

Abstract
Metal ion‐containing macromolecules have fundamental roles in essentially all biological processes throughout the evolutionary tree. For example, iron‐containing heme is a cofactor in enzyme catalysis and electron transfer and an essential hemoglobin constituent. To meet the intense demand for hemoglobin assembly in red blood cells, the cell type‐specific factor GATA‐1 activates transcription of Alas2, encoding the rate‐limiting enzyme in heme biosynthesis, 5‐aminolevulinic acid synthase‐2 (ALAS‐2). Using genetic editing to unravel mechanisms governing heme biosynthesis, we discovered a GATA factor‐ and heme‐dependent circuit that establishes the erythroid cell transcriptome. CRISPR/Cas9‐mediated ablation of two Alas2 intronic cis elements strongly reduces GATA‐1‐induced Alas2 transcription, heme biosynthesis, and surprisingly, GATA‐1 regulation of other vital constituents of the erythroid cell transcriptome. Bypassing ALAS‐2 function in Alas2 cis element‐mutant cells by providing its catalytic product 5‐aminolevulinic acid rescues heme biosynthesis and the GATA‐1‐dependent genetic network. Heme amplifies GATA‐1 function by downregulating the heme‐sensing transcriptional repressor Bach1 and via a Bach1‐insensitive mechanism. Through this dual mechanism, heme and a master regulator collaborate to orchestrate a cell type‐specific transcriptional program that promotes cellular differentiation.
Keywords: Bach1, GATA factor, heme, network, transcriptome
Subject Categories: Development & Differentiation, Transcription
Introduction
Metal ions regulate organismal development and homeostasis by controlling enzymes, sequence‐specific DNA‐binding proteins, and a plethora of cellular processes. While the role of metal ions in biochemical, physiological, and pathological regulation has received considerable attention, fundamental mechanistic and systems‐level questions remain unanswered. For example, how do changes in metal ion availability impact transcriptomes and epigenomes that govern the establishment/maintenance of cell type‐specific phenotypes and phenotypic plasticity?
Multiple transcriptional and posttranscriptional gene expression mechanisms are metal ion dependent. Through conformational transitions in the zinc‐ and heavy metal‐binding metal‐regulatory transcription factor 1 (MTF1), metal ions activate DNA binding 1. Iron‐sulfur clusters within certain transcription factors confer regulation by sensing oxidative stress and gases including nitric oxide 2. Iron is a requisite cofactor for dioxygenases/histone demethylases that mediate gene regulation through chromatin modification 3, 4. Though heme iron has been studied extensively, based on its functions as an enzyme cofactor and a hemoglobin component in erythroid cells, heme is an important transcriptional regulator. Heme binding to the transcriptional repressor Bach1 stimulates its degradation via the proteasome 5. In heme‐deficient erythroid cells, Bach1 accumulates, occupies chromatin, and represses target gene transcription. In erythroid cells synthesizing vast amounts of hemoglobin, Bach1 is limiting, which favors globin gene expression and contributes to a balance between globin chain and heme biosynthesis 6.
Posttranscriptionally, EIF2α kinase (HRI) inhibits globin and heme biosynthetic enzyme translation upon heme deficiency. Heme binding to HRI opposes this mechanism 7. In heme deficiency lacking HRI, globin chains accumulate, which elicits cytotoxicity. Iron‐dependent translational control is also achieved through the iron response element (IRE)/iron regulatory protein (IRP) system. IRP binding to IREs on mRNAs encoding ferritin and ALAS‐2 represses translation in low iron 8, thus contributing to a balance between iron levels and heme biosynthesis.
Heme biosynthesis in erythroid cells requires GATA‐1 9, 10, a master regulator of erythropoiesis 11, 12. GATA‐1 activates transcription of genes encoding hemoglobin subunits, heme biosynthetic enzymes, including ALAS‐2, and diverse red cell constituents 13, 14, 15, 16. Although heme downregulates the globin gene repressor Bach1 5, 17, 18, whether heme has a vital role in establishing/maintaining the erythroid transcriptome is unknown.
It is instructive to consider how GATA‐1‐ and heme‐dependent mechanisms might converge to establish the erythroid cell transcriptome and constellation of erythroid cell phenotypes. We hypothesized that Alas2 transcription is regulated by two GATA‐1‐occupied cis elements that we identified based on sequence and chromatin attributes. CRISPR/Cas9‐mediated excision of both Alas2 cis elements severely reduced Alas2 transcription, impairing heme biosynthesis. This system revealed GATA‐1/heme‐regulated genes that constitute an important sector of the erythroid cell transcriptome. While a subset of the GATA‐1/heme‐activated genes were Bach1 sensitive, a distinct cohort was Bach1 insensitive. GATA‐1 strongly upregulated Bach1 transcription, and Bach1 accumulated only in heme‐deficient cells. GATA‐1 induction of globin chains, ALAS‐2/heme biosynthesis, and Bach1, with heme repressing Bach1, constitutes a type I incoherent feed‐forward loop, an essential component of a complex network that establishes/maintains the erythroid cell transcriptome. Our results establish the cis regulatory mechanism governing heme biosynthesis, a complex network in which heme interfaces with a GATA factor to establish/maintain a cell type‐specific transcriptome, and a new molecular mechanism by which heme sculpts a transcriptome.
