

Abstract
The trans‐Golgi network (TGN) is the main secretory pathway sorting station, where cargoes are packed into appropriate transport vesicles targeted to specific destinations. Exomer is a cargo adaptor necessary for direct transport of a subset of cargoes from the TGN to the plasma membrane in yeast. Here, we show that unlike classical adaptor complexes, exomer is not recruited en bloc to the TGN, but rather assembles through a stepwise pathway, in which first the scaffold protein Chs5 and then the cargo‐binding units, the ChAPs, are recruited. Although all ChAPs are able to assemble functional exomer complexes, they do so with different efficiencies. The mutual relationship between ChAPs varies from cooperation to competition depending on their expression levels and affinities to Chs5 allowing an optimized and efficient cargo transport. The multifactorial assembly pathway results in an exquisitely fine‐tuned adaptor complex, enabling the cell to quickly respond and adapt to changes such as stress.
Keywords: cargo adaptor, FCS, FRAP, Golgi‐plasma membrane transport, quantitative analysis
Subject Categories: Membrane & Intracellular Transport
Introduction
The trans‐Golgi network (TGN) is the main cargo sorting station in the exocytic pathway of eukaryotic cells. Cargo adaptors interact with the cytosolic domains of transmembrane cargoes, and cargo receptors and collect them into appropriate transport carriers. The carriers are subsequently targeted to different destinations such as the plasma membrane, endosomal compartments, and lysosomes 1, 2. Most information about the function of cargo adaptors in protein transport has been obtained by studying COPI and COPII transport machineries, and AP adaptor complexes involved in clathrin‐dependent or independent transport 2, 3, 4. While those transport machineries presumably cover a large repertoire of cargoes, information about other transport platforms, which deliver the rest of the cargoes to the plasma membrane, is limited 4, 5, 6, 7.
The exomer complex is a putative coat/cargo adaptor involved in the direct transport of a subset of cargoes from the TGN to the plasma membrane in S. cerevisiae 8, 9, 10, 11. In contrast to other known adaptor complexes, exomer‐dependent cargoes are sorted at the TGN in a cell cycle‐dependent manner 8, 9, 11, 12. Although different direct AP‐independent transport pathways from the TGN to the plasma membrane have also been described in mammalian cells 13, 14, the nature of the transport machineries remains elusive to date. Exomer appears to be a specific export platform, which is modulated during the cell cycle and through environmental stress 11, 15. Although the cellular function of exomer cargoes is very diverse, they all respond exquisitely to changes in the environment and cell stress 11, 15, 16, 17. Notably, Chs3 and Pin2 are rapidly endocytosed after exposure to stress 11, 15. While Chs3 is then fast reexported in a non‐polarized fashion, presumably to strengthen the cell wall, Pin2 is retained at the TGN and is only efficiently transported once the stress has subsided. Exomer‐dependent transport may be particularly important to ensure localization of a subset of stress‐responsive proteins at the plasma membrane, a process, which is not understood at all in mammalian cells. Exomer therefore serves as a model to understand the regulation of the assembly of non‐classical coat/adaptor complexes, involved in cellular homeostasis.
Exomer consists of five peripheral TGN proteins, Chs5 and four paralogues—Chs6, Bud7, Bch1, and Bch2—collectively termed ChAPs 8, 9. The N‐terminal half of Chs5 resembles the appendage domains found in AP2 and COPI coats 18, and the full‐length protein serves as a scaffold for exomer assembly 12. Chs5 recruits ChAPs to the TGN membrane in a process that depends on its interaction with the activated form of Arf1, Arf1‐GTP 8, 9. The ChAPs, in turn, interact directly with cargoes 8, 11, 19 and lipids 18. Thus, as for other coat/adaptor complexes 3, 20, 21, productive binding to exit sites is dependent on an active small GTPase of the Arf1/Sar1 family, cargo, and lipid binding 22.
Crystal structures of the N‐terminal half of Chs5 with either Bch1 or Chs6 revealed a complex of two Chs5 molecules and two molecules of ChAPs 18, 23. Such exomer tetramers can exist theoretically in up to 10 distinct variants, because different combinations of ChAPs can bind to the Chs5 dimer 8, 10, 12. Which exomer complexes exist in vivo and are relevant for the cell cycle‐regulated cargo export has not been addressed to date. Complexes containing either Bch1 or Bch2 are sufficient to localize Pin2 at the plasma membrane 11. In contrast, Chs3 is absolutely dependent on Chs6, which probably is the only ChAP capable of Chs3 recognition 8, 24. However, Chs6 is not sufficient for Chs3 transport in the absence of other ChAPs 8. Thus, ChAPs differ not only in their cargo specificities, but might also have non‐redundant roles in exomer complex formation.
The possible versatility of exomer to form a number of distinct complexes may allow fine‐tuning of the delivery of a very distinct subset of cargoes to the plasma membrane in a context‐specific manner, let this be cell cycle stage or insult from the outside. The coexistence of all possible complexes would indicate that the cellular concentration of the ChAPs would be equal and that the total abundance of the ChAPs should not be greater than that of Chs5. In addition, the ChAPs should not exhibit any competitive behavior when binding to Chs5. Moreover, cargo availability would not influence the abundance of a particular exomer complex. Given these considerations, how could specificity in cargo transport be achieved? We assumed that not all complexes coexist in vivo, and hypothesized that in order to generate a distinct subset of functional exomer complexes, any, if not all, of the above‐mentioned parameters should be different and used to fine‐tune the assembly pathway.
We tested our hypothesis and found that exomer complexes are not preformed in the cytoplasm but are assembled on the TGN. Each ChAP has individual intrinsic properties that contribute to the assembly of specific exomer populations. Additional contributing factors are the relative expression levels of the ChAPs and their ability to interact with cargo. This controlled and biased assembly pathway renders exomer exquisitely versatile in responding to cellular regulatory processes and cargo availability.
Results
ChAPs and Chs5 exist as independent molecular species in the cytoplasm
Classical adaptor complexes are preformed in the cytoplasm and are recruited en bloc to the membrane where they are supposed to act. We wanted to test whether this would also be the case for exomer complexes. Therefore we analyzed the dynamic behavior of individual components of the exomer complex using two quantitative fluorescence live‐cell imaging techniques: fluorescence correlation spectroscopy (FCS) and fluorescence recovery after photobleaching (FRAP). While FCS was used to define diffusion properties and to discriminate monomeric exomer proteins from those engaged in various protein complexes in the cytoplasm, the FRAP approach allowed us to determine binding properties of exomer components at the TGN.
First, we individually tagged Chs5 and ChAPs chromosomally with GFP and confirmed that the GFP‐tag did not impair Chs5 and Chs6 function (Fig EV1A). As expected, all GFP‐tagged proteins were enriched at the TGN as they colocalized with the TGN marker Sec7 (Fig 1A) 8. While Chs5 was mostly found at the TGN, significant amounts of the ChAPs were present also in the cytoplasm. The ratio of membrane‐associated to cytoplasmic pool varied between different ChAPs (Fig 1A and B). Bch2 exhibited the highest level of TGN enrichment, followed by Bud7, Chs6, and Bch1, consistent with previously published results 8.
Figure EV1. Characterization of Chs5 and the ChAPs fusion proteins.
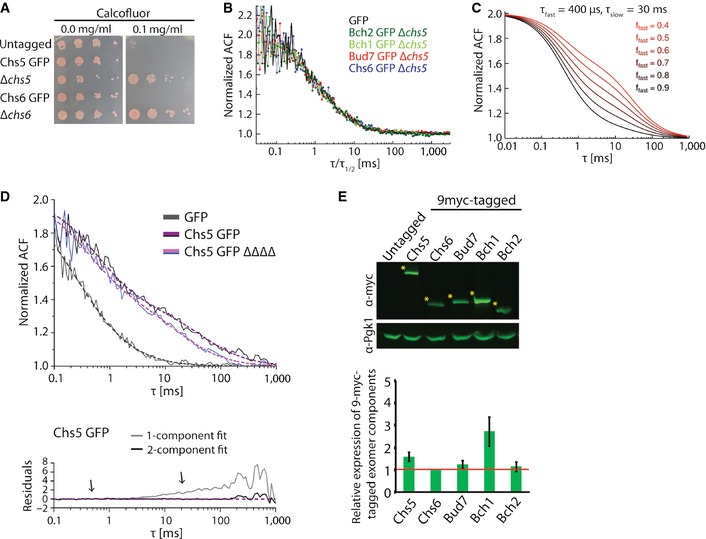
- GFP‐tag does not affect functionality of the GFP‐tagged exomer components. Chs5‐GFP‐ and Chs6‐GFP‐expressing strains were sensitive to calcofluor similarly as the WT strain. Plates were incubated at 30°C for 2–3 days.
- Comparison of the ACFs for GFP and ChAPs in the ∆chs5 strain rescaled by their respective half times.
- Simulated FCS traces of the fast and slow component with fixed diffusion correlation times (τfast, τslow) and variable fast component fractions (ffast).
- FCS measures the in vivo mobility of Chs5 complexes in the cytoplasm. FCS analysis was performed on the GFP and Chs5‐GFP in the presence or absence (ΔΔΔΔ) of the ChAPs. 10–20 FCS measurements were performed for each condition using different cells and representative curves are shown. The ACF of free GFP is fitted with a one‐component anomalous diffusion model. The ACF of Chs5‐GFP in both WT and ΔΔΔΔ background was fitted with a two‐component diffusion model. The residuals assess the quality of the fit. Note the ChAP deletion affects the diffusion of the slow component of Chs5 only mildly. Estimated diffusion correlation times, diffusion coefficients, and fractions are shown in Table 1.
- Quantification of the relative expression levels of the exomer components in 9‐myc‐tagged strains. Representative immunoblot of yeast lysates is shown; Pgk1 serves as a loading control. The asterisks depict the analyzed bands. The plot shows average and SD of three independent biological experiments. The relative expression levels are normalized to Chs6 (depicted by the red line).
Figure 1. Exomer assembles from its subunits on the trans‐Golgi network (TGN).
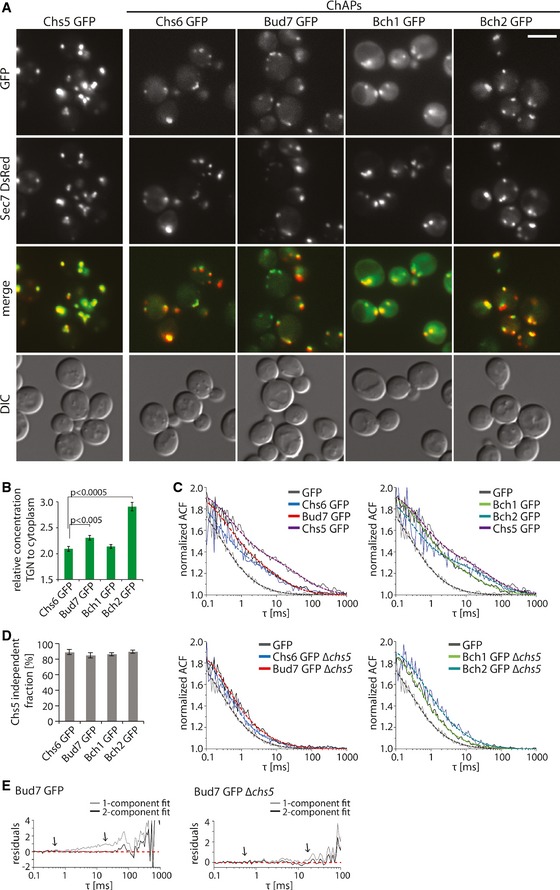
- Exomer components localize to different extent to the TGN and to the cytoplasm. Fluorescence images of cells expressing GFP‐tagged exomer components and TGN marker Sec7‐DsRed. The brightness and contrast were adjusted differently in the GFP and merge images. Scale bar, 5 μm.
- Quantification of the relative TGN to cytoplasm concentration of the GFP‐tagged ChAPs in the cell. The mean fluorescence at the TGN and cytoplasm was determined using the Icy imaging software. 20–30 individual cells were analyzed. Error bars represent SD. For statistical significance testing, Student's t‐test was used.
