

Abstract
NgBR is a transmembrane protein identified as a Nogo‐B‐interacting protein and recently has been shown to be a subunit required for cis‐prenyltransferase (cisPTase) activity. To investigate the integrated role of NgBR in vascular development, we have characterized endothelial‐specific NgBR knockout embryos. Here, we show that endothelial‐specific NgBR knockout results in embryonic lethality due to vascular development defects in yolk sac and embryo proper. Loss of NgBR in endothelial cells reduces proliferation and promotes apoptosis of the cells largely through defects in the glycosylation of key endothelial proteins including VEGFR2, VE‐cadherin, and CD31, and defective glycosylation can be rescued by treatment with the end product of cisPTase activity, dolichol phosphate. Moreover, NgBR functions in endothelial cells during embryogenesis are Nogo‐B independent. These data uniquely show the importance of NgBR and protein glycosylation during vascular development.
Keywords: cis‐prenyltransferase, dolichol, glycosylation, NgBR, vascular development
Subject Categories: Development & Differentiation, Vascular Biology & Angiogenesis
Introduction
Nogo‐B receptor (NgBR) was initially identified via expression cloning as an interacting protein necessary for Nogo‐B (also called reticulon‐4b)‐stimulated chemotaxis and tube formation of endothelial cells in vitro 1. Mechanistic studies using the C‐terminus of NgBR as bait in a yeast two‐hybrid screen identified an interaction of NgBR with Niemann Pick C2 protein (NPC2) and deficiency of NgBR destabilized NPC2 and promoted the lysosomal accumulation of free cholesterol 2. More recently, it is clear that NgBR is an evolutionary conserved subunit of cis‐prenyltransferase, a enzymatic activity critical for the synthesis of dolichol, an obligate carrier of oligosaccharides for protein glycosylation reactions 3, 4 and may serve as a nexus regulating intracellular cholesterol versus protein glycosylation. Several studies suggest crucial roles of NgBR in vivo for embryonic and vascular development in mice and zebrafish 4, 5, congenital disorders of glycosylation, and in cancer. Global deficiency of NgBR results in peri‐implantation embryonic lethality before embryonic day (E)6.5, suggesting its essential role in early embryogenesis 4. Patients harboring a mutation in the C‐terminus of NgBR present clinical features of a congenital disorder of glycosylation 4 and deletion within the NgBR locus may predispose patients to pediatric epilepsy 6. In addition, enhanced mRNA expression levels of NgBR have been shown in several human cancers including invasive ductal breast carcinoma and non‐small cell lung carcinoma 7, 8, 9.
The role of NgBR in the vascular development is of particular interest because it is crucial not only for all aspects of normal tissue function but also for tumor growth and survival. During embryonic development, endothelial cells (EC) start to form a primary vascular plexus in extraembryonic tissues via vasculogenesis 10 and the primary vascular plexus undergoes remodeling and organization via angiogenesis 11, 12. Significant defects on vasculogenesis or angiogenesis during development leads to embryonic lethality, and many critical molecular pathways have been identified to be essential for vascular development. For example, vascular endothelial growth factor (VEGF) and its cognate receptors are crucial for the development of vascular system 13.
Thus, the goal of the present study is to investigate the integrated role of NgBR in EC in vivo, using a conditional knockout system in mice. Here, we show that endothelial‐specific NgBR deletion markedly impairs vascular development by reducing endothelial cell growth during early mouse embryogenesis. Moreover, the loss of NgBR reduces glycosylation of VEGFR2 in vivo and in vitro, an effect that can be partially rescued by exogenous dolichol phosphate. This effect is independent of Nogo‐A/B since mice lacking Nogo‐A/B knockout did not recapitulate the phenotype in the NgBR endothelial‐specific knockout mice. Collectively, these results demonstrate that NgBR has a Nogo‐B‐independent role in vascular development by regulating protein N‐glycosylation of key endothelial cell proteins such as VEGFR2, VE‐cadherin, and CD31.
Results
Tie2‐Cre‐mediated ablation of NgBR leads to mid‐gestation embryonic lethality with abnormal extraembryonic vascular development
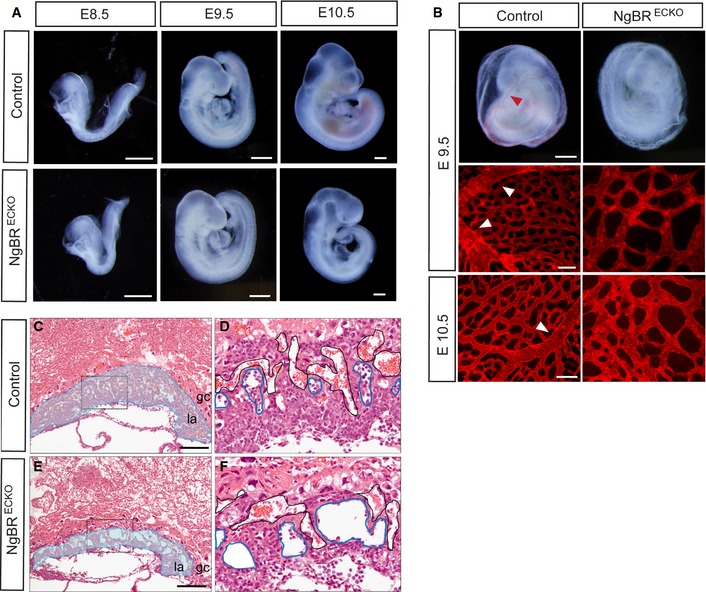
Previously, we have shown that global NgBR knockout mice are post‐implantation embryonic lethal before E6.5 4. To overcome early embryonic lethality and to study functions of NgBR during embryogenesis, we used conditional NgBR mice with a floxed NgBR gene 4. NgBR f/f mice were crossed to mice bearing a Tie2‐Cre transgene, which is active in EC and hematopoietic lineages starting as early as E7.0 14, 15, 16. As shown in Fig 1, NgBR f/f ;Tie2‐Cre (NgBRECKO) embryos at E8.5 and E9.5 were morphologically indistinguishable from the control littermates. At E10.5, NgBRECKO embryos were smaller and paler than controls and showed lethality between E10.5 and E11.5. To investigate defects in the vascular system in NgBRECKO embryos, whole mounts were stained with anti‐CD31 antibody to visualize the vasculature (Fig EV1). There was no obvious difference in the development of vascular structures among the NgBRECKO and control at E9.5.
Figure 1. Tie2‐Cre‐mediated ablation of NgBR in endothelial cells impairs extraembryonic vascular development.
-
AGross morphology of control and NgBRECKO embryos at E8.5, E9.5, and E10.5. Scale bar, 500 μm.
-
BGross morphology of whole yolk sac at E9.5 and CD31 staining on yolk sac at E9.5 and E10.5 in NgBRECKO and control embryos. The control yolk sacs exhibited large vitelline vessels indicated with red and white arrowheads. Scale bar, 500 μm for whole‐mount yolk sac and 100 μm for CD31 staining.
-
C–FHematoxylin and eosin stained sections of E9.5 placenta of control and NgBRECKO embryos. Labyrinthine layer of mutants is markedly thinner compared to that of the control. Magnification of the boxes in (C, E) is shown in (D, F), respectively. Embryonic vessels and maternal vessels are marked with blue and black lines, respectively. gc, giant trophoblast cell layer; la, labyrinthine layer. Scale bar, 200 μm.
