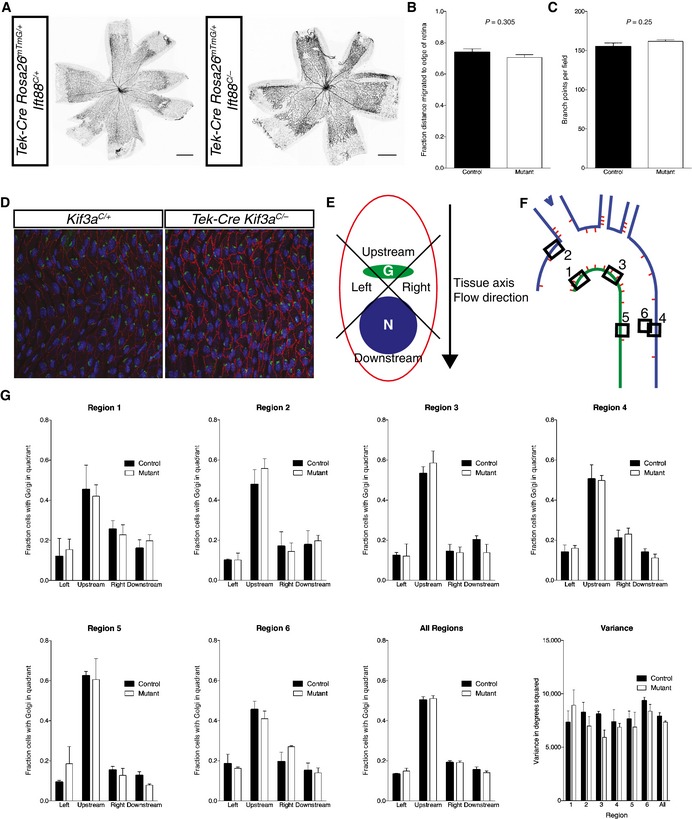
- The vasculature in P5 retinas from (A) mTmG reporter mice with and without EC cilia was visualized by EGFP fluorescence.
- The fractional distance migrated to the edge of the retina was quantified. There was no statistically significant difference (P = 0.305, Student's two‐tailed t‐test, n = 6 mice for each group). The average value of 8 measurements (4 retina leaflets measured per eye on both eyes) was used for each mouse.
- The number of vascular branch points was counted. There was no statistically significant difference (P = 0.25, Student's two‐tailed t‐test, n = 6 for control and n = 5 for mutant).
- Control and mutant adult mouse aortas were stained for GM130 (green) to mark the Golgi, CD144 (red) to mark ECs, and DAPI (blue) to label DNA.
- The angle between the Golgi, nucleus, and tissue axis was measured and binned as “left”, “right”, “upstream”, or “downstream”.
- Six areas in the aorta were analyzed.
- There was no difference in Golgi distribution at any single region or overall, nor was there a difference in the variance of the individual unbinned angles.
One well‐characterized response of ECs to fluid flow is polarization in the direction of flow. Because primary cilia are important for mechanosensation in several cell types, including ECs 5, 7, and are required for interpretation of planar polarity cues in the cochlea 20, we assessed whether EC cilia participate in flow‐induced polarization. We examined EC polarity in Tek‐Cre Kif3a C/− mice lacking endothelial cilia. The Golgi apparatus and centrosome are typically polarized upstream of the nucleus (toward the heart) in aortic ECs 21. We quantified the angle between the Golgi and nucleus relative to the axis of the tissue in multiple locations within aortas of mutant and control mice (Fig 2D–F). Interestingly, there was neither a difference in the average angle of polarization between mutant and control mice, nor was there a difference in the variance (Fig 2G). Thus, cilia are not required for either the accuracy or precision of EC polarization in the direction of blood flow in vivo.
Given the close correlation between the distribution of cilia and the atherosclerosis‐prone areas of the aorta, we investigated whether loss of EC cilia influences the development of atherosclerotic plaques. Apoe −/− mice lack apolipoprotein E, have increased levels of circulating LDL, VLDL, and cholesterol, and develop atherosclerosis 22. We fed experimental mice lacking EC cilia (Tek‐Cre Ift88 C/− Apoe −/−) and control mice (Tek‐Cre Ift88 C/+ Apoe −/−, Ift88 C/+ Apoe −/−, or Ift88 C/− Apoe −/−) a high‐fat, high‐cholesterol diet that accelerates the development of atherosclerosis 23. After 8 weeks on this diet, we quantified the amount of atherosclerotic plaques by oil red O staining (Fig 3B and C). Experimental mice lacking endothelial cilia displayed a 59% increase in atherosclerotic lesional surface area over control mice in females (P = 0.0135) (Fig 3D) and 67% in males (P = 0.0106) (Fig 3E). Loss of cilia did not change the distribution of plaque formation or induce ectopic plaques, but increased the size of plaques at atherosclerosis‐prone sites including the branch points of the great arteries and the inner curvature of the aorta. A cohort of Apoe −/− mice were also analyzed histologically at the aortic sinus for plaque size and composition. Although the plaque size of mice lacking EC cilia tended to be larger (Fig EV3A and B) and have increased CD68+ area (Fig EV3C and D), an indicator of macrophage infiltration, there was no statistically significant difference.
Figure 3. Loss of EC cilia accelerates atherosclerosis.
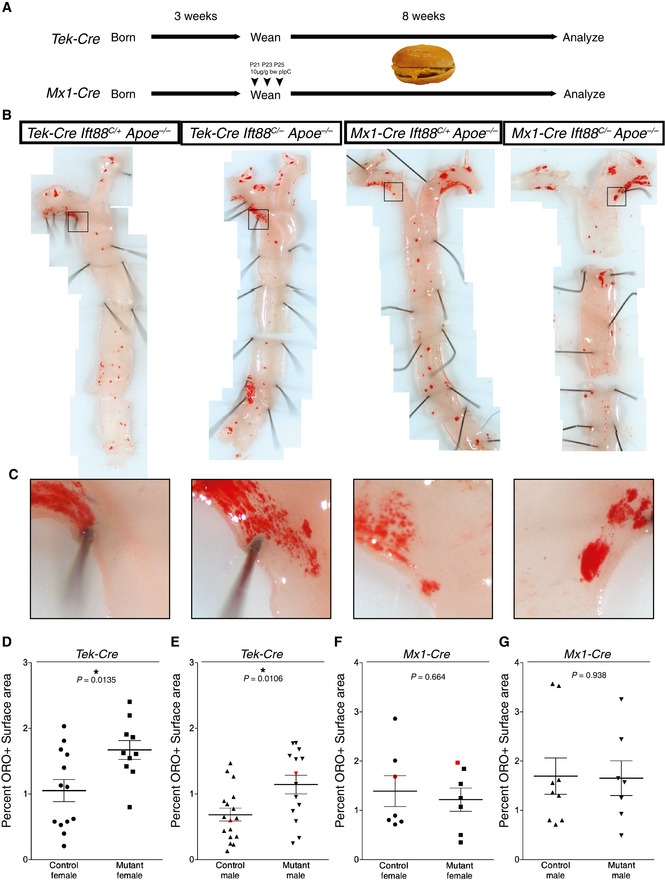
-
AScheme for induction of atherosclerosis Apoe −/− mice lacking Ift88 in the Tek‐Cre and Mx1‐Cre lineages (and control littermates). Following weaning, mice were placed on a high‐fat, high‐cholesterol diet for 8 weeks prior to analysis.