- FCS measures the in vivo mobility of exomer components in the cytoplasm. FCS analysis was performed on the GFP, Chs5‐GFP, and GFP‐tagged ChAPs in the wild‐type background (top) and on GFP‐tagged ChAPs in the Δchs5 background (bottom). 10–20 FCS measurements were performed for each condition using different cells. The ACF of free GFP is fitted with a one‐component anomalous diffusion model. The ACF of Chs5 in WT and ChAPs in WT and Δchs5 background is fitted with a two‐component diffusion model. Representative ACF curves (thin full lines) and fits (thick dashed lines) are shown. Estimated diffusion correlation times, diffusion coefficients, and fractions are shown in Table 1.
- Quantification of the Chs5 independent fraction of ChAPs present in the cytoplasm based on the measurements shown in (C). Error bars represent SD.
- Comparison of the residuals of the one‐component and two‐component fit (arrows).
To assess the diffusion properties of Chs5 and ChAPs in the cytoplasm, we performed an FCS analysis. FCS monitors fluctuations in fluorescence intensity caused by the movement of fluorescence molecules through the focal volume of a laser beam. The recorded fluorescence time series are correlated over time resulting in an autocorrelation function (ACF). Small molecules diffuse faster and stay in the focal volume for a shorter time than large complexes. Therefore, the ACF declines more steeply for small molecules than for large complexes. Fitting the ACF with an appropriate mathematical model can be used to determine diffusion coefficients and fractions of different molecular species of the analyzed protein; see 25 for an introduction.
The ChAPs and Chs5 diffused with different rate constants through the focal volume as indicated by the autocorrelation functions (ACFs) (Fig 1C, top panels), yet they were all slower than GFP. The slower diffusion behavior of the exomer components could either be due to differences in molecular weight or their ability to form complex with varying stoichiometries 8, 9, 10, 12. The latter possibility can readily be tested. Since Chs5 is the common component of all exomer complexes, we determined the diffusion behavior of the ChAPs in the absence of Chs5 (Fig 1C, bottom panels). Interestingly, the ChAPs diffusion in the cytoplasm became faster and more closely resembled the one of GFP. Moreover, rescaling the ACFs for GFP and ChAPs in the ∆chs5 strain by their respective half times yields a collapse of all ACFs to a single master curve (Fig EV1B). Hence, the ChAPs show a very similar diffusion pattern in the ∆chs5 strain to soluble GFP, whereas in wild‐type cells seemingly an additional, slow diffusing component is present.
Autocorrelation functions, which report on a diffusional behavior that is composed of more than one component (i.e., monomer and oligomeric complex) are difficult to interpret without appropriate mathematical models, since the relative fraction of faster to slower migrating species has a great influence on the shape of the curve. This effect is illustrated in Fig EV1C. Thus, we next fitted our ACF data with mathematical models that describe the anomalous diffusion of a single component or two components with different diffusion constants (cf. Eqs (1) and (2) in Materials and Methods). In line with previously published data 26, the ACF of free GFP is well described by a single component model with a slight diffusion anomaly (α ≈ 0.9) and a diffusion time τ 1 ≈ 290 μs, corresponding to a diffusion constant of 25 μm2/s (Table 1). In contrast, the anomalous one‐component model did not provide a good fit to the ACF data of the ChAPs: While a fast diffusing fraction of the ChAPs with τ 1 ~400 μs was well captured, analysis of the residuals revealed the existence of a second, slow component with τ 2 ~20 ms (see Fig 1E). Therefore, we employed a two‐component fit function for all FCS data, which resulted in very low residuals, that is, a very good overlap with experimental data (Fig 1E, arrows). From these fits, we extracted the diffusion times of the fast and slow component, τ 1 and τ 2, respectively, as well as the fraction of the fast component, F 1 (Table 1). As a result, we found that cytosolic ChAPs in the wild‐type background populate predominantly the fast fraction (F 1 = 70–80%) with diffusion constants that compare favorably to theoretical estimates for monomeric ChAPs (Table 1). Although we cannot exclude that the ChAPs interact with themselves or other factors in the cytoplasm, our data clearly indicates that all individuals in the fast fraction of all ChAPs are at least quite similar in size, presumably monomers. We would also like to emphasize that the ACF time of the fast fraction of the ChAPs only varied slightly within a 10% range, which is not significant variation in single‐beam FCS experiments 27. The fast fraction of the ChAPs was only enhanced moderately (F 1 = 80–90%) in the ∆chs5 strain (Table 1) despite differences in the form of the ACF (Fig EV1C). Moreover, due to the good overlap of the ACFs for GFP and ChAPs in the ∆chs5 strain (Fig EV1B), the very low residual fraction of slow diffusing particles in the ∆chs5 strain was deemed negligible. Thus, our FCS experiments revealed that the vast majority of the ChAPs move independent of Chs5 in the cytoplasm (Fig 1D). The minor fraction of cytoplasmic exomer complexes (~10%) is unlikely playing a major role in exomer‐dependent export from the TGN.
Table 1.
Calculated parameters derived from FCS and theoretical diffusion coefficients of exomer components
| Analyzed protein | Strain | τ 1 (μs) | Df1 (μm2/s) | F 1 (%) | τ 2 (ms) | Df2 (μm2/s) | Dfth (μm2/s) | Size including GFP (kDa) |
|---|---|---|---|---|---|---|---|---|
| GFP | WT | 290 ± 31 | n.d. | 100 | n.d. | n.d. | 25.0a | 25a |
| Chs6 GFP | WT | 369 ± 40* | 19.6 | 78 ± 2** | 29 ± 4 | 0.25 | 15.2 | 111 |
| Δchs5 | 334 ± 27 | 21.7 | 90 ± 3** | 18 ± 5 | 0.40 | |||
| Bud7 GFP | WT | 440 ± 27* | 16.5 | 71 ± 2** | 21 ± 4 | 0.35 | 15.3 | 110 |
| Δchs5 | 399 ± 19 | 18.2 | 86 ± 1** | 17 ± 5 | 0.43 | |||
| Bch1 GFP | WT | 442 ± 31* | 16.4 | 71 ± 2** | 15 ± 2 | 0.48 | 15.4 | 107 |
| Δchs5 | 393 ± 24 | 18.5 | 85 ± 1** | 17 ± 4 | 0.43 | |||
| Bch2 GFP | WT | 357 ± 56* | 20.3 | 67 ± 2** | 38 ± 4 | 0.19 | 15.1 | 113 |
| Δchs5 | 415 ± 38 | 17.5 | 78 ± 1** | 21 ± 3 | 0.35 | |||
| Chs5 GFP | WT | 621 ± 54* | 11.7 | 59 ± 2* | 32 ± 4 | 0.22 | 15.8 | 99 |
| ΔΔΔΔ | 524 ± 34 | 13.8 | 65 ± 5* | 19 ± 3 | 0.38 |
n.d., not determined; **P < 0.002; *P < 0.02; a, 26.
Diffusion correlation times τ1 and τ2 reflect diffusion of the free protein fraction (F 1) and the slow‐moving exomer subcomplex fraction, respectively. τ 1 and τ 2 were used to calculate the diffusion coefficients Df1 and Df2, respectively. The mean ± SEM is shown. P‐values in F 1 column depict the difference in F 1 between WT and Δchs5 or ΔΔΔΔ strain. P‐values in τ 1 column depict the difference in τ1 between Chs5 and all the ChAPs. P‐values were determined by Student's t‐test. The theoretical diffusion coefficients Dfth were calculated based on Einstein–Stokes relationship. For the detailed calculation procedures see Materials and Methods section.
A fraction of Chs5 is part of large, ChAPs‐independent structures in the cytoplasm
The results above suggest that the cytoplasmic pool of Chs5 diffuses independently of the ChAPs. To test this prediction, we compared the ACFs of Chs5 in wild type and in a strain in which all four ChAPs were deleted. Similarly to the ChAPs, the ACF curves of Chs5 were very well described by a two‐component fit (Figs 1C and EV1D; Table 1). About 60% of Chs5 diffused as a fast component through the cytoplasm. Interestingly, the associated diffusion coefficient was somewhat smaller than the one predicted theoretically, which might be due to the rather stretched and not globular shape of the protein 18. Deletion of the ChAPs affected the diffusion pattern of Chs5 only to a minor extent (Fig EV1D), indicating that the ChAP‐dependent fraction of Chs5 was only ~5% (Table 1). This result supports the hypothesis that formation of exomer complexes takes place at the TGN. The remaining, slow Chs5 fraction (~35%) was part of large structures, independent of the ChAPs. The estimated size of these structures was 100–180 nm in diameter, which would be in the range of transport containers. The approximately twofold slower diffusion of Chs5 as compared to the ChAPs and the ChAP‐independent diffusion behavior of Chs5 strongly indicate that exomer complexes are not stable entities in the cytoplasm but rather assemble on a platform, such as the TGN membrane.
Exomer complex assembles on TGN membranes in a unique manner
The apparent differences in the extent of the steady‐state TGN localization of the individual exomer components (Fig 1A and B) imply that their association and/or dissociation rates may vary. We tested this hypothesis by employing FRAP in strains in which the TGN was marked by Sec7‐DsRed and exomer components were tagged with GFP. The fluorescence recovery curve reflects the movement of unbleached molecules into the bleached region, and this movement is determined by the diffusion and binding characteristics of the analyzed molecules. Since the recorded fluorescence recoveries at the TGN occurred in larger time scales (20–30 s) (Fig 2A) than the fast diffusion in the cytoplasm (Table 1), we modeled the turnover of fluorescent proteins at the TGN as a simple reaction‐limited on‐off process neglecting the diffusional contribution from the cytoplasm. The FRAP data were fitted according to 28. By fitting single‐exponential curves to our FRAP data we obtained proteins' mean residence time on the TGN, which is simply the inverted value of the dissociation constant koff (Table 2). Likewise, we determined the immobile fraction, which reflects the amount of molecules that are not exchanged at the TGN within the time frame of the experiment. First, we compared the dynamic behavior of exomer components to components of the conventional COPI and clathrin‐AP coats (Fig 2A; Table 2). Two COPI coat subunits, Cop1 and Sec28, had almost identical residence times (about 5.7 s), consistent with them being recruited together as part of the coatomer complex (Fig 2A; Table 2). Similarly, clathrin light chain (Clc1) and the adaptor complex 1 component, Apm1, had comparable residence times (Fig 2A; Table 2). However, the residence times of the two exomer components Chs5 and Bch1 varied drastically. While the recovery kinetics of Bch1 was in a similar range as observed for the other coats/adaptors, Chs5 displayed the largest residence time (20.4 s) and the largest immobile fraction (~60%), indicating that it is much more strongly bound to the TGN and does not exchange as rapidly as the other components. These data are consistent with exomer complex assembly at the TGN, rather than being recruited en bloc like AP adaptor complexes and coatomer.
Figure 2. Exomer components display distinct dynamic behavior at the trans‐Golgi network (TGN) compared to classical coat components.
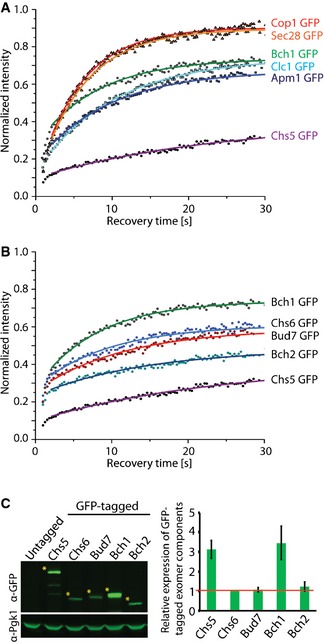
- Comparison of the dynamic behavior of the GFP‐tagged Chs5 and Bch1 with the GFP‐tagged Sec28, Cop1, Clc1, and Apm1. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2.
- Binding kinetics of the GFP‐tagged Chs5 and ChAPs at the TGN is different. Data processing was carried out as in (A).