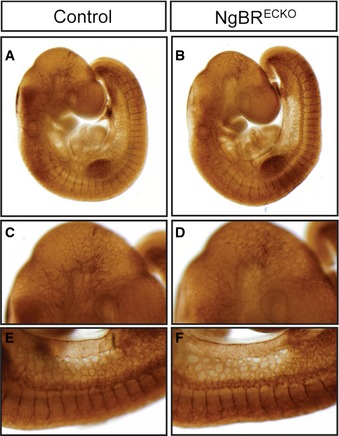
Figure EV1. NgBR endothelial KO embryos exhibit normal vascular development.
-
A–FWhole‐mount immunohistochemistry of vasculatures with CD31 antibody in NgBRECKO and control embryos at E9.5. Properly developed vasculature was present in both mutant and control embryos. Note the similar CD31 staining pattern on head vasculature (C, D) and intersomitic vessels (E, F).
Although there was no visible difference on NgBRECKO embryonic development until E10.5, the mutants were easily identified from controls because their yolk sacs were dimpled and wrinkled at E9.5. Yolk sacs of control mice exhibited a well‐organized vascular network consisting of both capillaries and large vitelline vessels (Fig 1B, Table EV1). In mutant littermates, however, yolk sacs were poorly organized with dilated primitive capillaries and had no large vitelline vessels. In addition to the yolk sac phenotype, the placental vasculature was examined in control and NgBRECKO embryos. In control placentas, the fetal vessels invaded the chorionic plate to establish the labyrinthine layer. However, the labyrinthine layers of mutants were markedly thinner compared to that of the control. Since fetal erythrocytes contain nuclei that stain with hematoxylin, fetal vessels can be distinguished from maternal vessels. Mutant placentas show dilated embryonic vessels and decreased numbers of embryonic vessels compared to controls (Fig 1C–F). These results clearly show severely impaired extraembryonic vascular development in NgBRECKO at E9.5, whereas embryonic vascular development was largely unaffected at this time point.
Inducible NgBR deletion in EC results in both embryonic and extraembryonic vascular defects
Deletion of NgBR using the Tie2‐Cre driver suggests that NgBR is essential for extraembryonic vascular development. However, it was unclear whether NgBR only functions in the extraembryonic vascular developmental program or whether NgBRECKO embryos died due to the yolk sac vascular defect prior to embryonic vascular defects. To investigate embryonic vascular phenotypes, we used Chd5‐CreERT2, a tamoxifen‐inducible Cre line to temporally control NgBR deletion in EC 17, 18. Deletion of NgBR was induced with tamoxifen (0.12 mg/g body weight) administration to pregnant females by oral gavage at E8.5 and E9.5, and embryos were harvested after 4 days. NgBR f/f ;Chd5‐CreERT2 (NgBRi∆EC) embryos displayed subcutaneous edema and extensive multifocal subcutaneous hemorrhages at E12.5 and E13.5 (Fig 2A, Table EV2), and mutant embryos were dead 5 days after tamoxifen injection. Examination of histological sections revealed dilation of subcutaneous microvessels and blood filled jugular lymph sacs at E13.5 in NgBRi∆EC embryos (Fig 2B). CD31 immunofluorescent staining of NgBRi∆EC embryonic tissues including hindbrain, skin, and hindlimb showed reductions in vascular density and disorganized vascular networks compared to controls (Fig 2C). In addition to embryonic tissues, yolk sacs of NgBRi∆EC display similar vascular defect that was shown in yolk sacs of NgBRECKO at E9.5 (Fig 2C). These findings suggest that NgBR is required for both extraembryonic and embryonic vascular development.
Figure 2. Hemorrhages and reduced capillary density in inducible NgBR endothelial‐specific knockout embryos.
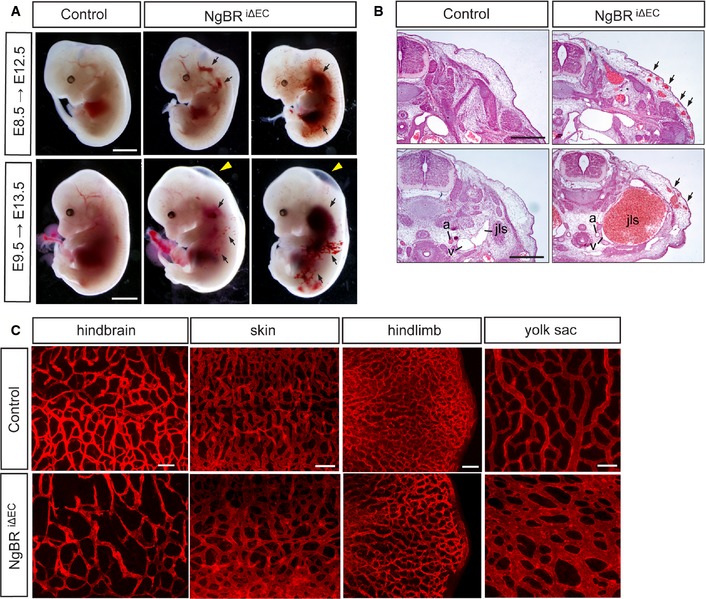
- Whole‐mount view of NgBR i∆ EC mutant and control embryos at E12.5 and E13.5 after tamoxifen administration at E8.5 and E9.5, respectively. Hemorrhagic lesions (arrows) and subcutaneous edema (yellow arrowheads) were observed in NgBR i∆ EC mutants. Scale bar, 2 mm.
- Histological analysis of NgBR i∆ EC mutant and control embryos at E13.5 after tamoxifen administration at E9.5. Cross sections of the embryos were stained with hematoxylin and eosin. a, aorta; v, vein; jls, jugular lymph sac. Scale bar, 1 mm.
- Immunofluorescence staining with CD31 on various tissues in NgBR i∆ EC mutant and control embryos at E12.5 after tamoxifen administration at E8.5. Scale bar, 100 μm.
NgBR deletion in EC results in decreased proliferation and increased cell death
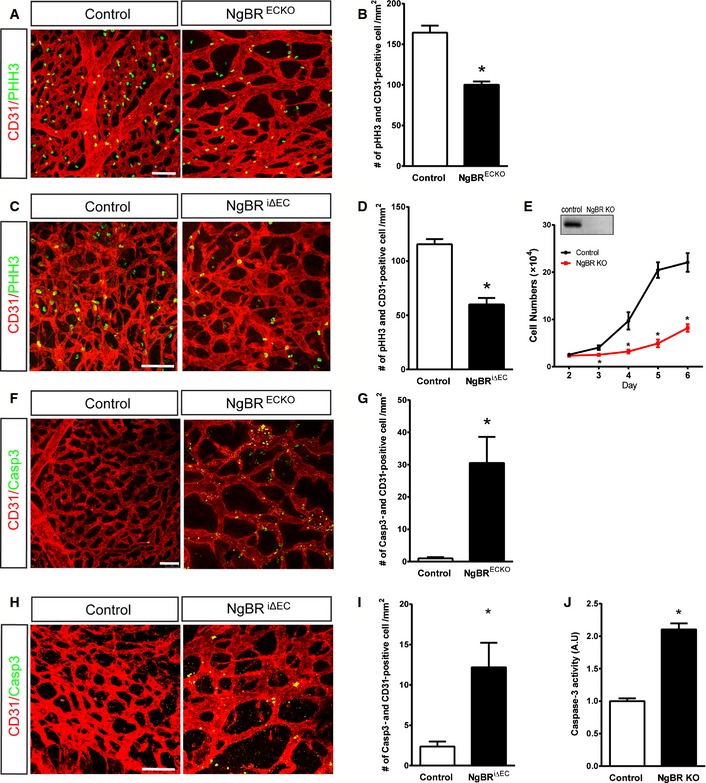
We next investigated the cause of impaired vessel development in NgBRECKO and NgBRi∆EC embryos. First, we analyzed endothelial cell proliferation in vivo by anti‐phospho‐histone H3 (pHH3) staining, which labels cells in all phases of mitosis. Both yolk sacs of NgBRECKO and back skins of NgBRi∆EC embryos were examined for the pHH3 staining. Yolk sacs of NgBRECKO at E9.5 (Fig 3A and quantification in B) and embryos of NgBRi∆EC at E12.5 (Fig 3C and quantification in D) showed 39% and 48% reductions in endothelial cell proliferation compared to controls, respectively. We also tested the proliferation rate of EC in vitro using isolated mouse lung EC (MLEC). MLEC were isolated from NgBR f/f mice and infected with adenoviruses encoding GFP (Ad‐GFP) as a control or Ad‐Cre for gene deletion and cell growth was quantified from days 2 to 6 post‐infection. Deletion of NgBR significantly reduced proliferation compared to Ad‐GFP‐infected cells (Fig 3E).