-
BLoss of EC cilia by Tek‐Cre increases atherosclerosis in the aortas of Apoe −/− mice on a high‐fat, high‐cholesterol diet as visualized by oil red O (ORO) staining (left panels), whereas loss of cilia genes in blood by Mx1‐Cre has no effect (right panels).
-
CCropped views of the boxed areas in (B) showing increased atherosclerosis in mice with Tek‐Cre‐ but not Mx1‐Cre‐mediated loss of Ift88 in the aortic arch.
-
D–GScatter plots of the percent ORO‐positive area of the thoracic aorta in (D, E) Tek‐Cre and (F, G) Mx1‐Cre mice, separated by (D, F) females and (E, G) males. Tek‐Cre Apoe −/− Ift88 C/− mice had a 58.9% (P = 0.0135, Student's two‐tailed t‐test, n = 13 control and n = 10 experimental mice) and 67% (P = 0.0106, Student's two‐tailed t‐test, n = 17 control and n = 14 experimental mice) increase in ORO‐positive area versus control littermates in females and males, respectively. Mx1‐Cre Apoe −/− Ift88 C/− mice had no significant difference in ORO‐positive area in females (P = 0.6643, Student's two‐tailed t‐test, n = 7 control and experimental mice) or males (P = 0.9381, Student's two‐tailed t‐test, n = 9 control and n = 7 experimental mice). Black bars represent the mean and error bars are ± 1 SEM. Aortas depicted in (B, C) are red in the scatter plots.
Figure EV3. Apoe −/− mice lacking EC cilia show increased atherosclerosis at the aortic sinus.
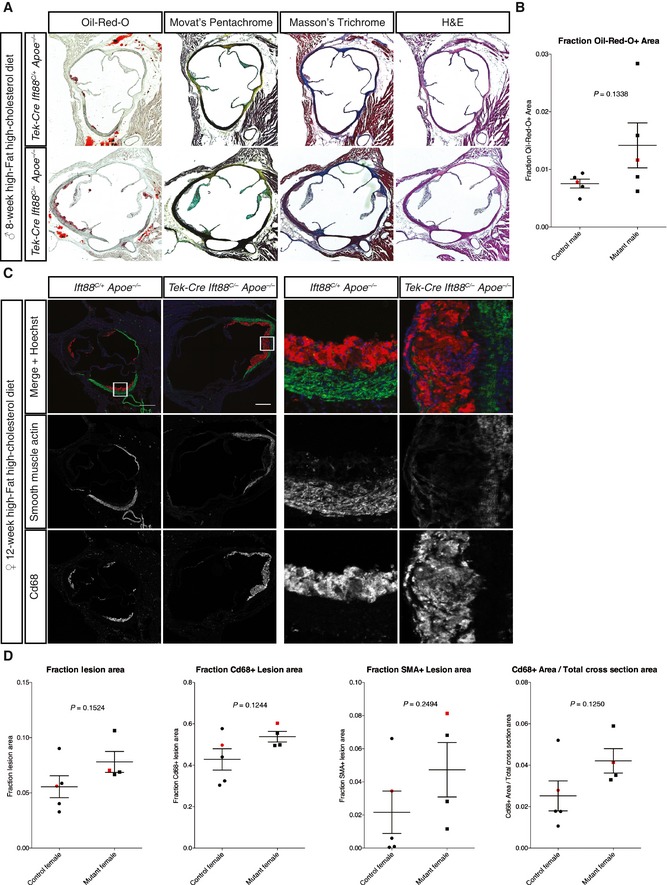
- The aortic sinuses from a cohort of male Apoe −/− mice with and without EC cilia were analyzed histologically with the indicated stains to determine plaque size and composition.
- Quantitation of the ORO+ lesion area showed a trend for increased lesion size in mice lacking EC cilia (88% increase over controls, P = 0.1338, Student's two‐tailed t‐test, n = 5 mice each genotype). Black bars represent the mean and error bars are ± 1 SEM. Data points for sinuses in (A) are indicated in red.
- The aortic sinuses of female Apoe −/− mice with and without EC cilia were stained for CD68 and smooth muscle actin (SMA) following 12 weeks of high‐fat, high‐cholesterol diet. Scale bars, 250 μm.
- Quantitation of plaque composition from (C) showed no statistically significant differences between mice with and without EC cilia, although there was a trend for increased macrophage (CD68+) area. Black bars represent the mean and error bars are ± 1 SEM. Data points for sinuses in (C) are indicated in red. Student's two‐tailed t‐test, n = 5 control and n = 4 mutant mice.
Because Tek‐Cre induces recombination in the angioblast, the common precursor of ECs and the hematopoietic lineage 14, the increased atherosclerosis observed in Tek‐Cre Ift88 C/− Apoe −/− mice could be attributable to loss of Ift88 in the ECs, the blood, or both. Although blood cells are not known to possess primary cilia, non‐ciliary roles for IFT proteins have been described in immune cells 24. To distinguish whether ciliogenic genes are required in the blood lineages or the endothelium to inhibit atherosclerosis, we removed Ift88 from blood using the inducible Mx1‐Cre. As expected, Mx1‐Cre removed Ift88 from blood cells as efficiently as Tek‐Cre, but showed little recombination in non‐hematopoietic tissues compared to Tek‐Cre, reflecting the lack of vascular recombination (Fig EV4A and B). Experimental mice lacking blood Ift88 (Mx1‐Cre Ift88 C/− Apoe −/−) and control mice (Mx1‐Cre Ift88 C/+ Apoe −/−, Ift88 C/+ Apoe −/−, Ift88 C/− Apoe −/−) displayed indistinguishable amounts of atherosclerosis (Fig 3E and F). Thus, loss of Ift88 in blood cells did not alter atherosclerosis. To confirm that ciliogenic genes do not function in the blood to affect atherosclerosis, we generated Mx1‐Cre Kif3a c/− mice to remove Kif3a from blood cells. As with Ift88, loss of Kif3a from the blood cells did not affect atherosclerosis (Fig EV4C). Thus, the anti‐atherosclerotic effect of ciliogenic genes is due to their role in ECs, not blood.