- Quantification of the relative expression levels of the exomer components in GFP‐tagged strains. A representative immunoblot of yeast lysates is shown; Pgk1 serves as a loading control. The asterisks depict the analyzed bands. The plot shows average and SD of three independent biological experiments. The relative expression levels are normalized to Chs6 (depicted by the red line).
Table 2.
Calculated parameters derived from FRAP analysis of the exomer components and components of the coat/adaptor machineries at the trans‐Golgi network (TGN)
| Analyzed protein | Strain background | koff (s−1) | 1/koff (s) | Fim (%) | kon (s−1) | 1/kon (s) |
|---|---|---|---|---|---|---|
| Chs5 GFP | WT | 0.049 ± 0.005 | 20.4 ± 1.9 | 60 ± 2 | 0.294 ± 0.037 | 3.4 ± 0.4 |
| CHS6 ΔΔΔ | 0.177 ± 0.017 | 5.6 ± 0.5 | 26 ± 2 | 0.287 ± 0.039 | 3.5 ± 0.4 | |
| BUD7 ΔΔΔ | 0.102 ± 0.009 | 9.8 ± 0.8 | 46 ± 2 | 0.229 ± 0.028 | 4.4 ± 0.5 | |
| BCH1 ΔΔΔ | 0.058 ± 0.005 | 17.2 ± 1.3 | 63 ± 1 | 0.279 ± 0.039 | 3.6 ± 0.4 | |
| BCH2 ΔΔΔ | 0.120 ± 0.009 | 8.3 ± 0.6 | 39 ± 2 | 0.254 ± 0.029 | 3.9 ± 0.4 | |
| ΔΔΔΔ | 0.122 ± 0.011 | 8.2 ± 0.7 | 40 ± 3 | 0.252 ± 0.031 | 4.0 ± 0.4 | |
| GPD‐BCH2 ΔΔΔ | 0.070 ± 0.011 | 14.3 ± 1.9 | 48 ± 2 | 0.392 ± 0.080 | 2.6 ± 0.4 | |
| GPD‐CHS6 ΔΔΔ | 0.086 ± 0.005 | 11.6 ± 0.6 | 35 ± 2 | 0.504 ± 0.051 | 2.0 ± 0.2 | |
| CHS6 BUD7 ΔΔ | 0.091 ± 0.007 | 11.0 ± 0.8 | 51 ± 1 | 0.242 ± 0.040 | 4.1 ± 0.6 | |
| CHS6 BCH2 ΔΔ | 0.109 ± 0.010 | 9.2 ± 0.8 | 42 ± 2 | 0.279 ± 0.035 | 3.6 ± 0.4 | |
| CHS6 BCH2 ΔΔ +Pin2‐CEN | 0.103 ± 0.010 | 9.7 ± 0.8 | 48 ± 1 | 0.335 ± 0.047 | 3.0 ± 0.4 | |
| CHS6 BCH2 ΔΔ +Pin2‐2μ | 0.098 ± 0.010 | 10.2 ± 0.9 | 49 ± 2 | 0.300 ± 0.038 | 3.3 ± 0.4 | |
| +p426 GPD | 0.052 ± 0.006 | 19.2 ± 2.0 | 56 ± 3 | 0.311 ± 0.048 | 3.2 ± 0.4 | |
| +p426 GPD‐CHS6 | 0.056 ± 0.006 | 17.9 ± 1.8 | 44 ± 2 | 0.310 ± 0.044 | 3.2 ± 0.4 | |
| Bch1 GFP | WT | 0.133 ± 0.008 | 7.5 ± 0.4 | 26 ± 3 | 0.285 ± 0.022 | 3.5 ± 0.3 |
| ΔΔΔ | 0.150 ± 0.009 | 6.7 ± 0.4 | 14 ± 2 | 0.336 ± 0.031 | 3.0 ± 0.3 | |
| CHS6 ΔΔ | 0.138 ± 0.007 | 7.2 ± 0.4 | 20 ± 1 | 0.355 ± 0.029 | 2.8 ± 0.2 | |
| BCH2 ΔΔ | 0.139 ± 0.007 | 7.2 ± 0.3 | 27 ± 2 | 0.335 ± 0.027 | 3.0 ± 0.2 | |
| BUD7 ΔΔ | 0.147 ± 0.009 | 6.8 ± 0.4 | 28 ± 3 | 0.352 ± 0.033 | 2.8 ± 0.2 | |
| +p426 GPD | 0.133 ± 0.011 | 7.5 ± 0.6 | 24 ± 1 | 0.277 ± 0.031 | 3.6 ± 0.4 | |
| +p426 GPD‐CHS6 | 0.273 ± 0.054 | 3.7 ± 0.6 | 7 ± 2 | 0.342 ± 0.080 | 2.9 ± 0.5 | |
| Bud7 GFP | WT | 0.089 ± 0.005 | 11.2 ± 0.6 | 37 ± 2 | 0.205 ± 0.016 | 4.9 ± 0.3 |
| ΔΔΔ | 0.083 ± 0.005 | 12.1 ± 0.7 | 47 ± 2 | 0.262 ± 0.024 | 3.8 ± 0.3 | |
| Chs6 GFP | WT | 0.097 ± 0.006 | 10.3 ± 0.6 | 35 ± 2 | 0.203 ± 0.017 | 4.9 ± 0.4 |
| ΔΔΔ | 0.125 ± 0.008 | 8.0 ± 0.5 | 50 ± 2 | 0.361 ± 0.035 | 2.8 ± 0.2 | |
| Bch2 GFP | WT | 0.066 ± 0.006 | 15.2 ± 1.3 | 47 ± 4 | 0.192 ± 0.023 | 5.2 ± 0.6 |
| ΔΔΔ | 0.078 ± 0.007 | 12.8 ± 1.0 | 46 ± 2 | 0.265 ± 0.032 | 3.8 ± 0.4 | |
| Arf1 GFP | WT | 0.113 ± 0.008 | 8.9 ± 0.6 | 20 ± 2 | 1.123 ± 0.146 | 0.9 ± 0.1 |
| Cop1 GFP | WT | 0.171 ± 0.008 | 5.8 ± 0.3 | 8 ± 1 | 0.425 ± 0.030 | 2.4 ± 0.2 |
| Sec28 GFP | WT | 0.176 ± 0.010 | 5.7 ± 0.3 | 9 ± 2 | 0.392 ± 0.041 | 2.6 ± 0.2 |
| Clc1 GFP | WT | 0.108 ± 0.008 | 9.2 ± 0.7 | 28 ± 2 | 0.347 ± 0.038 | 2.9 ± 0.3 |
| Apm1 GFP | WT | 0.096 ± 0.007 | 10.4 ± 0.7 | 32 ± 1 | 0.321 ± 0.038 | 3.1 ± 0.3 |
The mean ± SEM is shown for dissociation constant koff and immobile fraction Fim. The inverted value of the mean koff reflects the mean residence time at the TGN. The association constant kon was calculated according to the Eq. (2). The inverted value of the mean kon reflects the mean time of one binding event. For the detailed calculation procedures see Materials and Methods.
The next question is whether all possible complexes form at the TGN or whether there is a bias toward a subset of complexes? In the first case, we would expect relatively similar residence times and immobile fractions for all the ChAPs, while in the latter case they would be different. Bch2 had the largest immobile fraction and longest residence time at the TGN (~47%, 15.2 s), followed by Bud7 (~37%, 11.2 s), Chs6 (~35%, 10.3 s), and Bch1 (~26%, 7.5 s; Fig 2B; Table 2). The observed differences between individual ChAPs correlate well with their apparent TGN localization (Fig 1A and B). These data indicate that not all 10 possible exomer complexes exist with the same likelihood at any given time and that there is a bias toward a subset of complexes. Moreover, the different dynamic behavior of the ChAPs strongly supports the notion of exomer complex assembly on the TGN.
ChAPs may compete for Chs5 binding in a dynamic manner
We have shown previously that exomer can form complexes with varying stoichiometries 8, independently confirming the observations above. Moreover, purified exomer was obtained with a 5:4:1:1:1 (Chs5:Bch1:Chs6:Bch2:Bud7) stoichiometry 12. Since structural data indicate that exomer exists as tetramer in vitro, consisting of two molecules of Chs5 and two ChAPs 18, Bch1‐containing complexes should be overrepresented. We wondered whether this was also reflected in the expression levels of the individual exomer components in vivo. We determined the relative expression levels of GFP‐ and 9‐myc‐tagged exomer components by quantitative immunoblot (Figs 2C and EV1E). With either tag, we obtained comparable results, indicating that the tags did not greatly change the protein stability under these conditions. Bch1 and Chs5 are expressed at a similar level, which is about three times higher than the other ChAPs. Thus, the higher expression level of Bch1 could contribute to preferential integration into exomer complexes and hence provide a bias for exomer complex formation. The ChAPs are three times more abundant than Chs5, indicating that ChAPs might compete for binding sites on Chs5, which could be the basis for an additional bias.
Based on the values of the relative amounts and the immobile fractions determined above (Table 2), we estimated the ratio of individual exomer components present in the complexes at the TGN to be 5:2:1:1:1 for Chs5, Bch1, Chs6, Bud7, and Bch2. This result is comparable to the previously published 5:4:1:1:1 ratio 12. We expect that only 2:2 Chs5:ChAP exomer complexes are stably bound at the TGN and that they can be distinguished as immobile fraction from the entire exomer component pool by FRAP analysis. The mobile fraction would then reflect active turnover of exomer components at the TGN. The calculated ratio of Chs5, Bch1, Chs6, Bud7, and Bch2 in the mobile fraction is 2:4:1:1:1, indicating that the ChAPs indeed compete for Chs5 binding sites.
Chs5 is essential for ChAPs stabilization at the TGN
On the one hand, it appears as if the abundance of the individual ChAPs is important in determining which exomer complexes are predominantly present in the cell. On the other hand, ChAPs appear to compete for binding sites on Chs5, which may equally influence complex formation. In order to understand the rules that govern exomer complex assembly at the TGN, we need insights into how the individual exomer components influence each other. We had previously shown by immunofluorescence that ChAPs were not detectable at the TGN at steady state in ∆chs5 cells, while Chs5 was still TGN localized in the absence of all ChAPs 8. Likewise, GFP‐tagged ChAPs were not present at the TGN in ∆chs5 cells (Fig EV2A). We extended the analysis and performed differential centrifugation determining the membrane‐associated pool of the ChAPs in wild type and ∆chs5. Consistent with our previous data, the ChAPs lost TGN localization in the absence of Chs5 (Figs 3A and EV2B). Thus, Chs5 is critical for ChAPs TGN recruitment.
Figure EV2. ChAPs distribution is dependent on Chs5.
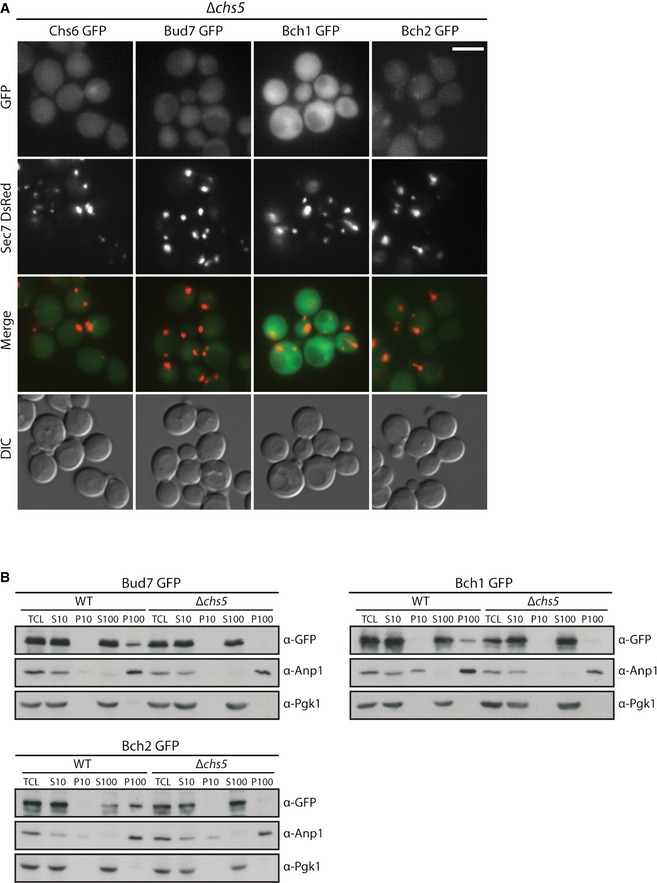
- Chs5 is necessary for recruitment and stable binding of ChAPs at the TGN. Fluorescence images of cells expressing GFP‐tagged ChAPs and TGN marker Sec7‐DsRed in Δchs5 background. Scale bar, 5 µm.