Figure 3. Loss of NgBR decreases proliferation and increases apoptosis of endothelial cells.
-
A–DImmunofluorescence (IF) staining with CD31 (red) and pHH3 (green) on E9.5 NgBRECKO yolk sac and (B) quantification of pHH3‐positive cells. (C) IF staining with CD31 (red) and pHH3 (green) on E12.5 back skin of NgBR i∆ EC and control embryos. (D) Quantification of (C).
-
EReduced proliferation of MLEC by NgBR deletion. MLEC isolated from NgBR f/f animals were infected with Ad‐GFP (Control) or Ad‐Cre (NgBR KO). Cells were counted for 5 days after infection (n = 3). NgBR deletion in MLEC was detected by Western blotting with NgBR antibody.
-
F–IIF staining for cleaved caspase‐3 on E9.5 NgBRECKO yolk sac (F) and E12.5 back skin of NgBR i∆ EC embryos (H) and respective quantification of Casp3‐positive cells (G, I).
-
JCaspase‐3 activity assay on control and NgBR KO MLEC.
Next, we examined apoptosis of EC in vivo by immunostaining tissues for cleaved caspase‐3 (Casp3) along with CD31. In NgBRECKO yolk sacs, Casp3‐positive cells were readily detected in EC (Fig 3F and quantification in G). The dorsal skin of NgBRi∆EC embryos also displayed more Casp3‐positive cells compared to controls (Fig 3H and quantification in I). We also confirmed increased apoptosis of EC using cultured MLEC (Fig 3J). The endothelial loss of NgBR, however, did not result in any significant changes in neuron‐glial antigen 2 (NG2)‐positive pericyte distribution (Fig EV2A). Vessel regression, determined with collagen IV staining, did not change in NgBR‐deleted EC compared to controls (Fig EV2B). Thus, the loss of NgBR in EC reduces growth and promotes apoptosis.
Figure EV2. Normal pericyte coverage and no vessel regression in the NgBR i∆ EC embryo.
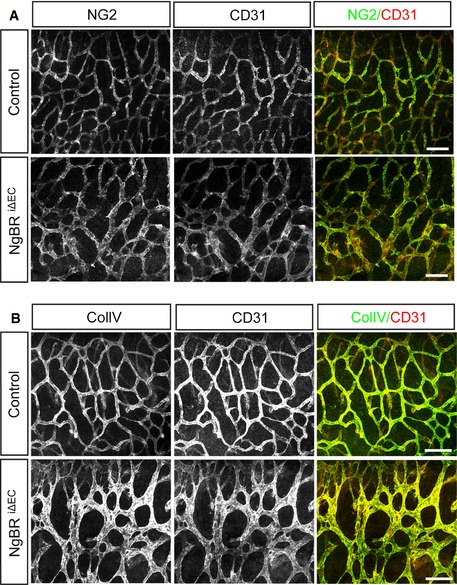
- NG2 immunostaining with CD31 on the hindbrain of NgBR i∆ EC embryos at E12.5. There was no obvious difference in NG2‐positive pericyte distribution between control and NgBR i∆ EC.
- Immunodetection of collagen IV and CD31 in the hindbrain of NgBR i∆ EC embryos at E12.5. Vessel regression that is associated with the presence of empty basement membrane sleeves was not detected in NgBR i∆ EC.
Data information: Scale bar, 100 μm.
NgBR deletion in EC causes hypo‐glycosylation of VEGFR2
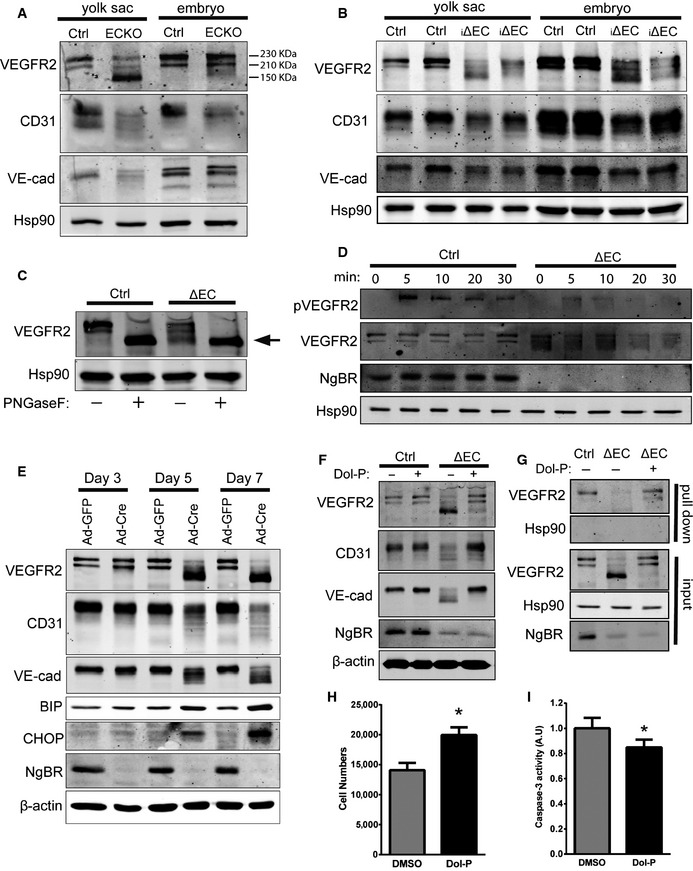
Recently, NgBR has been shown to serve as a subunit of cisPTase and is required for dolichol biosynthesis in yeast and man 3, 4. Dolichol is an obligate glycosyl carrier lipid in the biosynthesis of protein glycosylation reactions. To determine whether the vascular defects shown in NgBRECKO and NgBRi∆EC animals are due to defective dolichol biosynthesis and protein glycosylation, we examined protein glycosylation levels in embryo or yolk sac lysates. Since VEGF signaling through VEGFR2 is essential for vascular development and VEGFR2 glycosylation is critical for its function 19, 20, we initially examined VEGFR2 glycosylation. VEGFR2 is usually detected as two molecular weight species, ~230 and ~210 kDa on SDS–PAGE. We observed a molecular weight shift of VEGFR2 in yolk sacs of NgBRECKO (Fig 4A) and most VEGFR2 in the NgBRECKO yolk sacs was detected at ~150 KDa instead of at 230 or 210 KDa as in control lysates. Consistent with normal vascular phenotype in NgBRECKO embryos, there were no overt differences in the migration of VEGFR2 in the embryo lysates (Fig 4A). The change in MW in yolk sac lysates was also detected in yolk sacs and embryos of NgBRi∆EC at E12.5 (Fig 4B) with changes in the mobility of other glycosylated proteins, CD31 and VE‐cadherin (VE‐cad). To test this in vitro, EC were isolated from NgBR f/f mice and infected with Ad‐GFP or Ad‐CRE to induced excision of the NgBR locus (NgBR∆EC). As seen in Fig 4C, treatment of protein lysates from control and NgBR∆EC with PNGaseF, to cleave N‐linked oligosaccharides from glycoproteins, showed a complete band shift of VEGFR2 to ~150‐KDa form, implying that the band detected around 150 KDa is a non‐ or hypo‐glycosylated form of VEGFR2.