Figure EV4. Extrahematopoietic, but not blood recombination, differs between Tek‐Cre and Mx1‐Cre mice and loss of Kif3a via Mx1‐Cre does not affect atherosclerosis.
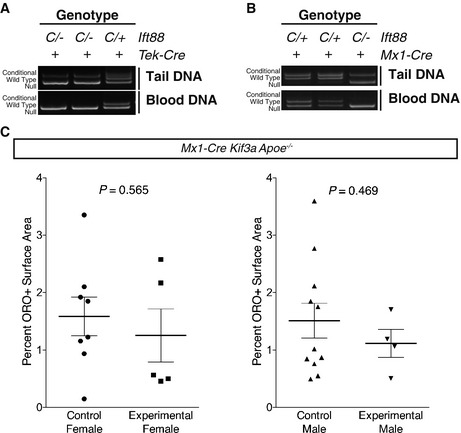
-
A, BTail and peripheral blood DNA was isolated from adult Ift88 mice of the indicated genotypes carrying either (A) Tek‐Cre or (B) Mx1‐Cre. Genotyping for Ift88 indicated total loss of the conditional allele in blood from both Cre lines. However, only Tek‐Cre caused noticeable recombination in tail DNA, reflecting vascular recombination in the Tek‐Cre but not Mx1‐Cre lines. In each panel, the same 3 mice were assayed for blood and tail recombination and are presented in the same order. “C” indicates conditional allele, “+” indicates the wild‐type allele, and “‐” indicates the recombined null allele. The listed genotypes are the germ line genotypes of each mouse.
-
CAtherosclerosis was induced in control (Mx1‐Cre Kif3a C/+ Apoe −/−, Kif3a C/+ Apoe −/−, Kif3a C/− Apoe −/−) and mutant (Mx1‐Cre Kif3a C/− Apoe −/−) mice and the percent atherosclerosis assessed by oil red O staining. No difference was observed between control and experimental mice in males (P = 0.4692, Student's two‐tailed t‐test, n = 11 control and n = 4 experimental mice) or females (P = 0.5653, Student's two‐tailed t‐test, n = 8 control and n = 5 experimental mice). Horizontal bars are the mean and error bars are ± 1 SEM.
To investigate how EC cilia limit atherosclerosis, we analyzed whole aortas for gene expression changes. As expected, deletion of Ift88 from ECs in Tek‐Cre Ift88 C/− mice caused a decrease in the aortic expression of Ift88 (Fig 4A). As cilia are required for vertebrate Hh signaling, we tested whether the Hh pathway was perturbed. Expression of Hh pathway signaling components, including readouts of pathway activation Ptc1, Ptc2, and Gli1, was not altered by removal of endothelial cilia (Fig 4A). Therefore, alterations in Hh signaling do not correlate with changes in atherosclerosis, suggesting that the function of cilia in protecting against atherosclerosis is independent of Hh signaling.
Figure 4. Loss of EC cilia activates pro‐atherosclerotic pathways in Apoe −/− mice.
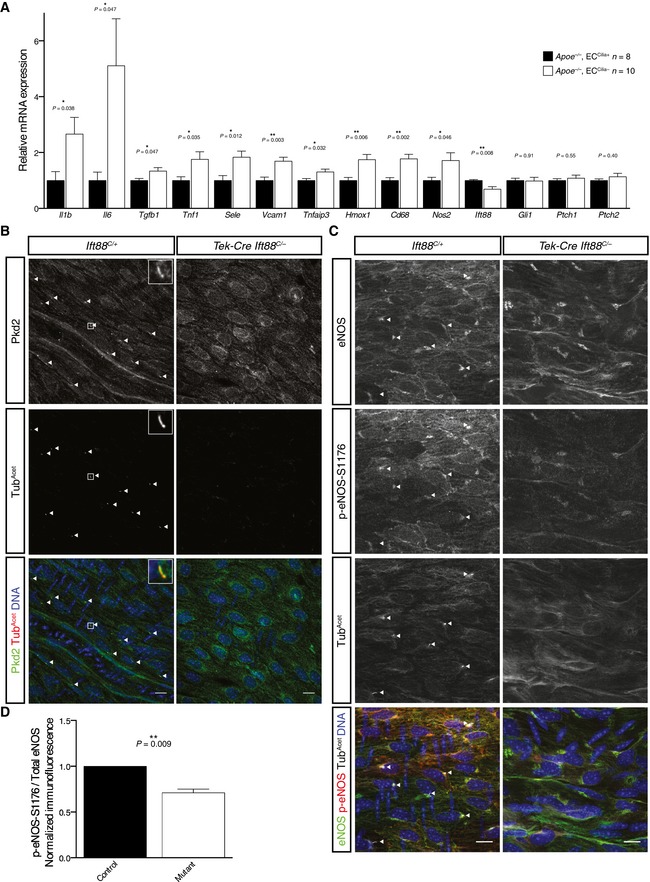
- Transcripts for the inflammatory cytokines Il1b, Il6, Tnf1, and Tgfb1, the adhesion molecules Vcam1 and Sele, the lymphocyte marker Nos2 and macrophage marker Cd68, and the NFκB responsive genes Tnfaip3 and Hmox1 were upregulated in aortas from Apoe −/− mice lacking EC cilia, as assessed by RT–qPCR, while transcripts of the Hh target genes Gli1, Ptc1, and Ptc2 were not. Error bars are +1 SEM. n = 8 control and n = 10 mutant mice of mixed sex. P‐values calculated by Student's two‐tailed t‐test.
- Pkd2 localizes to EC cilia in vivo. Adult mouse aortas were stained for Pkd2, acetylated α‐tubulin, and Hoechst to label DNA. Pkd2 localized strongly to primary cilia but also to other membranous structures in endothelial cells. Arrowheads indicate cilia. Representative cilium highlighted in inset. Scale bars, 10 μm.
- Phosphorylation at serine 1,176 of eNOS was reduced in the aortas of mice lacking EC cilia. Aortas from control and mutant mice were stained for eNOS, p‐eNOS‐S1176, acetylated α‐tubulin, and Hoechst to label DNA. Arrowheads point out cilia. Arrowheads indicate cilia. Scale bars, 10 μm.
- The ratio of the total fluorescence p‐eNOS‐S1176 to total eNOS in the upper thoracic aorta (near the first intercostal artery) was calculated for control and mutant mice. The relative p‐eNOS‐S1176 to eNOS ratio was reduced in the mutant to 72.7% the level of control siblings (P = 0.009, paired two‐tailed ratio t‐test, n = 7 sets of mice). Error bar is +1 SEM. Four sets consisted of one control mouse and one mutant mouse. Three sets consisted of two control or mutant mice and the average value of the two was used.