- Chs5 is necessary for stable binding of ChAPs at the Golgi membranes in vitro. Differential centrifugation of cell lysates obtained from Bud7‐GFP, Bch1‐GFP and Bch2‐GFP WT and Δchs5 strains. TCL, total cell lysate; S10, 10,000 g supernatant; P10, 10,000 g pellet; S100, 100,000 g supernatant; P100, 100,000 g pellet. Anp1 serves as the Golgi marker and Pgk1 as the cytoplasm marker. A representative immunoblot of three independent biological experiments is shown.
Source data are available online for this figure.
Figure 3. ChAPs affect differently Chs5 recruitment and stabilization at the trans‐Golgi network (TGN).
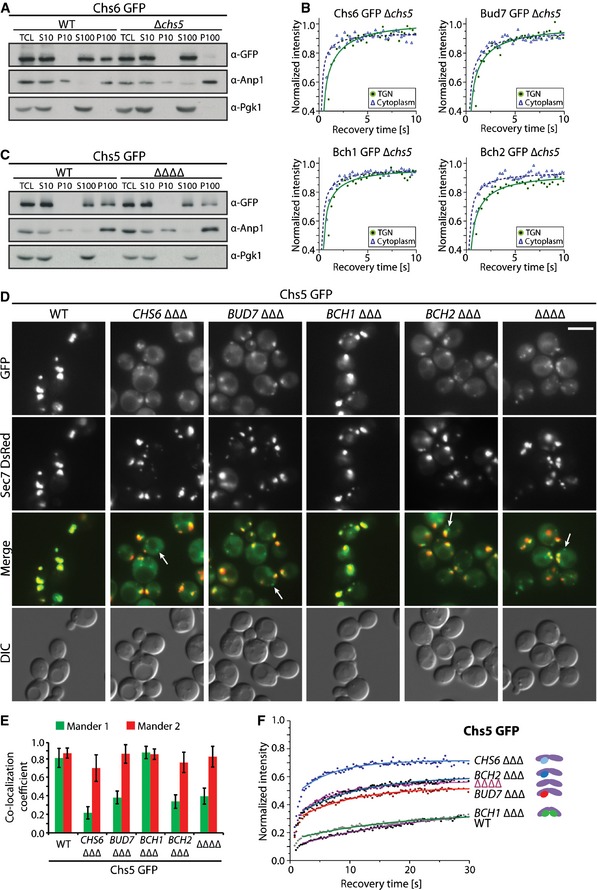
- Chs5 is necessary for recruitment and stable binding of ChAPs at the TGN. Differential centrifugation of cell lysates obtained from Chs6‐GFP WT and Δchs5 strain. TCL, total cell lysate; S10, 10,000 g supernatant; P10, 10,000 g pellet; S100, 100,000 g supernatant; P100, 100,000 g pellet. Anp1 serves as the Golgi marker and Pgk1 as the cytoplasm marker. A representative immunoblot of three independent biological experiments is shown.
- Diffusional behavior of GFP‐tagged ChAPs in the cytoplasm (open triangles, dashed line) and at the TGN (closed circles, full line). The mean of 10–20 FRAP measurements from different cells is shown. Calculated diffusion coefficients are shown in Appendix Table S1.
- Chs5‐GFP displays reduced membrane association in the absence of ChAPs. Differential centrifugation of cell lysates obtained from Chs5 GFP WT and ΔΔΔΔ strains. TCL, total cell lysate; S10, 10,000 g supernatant; P10, 10,000 g pellet; S100, 100,000 g supernatant; P100, 100,000 g pellet. Anp1 serves as the Golgi marker and Pgk1 as the cytoplasm marker. A representative immunoblot of three independent biological experiments is shown.
- Fluorescence images of cells expressing Chs5‐GFP and TGN marker Sec7‐DsRed in WT, ChAP ΔΔΔ, and ΔΔΔΔ strains. Merged images depict the differences in Chs5‐GFP localization to the TGN, cytoplasm, and distinct foci (arrows) in these strains. The brightness and contrast were adjusted differently in the merged images. Scale bar, 5 μm.
- Colocalization analysis of Chs5‐GFP and TGN marker Sec7‐DsRed in WT, ChAP ΔΔΔ, and ΔΔΔΔ strains. Mander 1 depicts the relative amount of the GFP signal colocalizing with the overall DsRed signal, and Mander 2 vice versa. Manders coefficients were obtained using the JACoP plug‐in for ImageJ after background subtraction (N = 20) and are given in Appendix Table S2. Error bars represent SD.
- Binding kinetics of Chs5‐GFP at the TGN in WT, ChAP ΔΔΔ, and ΔΔΔΔ strains. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2. The schematic cartoons depict the predicted majority of the analyzed complexes. The violet ovals represent Chs5 dimer and circles represent ChAPs: green, Bch1; red, Bud7; dark blue, Bch2; light blue, Chs6.
Source data are available online for this figure.
The ChAPs are the cargo‐interacting part of exomer. Moreover, Paczkowski and Fromme 2 provided evidence that Bch1 directly interacts with lipids and Arf1 at the TGN. Thus, we wanted to determine whether those factors contribute to retention of ChAPs at the TGN. In the ∆chs5 cells, the immobile fraction of ChAPs at the TGN is completely abolished and ChAPs exhibit rather diffusional behavior (Fig 3B). The diffusion of ChAPs at the TGN was slowed down approximately 2 times when compared to the movement in the cytoplasm (Appendix Table S1), indicating that the ChAPs may be able to very transiently interact with cargoes, lipids and Arf1 at the TGN, but by themselves these interactions are insufficient to stabilize the ChAPs at the membrane. The stabilization process appears to be strictly dependent on Chs5. This conclusion is likely to be correct as Chs6 and Chs5 stabilized each other on liposomes through cooperative binding in vitro 18.
The ChAPs contribute to Chs5 binding at the TGN
When we performed the differential centrifugation experiments, we realized that the Chs5 signal was reduced in the TGN‐containing fraction when all ChAPs had been deleted (Fig 3C, ∆∆∆∆). Likewise, the Chs5‐GFP TGN signal was diminished in ∆∆∆∆ cells as observed by live‐cell imaging (Fig 3D). In addition, a fraction of Chs5 associated with lipid droplets under these conditions (Fig EV3A). This led us to investigate how the individual ChAPs would influence Chs5 TGN localization. To this end, we generated Chs5‐GFP strains in which only one ChAP (Chs5‐ChAP∆∆∆) was present (Fig 3D). Bch1 by itself was the only ChAP that was able to promote Chs5 TGN association to the same level as in wild type (Fig 3D and E). The other ChAPs did not contribute to Chs5 TGN association. More importantly, Chs6 appeared to have even a slight negative effect on Chs5 localization (Fig 3D and E; Appendix Table S2).
Figure EV3. Analysis of Chs5‐GFP and ChAPs expression levels.
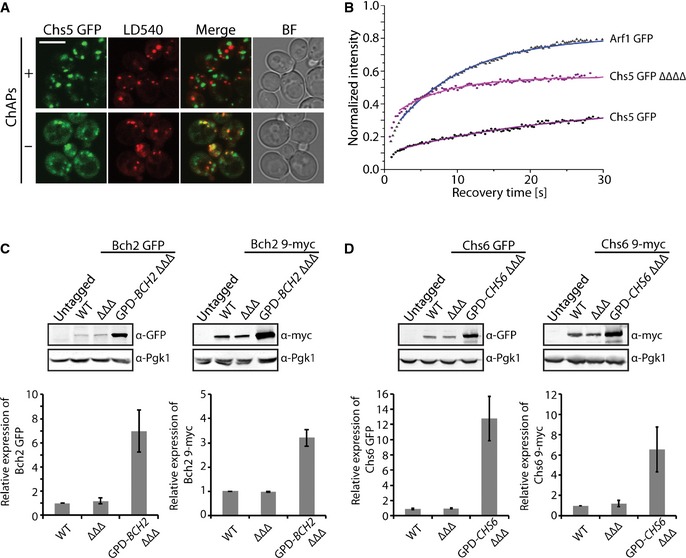
- In the absence of ChAPs, Chs5‐GFP localizes to the trans‐Golgi network, cytoplasm, and lipid droplets. Confocal images of strains expressing Chs5‐GFP with and without the ChAPs, stained with a lipid droplet staining dye LD540. Scale bar, 5 µm.
- Binding kinetics of Arf1‐GFP in the WT and Chs5‐GFP in the WT and ΔΔΔΔ strains. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2.
- Quantification of the relative expression levels of the GFP‐ and 9‐myc‐tagged Bch2 in WT, BCH2 ΔΔΔ, and GPD‐BCH2 ΔΔΔ strains. Representative immunoblots of yeast lysates are shown; Pgk1 serves as a loading control. The plots show average and SD of three independent biological experiments. The relative expression levels are normalized to WT.
- Quantification of the relative expression levels of the GFP‐ and 9‐myc‐tagged Chs6 in WT, CHS6 ΔΔΔ, and GPD‐CHS6 ΔΔΔ strains. Representative immunoblots of yeast lysates are shown; Pgk1 serves as a loading control. The plots show average and SD of three independent biological experiments. The relative expression levels are normalized to WT.
To corroborate these findings and to quantify Chs5 turnover at the TGN, we again performed FRAP (Fig 3F; Table 2). In Chs5‐∆∆∆∆, Chs5 recovered much faster but the immobile fraction was significantly reduced when compared to wild type. Thus, binding of Chs5 to the TGN was not abolished in the absence of ChAPs. The Chs5 immobile fraction was still higher than the one of Arf1 (Fig EV3B), indicating that other factors may contribute to Chs5 TGN retention. Consistent with the colocalization analysis, Bch1 by itself was sufficient to stabilize Chs5, which was reflected in a residence time (17.2 s) and an immobile fraction (~63%) similar to wild type (Table 2). Bud7 induced a mild stabilization (9.8 s, ~46%), while Bch2 had no effect on Chs5 dynamics (8.3 s, ~39%). Interestingly, when Chs6 was the sole ChAP, Chs5 was destabilized from the TGN even more strongly (5.6 s, ~26%) than in Chs5‐∆∆∆∆ cells. The observed effects are not related to the ability of Chs5 binding to the TGN, as the kon of Chs5 (Table 2) was independent of the presence of the ChAPs. Thus, in spite of being members of the same protein family, the ChAPs strikingly differed in their capability to stabilize the scaffold protein Chs5 on the TGN membrane.
Bch1 and Chs5 are present at similar cellular concentration, allowing the formation of 2:2 Chs5:Bch1 complexes at the TGN. In contrast, none of the other ChAPs is abundant enough, and hence, non‐occupied Chs5 dimers and 2:1 Chs5:ChAP complexes may arise that are less stably associated with the TGN. Thus, not only the relative abundance of the individual ChAPs is important, but also their different intrinsic properties such as their ability to either stabilize or destabilize Chs5 at the TGN, may play a critical role.
Exomer assembly on the TGN is not solely driven by abundance of the ChAPs
To test the notion of the different intrinsic properties, we overexpressed the less abundant ChAPs Bch2 and Chs6. If the exomer complex formation was solely driven by relative abundance, overexpression of either ChAP should stabilize Chs5 to wild‐type level. We overexpressed Bch2 about fivefold (Fig EV3C), which resulted in an almost 1:2 ratio of Chs5 to Bch2. Under these conditions Chs5 association with the TGN was improved (Fig 4A). Moreover, the immobile fraction of Chs5 at the TGN increased (about 10%) and also residence time of Chs5 was prolonged (from 8.3 s to 14.3 s) compared to the Chs5‐BCH2∆∆∆ strain (Fig 4B; Table 2). These data indicate that ChAPs expression levels contribute to Chs5 stabilization at the TGN, but are insufficient to explain the specific effects of individual ChAPs on Chs5 dynamics. Thus, ChAPs have different intrinsic properties that contribute to the overall Chs5 stabilization at the TGN.