Figure 4. NgBRECKO and NgBR i∆ EC embryos exhibit altered VEGFR2 glycosylation and partial rescue by Dol‐P supplementation.
-
AWestern blot analysis of NgBRECKO embryos and yolk sac lysates to detect cell surface glycoproteins. Most VEGFR2 in the mutant yolk sac was detected at ˜150 kDa instead of at 230 or 210 kDa as in the control.
-
BWestern blot analysis of NgBR i∆ EC embryos and yolk sac lysates to detect cell surface glycoproteins.
-
CAnalysis of N‐glycosidase F (PNGaseF)‐treated NgBR ∆ EC lysates. The arrow indicates non‐glycosylated VEGFR2. PNGaseF cleaves between the innermost GlcNAc and asparagine residues of high mannose, hybrid, and complex oligosaccharides from N‐linked glycoproteins.
-
DVEGFR2 phosphorylation in response to VEGF. pVEGFR2 Y1175 was detected after stimulation with VEGF (50 ng/ml) by immunoblot analysis.
-
EWestern blot analysis of endothelial cell and ER stress marker proteins in primary MLEC. Isolated MLEC from NgBR f/f were treated with Ad‐GFP or Ad‐Cre, and lysates were collected at day 3, 5, and 7 after virus infection.
-
FRescue of EC glycosylation defects with Dol‐P treatment. After 2 days of Ad‐GFP or Ad‐Cre infection of NgBR f/f MLEC, Dol‐P (50 μg/ml) was added for additional 72 h. Cell lysates were collected and EC protein glycosylation detected by Western blotting. Data are representative of 3–5 independent experiments.
-
GDol‐P rescues surface expression of VEGFR2. Surface protein biotinylation of MLEC infected with Ad‐GFP (Ctrl) incubated with DMSO and Ad‐Cre (i∆EC) incubated with DMSO or Dol‐P (50 μg/ml). HSP90, a control intracellular protein, was present only in the input fraction.
-
H, IProliferation (H) and caspase‐3 activity (I) of NgBR‐deleted MLEC with Dol‐P supplementation. *P < 0.005. P‐values were calculated by unpaired Student's t‐test. Data are mean ± SEM from 3 independent experiments.
To determine whether defective VEGFR2 glycosylation influenced VEGF receptor activation, the tyrosine phosphorylation on Y1175 was examined. The phosphorylation of VEGFR2 was reduced in NgBR∆EC MLEC compared to controls (Fig 4D). To evaluate whether the defects in glycosylation were due to reduced dolichol content in NgBR‐deleted EC, we measured dolichol levels in NgBR∆EC MLEC and control by mass spectrometry (Fig EV3A and B). Total dolichol levels were significantly reduced in NgBR∆EC MLEC compare to controls. This was confirmed in HeLa cells with stable knockdown of NgBR by shRNA (NgBR KD) compared to controls (NgBR NS) and rescue of dolichol level with NgBR re‐expression in NgBR KD cells (Fig EV3C and D). Since defects in protein glycosylation can induce the unfolded protein response (UPR), activation of the UPR pathway in yolk sacs from NgBRECKO and control mice were examined by RT–PCR for marker genes of the pathway including Chop and Chac. Both genes were markedly increased in NgBRECKO yolk sacs (Fig EV3E and F), implying that defects in protein glycosylation were activating the UPR pathway of ER stress in NgBR‐deficient EC. Collectively, loss of NgBR in EC results in hypo‐glycosylation of endothelial proteins including VEGFR2, CD31, and VE‐cad and induces ER stress during mouse vascular development.
Figure EV3. Dolichol level measurement and gene expression analysis in NgBR‐depleted tissues and cells.
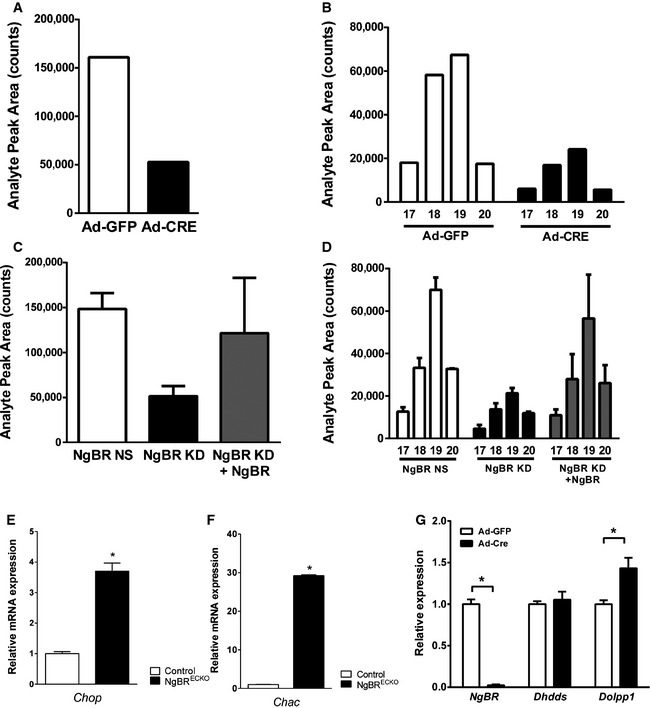
-
A, BTotal dolichol levels (A) and distribution of dolichol‐17, 18, 19, and 20 (B) measured from primary MLEC treated with Ad‐GFP or Ad‐Cre.
-
C, DTotal dolichol levels (C) and distribution of dolichol‐17, 18, 19, and 20 (D) measured from HeLa cells with stable knockdown of NgBR by shRNA (NgBR KD), control (NgBR NS), and pcDNA‐NgBR‐transfected NgBR KD cells showing conservation of the role of NgBR. Analysis of dolichol species by liquid chromatography and mass spectroscopy was performed as described in 4.
-
EmRNA expression levels of Chop in the NgBRECKO yolk sac detected by qPCR.
-
GmRNA expression levels of Chac in the NgBRECKO yolk sac detected by qPCR.
-
HPrimary MLEC were treated with Ad‐GFP or Ad‐Cre, and mRNA expression levels were determined after 5 days of infection by qPCR. In Ad‐Cre‐infected cells, almost no NgBR mRNA expression was detected. Dolpp1 mRNA expression was upregulated in NgBR‐deleted MLEC.