Atherosclerosis is driven by increased circulating lipids, such as LDL and cholesterol 25. To determine whether loss of EC cilia altered serum lipid composition, we measured serum cholesterol, HDL, LDL, triglyceride, and non‐esterified fatty acid levels in female control (Tek‐Cre Ift88 C/+ Apoe −/−, Ift88 C/+ Apoe −/−, Ift88 C/− Apoe −/−) and experimental (Tek‐Cre Ift88 C/− Apoe −/−) mice (Fig EV5A). We detected no differences in the levels of circulating lipids, suggesting that EC cilia do not restrict atherosclerosis through effects on circulating lipid levels. We also detected no difference in body weight between Apoe −/− mice with and without EC cilia (Fig EV5B).
Figure EV5. Serum lipid profiles and body weight are unchanged by loss of EC cilia in Apoe −/− mice.
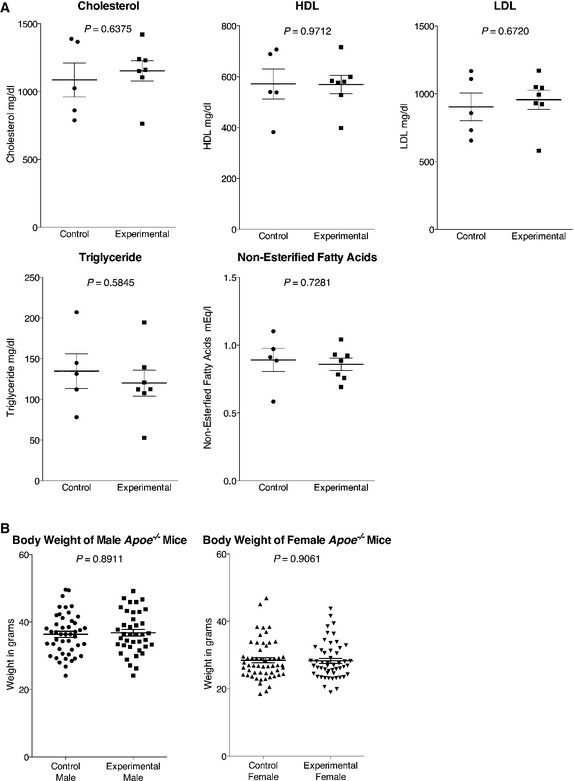
- Serum levels of cholesterol, HDL, LDL, triglyceride, and non‐esterified fatty acids were measured in n = 5 control (Tek‐Cre Ift88 C/+ Apoe −/−, Ift88 C/+ Apoe −/−, Ift88 C/− Apoe −/−) and n = 7 experimental (Tek‐Cre Ift88 C/− Apoe −/−) female mice. Large horizontal bars are the mean and error bars are ± 1 SEM.
- There is no difference in body weight in Apoe −/− mice lacking EC cilia among males (P = 0.8911, Kolmogorov–Smirnov test, n = 46 control and n = 39 mice) or females (P = 0.9061, Kolmogorov–Smirnov test, n = 55 control and n = 52 mutant mice). These data include mice used for other experiments, hence the increased n. Horizontal bars are the mean and error bars are ± 1 SEM.
In contrast to the Hh pathway and lipid levels, removing EC cilia changed the aortic expression of several inflammatory genes. Loss of cilia upregulated the expression of genes encoding the proinflammatory cytokines IL‐1β (Il1b), IL‐6 (Il6), TGF‐β (Tgfb), and TNFα (Tnfa), the NF‐κB target TNFAIP3 (Tnfaip3), as well as the inflammatory adhesion molecules VCAM‐1 (Vcam1) and E‐selectin (Sele) (Fig 4A). Upon removing EC cilia, there was an increase in the expression of genes encoding the macrophage marker CD68 (Cd68) and the lymphocyte marker iNOS (Nos2), suggesting increased immune cell infiltration. Mice lacking EC cilia also displayed an increase in expression of Hmox1, encoding the anti‐inflammatory enzyme Heme oxygenase 1 which is upregulated in response to stress pathways such as NF‐κB 26.
To explore the origins of the increased atherosclerosis caused by removing EC cilia, we examined whether the calcium channel Pkd2 associated with EC cilia in vivo. Pkd2 localized to endothelial primary cilia of mouse aortas (Fig 4B), consistent with observations in cultured endothelial cells 7 and femoral arteries 27. In addition to ciliary localization, substantial signal was detected on other membranous structures in the cell, indicating that Pkd2 could have roles outside cilia. To determine the role of Pkd2 in ECs, we generated Tek‐Cre Pkd2 C/− mice. Unlike mice lacking EC cilia, mice lacking EC Pkd2 were subviable (Fig EV2A and 28, 29, 30). The differences in phenotype caused by removing Pkd2 and cilia in ECs indicate either that Pkd2 has critical functions outside of cilia or that EC cilia restrain Pkd2 function, as has been recently suggested to be the case for kidney cilia 31.
Because Pkd proteins have been shown to participate in flow‐induced nitric oxide synthesis in cultured ECs 7 and nitric oxide signaling restrains atherosclerosis 25, 32, we examined whether EC cilia are required for eNOS activation. We analyzed eNOS activation by staining mouse aortas using antibodies against total eNOS and eNOS phosphorylated at serine 1,176, a marker of eNOS activation (Fig 4C). The ratio of p‐eNOS‐S1176 to total eNOS fluorescence in Ift88 C/− Tek‐Cre mice was reduced to 72.7% (P = 0.009) that of controls (Fig 4D), indicating less active eNOS in mice lacking EC cilia. Importantly, the reduction in eNOS activity was observed in Apoe +/+ mice, consistent with altered eNOS activity not being secondary to atherosclerosis but being a cause of the increased atherosclerosis observed in Apoe −/− mice lacking EC cilia.
Taken together, this investigation into the role of EC primary cilia in vivo demonstrated that that EC cilia are distributed non‐uniformly in the aorta, that mice lacking EC cilia develop normally, and that EC cilia inhibit the development of atherosclerosis. The observations that Tek‐Cre Ift88 C/− and Tek‐Cre Smo C/C conditional mutant mice are viable and that Tek‐Cre Ift88 C/− mice show no defects in angiogenesis indicate that neither cilia nor the Hh pathway is required in angioblasts for mouse development. Several studies show Hh ligands induce tube formation in cultured mouse ECs 33, 34. Taken together, these data suggest that, although Hh signals may be sufficient to induce tube formation in vitro, EC Hh signaling is not necessary for proper vascular development in vivo. Our findings are consistent with an indirect role of the Hh pathway in vascular patterning: Hh regulates the expression of vascular regulators including VEGF and Notch signals, but does not act on ECs directly 15, 35, 36. It has also been proposed that Hedgehog regulates vascular permeability in the brain, although whether cilia are involved is unclear 37. Our results differ from what has been reported in zebrafish, where the Hh pathway is required in angioblasts for arterial specification 38 and EC primary cilia are required to control angiogenesis 5. Thus, EC cilia and ciliary Hh signaling function distinctly in fish and mammals.