Figure 4. Formation of the exomer tetramer is dependent on ChAP concentration.

-
A, BOverexpression of Bch2 in absence of other ChAPs results in more efficient Chs5 recruitment and stabilization at the trans‐Golgi network (TGN). Fluorescence images of cells expressing Chs5‐GFP (A) and binding kinetics of Chs5‐GFP at the TGN (B) in WT, BCH2 ΔΔΔ, and GPD‐BCH2 ΔΔΔ strains. Scale bar, 5 μm. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2. The schematic cartoons depict the predicted majority of the analyzed complexes: Chs5 dimer, violet ovals; Bch2, dark blue circles.
-
C, DOverexpression of Chs6 in absence of other ChAPs results in more efficient Chs5 recruitment and stabilization at the TGN. (C) Fluorescence images of cells expressing Chs5‐GFP in WT, CHS6 ΔΔΔ, and GPD‐CHS6 ΔΔΔ strains. Scale bar, 5 μm. (D) Binding kinetics of Chs5‐GFP at the TGN in WT, ΔΔΔΔ, CHS6 ΔΔΔ, and GPD‐CHS6 ΔΔΔ strains. Data processing was carried out as in (A and B). The schematic cartoons depict the predicted majority of the analyzed complexes; Chs5 dimer: violet ovals, Chs6: light blue circles.
-
E–GOverexpression of Chs6 rescues Chs3 export in absence of other ChAPs. (E) Fluorescence images of cells expressing Chs3‐GFP in WT, CHS6 ΔΔΔ, and GPD‐CHS6 ΔΔΔ strains. Arrows point to Chs3‐GFP signals at the bud neck. Scale bar, 5 μm. The inset represents a twofold magnification. (F) Quantification of phenotypes in (E); 100 small‐budded and 100 large‐budded cells were quantified per experiment. Average and SD of three independent biological experiments are shown. (G) Chs3 GFP GPD‐CHS6 ΔΔΔ strain was sensitive to calcofluor to a similar extent as the WT strain. Plates were incubated at 30°C for 2–3 days. A representative drop test of three independent biological experiments is shown.
To corroborate this notion, we overexpressed Chs6 by about tenfold in Chs5‐CHS6∆∆∆ cells (Fig EV3D). Similar to Bch2 overexpression, increasing Chs6 levels caused more efficient recruitment of Chs5 to the TGN (Fig 4C), presumably through the formation of 2:2 Chs5:Chs6 complexes. The residence time of Chs5 increased (from 5.6 s to 11.6 s), yet the immobile fraction of Chs5 only reached a similar level as in the Chs5‐∆∆∆∆ strain (Fig 4D; Table 2), suggesting that the 2:2 Chs5:Chs6 complex may adopt a conformation that is not efficiently retained at the TGN. Alternatively, the intrinsic tendency of Chs5 and Chs6 to assemble into a 2:2 exomer complex might be relatively low. Taken together, exomer complex formation at the TGN is dependent on the relative concentration and intrinsic properties of the ChAPs.
A mixed complex of two ChAPs and Chs5 is required for efficient export of Chs3
The lack of ability of Chs6 to stabilize Chs5 on the TGN might explain why Chs3 is not exported from the TGN under those conditions, albeit Chs6 being essential for Chs3 transport 8, 12, 24. Only when either Bch1 or Bud7 is also expressed, Chs3 reaches the bud neck 12. Since Chs6 overexpression increased Chs5 stabilization at the TGN and possibly the formation of the 2:2 Chs5:Chs6 exomer complexes, we wondered whether Chs3 export would be rescued when the only ChAP present overexpressed was Chs6. Localization of Chs3 at the plasma membrane was partially restored upon Chs6 overexpression (Fig 4E and F), which supports the presence of 2:2 Chs5:Chs6 exomer complexes under this condition. Furthermore, Chs6 overexpression rendered cells calcofluor‐sensitive again (Fig 4G). Moreover, only Bch1 appears to have the necessary concentration to assemble with Chs5 into a 2:2 exomer complex in wild type. Thus, it seems as if the relative abundance and the intrinsic properties of the ChAPs would preferentially promote the formation of 2:1:1 Chs5:ChAP1:ChAP2 complexes. We will refer from now on to complexes consisting of two Chs5 and two different ChAPs as mixed exomer complexes.
ChAPs assemble into a mixed exomer complex to stabilize Chs6‐containing complexes on the TGN
Our data above indicate that export of Chs3 from the TGN requires mixed exomer complexes consisting of a dimer of Chs5 and one molecule of Chs6 and one of Bch1 or Bud7. As a consequence, Chs5 residence time on the Golgi should be increased in the presence of Chs6 and Bud7 over the residence time when only Chs6 is present. Chs5 localized more efficiently to the TGN in Chs5‐CHS6 BUD7∆∆ than in Chs5‐CHS6∆∆∆ cells (Fig 5A). Importantly, Chs5 residence time and the immobile fraction at the TGN (11 s, ~51%) was even higher than in a Chs5‐BUD7∆∆∆ strain (Fig 5B; Table 2). Thus, mixed Chs6 containing exomer complex may reside long enough at the TGN to allow efficient recognition and sorting of the Chs6‐specific cargo Chs3.
Figure 5. ChAPs cooperate during the assembly of the exomer tetramer in vivo .
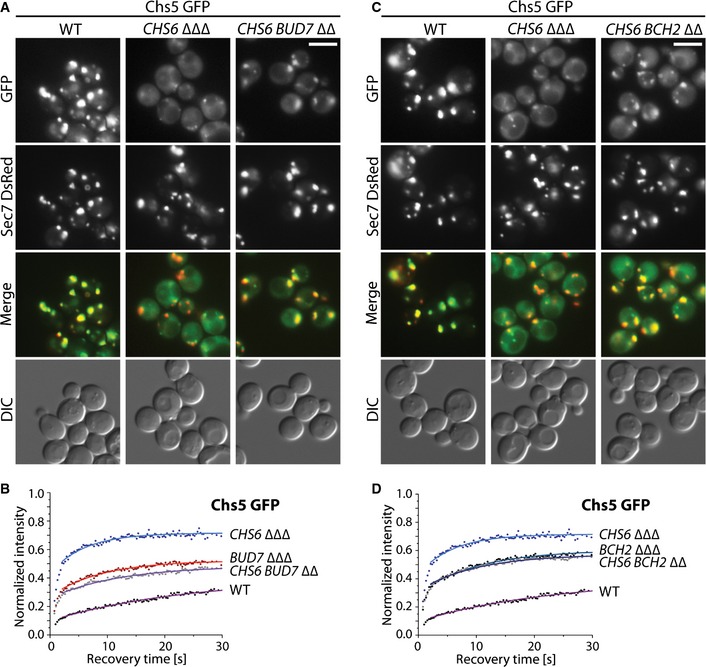
-
A–DFluorescence images of cells expressing Chs5‐GFP and trans‐Golgi network (TGN) marker Sec7 DsRed (A, C) and binding kinetics of Chs5‐GFP at the TGN (B, D) in WT, CHS6 ΔΔΔ and CHS6 BUD7 ΔΔ (A, B) or CHS6 BCH2 ΔΔ (C, D) strains. The brightness and contrast were adjusted differently in the merged images. Scale bar, 5 μm. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2.
We have shown previously that Chs6 and Bch2 are insufficient to promote export of Chs3 (Fig EV4A) 8. If our assumption above was correct, Chs6 and Bch2 should only inefficiently, if at all, cooperate to increase Chs5 residence time at the TGN. Indeed, the residence time and immobile fraction of Chs5 increased only marginally in Chs5‐CHS6 BCH2 ∆∆ (Fig 5C and D; Table 2). Our data suggest that mixed exomer complexes containing Chs6 and either Bch1 or Bud7 are the physiologically relevant ones for Chs3 export. Moreover, they also indicate that there is a bias which mixed exomer complexes might be formed in vivo.
Figure EV4. ChAPs influence each others behavior.
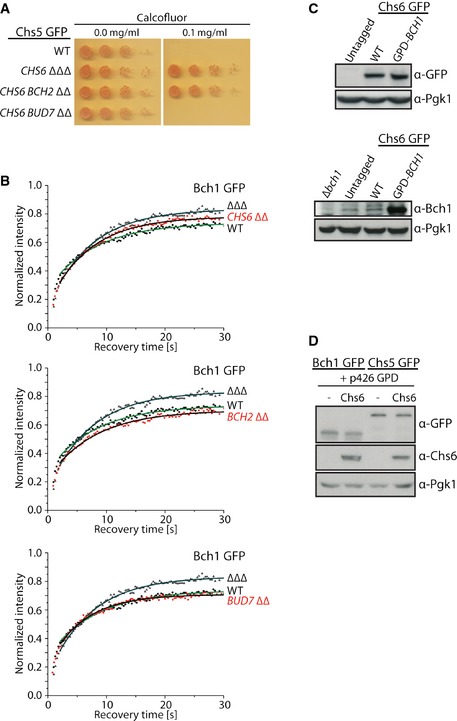
- Chs6 with Bud7 but not Bch2 promotes export of Chs3 to the plasma membrane. Cells bearing Chs6 alone or in combination with Bch2 are resistant to calcofluor, while the combination of Chs6 and Bud7 is sensitive to calcofluor. Plates were incubated at 30°C for 2–3 days.
- Bch1 requires the presence of at least one other ChAP to be stabilized at the trans‐Golgi network. Binding kinetics of Bch1‐GFP in WT, ΔΔΔ, and ChAP ΔΔ strains. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2.
- Comparison of endogenous (WT) and overexpressed (GPD‐BCH1) Bch1 expression levels in Chs6‐GFP cells. Immunoblot of yeast lysates is shown; Pgk1 serves as a loading control.
- Overexpression of Chs6 from GPD‐p426 plasmid in Bch1‐GFP and Chs5‐GFP strains analyzed by immunoblot; Pgk1 serves as a loading control.
Source data are available online for this figure.
ChAPs compete and cooperate during exomer assembly
If this assumption was correct, we would expect that the individual ChAPs would either cooperate or compete with each other. To test this prediction, we analyzed the dynamics of the individual ChAPs at the TGN in the presence or absence of other ChAPs (Fig 6). The immobile fraction of Chs6 and Bud7 was higher when they were the sole ChAPs (Fig 6A and B), consistent with the notion that ChAPs can compete for Chs5 binding. These data also confirmed that a fraction of Chs5 and Chs6 could form 2:2 exomer complexes at the TGN (F i ~26%, Table 2). These complexes were however not sufficient to promote Chs3 export (Fig 4E–G) 12. The residence time of Chs6 decreased from 10.3 s in the wild type to 8.0 s in CHS6∆∆∆. Likewise, Chs5 in CHS6∆∆∆ was also rapidly removed from the TGN (residence time 5.6 s, Fig 3F; Table 2). These data suggest that a fraction of Chs6 could leave the TGN together with Chs5 as a 2:1 Chs5:Chs6 intermediate and provide an explanation why Chs5 is even destabilized at the TGN in the presence of Chs6 alone. The immobile fraction of Bch2, which has the highest affinity to Chs5, remained unaffected by the absence of the other ChAPs (Fig 6C; Table 2). However, similarly to what we observed for Chs6, the residence time of Bch2 at the TGN decreased from 15.2 s to 12.8 s under these conditions. In contrast, Bch1, which has the lowest affinity for Chs5, benefitted from the presence of other ChAPs for the residence time and immobile fraction at the TGN (Fig 6D; Table 2). We therefore analyzed next Bch1 dynamics at the TGN when at least one other ChAP is present. We detected partial or total rescue to the wild‐type dynamic behavior (Fig EV4B; Table 2). These data establish that similar to Chs6, productive Bch1 exomer assemblies are mostly mixed exomer complexes. Moreover, they demonstrate that ChAPs display either cooperative or competitive behavior.