Results
Exploiting cis regulatory mechanisms to reengineer heme biosynthesis
A GATA‐2‐activated cis element (+ 9.5) within a Gata2 intron consists of an E‐box‐8‐bp spacer‐GATA motif 19, 20, 21. Targeted disruption of the + 9.5 in the mouse revealed its importance for activating Gata2 transcription in hemogenic endothelium and hematopoietic stem/progenitor cells (HSPCs), promoting hematopoietic stem cell (HSC) emergence in the aorta gonad mesonephros (AGM) region of the embryo, establishing the fetal liver HSPC compartment, and conferring vascular integrity 22, 23. A conditional Gata2 knockout using a + 9.5 site‐containing DNA segment driving Cre recombinase yielded similar fetal liver HSPC and vascular phenotypes 24. “+ 9.5‐like” cis elements share + 9.5 sequence/chromatin attributes and mediate GATA‐2‐dependent activation of the associated gene 25.
Alas2 intron 8 contains a + 9.5‐like cis element (Fig 1A), and Alas2 is expressed in erythroid cells containing GATA‐1, but not GATA‐2. Although GATA‐1 occupies + 9.5‐like elements 13, 25, we are unaware of nonredundant GATA‐1 function through such endogenous sites. As millions of GATA motifs reside in genomes 26, 27, 28, GATA motif function is not predictable based on established parameters, including chromatin occupancy. Since GATA‐1 directly activates Alas2 transcription 29, 30, and cis elements mediating GATA‐1‐dependent Alas2 activation were unknown, we tested whether GATA‐1 functions through the Alas2 + 9.5‐like element in erythroid cells, analogous to GATA‐2 function through the + 9.5 in hematopoietic precursor cells. Another GATA binding cis element in ALAS2 intron 1 contains a GATA motif, but lacks a + 9.5‐like composite element, and is associated with sideroblastic anemia 31, 32. ChIP‐seq data revealed GATA‐1 occupancy of intron 1 and 8 cis elements in erythroid cells, which harbor enhancer attributes (DNase hypersensitivity, histone H3 monomethylation at lysine 4, and Pol II occupancy) (Fig 1B).
Figure 1. CRISPR/Cas9‐mediated deletion of two GATA motif‐containing intronic sites in Alas2 .
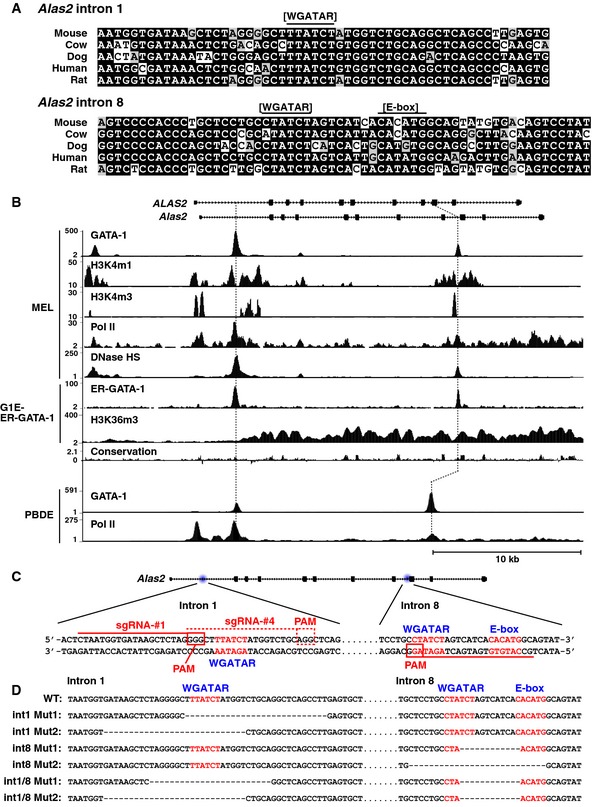
- Sequence alignment of GATA and E‐box motifs in Alas2 intron 1 or 8 demonstrating conservation among mammals.
- DNase hypersensitivity and ChIP‐seq profiles for GATA‐1 or Pol II occupancy and histone modifications at Alas2 and ALAS2 (accession numbers: GSM912907, GSM912895, GSM1003744, GSM1003753, GSM1014191, GSM923572, GSM946526, GSM935465, and GSM935462).
- CRISPR/Cas9 strategy to delete GATA motifs in intron 1 or 8 of Alas2 gene in G1E‐ER‐GATA‐1 cells. PAM: Protospacer adjacent motif.
- DNA sequences at Alas2 introns of wild‐type (WT) and mutant clones.
To rigorously test whether the Alas2 cis elements are functionally important, we used CRISPR/Cas9 to generate erythroid cells lacking one or both cis elements. This analysis was conducted in G1E‐ER‐GATA‐1 cells, normal proerythroblast‐like cells derived from murine GATA‐1‐null embryonic stem cells 33. G1E‐ER‐GATA‐1 cells stably express physiological levels of a conditional GATA‐1 allele (ER‐GATA‐1) encoding an estrogen receptor hormone binding domain fused to GATA‐1 34, 35. Estradiol activation of ER‐GATA‐1 induces rapid and synchronous erythroid maturation and recapitulates a physiological program in which proerythroblasts mature to orthochromatic erythroblasts prior to enucleation 16, 33, 35. Alas2 resides on chromosome X, and G1E‐ER‐GATA‐1 cells harbor one Alas2 allele. One sgRNA targeting intron 8 and two sgRNAs targeting intron 1 were designed and co‐expressed in G1E‐ER‐GATA‐1 cells with a Cas9‐expression vector (Fig 1C). Clonal lines harboring intron 1 (int1 Mut1 and 2 obtained with sgRNA vector #4 and #1, respectively) (Fig 1D) or intron 8 mutations were generated (int8 Mut1 and 2). Double‐mutant lines harboring deletions of intron 1 and intron 8 GATA motifs (int1/8 Mut1 and 2) were derived from int8 Mut1 cells transfected with intron 1‐targeting sgRNA vector #4 and #1, respectively. Mutations were identified by sequencing genomic DNA amplicons (Fig EV1).