High shear stress disassembles EC cilia in vitro 39 and high shear stress inversely correlates with EC cilia distribution in vivo (Fig 1A and B and 10). It is unknown whether cilia sense flow directly to regulate their own presence on ECs or whether ciliogenesis is instead regulated by a distinct upstream mechanosensor. One possibility that could explain their observed distribution would be the induction of EC cilia in response to a particular regime of flow, such as weak flow, or their inhibition by strong laminar flow 5. Consistent with the idea that ECs respond differently to distinct flow regimens, ECs have multiple means for mechanosensation, including a variety of adhesion molecules 40. For example, Pecam1 is implicated in EC mechanosensation 41. We tested whether cilia and Pecam1 might have a synthetic relationship, but found that Tek‐Cre Kif3a C/C Pecam1 Gt/Gt mice are viable and display no obvious vascular defects, indicating that these two pathways do not have overlapping roles critical for vascular development (Fig EV2A and B).
Like cilia, atherosclerosis does not occur uniformly throughout the vasculature. Plaques occur most commonly at areas of high curvature and branch points, where blood flow is more disturbed 11, 12. Elegant studies have shown that disturbed, non‐laminar flow is sufficient to drive inflammatory EC gene expression and atherosclerosis 42. The more frequent occurrence of cilia on ECs in areas of disturbed flow is consistent with the observation that laminar flow disassembles EC cilia in vitro 39. Furthermore, primary cilia are found in association with atherosclerotic plaques in mice 10 and humans 43, 44, 45. Previous studies have alternately proposed anti‐inflammatory 7, 46, 47, 48 and proinflammatory 49, 50, 51 roles for primary cilia in different systems, including a recent report showing ECs lacking cilia are sensitized to endothelial‐to‐mesenchymal transition and calcification 46, 48, but whether EC cilia affect the development of atherosclerosis was unknown.
We found that mice lacking EC primary cilia are more susceptible to atherosclerosis on an Apoe −/− background and placed on a high‐fat, high‐cholesterol diet. Consistent with this, expression of several known inflammatory cytokines and adhesion molecules is elevated in mutant aortas. Multiple signaling pathways modulate athero‐sclerosis to varying degrees 25. For instance, removing Caveolin‐1, an inhibitor of eNOS, on an Apoe −/− background causes an approximately 50% reduction in atherosclerotic lesional area 52. Conversely, removing eNOS on an Apoe −/− background causes a 59.2% increase in females and 93.6% increase in males in atherosclerotic lesional surface area 32. The 59–67% increase in atherosclerosis in Apoe −/− mice caused by removing EC cilia suggests that primary cilia are significant regulators of atherosclerosis.
We found that aortic ECs express Pkd2, a proposed mechanosensitive calcium channel. One potential insight into the connection between Pkd2 function, EC cilia, and vascular disease comes from autosomal dominant polycystic kidney disease (ADPKD), a disease caused by mutation in either PKD1 or PKD2. Cardiovascular manifestations of ADPKD include hypertension, left ventricular hypertrophy, increased carotid intima‐media thickness, and, most clinically important, aneurysm 53. Some of these effects are likely to be secondary to increased renin–angiotensin activity caused by renal insufficiency 53. However, many patients with ADPKD exhibit endothelial dysfunction and increased carotid intima‐media thickness, both indicators of atherosclerosis, before signs of renal dysfunction or hypertension 53. Thus, PKD1 or PKD2 dysfunction within endothelial cells may contribute to atherosclerosis and aneurysm in a way that may partially depend upon cilia.
The reduced activated eNOS levels in mice lacking EC cilia may be a downstream consequence of altered Pkd2 activity and may be the cause of the observed increase in inflammatory gene expression and atherosclerosis. There are multiple pathways that can activate eNOS and loss of EC cilia may remove one of several eNOS inputs 54. Loss of cilia may remove a brake on EC activation and lead to earlier or more rapid development of atherosclerosis, particularly in areas of the vasculature with more cilia. However, our data do not exclude effects of cilia on atheroprotective mechanisms other than eNOS. Our finding that EC primary cilia serve an anti‐inflammatory role contrasts with several recent studies reporting that cilia potentiate the cellular response to IL‐1β, a proinflammatory cytokine, in chondrocytes 49, 50, 51. Therefore, the effects of cilia on inflammation may depend on cell type.
Our results revealing a novel role for EC cilia in inhibiting the development of atherosclerosis in mouse models raise the question of whether human EC cilia function similarly. Primary cilia occur near atherosclerotic plaques in humans 43, 44, 45, suggesting that EC cilia may modulate human atherosclerosis, a leading cause of mortality.
Materials and Methods
Mouse lines
Tek‐Cre (MGI: 5052345), Mx1‐Cre (MGI: 2176073), Pecam1 (MGI: 1888376), Ift88 (MGI: 3710185), Kif3a (MGI: 2386464), and Smo (MGI: 2176256) mice have been described elsewhere and were gifts of Rong Wang, Ben Barres, Mark Ginsberg, Bradley Yoder, Larry Goldstein, and Andrew McMahon, respectively. Apoe (MGI: 1857129), Pkd2 (MGI: 4843125), and mTmG (MGI: 3716464) mice were purchased from Jackson Labs. Mice were housed in a barrier facility with veterinary supervision and given food and water ad libitum. All mouse protocols were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco. Mice were maintained on a mixed background. Genotyping primers are listed in Table EV1.
Conditional Cre induction
Mx1‐Cre mice were administered three doses of 10 μg/g body weight polyinosinic:polycytidylic acid (pIpC, Sigma‐Aldrich) by intraperitoneal injection at 48‐h intervals at approximately 3 weeks of age. pIpC was dissolved in D‐PBS to a concentration of 1 mg/ml and sterile filtered.