Figure 6. ChAPs compete and cooperate for binding to Chs5 in vivo .
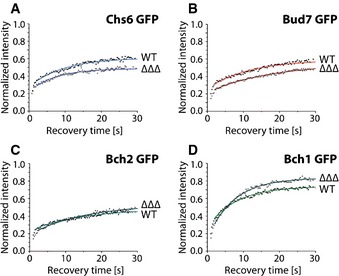
-
A–DChAPs are differently dependent on each other in terms of binding at the trans‐Golgi network (TGN). Binding kinetics of GFP‐tagged Chs6 (A), Bud7 (B), Bch2 (C), and Bch1 (D) at the TGN in the presence (WT) or absence of the other ChAPs (ΔΔΔ). The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2.
Relative concentration and intrinsic behavior of the ChAPs drive exomer complex assembly
We have used deletion strains to determine the contributions of the individual ChAPs in exomer assembly. Next we wanted to test our predictions in the presence of all ChAPs. Bch1 showed the lowest affinity for but the highest ability to stabilize Chs5 at the TGN. Increasing the amount of Bch1 might therefore displace the other ChAPs from the TGN. Overexpressed Bch1 (Fig EV4C) displaced Chs6 from the TGN (Fig 7A) and strongly reduced Chs3 export (Fig 7B and C). Moreover, less chitin was produced in the bud neck as indicated by staining with calcofluor (Fig 7D). However, overexpression of Bch1 did not inhibit exomer‐dependent transport per se, because another exomer‐dependent cargo, Pin2 11, was properly localized (Fig 7E). Bch1 by itself is sufficient to promote Pin2 export 11. Although Bch1 was highly overexpressed (Fig EV4C), Chs3 export was not completely blocked. We attribute this result to the preferred cooperative behavior of Bch1.
Figure 7. The relative expression levels of ChAPs are set to fine‐tune the exomer assembly pathway.
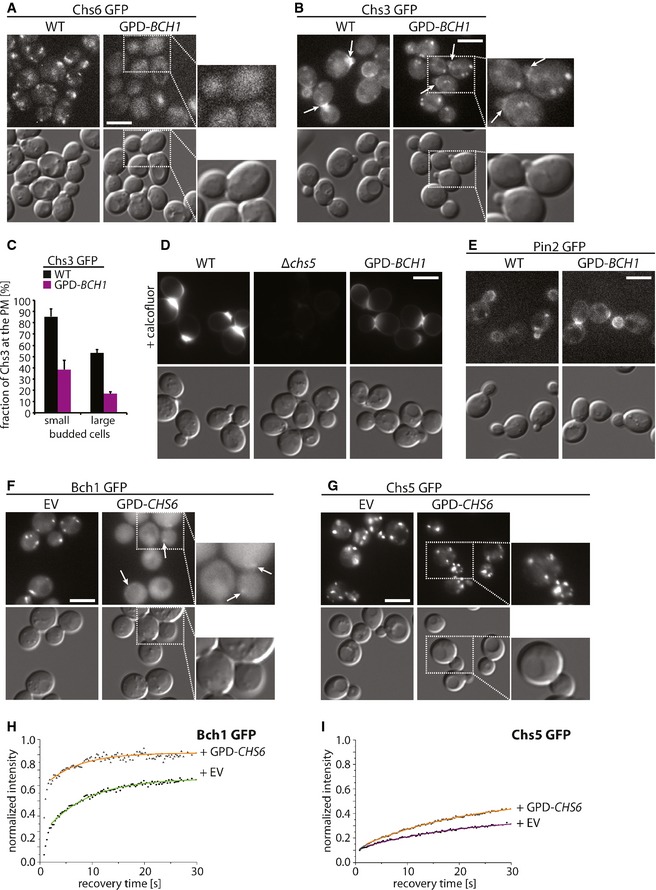
-
A, BOverexpression of Bch1 displaces Chs6 from the trans‐Golgi network (TGN) and impairs plasma membrane localization of Chs6‐dependent cargo Chs3. Fluorescence images of Chs6‐GFP (A) and Chs3‐GFP (B) in WT and GPD‐BCH1 strains. Scale bars, 5 μm. Arrows point to Chs3‐GFP signals at the bud neck. The inset represents a twofold magnification.
-
CQuantification of the phenotypes in (B); 100 small‐budded and 100 large‐budded cells were quantified per experiment. Average and SD of three independent biological experiments are shown.
-
DChitin production is reduced upon Bch1 overexpression. Fluorescence images of chitin stained with calcofluor in Chs3‐GFP WT, Δchs5, and GPD‐BCH1 strains. Scale bar, 5 μm.
-
EOverexpression of Bch1 does not impair plasma membrane localization of the Bch1‐dependent cargo Pin2. Fluorescence images of Pin2‐GFP in WT and GPD‐BCH1 strains. Scale bar, 5 μm.
-
F–IOverexpression of Chs6 displaces Bch1 and Chs5 from the TGN. Fluorescence images (F–G) and binding kinetics at the TGN (H–I) of cells expressing Bch1‐GFP (F, H) and Chs5‐GFP (G, I) upon overexpression of Chs6 under GPD promoter from 2μ plasmid. The insets in (F, G) represent two‐fold magnifications. The arrows in (F) point to residual Bch1 staining at the TGN. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2. Scale bars represent 5 μm.
Chs6 destabilized Chs5 at the TGN and displayed competitive behavior with respect to other ChAPs (Figs 3F and 6A). Thus, Chs6 overexpression should reduce the amount of steady‐state TGN localization of Chs5 and Bch1. Not only did Chs6 overexpression (Fig EV4D) reduce Chs5 and Bch1 levels at the TGN (Fig 7F and G), it also decreased the immobile fraction and the residence time of both proteins (Fig 7H and I). Thus, the intrinsic properties and the relative concentration of the ChAPs are key determinants in driving biased exomer complex assembly.
Cargo supports exomer assembly
The ChAPs act as cargo receptors in the exomer complex. Thus, the next question was whether the cargo would contribute to complex assembly. Either Bch1 or Bch2 is sufficient for Pin2 export from the TGN 11. We overexpressed Pin2 in Chs5 GFP ∆bud7∆bch1 cells, leaving only Bch2 for Pin2 recognition (Fig 8A and B). Chs5 stabilization at the TGN in ∆bud7∆bch1 cells is comparable to that in ∆∆∆∆ cells. Both moderate and strong overexpression of Pin2 increased the residence time and the immobile fraction of Chs5 (Fig 8A; Table 2). The stabilization was not complete because Bch2 becomes limiting under those conditions. Still, the cargo Pin2 contributed to the stabilization of exomer complexes at the membrane. Thus, exomer complex assembly on the TGN is regulated by the availability, and intrinsic properties of the ChAPs, their interaction with Chs5, and the presence of cargo proteins.
Figure 8. Cargo promotes assembly of the exomer tetramer.
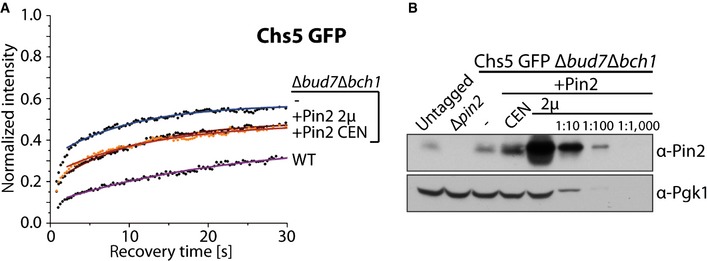
- Overexpression of Pin2 enhances Chs5 stabilization at the trans‐Golgi network. Binding kinetics of Chs5 GFP in WT, Δbud7Δbch1, and Δbud7Δbch1 strain expressing Bch2‐dependent cargo Pin2 from a centromeric or a 2μ plasmid. The mean of 20–30 FRAP measurements from different cells is shown. Calculated parameters are shown in Table 2.
- Comparison of the expression levels of the endogenous Pin2 with the levels of Pin2 expressed from a centromeric or a 2μ plasmid. Pgk1 serves as a loading control. A representative immunoblot of three independent biological experiments is shown.
Source data are available online for this figure.
Discussion
Exomer is a coat/adaptor complex essential for sorting a subset of proteins into secretory vesicles at the TGN. Through the analysis of the dynamic behavior of the different exomer components, we were able to decipher parameters that will drive efficient exomer assembly. Importantly, exomer complexes are not preformed in the cytoplasm but assemble on the TGN. This assembly pathway is notably different from the en bloc recruitment of AP complexes and coatomer 2, 21. In the COPII coat assembly pathway, a complex of the GAP Sec23 together with the cargo adaptor Sec24 is first recruited by Sar1 and then the scaffold complex Sec13/31, which promotes membrane deformation docks on a complex consisting of Sar1, Sec23/24, and cargo 20. Exomer assembly is different as first the scaffold is recruited and then two cargo receptor proteins are selected. Moreover, the individual cargo‐binding units compete and cooperate for binding to the scaffold, which is not the case for COPII coat assembly. The assembly on the TGN allows for greater versatility in selecting cargo, which is deposited in a cell cycle‐dependent manner and which is highly sensitive and responsive to stress.
Although the ChAPs are homologous proteins and share an overall three‐dimensional structure 18, 23, they vary in their contribution to exomer assembly. Our data indicate that Chs6 is strongly involved in specific cargo selection but does not contribute at all to Chs5 stabilization at the TGN, in fact it even destabilizes Chs5 (Fig 9A). At the other end of the spectrum is Bch1, which might be more promiscuous as far as cargo selection is concerned but has the highest ability to stabilize Chs5 at the TGN. Likewise, Bch2 has the highest and Bch1 the lowest affinity for Chs5, yet Bch2 shows only an intermediate ability to retain Chs5 at the TGN. This is most likely due to the threefold lower abundance of Bch2 compared to Chs5. We provide evidence that relative abundance of the ChAPs is an important component in driving exomer complex assembly. Finally, the ChAPs do not only influence the Chs5 residence time at the TGN, they also contribute to the selection of which ChAP can bind to the second binding site in the exomer complex. Again, we observed a range from competition (Bud and Chs6) to cooperativity (Bch1). Thus, of the 10 possible exomer complexes, only a fraction is present in the cell at any given time. The cooperative binding behavior of the most abundant ChAP Bch1, the relative abundance of the ChAPs, and the unstable Chs5:Chs6 complex will promote the formation of mixed Chs5:ChAP1:ChAP2 complexes over Chs5:ChAP complexes containing the same ChAP.
Figure 9. Model of exomer assembly at the trans‐Golgi network (TGN).

- Dual role of ChAPs in formation of the exomer complexes at the TGN. ChAPs differently stabilize Chs5 at the trans‐Golgi network TGN and also have variable specificities for different cargoes.
- Model of the dynamic assembly of the exomer cargo adaptor. (1) Chs5 and Arf1 initiate the complex formation. (2) Chs6 recruitment promotes assembly of the exomer intermediate. (3) Bch1 enhances stabilization of the exomer tetramer at the TGN membrane. (4) Arf1, exomer tetramer, and the cargoes act together as the primer for the exomer coat mediated vesicle budding.
Our data indicate that exomer assembly at the TGN is dependent on: (i) the relative abundance of the individual exomer components; (ii) their binding affinities; (iii) the ability of the ChAPs to stabilize Chs5; and (iv) the competitive and cooperative behavior of the ChAPs when binding to Chs5. Finally, the relative abundance of exomer‐dependent cargo, will also contribute, which of the possible exomer complexes assemble at the TGN. The activation of Arf1 at the TGN, which leads to Chs5 recruitment 8, 18, is likely to be upstream of the exomer assembly process. Likewise, the lipid environment presumably plays a role in both exomer stabilization and cargo segregation and recruitment at the TGN, yet may only be indirectly involved in determining which exomer complex will form at any particular time.