Figure EV1. DNA sequences at Alas2 intron 1 or 8 of mutant clonal cell lines.
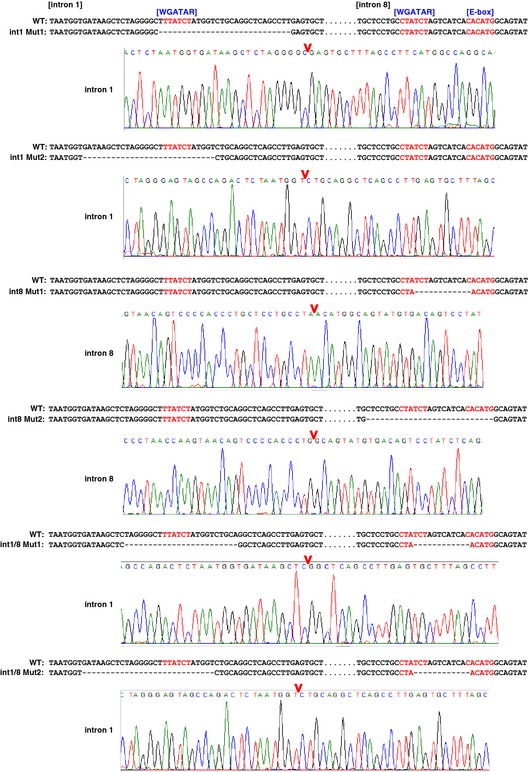
CRISPR/Cas9‐mediated mutations were detected by direct sequencing of the PCR amplicons of genomic DNA.
ER‐GATA‐1 activation in wild‐type and intron 8 mutant cells induced erythroid maturation within 48 h, which involved a considerable reduction in cell size, concomitant with development of pink/red color, reflecting hemoglobinization (Fig 2A). β‐estradiol treatment of double‐mutant cells decreased cell size similar to wild‐type cells, suggesting that the double‐mutant cells were competent to undergo at least certain steps of erythroid maturation (Fig 2B). However, the intron 1 and double‐mutant cells remained pale (Fig 2A). Since β‐estradiol‐treated double‐mutant cells had a slightly different cytoplasmic morphology (Fig 2B), we quantitated cell proliferation in WT and double‐mutant cells. The proliferative rate of untreated and β‐estradiol‐treated double‐mutant and WT cells was indistinguishable (Fig 2C). Despite little or no difference in ER‐GATA‐1 protein levels (Fig 2D), induction of erythroid maturation with intron 1 and intron 1/8 double mutants was not associated with the expected large increase in Alas2 expression (Fig 2E, left). By contrast, the intron 8 mutation alone did not significantly alter ER‐GATA‐1‐mediated induction of Alas2 expression (Fig 2E, left). However, in the context of the double cis element mutant, the intron 8 GATA motif contributed to the very low level Alas2 expression (Fig 2E, right), suggesting a cooperative function of the enhancers.
Figure 2. GATA motif deletions abrogate Alas2 expression during erythroid maturation.
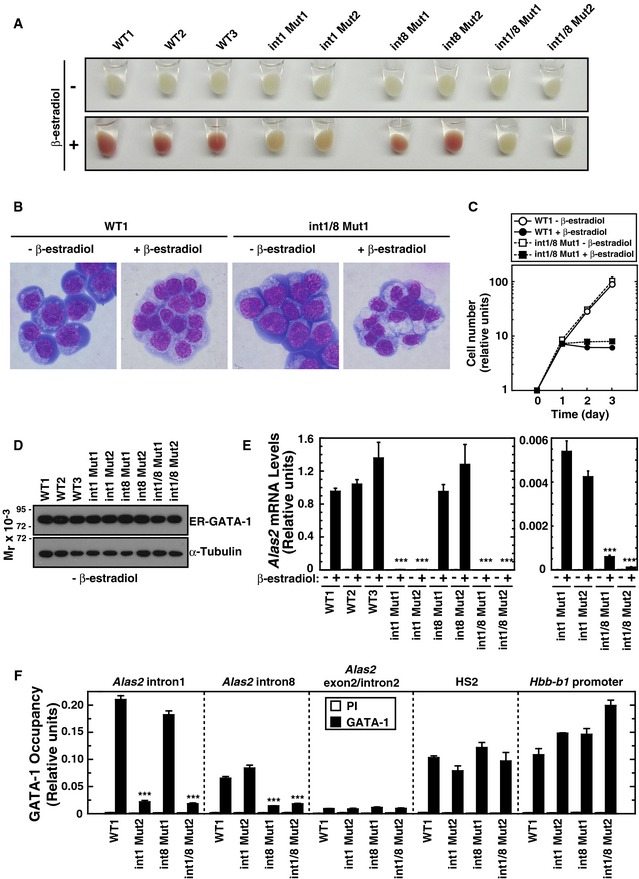
- Representative pellets of WT and mutant G1E‐ER‐GATA‐1 cell clones treated with or without β‐estradiol for 48 h.
- Wright–Giemsa staining of untreated and β‐estradiol‐treated (48 h) WT clone 1 (WT1) as well as double‐mutant clone 1 (int1/8 Mut1).