Data information: *P < 0.05. P‐values were calculated by unpaired Student's t‐test. Data are mean ± SEM from 3–5 independent experiments.
Loss of NgBR in EC results in a time‐dependent reduction in protein glycosylation that can be rescued by dolichol phosphate supplementation
In addition to the in vivo evidence showing that NgBR affects VEGFR2, CD31, and VE‐cad glycosylation, we also utilized primary MLEC to examine the temporal changes in protein glycosylation. Ad‐Cre infection reduced NgBR to undetectable levels after 3 days and markedly reduced the glycosylation of VEGFR2, CD31, and VE‐cad and increased ER stress markers 5 and 7 days later implying a kinetic delay in dolichol synthesis, N‐glycosylation, and protein turnover (Fig 4E). Previous work has shown that the complex of NgBR and hCIT is required for polyprenol and dolichol synthesis 4. Dolichol is phosphorylated by dolichol kinase to make dolichol phosphate (Dol‐P), and Dol‐P acts as the carrier in the assembly of pyrophosphate‐linked oligosaccharides for protein N‐glycosylation. There is evidence that [H3]Dol‐P can be processed into lipid‐linked oligosaccharides 21; however, the effects of Dol‐P on protein glycosylation in the cells with glycosylation defects have never been addressed. To investigate whether Dol‐P could rescue defects in NgBR KO cells, Dol‐P was added to culture media and protein lysates were collected at 72 h after supplementation. As shown in Fig 4F, Ad‐GFP‐infected cells show no differences in glycosylation of VEGFR2, CD31, and VE‐cad between vehicle (DMSO) and Dol‐P treatment. Remarkably, Dol‐P supplementation in the media increased the levels of glycosylated endothelial proteins and reduced the levels of non‐glycosylated forms, suggesting that Dol‐P supplement can rescue the glycosylation defects in NgBR‐deficient cells. Next, we examined whether the glycosylation and trafficking of the VEGFR2 to the cell surface were affected in NgBR‐deficient cells in a Dol‐P‐dependent manner. Cell surface proteins of NgBR∆EC and control MLEC were biotinylated and captured on NeutrAvidin beads. NgBR deletion strongly reduced the levels of VEGFR2 on the cell surface, and this effect was rescued by Dol‐P supplementation (Fig 4G). Finally, to test whether rescue of glycosylation in Dol‐P‐treated cells can improve cellular functions in cells lacking NgBR, we examined cell proliferation and apoptosis with or without Dol‐P supplement to NgBR KO cells (Fig 4H and I). Both proliferation and apoptosis defects in NgBR KO EC were partially rescued by Dol‐P supplementation.
NgBR functions in EC during development are Nogo‐B independent
NgBR was discovered as a protein that interacted with reticulon 4B, also called Nogo‐B 1. In zebrafish embryos, the loss of zNogo‐B using MO injection reduced intersomitic vessel formation during embryogenesis 5. Therefore, we investigated whether the loss of Nogo‐B affected NgBR function in vivo. Nogo‐A/B−/− females 22 were crossed with NgBR f/f;NogoA/B −/−;Chd5‐CreERT2 to generate NgBR f/+;NogoA/B −/− and NgBR f/+;NogoA/B −/−;Chd5‐CreERT2 embryos. Cre‐mediated recombination was induced by tamoxifen administration at E8.5, and embryos were harvested at E12.5. Both NgBR f/+;NogoA/B −/− and NgBR f/+;NogoA/B −/−;Chd5‐CreERT2 embryos were macroscopically indistinguishable compared with CD31 staining in hindbrain of those embryos that also did not show significant vascular defects (Fig EV4A). To address whether or not the levels of Nogo‐B influenced EC glycosylation levels, MLEC were isolated from control, NgBR f/f and Nogo‐A/B KO mice, and VEGFR2 glycosylation levels were compared (Fig EV4B). Unlike NgBR KO MLEC, the loss of Nogo‐A/B in MLEC had no effect on VEGFR2 glycosylation. Also, the genetic deletion of Nogo‐A/B in fibroblasts had no effect on cisPTase activity (Fig EV4C). Collectively, these results show that there is no genetic epistasis between Nogo‐B and NgBR during vascular development, and NgBR functions on cisPTase activity and protein glycosylation are Nogo‐B‐independent.
Figure EV4. Nogo‐B is not required for NgBR functions in endothelial cells during embryogenesis.
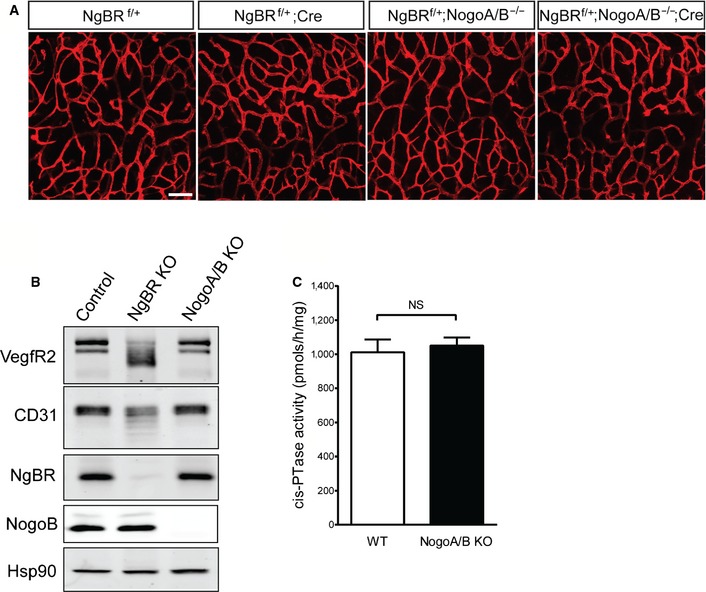
- Staining with anti‐CD31 on hindbrain of E12.5 embryos after tamoxifen injection at E8.5. Scale bar, 100 μm.
- Western blot analysis for protein glycosylation in MLEC. Nogo‐A/B KO MLEC were isolated from Nogo‐A/B KO animals. Expression of VEGFR2 and CD31 was detected. Knockout cells were confirmed by detection with anti‐NgBR and anti‐Nogo‐B antibodies. Hsp90 was used as a loading control.
- Microsomal cisPTase activity assay for Nogo‐A/B KO MEF. No significant difference was detected between control and NogoA/B KO cells. cisPTase activity assay was performed as described in 4. NS, not significant (P > 0.05). P‐value was calculated by unpaired Student's t‐test. Data are mean ± SEM, n = 3 per group.
Discussion
Our results reveal an essential role of NgBR in vascular development during mouse embryogenesis. Here, we show that the loss of NgBR in EC results in early embryonic lethality due to failure to form proper vascular structures in both extraembryonic and embryonic tissues by regulating protein N‐glycosylation. Furthermore, we have shown that during early embryonic development, NgBR has a Nogo‐B‐independent role in vascular development in the mouse. These data uniquely define the role of NgBR in the glycosylation of endothelial proteins, essential for vascular patterning.
A main function of NgBR in EC during embryogenesis
Previously, we have shown that ablation of NgBR in the mouse results in early embryonic lethality before E6.5 4, 23 and vascular defects in zebrafish 5. Due to early embryonic lethality, detailed studies on NgBR functions have not been done. To address in vivo functions of NgBR during embryogenesis in EC, we utilized a conditional knockout approach in mice. In both NgBRECKO and NgBRi∆EC embryos, vascular developmental defects were observed and the phenotypes were correlated with defective protein glycosylation. The defective glycosylation in the EC was clearly shown by the accumulation of a low apparent molecular weight VEGFR2 species and increased ER stress markers both in vivo embryos and in vitro cell culture system. These novel data supports that a main function of NgBR during development is as a subunit of cisPTase, which is required for polyprenol and dolichol biosynthesis and ultimately, protein glycosylation.