Immunofluorescence
Mice were anesthetized with 2.5% w/v 2,2,2‐tribromoethanol (Sigma‐Aldrich) in D‐PBS and perfused via the left ventricle with 10 ml of D‐PBS containing 1 U/ml heparin (Sigma‐Aldrich) followed by 10 ml 4% formaldehyde in D‐PBS. Aortas were dissected and postfixed for 15 min in 4% formaldehyde, rinsed in D‐PBS, cleaned of fat and connective tissue, and opened longitudinally with microscissors. Samples were incubated in blocking buffer (D‐PBS without Ca2+ and Mg2+, 0.1% Triton X‐100, 2% BSA, 1% donkey serum) for 1 h. For samples to be stained with mouse or rat primary antibodies, Mouse on Mouse IgG blocking reagent (Vector Labs) was included in the blocking buffer. Samples were incubated with primary antibodies in blocking buffer at 4°C overnight. The next day samples were rinsed 3 × 5 min in D‐PBS without Ca2+ and Mg2+, stained with appropriate Alexa Fluor 488‐, 555‐, 568‐, or 647‐conjugated secondary antibodies (Life Technologies) at 1:1,000 and Hoechst or DAPI in blocking buffer for 1 h, rinsed 3 × 5 min in D‐PBS without Ca2+ and Mg2+, and mounted in Gelvatol or Prolong Diamond Antifade (Life Technologies). All steps were at room temperature unless otherwise noted. Stained samples were imaged on a Leica SP‐5 or SPE confocal microscope.
Antibodies
Primary antibodies used were mouse anti‐acetylated α‐tubulin (Sigma, 1:1,000), rabbit anti‐Arl13B (gift Tamara Caspary and ProteinTech Labs 17711‐1‐AP, 1:5,000), rat anti‐CD144 (BD 550548, 1:100), rat anti‐CD31 (BD 550274, 1:100), rabbit anti‐CD31 (ProteinTech Labs 11265‐1‐AP 1:100), rabbit anti‐Pkd2 (gift Feng Qian, 1:200), rabbit anti‐pericentrin (Covance PRB432‐C, 1:500), mouse anti‐GM130 (BD 610822, 1:200), mouse anti‐eNOS (BD 610297, 1:100), and rabbit anti‐p‐eNOS‐S1177 (Cell Signaling 9570, 1:100).
Cilia quantification
Male and female wild‐type adult mouse aortas were stained for Arl13B, CD31, and Hoechst as described above. Two fields in each region indicated in Fig 1 were imaged and the number of cilia and number of endothelial cells in each field were quantified. The two fields were summed so that at least 100 cells were counted per region per mouse. The percent of ciliated cells was then averaged across the mice (n = 7). Specifically, the six regions quantified are the proximal arch, mid‐arch, distal arch, the first two pairs of aortic intercostal arteries, the fifth and sixth pairs of intercostal arteries, and the subcostal arteries.
Polarity analysis
Adult mouse aortas were stained for GM130 to mark the Golgi, CD144 to mark ECs, and DAPI to mark the nucleus and imaged as described above. Images were oriented so that upstream (toward the heart) was up. The nucleus and Golgi of each cell in the field were manually traced in MetaMorph and the angle between the center of mass of the organelles relative to the axis of the tissue was computed. Three mice of each genotype were used, although suboptimal staining resulted in an n = 2 for the mutant data in region 2. At least 50 cells were measured per field.
eNOS analysis
Adult mouse aortas were prepared for immunofluorescence as above, with the modification that they were treated for 5 min with 1% SDS in D‐PBS without Ca2+ and Mg2+ following fixation, which improved consistency of the staining. Following blocking, samples were stained for eNOS, acetylated α‐tubulin, and p‐eNOS‐S1176 (the mouse residue corresponding to human eNOS‐S1177, which the antibody was raised against). Because eNOS and acetylated α‐tubulin are both mouse monoclonal antibodies, IgG isoform‐specific secondaries were used: anti‐IgG1‐Alexa 488 for eNOS and anti‐IgG2b‐Alexa 555 for acetylated α‐tubulin. Samples were mounted together and tiled confocal stacks encompassing the endothelium acquired across the aorta at the level of the first intercostal artery. Laser power, gain, and offset were adjusted to minimize saturated pixels and set the background level just above black (i.e., only a few pixels of intensity = 0 in regions of the image with no sample present), ensuring the acquired pixel values were representative of actual fluorescence intensity. Samples stained in parallel were imaged using identical settings. The resulting stitched z‐stacks were projected into a single image using the Z project, Sum function in Fiji, the region of interest was outlined, and the average pixel intensity in the eNOS and p‐eNOS‐S1176 channels was measured (Analyze, Measure, Mean function in Fiji). The values were then corrected by subtracting the average background pixel intensity, calculated by measuring an area of the image with no tissue. The ratio of p‐eNOS‐S1176 to eNOS intensity was calculated and statistics performed using a paired two‐tailed ratio t‐test for seven sets of mice (GraphPad Prism 6).
Atherosclerosis studies
Mice were crossed onto an Apoe −/− background. Following weaning at 3 weeks of age, mice were placed on a high‐fat, high‐cholesterol diet (TestDiet 5TJN “Western Diet for Rodents”; 39.1% calories from fat, 1,701 ppm cholesterol) for 8 weeks before being analyzed. Mice were perfused as for immunofluorescence, but were postfixed in 10% formalin overnight at 4°C. Aortas were rinsed in D‐PBS, equilibrated twice in 60% isopropanol, stained 15 min in freshly prepared and filtered 0.3% oil red O (Sigma‐Aldrich) in 60% isopropanol, destained 5 min in 60% isopropanol, then stored in dH20 until imaging. Samples were opened longitudinally, pinned onto a silicon dish with insect mounting pins, and imaged using a Zeiss Discovery V12 Stereo dissecting scope. Individual images from the same aorta were stitched together using Photoshop (Adobe) and oil red O‐positive area quantified using Fiji. The aorta was outlined and its area measured in pixels. Then, the oil red O‐positive area was measured using the RGB threshold function followed by the select and measure commands.
Aortic sinus analysis
Following overnight fixation, the top 30% of the heart was removed, incubated in 30% sucrose at 4°C overnight, and embedded in OCT. 10‐μm sections were stained for immunofluorescence as above or stained for histology. For histological staining, 4 sequential sections were collected, 11 discarded, another 4 collected, etc., through the extent of the sinus. ORO+ area was quantified as above. Only sections where all three leaflets of the aortic valve included were quantified and 2–4 sections were used per mouse. For immunofluorescence, the lesional area and cross‐sectional area of the sinus were outlined and measured in ImageJ. The Cd68+ and SMA+ staining within the lesion was thresholded and the positive area measured in ImageJ. Identical settings were used for all sections.
Retina analysis
P5 mice were euthanized and their eyes were enucleated and fixed for 45 min in 4% formaldehyde in D‐PBS. The lens and RPE layer were removed, hyaloid vasculature cut away, and several incisions made along the retina so it could lay flat. Retinas were then mounted and imaged. To calculate the migration distance, the distance from the center of the optic nerve to the leading edge of the vascular front and to the edge of the retina was measured on at least 4 leaflets per mouse for n = 6 control and mutant mice. The ratio of the two values was calculated and averaged over the four leaflets for each mouse. To calculate branch points, identically sized fields (423 × 423 μm) located behind the vascular front between a vein and artery (typically about halfway between the leading vascular edge and the center of the retina) were selected for each mouse. The number of vascular junctions, points at which at least 3 distinct vascular branches clearly came together, was counted (n = 6 control and n = 5 experimental mice).