- Growth curves in WT1 and double‐mutant clone 1 cells treated with or without β‐estradiol (n = 3, mean ± SE).
- Western blotting of ER‐GATA‐1 and α‐tubulin in WT and mutant clones.
- Real‐time RT–PCR of Alas2 mRNA in untreated or β‐estradiol‐treated WT and mutant clones. ***P < 0.001 with respect to all values of WT1‐3 (n = 4, mean ± SE) (left panel). Alas2 mRNA in intron 1 mutant clones was compared to that in intron 1/8 double‐mutant clones. ***P < 0.001 with respect to all values of intron 1 mutants 1 and 2 (right panel).
- Quantitative ChIP analysis of ER‐GATA‐1 occupancy at the GATA sites of Alas2 introns, HS2 of the β‐globin locus control region, and the Hbb‐b1 promoter in WT clone 1 and mutant clones. Alas2 exon 2/intron 2 junction site was used as a negative control. ***P < 0.001 with respect to WT1 (n = 4, mean ± SE).
Although ChIP analyses indicated that GATA‐1 occupied intron 1 and 8 sites, the intronic cis elements mediating GATA‐1 occupancy of the Alas2 locus and induction of Alas2 transcription were unknown. We tested whether the cis element mutations abrogated ER‐GATA‐1 occupancy (Fig 2F). ER‐GATA‐1 occupied both sites in wild‐type cells. The intron 1 GATA motif deletion abrogated ER‐GATA‐1 occupancy at intron 1, but not intron 8. The intron 8 GATA motif deletion abrogated intron 8, but not intron 1, occupancy. The double mutation abrogated ER‐GATA‐1 occupancy at both sites. As both cis elements mediated ER‐GATA‐1 occupancy, but only the intron 1 cis element contributed greatly to ER‐GATA‐1‐dependent transcriptional activation, this analysis decoupled ER‐GATA‐1 occupancy from function.
Heme amplifies GATA‐1‐mediated transcriptional activation
GATA‐1 activation of genes encoding heme biosynthetic enzymes and globin chains 36 illustrates the interconnectedness of mechanisms governing heme biosynthesis and globin expression. Sustaining globin production in a heme‐deficient state and presumably sustaining heme biosynthesis with a dearth of globin chains elicit cytotoxicity 37. Given the vital need to coordinate heme and globin production, the GATA‐1 requirement for hemoglobin biosynthesis, and GATA‐1 regulation of other erythroid cell constituents, we reasoned that heme biosynthesis may be more broadly linked to establishment/maintenance of the erythroid cell transcriptome. Defective heme biosynthesis would therefore alter diverse erythroid phenotypes. One facet of this link involves Bach1 upregulation upon heme deficiency and Bach1‐mediated repression of β‐like globin transcription 17. Whether Bach1 counteracts GATA‐1‐mediated activation and whether this mechanism is restricted to β‐like globin gene regulation or broadly impacts the erythroid cell transcriptome are unknown.
Alas2 intron 1 and intron 1/intron 8 cis element mutations, which severely reduced ER‐GATA‐1‐mediated activation of Alas2 expression (Fig 2E), decreased expression of Hbb‐b1 and Hba‐a1 (Fig 3), encoding major forms of β‐ and α‐globin, respectively. We assessed whether these mutations influenced expression of GATA‐1 target genes encoding other erythroid cell constituents. ER‐GATA‐1‐mediated activation of Slc4a1, encoding an erythroid cell anion transporter, was slightly less in single‐mutant vs. control cells and greatly reduced (9‐fold) in double‐mutant cells (Fig 3). By contrast, intron 1 and the double mutations did not alter ER‐GATA‐1‐mediated repression of Gata2 nor ER‐GATA‐1‐mediated activation of Epb4.9 and Ahsp expression, encoding the erythroid cytoskeletal protein dematin 38 and α‐globin chaperone 39, respectively. As Alas2 cis element mutations influenced a subset of GATA‐1 target genes, heme deficiency compromised only select GATA‐1 functions.
Figure 3. Deregulation of select GATA‐1 target genes in Alas2 cis element double‐mutant cells.
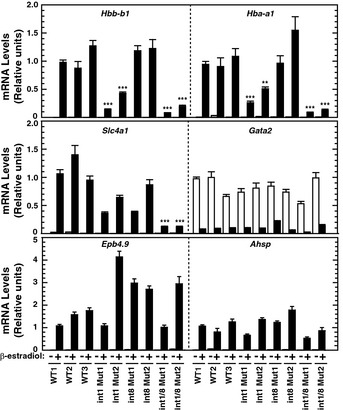
Real‐time RT–PCR analysis of mRNA levels of ER‐GATA‐1 target genes in untreated or β‐estradiol‐treated (48 h) WT and mutant clones. **P < 0.01; ***P < 0.001 with respect to all values of WT1‐3 (n = 4, mean ± SE). P‐values were calculated by a two‐tailed unpaired Student's t‐test using Microsoft Excel.