In Saccharomyces cerevisiae, deletion of ALG genes which add monosaccharides to dolichol for protein N‐glycosylation reactions promotes cell cycle arrest 24. Moreover, tunicamycin, a well‐characterized inhibitor of the early steps in glycoprotein synthesis, inhibits cell proliferation and induces apoptosis of bovine microvascular EC 25, 26 consistent with studies in EC lacking NgBR. During vasculogenesis and remodeling, EC undergo extensive proliferation and reorganization to form a final vascular network and these signals are highly dependent on EC growth factors, VEGF and FGF. Previous work has shown that only the 230‐kDa fully glycosylated form of VEGFR2 is expressed on the cell surface and transiently phosphorylated in the presence of VEGF 19. Interestingly, recent data has shown that the level of galectin‐1 in association with glycosylated VEGFR2 regulates VEGFR2 trafficking, signaling, vascular remodeling and angiogenesis in the absence of endogenous VEGF. These data in part may explain resistance to anti‐VEGF therapy and support the crucial role of glycoproteins in general and VEGFR2 glycosylation in endothelial cell functions 27. Thus, defective glycosylation of VEGFR2 and other important EC proteins such as CD31 and VE‐cad in NgBR‐deleted EC can explain the defects in vascular development and EC functions in NgBR‐deficient mice.
Nogo‐B‐independent NgBR function during embryonic vessel development
Since NgBR was identified as an interacting protein for amino portion of Nogo‐B 1, we considered the interrelationship between NgBR and Nogo‐B in EC during vasculogenesis. In zebrafish, either zNogo‐B or zNgBR morpholino silencing caused intersomitic vessel formation defects at 24 hpf during embryogenesis 5. In contrast, others have shown that zNogo‐B depletion using morpholinos does not lead to an embryonic lethal phenotype 28, 29. In mice, Nogo‐A/B knockouts are viable, fertile, and morphologically indistinguishable from their wild‐type littermates and typically exhibit phenotypes after a stressful insult 30, 31, 32. Unlike zebrafish, NogoA/B knockout mice did not recapitulate the phenotype shown in NgBR‐depleted embryos. This in vivo study suggests that Nogo‐B is not required for vascular development during mouse embryogenesis. In addition, it suggests that endothelial NgBR and Nogo‐B do not act as a crucial receptor–ligand pair during mouse embryogenesis. Moreover, in cell‐based assays, no appreciable differences in protein glycosylation or cisPTase activity were observed in Nogo‐A/B knockout cells, suggesting that the NgBR function in controlling protein N‐glycosylation during embryogenesis is independent of Nogo‐B.
Residual glycosylation in NgBR‐deleted EC
In NgBRECKO and NgBRi∆EC embryos, protein glycosylation defects in VEGFR2 and, to a lesser extent, in CD31 and VE‐cad were clear. In addition, there was a kinetic lag between the loss of NgBR in EC, dolichol content and overt defects in VEGFR2, CD31, and VE‐cad glycosylation. Direct measurement of dolichol by MS documents a marked reduction (> 70%) in NgBR∆EC cells, and the residual dolichol is likely due to the relatively long half‐lives of dolichol and dolichol phosphate. Previous work has shown that the half‐life of dolichol and dolichol phosphate in lysosomes isolated from rat liver is about 120 and 32 h, respectively 33. The half‐lives for dolichol and dolichol phosphate were 100 and 171 h in brain and 52.9 and 31.0 h in liver, respectively 34. These data indicate that dolichol and dolichol phosphate have a prolonged residence time in cells and can be utilized differentially in different cells/organs. Thus, residual dolichol after NgBR depletion in EC likely contributes to the dolichol‐linked sugars required for protein glycosylation. In addition to the novo synthesis of dolichol phosphate, dolichol‐P and dolichol‐PP can be recycled and utilized again as lipid carriers for protein glycosylation reactions 35, 36. In NgBR‐deleted MLEC, the mRNA levels of dolichol‐pyrophosphatase 1, an enzyme required for the conversion of dolichol‐PP to dolichol a pivotal step in the dolichol‐P recycling (Fig EV3G), was increased, suggesting that dolichol‐P recycling may be enhanced in the absence of NgBR to overcome protein glycosylation defects attributable to the loss of NgBR.
Interestingly, no detectable vascular defects were observed in NgBRECKO embryos at E9.5, whereas yolk sacs of the embryos show significantly impairments in vascular development. In parallel with the phenotype shown in NgBRECKO, glycosylation defects in VEGFR2 were only detectable in yolk sac tissue. Many gene deletion studies reported embryonic lethality with vascular development defects, and most reports of the targeted gene deletion show both embryonic and extraembryonic vascular defects simultaneously 37. The blood vessels of the mouse start to form in the yolk sac around at E6.5, and vasculogenesis begins within the embryo proper around at E7.5 38. Normal vascular structure within NgBRECKO embryo proper could be explained by temporal difference in vasculogenesis and residual dolichol or dolichol phosphate after NgBR deletion in the cells. Despite the temporal discordance, experiments in NgBRi∆EC clearly show both embryonic and extraembryonic vessels require NgBR for normal vascular development.
Partial rescue of NgBR KO phenotypes with Dol‐P
Since NgBR can influence cellular cholesterol and dolichol levels, it was interesting that exogenous Dol‐P rescued hypo‐glycosylation of VEGFR2, CD31, and VE‐cad levels and the cell surface expression of VEGFR2 and partially rescued growth and survival in NgBR‐depleted EC. The discrepancy between almost complete correction of protein glycosylation and partial rescue of EC functions by exogenous Dol‐P implies that there must be a kinetic lag between correction of glycosylation and rescue of complex integrated cellular functions such as growth and apoptosis. Despite these kinetic differences, the data strongly supports the critical role of NgBR in dolichol synthesis since the product of the cisPTase reaction, Dol‐P, indeed rescued key phenotypes. Consistent with the previous results in CHO cells 21, Dol‐P supplementation of control EC did not affect protein glycosylation, implying that Dol‐P is not rate limiting for N‐glycosylation reactions. Little is known about the uptake and utilization of exogenous Dol‐P, and additional experiments are clearly warranted to elucidate the mechanisms involved. In light of the results of our study, optimization of Dol‐P delivery may be a new approach to the treatment of congenital disorders due to defects in dolichol biosynthesis.
Thus, our results demonstrate an essential role of NgBR in EC during mouse embryogenesis. We propose that the primary function of NgBR in EC of developing embryos is to serve as an essential subunit for cisPTase, generating dolichol for synthesis of N‐glycosylation rather than Nogo‐B‐related functions. Understanding the hierarchy of glycoprotein synthesis in relation to the cellular pools of dolichol and the importance of dolichol metabolism and functions in mammalian cells is understudied area that may yield basic insights into this highly conserved lipid.