RT–qPCR
Aortas were perfused with 10 ml cold PBS + 1 U/ml heparin, dissected into cold PBS, cleaned of excess fat, and lysed in 200 μl RLT buffer in a 0.2‐ml glass tissue grinder. RNA was prepared using the RNEasy Plus Mini Kit (Qiagen) according to the manufacturer's instructions. cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio‐Rad) according to the manufacturer's instructions. cDNA was diluted 1:4 in dH20 and stored at −20°C. qPCR was performed using EXPRESS SYBR GreenER with premixed ROX (Invitrogen) and run on a 7900HT thermocycler (Applied Biosystems). 5 μl total reaction volume, 0.4 μl diluted cDNA and a concentration of 200 nM for each primer were used, with four technical replicates per sample. Expression levels were normalized to the geometric mean of four control genes (Actb, Hmbs, Hprt and Ubc), average normalized Ct values for control and experimental groups determined, and relative expression levels determined by ΔΔCt. Significance was determined by Student's two‐tailed t‐test. Primers are listed in Table EV2.
Statistics
P‐values were calculated in GraphPad Prism 6 using Student's two‐tailed homoscedastic t‐test. In all cases, n represents the number of individual mice used in the experiment except for Fig 3D where n is the number of mouse cohorts from which a ratio was calculated. Error bars in all graphs represent the standard error of the mean. Atherosclerosis data were subject to a D'Agostino–Pearson omnibus test of normality and F‐test for unequal variance to ensure the suitability Student's t‐test (GraphPad Prism 6). Unpaired t‐tests were utilized in all data except the p‐eNOS‐S1176 to eNOS ratio (Fig 3D), where a paired ratio t‐test was used to control for day‐to‐day variations in staining. The control female body weights (Fig EV5B) failed a normality test and so body weights were compared using a nonparametric Kolmogorov–Smirnov test (KS test). Observed versus expected birth ratios were subject to a χ2‐test (GraphPad Prism 6), except for the Smo cross, where only 2 possible outcomes allowed it to be tested with the more powerful two‐tailed binomial test. For all quantitation, all data were analyzed with only the index number of the mouse, which was not associated with a genotype until after the analysis.
Lipid analysis
Mice were anesthetized for perfusion as described above, but were exsanguinated via cardiac puncture using a 1‐ml syringe and 25‐G needle. Blood was transferred to gold‐capped serum separator tubes (BD) and allowed to clot at room temperature for 30 min. The serum was separated by centrifugation for 2 min at 6,000 × g. Serum was shipped overnight on ice to the UC Davis Comparative Pathology Laboratory for analysis.
Author contributions
JFR and CD conceived the study. CD performed the experiments. JFR and CD wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
We thank Mark Ginsburg, Per Fogelstrand, Tomoki Hashimoto for guidance, and Kevin Corbit for initial generation of some of the experimental strains. This work was supported by grants from the NIH (AR054396 and GM095941).
EMBO Reports (2016) 17: 156–166
See also: B Schermer & T Benzing (February 2016)
References
- 1. Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11: 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Praetorius HA (2015) The primary cilium as sensor of fluid flow: new building blocks to the model. Am J Physiol Cell Physiol 308: C198–C208 [DOI] [PubMed] [Google Scholar]
- 3. Praetorius HA, Spring KR (2001) Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 184: 71–79 [DOI] [PubMed] [Google Scholar]
- 4. Praetorius HA, Frokiaer J, Nielsen S, Spring KR (2003) Bending the primary cilium opens Ca2 + ‐sensitive intermediate‐conductance K+ channels in MDCK cells. J Membr Biol 191: 193–200 [DOI] [PubMed] [Google Scholar]
- 5. Goetz JG, Steed E, Ferreira RR, Roth S, Ramspacher C, Boselli F, Charvin G, Liebling M, Wyart C, Schwab Y et al (2014) Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep 6: 799–808 [DOI] [PubMed] [Google Scholar]
- 6. Hierck BP, Van Der Heiden K, Alkemade FE, Van de Pas S, Van Thienen JV, Groenendijk BCW, Bax WH, Van der Laarse A, Deruiter MC, Horrevoets AJG et al (2008) Primary cilia sensitize endothelial cells for fluid shear stress. Dev Dyn 237: 725–735 [DOI] [PubMed] [Google Scholar]
- 7. Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J (2008) Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin‐1. Circulation 117: 1161–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Praetorius HA, Spring KR (2003a) Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol 191: 69–76 [DOI] [PubMed] [Google Scholar]
- 9. Praetorius HA, Spring KR (2003b) The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens 12: 517–520 [DOI] [PubMed] [Google Scholar]
- 10. Van Der Heiden K, Hierck BP, Krams R, de Crom R, Cheng C, Baiker M, Pourquie MJBM, Alkemade FE, Deruiter MC, Gittenberger‐De Groot AC et al (2008) Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 196: 542–550 [DOI] [PubMed] [Google Scholar]
- 11. Caro CG (2009) Discovery of the role of wall shear in atherosclerosis. Arterioscler Thromb Vasc Biol 29: 158–161 [DOI] [PubMed] [Google Scholar]
- 12. Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP (2007) Hemodynamic shear stresses in mouse aortas: implications for atherogenesis. Arterioscler Thromb Vasc Biol 27: 346–351 [DOI] [PubMed] [Google Scholar]
- 13. Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK (2007) Intraflagellar transport is essential for endochondral bone formation. Development 134: 307–316 [DOI] [PubMed] [Google Scholar]
- 14. Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R (2006) Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol 172: 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. White AC, Lavine KJ, Ornitz DM (2007) FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network. Development 134: 3743–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LEH, Ingber DE (2009) A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457: 1103–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yashiro K, Shiratori H, Hamada H (2007) Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 450: 285–288 [DOI] [PubMed] [Google Scholar]
- 18. Nicoli S, Standley C, Walker P, Hurlstone A, Fogarty KE, Lawson ND (2010) MicroRNA‐mediated integration of haemodynamics and Vegf signalling during angiogenesis. Nature 464: 1196–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia‐Cardena G (2000) Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci 902: 230–239 discussion 239–40. [DOI] [PubMed] [Google Scholar]
- 20. Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P (2008) Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 40: 69–77 [DOI] [PubMed] [Google Scholar]