ALAS‐2 catalyzes the production of the heme precursor 5‐aminolevulinic acid (5‐ALA) 40. As 5‐ALA is cell permeable 41, 5‐ALA treatment of cells can replace the metabolite missing from double‐mutant cells, thus bypassing the heme biosynthetic defect. 5‐ALA induced double‐mutant clone 1 to turn pink/red 48 h post‐β‐estradiol treatment (Fig 4A). To quantitate the impact of the double mutation and 5‐ALA on heme biosynthesis, we analyzed the electronic absorption spectrum of the cytosolic fraction of β‐estradiol‐treated wild‐type or double‐mutant cells treated with or without 5‐ALA. An intense oxyhemoglobin spectrum was observed in cytosolic lysate from β‐estradiol‐treated wild‐type cells, with or without 5‐ALA, and in β‐estradiol‐treated double‐mutant cells with 5‐ALA (Fig 4B and C). Very little oxyhemoglobin was detected in β‐estradiol‐treated double‐mutant cells without 5‐ALA. The oxyhemoglobin and heme concentrations per cell were calculated; oxyhemoglobin and heme levels were 30‐fold lower in β‐estradiol‐treated double‐mutant cells without 5‐ALA than in β‐estradiol‐treated wild‐type cells. 5‐ALA rescued heme biosynthesis in double‐mutant cells upon induction of erythroid maturation; oxyhemoglobin and heme levels were 20‐fold higher in ALA‐ and β‐estradiol‐treated double‐mutant cells than in β‐estradiol‐treated double‐mutant cells. The total heme concentration measured by pyridine hemochromogen assay was comparable to the oxyhemoglobin concentration in all cells, consistent with hemoglobin constituting the majority of erythroid cell heme (Fig 4C, lower graph).
Figure 4. 5‐ALA rescues heme biosynthesis in Alas2 double‐mutant cells.
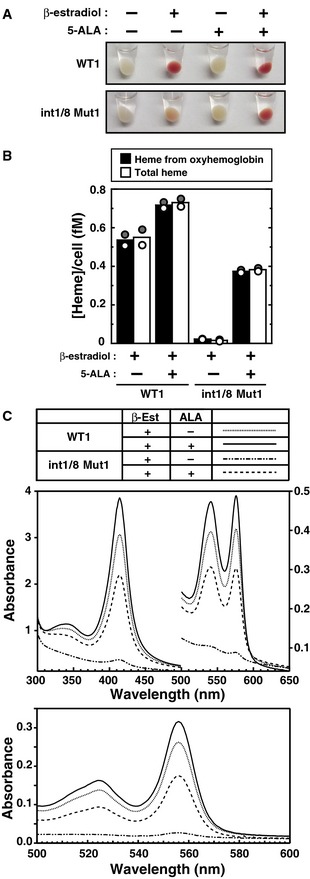
- Representative cell pellets of untreated or β‐estradiol‐treated (48 h) WT clone 1 and double‐mutant clone 1, incubated with or without 5‐ALA.
- Concentration of heme per cell expressed in femtomolar (fM) quantities. The concentration of heme from oxyhemoglobin was quantitated using known extinction coefficients for human hemoglobin monomers. The total heme concentration was quantified using the pyridine hemochrome assay (n = 2; bars show the mean; individual data points are shown; each data point was based on two measurements).
- Visible absorption spectra of cleared cytosolic fraction (upper panel) and the pyridine hemochromogen (lower panel) of β‐estradiol‐treated WT clone 1 or double‐mutant clone 1, incubated with or without 5‐ALA.
Given that 5‐ALA rescued heme biosynthesis in double‐mutant cells, we asked whether 5‐ALA rescued defective gene expression. 5‐ALA treatment of double‐mutant cells restored Hbb‐b1 and Hba‐a1 mRNA levels (Fig 5A). The mRNA levels of other ER‐GATA‐1 target genes and control genes were quantitated, and the fold changes upon 5‐ALA/β‐estradiol‐treated cells relative to β‐estradiol‐treated cells were calculated (Fig 5A and B). Whereas the fold changes of Alas2, Hbb‐b1, and Hba‐a1 mRNAs in response to 5‐ALA treatment of double‐mutant cells were significantly higher than wild‐type cells, the fold changes of Slc4a1, Epb4.9, Ahsp, Gata2, Eif2s1, and Eif3k mRNAs in double‐mutant cells were unaffected. We also quantitated Alas2, Hbb‐b1, and Hba‐a1 primary transcripts, using constitutively expressed Eif2s1 and Eif3k as controls. In double‐mutant cells, β‐estradiol and 5‐ALA did not increase Alas2 primary transcripts as expected (Fig 5C). 5‐ALA significantly increased Hbb‐b1 and Hba‐a1 primary transcripts in double‐mutant vs. wild‐type cells, whereas Eif2s1 and Eif3k primary transcripts were unaffected (Fig 5C and D). These results demonstrate that 5‐ALA and heme confer expression of key constituents of the erythroid cell transcriptome.
Figure 5. 5‐ALA rescues GATA‐1 activity in Alas2 double‐mutant cells.
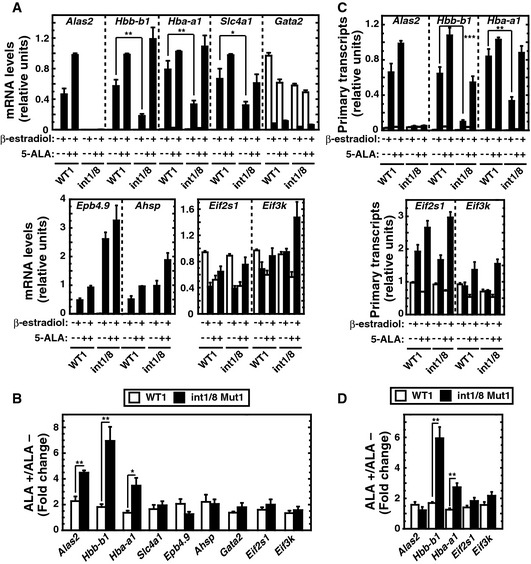
- Real‐time RT–PCR analysis of mRNA levels of ER‐GATA‐1 target genes and two housekeeping genes, Eif2s1 and Eif3k (n = 4, mean ± SE).