Based on our data we propose the following model of exomer tetramer formation at the TGN (Fig 9B). Arf1 is activated and together with Chs5 initiate assembly (i). To stabilize this initial complex, ChAPs must be recruited. Our data indicate that tetrameric exomer assembles through an intermediate (Chs5:ChAP 2:1 complex) (ii). Here, the relative ChAP to Chs5 expression levels and ChAP affinities for Chs5 play a significant role. The ChAPs with higher affinity (Chs6, Bud7, and Bch2), but lower expression, favor formation of intermediates, to which Bch1 would be recruited through cooperative binding. Since Bch1 is more abundant than the other ChAPs and has the best ability to stabilize Chs5 at the TGN, exomer tetramers will be formed efficiently (iii). The ChAPs with higher affinity for Chs5 may also be the preferred cargo receptors. Thus, this ensemble of cargo, exomer, and Arf1 could serve as a priming complex in vesicle formation (iv) 20, 21. Although exomer cargoes identified so far are not very abundant proteins, the primer complexes could be stabilized and coat polymerization would be driven in part through the oligomerization of cargo. At least Pin2 and Chs3 can form higher order oligomers 11, 29. Consistently, overexpression of cargo boosts formation of exomer complexes on the TGN in vivo. Moreover, increasing amounts of the exomer complexes augments the efficiency of vesicular budding in vitro 10.
Why would exomer assemble on the TGN in this unique fashion? We suggest that this provides a mechanism for temporally and spatially controlled localization of cargoes and to quickly respond to changes in the cellular environment. The so far known exomer cargoes all are superbly regulated in terms of plasma membrane localization 11, 15, 19, 29, 30. For example Chs3 and Pin2 localization is dependent on exomer, AP1‐dependent retrieval from endosomes, ubiquitination, endocytosis, and palmitoylation. Moreover, the localization of Chs3 and Pin2 is intricately sensitive to cellular stress, under which both proteins are rapidly endocytosed 7, 11, 30. Having only a few cargoes might be an advantage if the localization of these cargoes needs to be well regulated. The low abundance of the exomer subunits and the competitive and cooperative behavior of the ChAPs may ensure that proper amount of an individual cargo is localized at the plasma membrane.
We propose that the dynamic assembly of exomer, the distinct intrinsic properties and individual concentrations of the ChAPs, and the availability and oligomeric state of cargo molecules are key determinants to control the temporally and spatially regulated localization of exomer‐dependent cargoes.
Materials and Methods
Strains and growth conditions
Yeast strains used in this study are listed in Appendix Table S3. Standard yeast media were prepared as described 31. All strains were grown at 30°C. HC medium selective for the plasmid was used to grow strains. Calcofluor plates were based on minimal medium containing additionally 0.1% yeast extract, 1% MES buffer pH 6.0, and indicated concentrations of Calcofluor White (Sigma).
Yeast genetic methods and plasmids
Standard genetic techniques were used throughout 31. Chromosomal tagging and deletions were performed as described 32. PCR‐based chromosomal manipulations were confirmed by colony PCR.
The Sec7‐DsRed plasmid (pTPQ128) was described previously 33. For overexpression of PIN2, the p426GPD PIN2 plasmid 11 was used. The PIN2 ORF was cloned into p416 plasmid using BamHI/EcoRI restriction sites. The endogenous PIN2 promoter was amplified (500 bp upstream of the PIN2 ATG) from genomic DNA and cloned into p416PIN2 using SpeI/BamHI restriction sites. For overexpression of CHS6, CHS6 was cloned into the p426GPD plasmid using BamHI/XhoI restriction sites. Cloning was verified by sequencing.
Subcellular fractionation
Fifteen OD600 units of mid‐log cells were incubated in 2 ml DTT buffer (100 mM Tris pH 9.4, 10 mM DTT) for 5 min at 30°C, spun down at 1,000 g for 2 min, and resuspended in 2 ml SP buffer (75% YP medium, 0.7 M sorbitol, 0.5% glucose, 10 mM Tris pH 7.5). A total of 45 μl of zymolyase T20 (10 mg/ml) was added, and the cells were spheroplasted at 30°C for 40 min. Cells were washed once in zymolyase‐free SP buffer, resuspended in the same buffer, and incubated at 30°C for 30 min. Regenerated cells were spun down at 1,000 g for 2 min and lysed in 1 ml 50 mM Tris pH 7.5, 1 mM EDTA, 50 mM NaCl plus protease inhibitors by pipetting up and down. The lysate was cleared at 500 g for 2 min. The supernatants were adjusted with the lysis buffer to the same protein concentration (TCL) using the Bradford assay (Bio‐Rad) and subjected to centrifugation at 13,000 g for 10 min. The supernatant (S13) was carefully taken off with a pipette and subjected to centrifugation at 100,000 g (1 h). Both pellets (P13 and P100) were washed once in lysis buffer and then resuspended in the respective volume of lysis buffer corresponding to the amount of supernatant before the centrifugation step. All steps were carried out at 4°C. Samples were taken from all final fractions and subjected to immunoblot analysis.
Yeast lysates and Western blot detection
Ten OD600 units of cells were harvested (2 min, 1,000 g) and resuspended in 250 μl lysis buffer (50 mM Tris pH 7.5, 1 mM EDTA, 50 mM NaCl, and protease inhibitors), and 130 μl glass beads were added. Fastprep lysis was performed 2× at speed 6.5 for 30 s with 5‐min incubation intervals on ice. The lysate was cleared at 2,500 g for 1 min at 4°C, and the protein concentration was measured using the Bradford assay (Bio‐Rad).
Epitope tags and proteins were detected using the following antibodies: anti‐myc (Sigma 9E10; 1:1,000); anti‐GFP (Torrey Pines Biolabs, Secaucus, NJ; 1:5,000); anti‐Pgk1 (Invitrogen #A‐6457; 1:1,000); anti‐Bch1 serum (1:500); anti‐Chs6 serum (1:500); anti‐Pin2 serum 11 (1:2,000). ECL (GE Healthcare) was used for detection.
Microscopy
Cells were grown to log phase in YPD or selective medium supplemented with adenine, and then harvested, washed, and mounted. Images were acquired with an Axiocam mounted on a Zeiss Axioplan 2 fluorescence microscope, using filters for GFP and DsRed.
For colocalization analysis, cells were grown overnight in selective media, diluted, and grown for 4–6 h to log phase in rich media supplemented with adenine, washed, and mounted in HC‐complete onto 1.6% agarose pad. Data acquisition was performed on Leica SP5‐II‐Matrix confocal microscope equipped with an oil immersion objective (HCX Plan‐Apochromat 63× NA 1.40–0.6 oil, Lbd Blue CS) at 27°C. Z‐stacks were acquired in 576 × 576 pixel format with pixel size in XY of 43 nm and Z‐step size of 131 nm, with pinhole 1 Airy, zoom 10, at speed 1,000 Hz with line averaging 3 and 8‐bits resolution. Used Ar‐laser lines were 488 nm for GFP (detection range 493–550 nm) and 561 nm for DsRed (detection range 570–650 nm). HyD detectors were used for detection in both channels. Z‐stacks were subjected to Huygens deconvolution procedure. The Manders colocalization analysis was performed using JACoP plug‐in for ImageJ after background subtraction 34.
Staining of lipid droplets with LD540 was performed as described in 35 with minor modifications. Briefly, cells were grown to log phase in rich medium supplemented with adenine, washed, and incubated with 0.5 μg/ml LD540 in HC‐complete at 30°C for 10 min. Cells were then washed and mounted in HC‐complete onto 1.6% agarose pad. Data acquisition was performed on Leica SP5‐II‐Matrix confocal microscope equipped with an oil immersion objective (HCX Plan‐Apochromat 63× NA 1.40–0.6 oil, Lbd Blue CS) at room temperature. Images were acquired in 512 × 512 pixel format with pinhole 2.62 Airy, zoom 10, at speed 400 Hz with line averaging 4 and 8‐bits resolution. Laser lines were used in sequential mode; 488 nm for GFP (PMT range: 493–550 nm) and 561 nm for LD540 (PMT range: 570–700 nm).
To stain the chitin, cells in the mid‐log phase were incubated with 15 μg/ml calcofluor for 30 min in YPD, and washed twice in HC‐complete prior to analysis. Images were acquired with an Axiocam mounted on a Zeiss Axioplan 2 fluorescence microscope, using the DAPI filter.
All images were adjusted for brightness and contrast in the same way, if not stated otherwise.
Fluorescence correlation spectroscopy, data processing, and analysis
Cells were grown into log phase in rich media, washed, and mounted in HC‐complete onto 1.6% agarose pads. FCS measurements were performed at room temperature with a Zeiss LSM 710‐FCS confocal microscope using a 63×/1.4NA Oil DIC Plan‐Apochromat objective. While oil immersion objectives are known to introduce optical aberrations when performing FCS in aqueous media—that is, error bounds for determining absolute diffusion coefficients are large—correct ratios of diffusion coefficients can still be derived 36. Samples were illuminated with a 488‐nm laser line; for fluorescence detection, a 500‐ to 550‐nm band pass and a pinhole width of 1 Airy unit were used. For each sample, 10–20 FCS measurements with an acquisition time of 20 s were taken.
The instrument alignment was tested with 50 ng/ml FITC‐dextran (10 kDa, D1820; Invitrogen) calibration dye. The focal volume radius ω0 was estimated from Df = ω0 2/4*τD, where Df is the theoretical diffusion coefficient of the calibration dye and τD is the experimental correlation time of the calibration dye. The theoretical value of the diffusion coefficient Df of the calibration dye in PBS applied according to 37 was 75 μm2/s. The theoretical value of ω0 was calculated as 0.61*γ/NA, where γ is the laser wavelength and NA is the numerical aperture of the objective. The experimental value of the focal volume radius was estimated to be ω0 exp = 187 ± 13 nm, and the theoretical value was ω0 theor = 213 nm.
Fluorescence autocorrelation functions (ACFs), defined as with index “t” denoting temporal averaging, were fitted with XMGRACE (http://plasma-gate.weizmann.ac.il/Grace/) with a standard formula for anomalous diffusion of a single component 38:
| (1) |
or with an expression for unconstrained diffusion of two particle species 25:
| (2) |
Here, C 0 denotes the inverse mean particle number of fluorescent particles in the confocal volume and q = 0.04 reflects the unavoidable focus elongation along the optical axis; triplet fraction and times were considered via f T and t T, respectively. For better comparability of FCS curves, the offset C0 was rescaled to unity in Figs 1 and EV1. An exponent α ≈ 0.9 in Eq. (1) takes care of potential diffusion anomalies due to crowding 38 or optical effects 27 while fast and slow particles (“components”) undergoing normal diffusion enter Eq. (2) as fractions F 1 and 1−F 1, respectively. Diffusion coefficients Df 1 and Df 2 of the fast and slow components were extracted from the diffusion times τ1 and τ2 by using free GFP as a reference (τD ≈ 290 μs (Fig 1C), Df ≈ 25 μm2/s) 26. Theoretical diffusion coefficients were calculated using the Einstein–Stokes relation, Df = k B T/(3πηd); here, k B T is thermal energy, η is the viscosity of the fluid and d is the diameter of the protein. For simplicity, the protein's diameter was related to its molecular weight, m, as d 3 ∼ m; the molecular weight of GFP (25 kDa) and its known Df in cytoplasm 26 served again as a reference.
Fluorescence recovery after photobleaching, data processing, and analysis
The FRAP analysis was performed on the TGN close to the bud neck because this TGN moves less compared to other Golgi elements, allowing fluorescence recovery measurements for 60 s. Strains used for FRAP analysis were always transformed with the Sec7 DsRed plasmid to be able to monitor the TGN localization and turnover over the time of recovery. The TGN marker Sec7 was shown to label the Golgi cisternae for approximately 2 min 39. In case the cisternae moved out of focus, underwent fusion or fission, the respective measurement was discarded.