- 21. Tkachenko E, Gutierrez E, Saikin SK, Fogelstrand P, Kim C, Groisman A, Ginsberg MH (2013) The nucleus of endothelial cell as a sensor of blood flow direction. Biol Open 2: 1007–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zadelaar S, Kleemann R, Verschuren L, de Vries‐Van der Weij J, van der Hoorn J, Princen HM, Kooistra T (2007) Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol 27: 1706–1721 [DOI] [PubMed] [Google Scholar]
- 23. Breslow JL (1996) Mouse models of atherosclerosis. Science 272: 685–688 [DOI] [PubMed] [Google Scholar]
- 24. Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT (2009) Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol 11: 1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hopkins PN (2013) Molecular biology of Atherosclerosis. Physiol Rev 93: 1317–1542 [DOI] [PubMed] [Google Scholar]
- 26. Alam J, Cook JL (2007) How many transcription factors does it take to turn on the heme oxygenase‐1 gene? Am J Respir Cell Mol Biol 36: 166–174 [DOI] [PubMed] [Google Scholar]
- 27. AbouAlaiwi WA, Takahashi M, Mell BR, Jones TJ, Ratnam S, Kolb RJ, Nauli SM (2009) Ciliary polycystin‐2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res 104: 860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garcia‐Gonzalez MA, Outeda P, Zhou Q, Zhou F, Menezes LF, Qian F, Huso DL, Germino GG, Piontek KB, Watnick T (2010) Pkd1 and Pkd2 Are required for normal placental development. PLoS ONE 5: e12821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coxam B, Sabine A, Bower NI, Smith KA, Pichol‐Thievend C, Skoczylas R, Astin JW, Frampton E, Jaquet M, Crosier PS et al (2014) Pkd1 regulates lymphatic vascular morphogenesis during development. Cell Rep 7: 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Outeda P, Huso DL, Fisher SA, Halushka MK, Kim H, Qian F, Germino GG, Watnick T (2014) Polycystin signaling is required for directed endothelial cell migration and lymphatic development. Cell Rep 7: 634–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S (2013) Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45: 1004–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuhlencordt PJ, Gyurko R, Han F, Scherrer‐Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL (2001) Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double‐knockout mice. Circulation 104: 448–454 [DOI] [PubMed] [Google Scholar]
- 33. Vokes SA, Yatskievych TA, Heimark RL, McMahon J, Mcmahon AP, Antin PB, Krieg PA (2004) Hedgehog signaling is essential for endothelial tube formation during vasculogenesis. Development 131: 4371–4380 [DOI] [PubMed] [Google Scholar]
- 34. Chinchilla P, Xiao L, Kazanietz MG, Riobo NA (2010) Hedgehog proteins activate pro‐angiogenic responses in endothelial cells through non‐canonical signaling pathways. Cell Cycle 9: 570–579 [DOI] [PubMed] [Google Scholar]
- 35. Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, Shapiro R, Taylor FR, Baker DP, Asahara T et al (2001) The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med 7: 706–711 [DOI] [PubMed] [Google Scholar]
- 36. Lawson ND, Vogel AM, Weinstein BM (2002) sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 3: 127–136 [DOI] [PubMed] [Google Scholar]
- 37. Alvarez JI, Dodelet‐Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M et al (2011) The Hedgehog pathway promotes blood‐brain barrier integrity and CNS immune quiescence. Science 334: 1727–1731 [DOI] [PubMed] [Google Scholar]
- 38. Williams C, Kim S‐H, Ni TT, Mitchell L, Ro H, Penn JS, Baldwin SH, Solnica‐Krezel L, Zhong TP (2010) Hedgehog signaling induces arterial endothelial cell formation by repressing venous cell fate. Dev Biol 341: 196–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iomini C, Tejada K, Mo W, Vaananen H, Piperno G (2004) Primary cilia of human endothelial cells disassemble under laminar shear stress. J Cell Biol 164: 811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hahn C, Schwartz MA (2009) Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 10: 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tzima E, Irani‐Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, Delisser H, Schwartz MA (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431 [DOI] [PubMed] [Google Scholar]
- 42. Hoi Y, Zhou Y‐Q, Zhang X, Henkelman RM, Steinman DA (2011) Correlation between local hemodynamics and lesion distribution in a novel aortic regurgitation murine model of atherosclerosis. Ann Biomed Eng 39: 1414–1422 [DOI] [PubMed] [Google Scholar]
- 43. Haust MD (1987) Endothelial cilia in human aortic atherosclerotic lesions. Virchows Arch A Pathol Anat Histopathol 410: 317–326 [DOI] [PubMed] [Google Scholar]
- 44. Bystrevskaya VB, Lichkun VV, Antonov AS, Perov NA (1988) An ultrastructural study of centriolar complexes in adult and embryonic human aortic endothelial cells. Tissue Cell 20: 493–503 [DOI] [PubMed] [Google Scholar]
- 45. Bystrevskaya VB, Lichkun VV, Krushinsky AV, Smirnov VN (1992) Centriole modification in human aortic endothelial cells. J Struct Biol 109: 1–12 [DOI] [PubMed] [Google Scholar]
- 46. Egorova AD, Khedoe PPSJ, Goumans M‐JTH, Yoder BK, Nauli SM, Ten Dijke P, Poelmann RE, Hierck BP (2011) Lack of primary cilia primes shear‐induced endothelial‐to‐mesenchymal transition. Circ Res 108: 1093–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ubil E, Duan J, Pillai ICL, Rosa‐Garrido M, Wu Y, Bargiacchi F, Lu Y, Stanbouly S, Huang J, Rojas M et al (2014) Mesenchymal–endothelial transition contributes to cardiac neovascularization. Nature 514: 585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sánchez‐Duffhues G, de Vinuesa AG, Lindeman JH, Mulder‐Stapel A, Deruiter MC, Van Munsteren C, Goumans MJ, Hierck BP, Ten Dijke P (2015) SLUG is expressed in endothelial cells lacking primary cilia to promote cellular calcification. Arterioscler Thromb Vasc Biol 35: 616–627 [DOI] [PubMed] [Google Scholar]
- 49. Wann AK, Knight MM (2012) Primary cilia elongation in response to interleukin‐1 mediates the inflammatory response. Cell Mol Life Sci 69: 2967–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wann AK, Thompson CL, Chapple JP, Knight MM (2013) Interleukin‐1β sequesters hypoxia inducible factor 2α to the primary cilium. Cilia 2: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wann AK, Chapple JP, Knight MM (2014) The primary cilium influences interleukin‐1β‐induced NFκB signalling by regulating IKK activity. Cell Signal 26: 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fernandez‐Hernando C, Yu J, Suarez Y, Rahner C, Davalos A, Lasuncion MA, Sessa WC (2009) Genetic evidence supporting a critical role of endothelial caveolin‐1 during the progression of atherosclerosis. Cell Metab 10: 48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ecder T, Schrier RW (2009) Cardiovascular abnormalities in autosomal‐dominant polycystic kidney disease. Nat Rev Nephrol 5: 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dudzinski DM, Michel T (2007) Life history of eNOS: partners and pathways. Cardiovasc Res 75: 247–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File