- Fold changes of mRNA levels after 5‐ALA treatment relative to no treatment in β‐estradiol‐treated WT clone 1 or double‐mutant clone 1 (n = 4, mean ± SE).
- Real‐time RT–PCR analysis of primary transcripts of ER‐GATA‐1 target genes (n = 4, mean ± SE).
- Fold changes of primary transcripts with 5‐ALA treatment in comparison with no treatment in β‐estradiol‐treated cells (n = 4, mean ± SE).
Dual mechanism of integrating heme and GATA factor functions to establish a cell type‐specific transcriptome
Although heme deficiency deregulated select GATA‐1‐regulated genes, the extent to which heme influences GATA‐1 function was unclear. We used RNA‐seq to compare transcriptomes of untreated WT1, β‐estradiol‐treated WT1, β‐estradiol‐treated double‐mutant, and β‐estradiol/5‐ALA‐treated double‐mutant cells. GATA‐1‐regulated genes were identified by comparing untreated and β‐estradiol‐treated WT1 transcriptomes. Two strategies delineated heme‐regulated genes: (i) Comparing gene expression in β‐estradiol‐treated WT1 cells with β‐estradiol‐treated double‐mutant cells identified Alas2‐enhancer‐regulated genes and (ii) comparing gene expression in β‐estradiol‐treated double‐mutant cells with β‐estradiol/5‐ALA‐treated double‐mutant cells identified 5‐ALA‐regulated genes. Genes with FDR < 0.05 were deemed significant and subjected to subsequent analyses. The GATA‐1 regulatory effect (number of GATA‐1‐regulated genes) was considerably greater than Alas2‐enhancer and 5‐ALA regulatory effects, and the Alas2‐enhancer regulatory effect correlated well with the 5‐ALA regulatory effect (Coefficient of determination (r 2) = 0.57; Fig 6A). Using genes differentially expressed >2‐fold, each regulatory mode was subdivided into activated and repressed genes (Fig 6B). GATA‐1/Alas2‐enhancer/5‐ALA co‐regulated 94 genes (Fig 6C). The dysregulation of these genes in the Alas2 double‐mutant cells, and rescue by 5‐ALA, provides strong evidence that these genes are heme‐regulated. Thus, Alas2‐enhancer/5‐ALA‐activated or ‐repressed genes were deemed heme‐activated and heme‐repressed, respectively, and heme amplifies GATA‐1 activity to regulate these genes.
Figure 6. GATA‐1/heme regulation of the erythroid cell transcriptome.
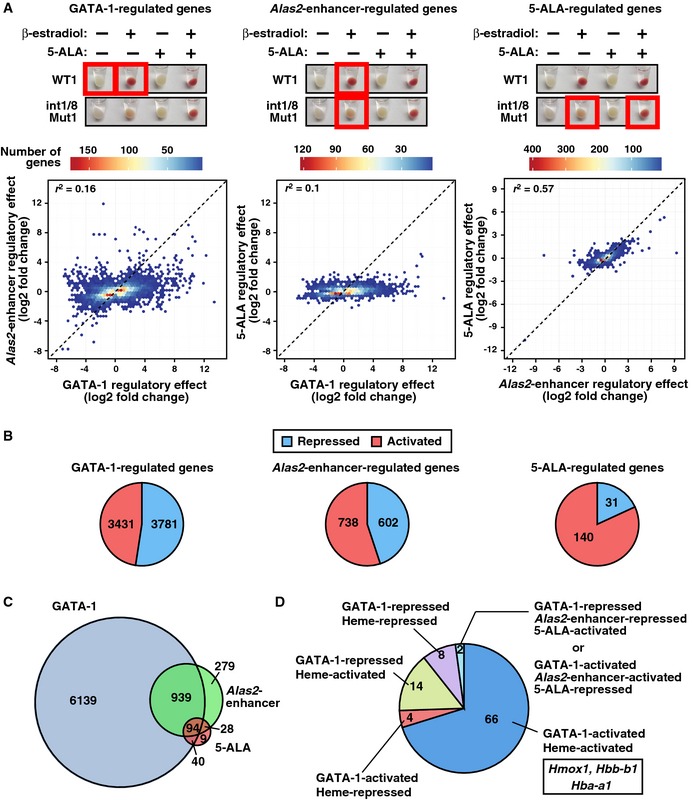
- GATA‐1‐, Alas2‐enhancer‐, and 5‐ALA‐regulated genes were identified by comparing conditions depicted by red boxes. Hexbin plots comparing the regulatory effects (number of regulated genes) between GATA‐1, Alas2‐enhancer, and 5‐ALA. All upregulated genes (FDR < 0.05 and log2 fold change > 0) and downregulated genes (FDR < 0.05 and log2 fold change < 0) were tested. Coefficient of determination (r 2) was shown in each plot.
- Pie charts display the percentage of activated and repressed genes (FDR < 0.05 and expressed differentially > 2‐fold) in GATA‐1‐, Alas2‐enhancer‐, or 5‐ALA‐regulated genes determined by RNA‐seq analysis.
- Venn diagram depicting relationships between GATA‐1‐, Alas2‐enhancer‐, and 5‐ALA‐regulated genes.
- Pie chart depicting the gene numbers in 4 categories for genes regulated by GATA‐1, Alas2‐enhancer, and 5‐ALA.