Cells were grown in selective media overnight, diluted, and grown for 4–6 h to log phase in rich media supplemented with adenine. Cells were washed and mounted in HC‐complete onto 1.6% agarose pad. FRAP measurements were performed using Leica SP5‐II‐Matrix confocal microscope equipped with an oil immersion objective (HCX Plan‐Apochromat 63× NA 1.40–0.6 oil, Lbd Blue CS) at 27°C. Data acquisition was performed in 512 × 512 pixel format with pinhole 2.62 Airy, zoom 20, at speed 1,000 Hz in bidirectional mode and 8‐bits resolution. Bleaching (0.3 s) was performed with a circular spot 1.0 μm in diameter using the 488‐nm Ar line at 100% laser power. Fluorescence recovery was monitored at low laser intensity (5–10%) at 0.26‐s intervals at the beginning of the recovery and at 0.8‐s intervals when reaching the plateau of recovery, in total for 60 s after bleach. Twenty to thirty separate FRAP measurements were performed for each sample. In case of FRAP analysis in Δchs5 background, the recovery was recorded for 15 s after bleach and 10–20 separate FRAP measurements were performed. All FRAP curves were double normalized to whole cell fluorescence loss during acquisition and background.
Normalized FRAP curves obtained in the Δchs5 background were fitted with the pure diffusion model according to 40. FRAP data obtained for protein (un)binding at the TGN were analyzed according to 28; that is, we assumed the protein turnover at the TGN to be an elementary association/dissociation process that is described by a single‐exponential recovery:
| (3) |
Here, gF 0 denotes the final fluorescence with g ≤ 1 considering an immobile fraction (1‐g); the value of f sets the initial value for the recovery in the first post‐bleach image. Since the initial recovery after the bleaching is contaminated by diffusion toward the TGN whereas Eq. (3) assumes a reaction‐limited event, we restricted fitting of our FRAP data to times t > 2 s.
The TGN is a small region as compared to the surrounding cytoplasm; that is, the recovery time directly determines the dissociation rate, k off = 1/τ. Following 28, the association rate was determined as with F TGN and F c being the total fluorescence in the TGN and in the cytoplasm, respectively; ATGN and Ac denote the areas of the respective regions. The total fluorescence and area of both the TGN and cytoplasm were determined with the Golgi segmentation protocol using Icy imaging software (more detailed description of the use of the Icy software is found in the Appendix Supplementary Methods).
Golgi segmentation protocol for Icy imaging software
The Golgi segmentation protocol consists of two independent sections. In the first part, the trans‐Golgi cisternae were detected using the signal from the DsRed channel (Sec7 DsRed‐TGN marker). A k‐means threshold was used to segment the image into three classes: background, cell body, Golgi; after smoothing the data with a Gaussian filter using a sigma value of 4 to avoid oversegmentation due to spot noise. In the second part, the cell body was detected in the GFP channel. The Huang method was used to segment the image, again after smoothing the data with a Gaussian filter of sigma size 4. The mask of the detected Golgi cisternae was copied from the DsRed channel to the GFP channel obtaining the mean intensity and area of the TGN (ITGNmean, ATGN) and whole cell (ITOTAL, ATOTAL). From this the mean intensity and area of the cytoplasm were extracted (ICYTmean, ACYT), and then, the TGN to cytoplasm intensity ratio was calculated as ITGNmean/ICYTmean.
Author contributions
MH and AS conceived the project. MH performed the experiments. MH, MW, and AS analyzed the data. AS and MH wrote the manuscript with input from MW and GM.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 8
Acknowledgements
We thank I. G Macara and C. Roncero for critical reading of the manuscript. We acknowledge the Imaging Core Facility (IMCF) at the Biozentrum and the Facility for Advanced Imaging of the Friedrich Miescher Institute for technical support. This work was supported by an EMBO long‐term fellowship to M.H., the Swiss National Science Foundation (31003_141207 to A.S.), and the University of Basel.
EMBO Reports (2016) 17: 202–219
References
- 1. De Matteis MA, Luini A (2008) Exiting the Golgi complex. Nat Rev Mol Cell Biol 9: 273–284 [DOI] [PubMed] [Google Scholar]
- 2. Paczkowski JE, Richardson BC, Fromme JC (2015) Cargo adaptors: structures illuminate mechanisms regulating vesicle biogenesis. Trends Cell Biol 25: 408–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kirchhausen T (2000) Three ways to make a vesicle. Nat Rev Mol Cell Biol 1: 187–198 [DOI] [PubMed] [Google Scholar]
- 4. Guo Y, Sirkis DW, Schekman R (2014) Protein Sorting at the trans‐Golgi network. Annu Rev Cell Dev Biol 30: 169–206 [DOI] [PubMed] [Google Scholar]
- 5. Bard F, Malhotra V (2006) The formation of TGN‐to‐plasma‐membrane transport carriers. Annu Rev Cell Dev Biol 22: 439–455 [DOI] [PubMed] [Google Scholar]
- 6. Kienzle C, von Blume J (2014) Secretory cargo sorting at the trans‐Golgi network. Trends Cell Biol 24: 584–593 [DOI] [PubMed] [Google Scholar]
- 7. Spang A (2015) The Road not Taken: Less Traveled Roads from the TGN to the Plasma Membrane. Membranes 5: 84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trautwein M, Schindler C, Gauss R, Dengjel J, Hartmann E, Spang A (2006) Arf1p, Chs5p and the ChAPs are required for export of specialized cargo from the Golgi. EMBO J 25: 943–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang CW, Hamamoto S, Orci L, Schekman R (2006) Exomer: a coat complex for transport of select membrane proteins from the trans‐Golgi network to the plasma membrane in yeast. J Cell Biol 174: 973–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paczkowski JE, Fromme JC (2014) Structural basis for membrane binding and remodeling by the exomer secretory vesicle cargo adaptor. Dev Cell 30: 610–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ritz AM, Trautwein M, Grassinger F, Spang A (2014) The prion‐like domain in the exomer‐dependent cargo Pin2 serves as a trans‐Golgi retention motif. Cell Rep 7: 249–260 [DOI] [PubMed] [Google Scholar]
- 12. Sanchatjate S, Schekman R (2006) Chs5/6 complex: a multiprotein complex that interacts with and conveys chitin synthase III from the trans‐Golgi network to the cell surface. Mol Biol Cell 17: 4157–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. von Blume J, Duran JM, Forlanelli E, Alleaume AM, Egorov M, Polishchuk R, Molina H, Malhotra V (2009) Actin remodeling by ADF/cofilin is required for cargo sorting at the trans‐Golgi network. J Cell Biol 187: 1055–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wakana Y, van Galen J, Meissner F, Scarpa M, Polishchuk RS, Mann M, Malhotra V (2012) A new class of carriers that transport selective cargo from the trans Golgi network to the cell surface. EMBO J 31: 3976–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zanolari B, Rockenbauch U, Trautwein M, Clay L, Barral Y, Spang A (2011) Transport to the plasma membrane is regulated differently early and late in the cell cycle in Saccharomyces cerevisiae. J Cell Sci 124: 1055–1066 [DOI] [PubMed] [Google Scholar]
- 16. Osmond BC, Specht CA, Robbins PW (1999) Chitin synthase III: synthetic lethal mutants and “stress related” chitin synthesis that bypasses the CSD3/CHS6 localization pathway. Proc Natl Acad Sci USA 96: 11206–11210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ragni E, Piberger H, Neupert C, Garcia‐Cantalejo J, Popolo L, Arroyo J, Aebi M, Strahl S (2011) The genetic interaction network of CCW12, a Saccharomyces cerevisiae gene required for cell wall integrity during budding and formation of mating projections. BMC Genom 12: 107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paczkowski JE, Richardson BC, Strassner AM, Fromme JC (2012) The exomer cargo adaptor structure reveals a novel GTPase‐binding domain. EMBO J 31: 4191–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barfield RM, Fromme JC, Schekman R (2009) The exomer coat complex transports Fus1p to the plasma membrane via a novel plasma membrane sorting signal in yeast. Mol Biol Cell 20: 4985–4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Springer S, Spang A, Schekman R (1999) A primer on vesicle budding. Cell 97: 145–148 [DOI] [PubMed] [Google Scholar]
- 21. Spang A (2002) ARF1 regulatory factors and COPI vesicle formation. Curr Opin Cell Biol 14: 423–427 [DOI] [PubMed] [Google Scholar]
- 22. Pucadyil TJ, Schmid SL (2009) Conserved functions of membrane active GTPases in coated vesicle formation. Science 325: 1217–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richardson BC, Fromme JC (2013) The exomer cargo adaptor features a flexible hinge domain. Structure 21: 486–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ziman M, Chuang JS, Tsung M, Hamamoto S, Schekman R (1998) Chs6p‐dependent anterograde transport of Chs3p from the chitosome to the plasma membrane in Saccharomyces cerevisiae . Mol Biol Cell 9: 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rigler R (2001) Fluorescence Correlation Spectroscopy Theory and Applications. Berlin: Springer Verlag; [Google Scholar]
- 26. Elsner M, Hashimoto H, Simpson JC, Cassel D, Nilsson T, Weiss M (2003) Spatiotemporal dynamics of the COPI vesicle machinery. EMBO Rep 4: 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hess ST, Webb WW (2002) Focal volume optics and experimental artifacts in confocal fluorescence correlation spectroscopy. Biophys J 83: 2300–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoffmann J, Fickentscher R, Weiss M (2015) Influence of organelle geometry on the apparent binding kinetics of peripheral membrane proteins. Phys Rev E Stat Nonlin Soft Matter Phys 91: 022721 [DOI] [PubMed] [Google Scholar]
- 29. Sacristan C, Manzano‐Lopez J, Reyes A, Spang A, Muniz M, Roncero C (2013) Oligomerization of the chitin synthase Chs3 is monitored at the Golgi and affects its endocytic recycling. Mol Microbiol 90: 252–266 [DOI] [PubMed] [Google Scholar]
- 30. Valdivia RH, Schekman R (2003) The yeasts Rho1p and Pkc1p regulate the transport of chitin synthase III (Chs3p) from internal stores to the plasma membrane. Proc Natl Acad Sci USA 100: 10287–10292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sherman F (1991) Getting started with yeast. Methods Enzymol 194: 3–21 [DOI] [PubMed] [Google Scholar]
- 32. Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E (1999) Epitope tagging of yeast genes using a PCR‐based strategy: more tags and improved practical routines. Yeast 15: 963–972 [DOI] [PubMed] [Google Scholar]
- 33. Proszynski TJ, Klemm RW, Gravert M, Hsu PP, Gloor Y, Wagner J, Kozak K, Grabner H, Walzer K, Bagnat M et al (2005) A genome‐wide visual screen reveals a role for sphingolipids and ergosterol in cell surface delivery in yeast. Proc Natl Acad Sci USA 102: 17981–17986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bolte S, Cordelieres FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232 [DOI] [PubMed] [Google Scholar]
- 35. Spandl J, White DJ, Peychl J, Thiele C (2009) Live cell multicolor imaging of lipid droplets with a new dye, LD540. Traffic 10: 1579–1584 [DOI] [PubMed] [Google Scholar]
- 36. Enderlein J, Gregor I, Patra D, Fitter J (2004) Art and artefacts of fluorescence correlation spectroscopy. Curr Pharm Biotechnol 5: 155–161 [DOI] [PubMed] [Google Scholar]
- 37. Dauty E, Verkman AS (2004) Molecular crowding reduces to a similar extent the diffusion of small solutes and macromolecules: measurement by fluorescence correlation spectroscopy. J Mol Recognit 17: 441–447 [DOI] [PubMed] [Google Scholar]
- 38. Weiss M (2014) Crowding, diffusion, and biochemical reactions. Int Rev Cell Mol Biol 307: 383–417 [DOI] [PubMed] [Google Scholar]
- 39. Losev E, Reinke CA, Jellen J, Strongin DE, Bevis BJ, Glick BS (2006) Golgi maturation visualized in living yeast. Nature 441: 1002–1006 [DOI] [PubMed] [Google Scholar]
- 40. Huranova M, Ivani I, Benda A, Poser I, Brody Y, Hof M, Shav‐Tal Y, Neugebauer KM, Stanek D (2010) The differential interaction of snRNPs with pre‐mRNA reveals splicing kinetics in living cells. J Cell Biol 191: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 8