Materials and Methods
Generation of NgBR conditional deletion in the EC
NgBR conditional knockout targeting strategy was previously reported 4. Generation and characterization of Tie2Cre and Chd5‐CreERT2 alleles have also been reported previously 14, 39. NgBRECKO embryos were generated by crossing NgBR f/f females with NgBR f/+ ;Tie2Cre males. NgBRi∆EC embryos were generated by breeding NgBR f/f females with NgBR f/f ;Chd5‐CreERT2 male. To induce Cre activity, pregnant females were given tamoxifen (0.12 mg/g body weight, Sigma T5648) by oral gavage. The morning in which a vaginal plug was found designated as E0.5. All experimental procedures were approved by the Yale University Institutional Animal Care Use Committee.
Immunofluorescence staining and histology
Embryos and yolk sacs were dissected in ice‐cold PBS, fixed in 4% PFA, and permeabilized in blocking buffer (PBS with 0.1% Triton X‐100, 2% BSA). Samples were stained with anti‐CD31 (Pharmingen, 553370), phospho‐histone H3 (Ser10) (Cell signaling 9701), cleaved caspase‐3 (Cell signaling, 9661), anti‐collagen IV (AbD Serotech 2150‐1470), and anti‐NG2 (Millipore AB5320). Images were acquired using a Leica SP5 confocal microscope with the Leica Application Suite (LAS) software.
Mouse lung EC (MLEC) isolation and culture
MLEC were isolated from 4‐ to 7‐week‐old NgBR f/f animals. Mouse lungs were excised, minced, and digested in 2 mg/ml collagenase in PBS for 45 min at 37°C. The digested lungs was passed multiple times through a 14‐gauge needle and filtered through a 70‐μm cell strainer. Collected cells were incubated with Dynabeads (Dynal USA) conjugated with anti‐mouse CD31 antibody (Pharmingen, 553370) followed by cell sorting using a magnetic cell separator. Isolated cells were plated on 0.1% gelatin‐coated dishes. When cells reached 70–80% confluency, a second immune selection was performed. Cells were cultured in 20% FBS, supplemented with MEM non‐essential amino acids, gentamicin and amphotericin B, penicillin streptomycin, L‐glutamine, endothelial mitogen (Biomed Tech Inc. BT‐203), and heparin 100 μg/ml (Sigma, H3393) in DMEM (Lonza 12‐709F). All in vitro experiments were done using MLEC between passages 2 and 4. 100 MOI of Ad5CMVCre‐eGFP (VVC‐U of Iowa‐1174) and Ad5CMVeGFP (VVC‐U of Iowa‐4) was infected into NgBR f/f MLCEs to generate NgBR knockout and control cells, respectively.
Proliferation and apoptosis assay on cultured MLEC
For cell proliferation, cells (1 × 104 cells/well) were seeded in 12‐well palates and cultured. The numbers of cells were counted with a hemocytometer after trypsinization. Three or four replicates of each condition were performed in each 3–5 independent experiments.
For apoptosis assays, caspase‐3 activity was measured by using caspase‐3 Colorimetric Assay Kit (R&D Systems, cat. n. BF3100), according to the manufacturer's instruction.
Immunoblot analysis
Cells were washed twice with ice‐cold PBS and lysed in lysis buffer (50 mM Tris–HCl, 1% NP‐40, 0.1% SDS, 0.1% deoxycholic acid, 0.1 mM EDTA, 0.1 mM EGTA, protease and phosphatase inhibitors). Protein extracts were separated by SDS–PAGE and then transferred to nitrocellulose membrane. Primary antibodies against NgBR (Abcam, ab168351), VEGFR2 (Cell Signaling, 2479), phospho‐VEGFR2 Y1175 (Cell Signaling, 2478), CD31 (R&D system, AF3628), VE‐cadherin (Santa Cruz, sc‐6458), Bip (BD Bioscience, 610978), Chop (Santa Cruz, Sc575), β‐actin (Sigma, A5441), and Hsp90 (BD, 610419) were used.
Cell surface protein biotinylation and isolation
Cell surface protein labeling was performed on MLEC. Cells were incubated with a 1‐mM EZ‐link‐sulfo‐NHS‐S‐S‐biotin solution (Pierce 21331) for 30 min on ice and then washed with 50 mM glycine/PBS. Subsequently, cells were harvested in lysis buffer and cell lysates were incubated with NeutrAvidin Protein agarose beads (Pierce 29200) at 4°C for 2 h. The beads were then washed three times in lysis buffer prior to addition of Laemmli buffer for immunoblot analysis.
Quantitative RT–PCR
E9.5 yolk sac was dissected and lysed in RLT buffer (Qiagen). Total RNA was extracted using RNeasy mini kit (Qiagen). RNA was reverse‐transcribed to cDNA using the SuperScript III First‐Strand Synthesis System (Invitrogen). Quantitative RT–PCR was performed with iQ SYBR Green Supermix on the iCycler (Bio‐Rad). Gapdh transcript was used as an internal control.
Dol‐P supplementation
C90‐dolichyl monophosphate was originally dissolved in chloroform/methanol (2:1, v/v). The organic solvents were evaporated under nitrogen, and the remaining Dol‐P was dissolved in DMSO with 5 mg/ml stock concentration. About 50 μg/ml Dol‐P in DMSO was mixed in culture media and added to cells.
Author contributions
EJP was responsible for all experiments, data analysis, and writing of the manuscript; KAG was responsible for cisPTase activity measurement, ZG contributed to MS analysis of dolichol levels, and WCS was responsible for the overall project and writing of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
This work was supported by Grants R01 HL64793, R01 HL61371, R01 HL081190 from the National Institutes of Health (W.C.S) and by postdoctoral fellowship from the American Heart Association (E.J.P) We acknowledge the generous gift of dolichyl monophosphate from Dr. Ewa Kula‐Swiezewska (Polish Academy of Science).