A large cohort of Alas2‐enhancer‐regulated genes were not rescued by 5‐ALA (Fig 6C). It is instructive to consider the following mechanisms to explain the lack of rescue of these genes. First, 5‐ALA only partially rescues heme levels in double‐mutant cells (Fig 4B), and full restoration of heme might be required to restore the normal gene expression pattern. Second, considering that the Alas2 enhancers function to establish and/or maintain transcription of these genes, loss of the enhancers would abrogate one or both of these mechanisms. It is reasonable to postulate that this would lead to irreversible epigenetic changes that hinder heme‐mediated re‐activation. Third, the enhancer deletions might perturb higher‐order locus positioning in nuclear subdomains, thus indirectly impacting upon transcriptional activity. Regardless of the specific mechanism responsible for the lack of rescue of a cohort of the Alas2 enhancer‐regulated genes, the genomic analysis provided strong evidence that heme amplifies GATA‐1 function at a substantial gene ensemble (94) in erythroid cells.
In the 94 GATA‐1/Alas2‐enhancer/5‐ALA co‐regulated genes, only two genes were Alas2‐enhancer‐repressed but 5‐ALA‐activated, or Alas2‐enhancer‐activated but 5‐ALA‐repressed. The remaining 92 GATA‐1/Alas2‐enhancer/5‐ALA co‐regulated genes were parsed into 4 groups: (i) GATA‐1 activated/heme activated; (ii) GATA‐1 activated/heme repressed; (iii) GATA‐1 repressed/heme activated; (iv) GATA‐1 repressed/heme repressed (Fig 6D). GATA‐1/heme activated Hmox1, Hbb‐b1, and Hba‐a1, consistent with the real‐time RT–PCR data (Figs 5A and 7C). The GATA‐1/heme‐activated genes were subjected to Gene Ontology (GO) analysis. This revealed functional classifications of “hemoglobin complex” (P = 5.0 × 10−9), “hematopoiesis” (P = 0.0073), “purine nucleotide binding” (P = 0.0024), and “microtubule‐based movement” (P = 0.0037) in the GATA‐1/heme‐activated cohort (Table EV1) suggesting a propensity for these genes to be involved in erythroid cell function and differentiation. Whereas Slc48a1 (HRG‐1) and Mfsd7b (FLVCR1) encoding heme transporters were heme‐activated, most heme and iron transporter genes and genes mediating heme biosynthesis were not heme‐regulated (Table EV2). Thus, heme deficiency influenced a subset of genes dictating heme or iron homeostasis.
Figure 7. Bach1‐dependent and Bach1‐independent modes of heme‐dependent GATA‐1 function.
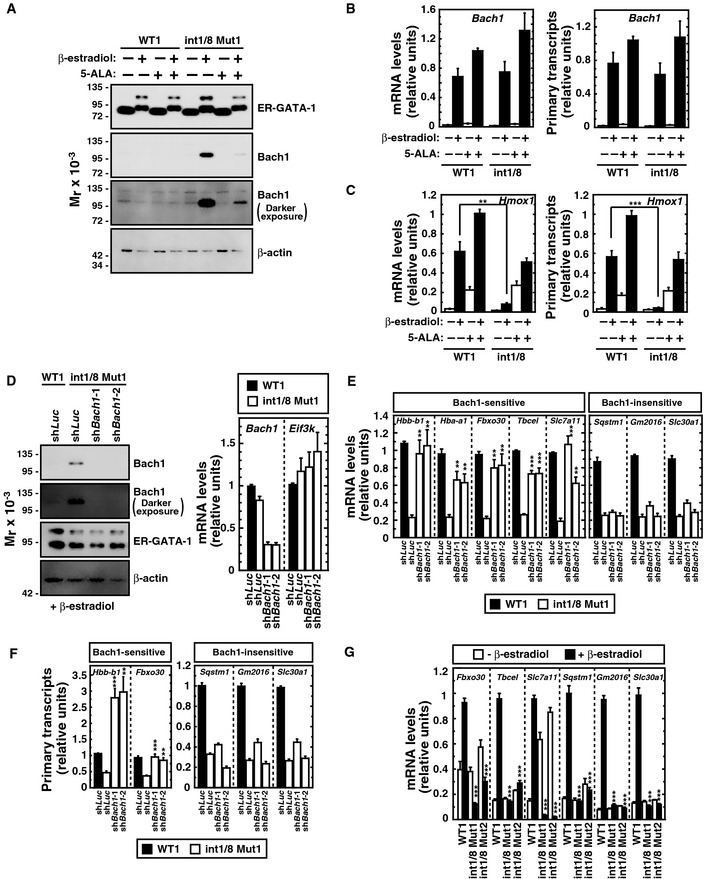
- Western blot of ER‐GATA‐1, Bach1, and β‐actin in untreated or β‐estradiol‐treated (48 h) WT clone 1 and double‐mutant clone 1 incubated with or without 5‐ALA. The upper band of the ER‐GATA‐1 blot represents sumoylated ER‐GATA‐1, which increases as active ER‐GATA‐1 becomes nuclear‐localized. A darker exposure of the Bach1 blot is also shown.
- Real‐time RT–PCR analysis of Bach1 mRNA levels (left panel) and primary transcripts (right panel) (n = 4, mean ± SE).
- Real‐time RT–PCR quantitation of Hmox1 mRNA levels (left panel) and primary transcripts (right panel) (n = 4, mean ± SE).