EMBO Reports (2016) 17: 167–177
References
- 1. Miao RQ, Gao Y, Harrison KD, Prendergast J, Acevedo LM, Yu J, Hu F, Strittmatter SM, Sessa WC (2006) Identification of a receptor necessary for Nogo‐B stimulated chemotaxis and morphogenesis of endothelial cells. Proc Natl Acad Sci USA 103: 10997–11002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harrison KD, Miao RQ, Fernandez‐Hernando C, Suarez Y, Davalos A, Sessa WC (2009) Nogo‐B receptor stabilizes Niemann‐Pick type C2 protein and regulates intracellular cholesterol trafficking. Cell Metab 10: 208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harrison KD, Park EJ, Gao N, Kuo A, Rush JS, Waechter CJ, Lehrman MA, Sessa WC (2011) Nogo‐B receptor is necessary for cellular dolichol biosynthesis and protein N‐glycosylation. EMBO J 30: 2490–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park EJ, Grabinska KA, Guan Z, Stranecky V, Hartmannova H, Hodanova K, Baresova V, Sovova J, Jozsef L, Ondruskova N et al (2014) Mutation of Nogo‐B receptor, a subunit of cis‐prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab 20: 448–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao B, Chun C, Liu Z, Horswill MA, Pramanik K, Wilkinson GA, Ramchandran R, Miao RQ (2010) Nogo‐B receptor is essential for angiogenesis in zebrafish via Akt pathway. Blood 116: 5423–5433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szafranski P, Von Allmen GK, Graham BH, Wilfong AA, Kang SH, Ferreira JA, Upton SJ, Moeschler JB, Bi W, Rosenfeld JA et al (2015) 6q22.1 microdeletion and susceptibility to pediatric epilepsy. Eur J Hum Genet 23: 173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pula B, Olbromski M, Owczarek T, Ambicka A, Witkiewicz W, Ugorski M, Rys J, Zabel M, Dziegiel P, Podhorska‐Okolow M (2014) Nogo‐B receptor expression correlates negatively with malignancy grade and ki‐67 antigen expression in invasive ductal breast carcinoma. Anticancer Res 34: 4819–4828 [PubMed] [Google Scholar]
- 8. Pula B, Werynska B, Olbromski M, Muszczynska‐Bernhard B, Chabowski M, Janczak D, Zabel M, Podhorska‐Okolow M, Dziegiel P (2014) Expression of Nogo isoforms and Nogo‐B receptor (NgBR) in non‐small cell lung carcinomas. Anticancer Res 34: 4059–4068 [PubMed] [Google Scholar]
- 9. Wang B, Zhao B, North P, Kong A, Huang J, Miao QR (2013) Expression of NgBR is highly associated with estrogen receptor alpha and survivin in breast cancer. PLoS ONE 8: e78083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Risau W, Flamme I (1995) Vasculogenesis. Annu Rev Cell Dev Biol 11: 73–91 [DOI] [PubMed] [Google Scholar]
- 11. Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8: 464–478 [DOI] [PubMed] [Google Scholar]
- 12. Semenza GL (2007) Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J Cell Biochem 102: 840–847 [DOI] [PubMed] [Google Scholar]
- 13. Olsson AK, Dimberg A, Kreuger J, Claesson‐Welsh L (2006) VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol 7: 359–371 [DOI] [PubMed] [Google Scholar]
- 14. Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA (2001) Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193: 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Srinivasan RS, Dillard ME, Lagutin OV, Lin FJ, Tsai S, Tsai MJ, Samokhvalov IM, Oliver G (2007) Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev 21: 2422–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R (2006) Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol 172: 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sorensen I, Adams RH, Gossler A (2009) DLL1‐mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113: 5680–5688 [DOI] [PubMed] [Google Scholar]
- 18. Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U et al (2010) Ephrin‐B2 controls VEGF‐induced angiogenesis and lymphangiogenesis. Nature 465: 483–486 [DOI] [PubMed] [Google Scholar]
- 19. Takahashi T, Shibuya M (1997) The 230 kDa mature form of KDR/Flk‐1 (VEGF receptor‐2) activates the PLC‐gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene 14: 2079–2089 [DOI] [PubMed] [Google Scholar]
- 20. Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC (1995) Failure of blood‐island formation and vasculogenesis in Flk‐1‐deficient mice. Nature 376: 62–66 [DOI] [PubMed] [Google Scholar]
- 21. Yuk IH, Wang DI (2002) Glycosylation by Chinese hamster ovary cells in dolichol phosphate‐supplemented cultures. Biotechnol Appl Biochem 36: 141–147 [DOI] [PubMed] [Google Scholar]
- 22. Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier‐Lavigne M (2003) Lack of enhanced spinal regeneration in Nogo‐deficient mice. Neuron 38: 213–224 [DOI] [PubMed] [Google Scholar]
- 23. Marek KW, Vijay IK, Marth JD (1999) A recessive deletion in the GlcNAc‐1‐phosphotransferase gene results in peri‐implantation embryonic lethality. Glycobiology 9: 1263–1271 [DOI] [PubMed] [Google Scholar]
- 24. Lennon K, Pretel R, Kesselheim J, te Heesen S, Kukuruzinska MA (1995) Proliferation‐dependent differential regulation of the dolichol pathway genes in Saccharomyces cerevisiae . Glycobiology 5: 633–642 [DOI] [PubMed] [Google Scholar]
- 25. Martinez JA, Torres‐Negron I, Amigo LA, Roldan RA, Mendez A, Banerjee DK (2000) Tunicamycin inhibits capillary endothelial cell proliferation by inducing apoptosis. Targeting dolichol‐pathway for generation of new anti‐angiogenic therapeutics. Adv Exp Med Biol 476: 197–208 [PubMed] [Google Scholar]
- 26. Martinez JA, Torres‐Negron I, Amigo LA, Banerjee DK (1999) Expression of Glc3Man9GlcNAc2‐PP‐Dol is a prerequisite for capillary endothelial cell proliferation. Cell Mol Biol 45: 137–152 [PubMed] [Google Scholar]
- 27. Croci DO, Cerliani JP, Dalotto‐Moreno T, Mendez‐Huergo SP, Mascanfroni ID, Dergan‐Dylon S, Toscano MA, Caramelo JJ, Garcia‐Vallejo JJ, Ouyang J et al (2014) Glycosylation‐dependent lectin‐receptor interactions preserve angiogenesis in anti‐VEGF refractory tumors. Cell 156: 744–758 [DOI] [PubMed] [Google Scholar]
- 28. Pinzon‐Olejua A, Welte C, Abdesselem H, Malaga‐Trillo E, Stuermer CA (2014) Essential roles of zebrafish rtn4/Nogo paralogues in embryonic development. Neural Dev 9: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brosamle C, Halpern ME (2009) Nogo‐Nogo receptor signalling in PNS axon outgrowth and pathfinding. Mol Cell Neurosci 40: 401–409 [DOI] [PubMed] [Google Scholar]
- 30. Di Lorenzo A, Manes TD, Davalos A, Wright PL, Sessa WC (2011) Endothelial reticulon‐4B (Nogo‐B) regulates ICAM‐1‐mediated leukocyte transmigration and acute inflammation. Blood 117: 2284–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang D, Utsumi T, Huang HC, Gao L, Sangwung P, Chung C, Shibao K, Okamoto K, Yamaguchi K, Groszmann RJ et al (2011) Reticulon 4B (Nogo‐B) is a novel regulator of hepatic fibrosis. Hepatology 53: 1306–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu J, Fernandez‐Hernando C, Suarez Y, Schleicher M, Hao Z, Wright PL, DiLorenzo A, Kyriakides TR, Sessa WC (2009) Reticulon 4B (Nogo‐B) is necessary for macrophage infiltration and tissue repair. Proc Natl Acad Sci USA 106: 17511–17516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edlund C, Brunk U, Chojnacki T, Dallner G (1988) The half‐lives of dolichol and dolichyl phosphate in rat liver. Biosci Rep 8: 139–146 [DOI] [PubMed] [Google Scholar]
- 34. Andersson M, Elmberger PG, Edlund C, Kristensson K, Dallner G (1990) Rates of cholesterol, ubiquinone, dolichol and dolichyl‐P biosynthesis in rat brain slices. FEBS Lett 269: 15–18 [DOI] [PubMed] [Google Scholar]
- 35. Schenk B, Fernandez F, Waechter CJ (2001) The ins(ide) and out(side) of dolichyl phosphate biosynthesis and recycling in the endoplasmic reticulum. Glycobiology 11: 61R–70R [DOI] [PubMed] [Google Scholar]
- 36. Denecke J, Kranz C (2009) Hypoglycosylation due to dolichol metabolism defects. Biochim Biophys Acta 1792: 888–895 [DOI] [PubMed] [Google Scholar]
- 37. Argraves WS, Drake CJ (2005) Genes critical to vasculogenesis as defined by systematic analysis of vascular defects in knockout mice. Anat Rec A Discov Mol Cell Evol Biol 286: 875–884 [DOI] [PubMed] [Google Scholar]
- 38. Drake CJ, Fleming PA (2000) Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood 95: 1671–1679 [PubMed] [Google Scholar]
- 39. Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH (2009) The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137: 1124–1135 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File