- Bach1 knockdown in β‐estradiol‐treated double‐mutant clone 1 (n = 4, mean ± SE). Western blot analysis of ER‐GATA‐1, Bach1, and β‐actin. A darker exposure of the Bach1 blot is also shown.
- Real‐time RT–PCR analysis of mRNA levels of GATA‐1/heme‐activated genes in β‐estradiol‐treated WT1 or double‐mutant clone 1 expressing shRNA targeting Bach1. P‐values were calculated with respect to β‐estradiol‐treated control double‐mutant clone 1 (n = 4, mean ± SE).
- Real‐time RT–PCR analysis of primary transcripts of GATA‐1/heme‐activated genes in β‐estradiol‐treated WT1 or Bach1‐knockdown double‐mutant clone 1. P‐values were calculated with respect to β‐estradiol‐treated control double‐mutant clone 1 (n = 4, mean ± SE).
- Real‐time RT–PCR analysis of mRNA levels of GATA‐1/heme‐activated genes in untreated or β‐estradiol‐treated WT1 and double‐mutant clones. P‐values were calculated with respect to β‐estradiol‐treated WT1 (n = 4, mean ± SE).
Although Hbb‐b1 expression decreased in double‐mutant cells, ER‐GATA‐1 occupancy at β‐globin locus control region HS2 and the Hbb‐b1 promoter was unaffected (Fig 2F). Thus, repression did not involve reduced ER‐GATA‐1 occupancy. Prior reports indicated that Bach1 represses Hbb‐b1 and Hba‐a1 expression in a heme‐dependent manner 17, 42. Whereas little to no Bach1 was detected in wild‐type cells, Bach1 was detected in β‐estradiol‐treated double‐mutant cells (Fig 7A). In β‐estradiol/5‐ALA‐treated double‐mutant cells, Bach1 decreased greatly. Bach1 expression was upregulated in β‐estradiol‐treated wild‐type and double‐mutant cells, with or without 5‐ALA (Fig 7B). Thus, GATA‐1 activates Bach1 expression upon erythroid differentiation, and heme controls Bach1 protein levels.
To assess the influence of GATA‐1 and heme on Bach1 function, we quantitated expression of the Bach1‐repressed gene Hmox1. GATA‐1 induced Hmox1 expression in WT1 cells, but not double‐mutant cells (Fig 7C). Heme deficiency, which upregulated Bach1, repressed Hmox1 expression (Fig 7C). 5‐ALA‐mediated rescue of heme biosynthesis, which downregulated Bach1 (Fig 7A), induced Hmox1 expression (Fig 7C). To test whether elevated Bach1 in β‐estradiol‐treated double‐mutant cells suppresses expression of heme‐regulated GATA‐1 target genes identified by the RNA‐seq, cells were infected with retrovirus expressing shRNA targeting luciferase or Bach1 (Fig 7D). In Bach1‐knockdown double‐mutant cells, Fbxo30, Tbcel, and Slc7a11 mRNA (Fig 7E) and primary transcript (Fig 7F) levels increased. Despite being GATA‐1/heme‐activated, surprisingly, Sqstm1, Gm2016, and Slc30a1 mRNA levels were unaffected (Fig 7E). Hbb‐b1 and Hba‐a1 mRNA levels were upregulated 4.4‐ and 2.8‐fold, respectively, by Bach1 knockdown, implicating Bach1 in globin gene repression upon heme deficiency (Fig 7E). Bach1 suppression of GATA‐1‐mediated activation of these genes illustrates a new mode of GATA factor function. Hbb‐b1, Hba‐a1 Fbxo30, Tbcel, and Slc7a11 were deemed Bach1 sensitive, and Sqstm1, Gm2016, and Slc30a1 Bach1 insensitive. As the Bach1 knockdown did not alter Sqstm1, Gm2016, and Slc30a1 primary transcripts, these genes were transcriptionally repressed in a Bach1‐independent manner (Fig 7F). To determine whether Bach1 sensitivity related to the magnitude of the GATA‐1‐dependent transcriptional response, we quantitated expression of Bach1‐sensitive and Bach1‐insensitive GATA‐1 target genes. The magnitude of GATA‐1‐mediated activation of the genes in β‐estradiol‐treated WT1 and double‐mutant cells did not predict Bach1 sensitivity (Fig 7G).
To further evaluate the mechanism of Bach1 sensitivity, we analyzed GATA‐1 and Bach1 occupancy at five GATA‐1/heme‐activated and Bach1‐sensitive genes. ChIP‐seq data from human peripheral blood‐derived erythroblasts (PBDE) 43 and K562 cells 13 revealed that GATA‐1 and Bach1 occupied similar sites at three genes (HBB, HBA1, and SLC7A11), Only Bach1 occupied FBXO30, and only GATA‐1 occupied TBCEL (Fig EV2A). These results indicated that GATA‐1 and Bach1 directly regulate four out of five GATA‐1/heme‐activated and Bach1‐sensitive genes. A genomewide comparison between GATA‐1 and Bach1 occupancy in K562 cells was also conducted (Fig EV2B and Table EV3). GATA‐1 and Bach1 occupied 3,132 and 2,976 genes, respectively, among a total of 57,820 GENCODE‐annotated genes. GATA‐1 and Bach1 co‐occupied 778 genes, which was highly significant (P < 2.2 × 10−16; naïve Fisher's exact test). GATA‐1 and Bach1 co‐occupancy of numerous chromatin sites suggests that Bach1 function at GATA‐1 target genes represents a common mechanism.
Figure EV2. GATA‐1 and Bach1 ChIP‐seq profiles.
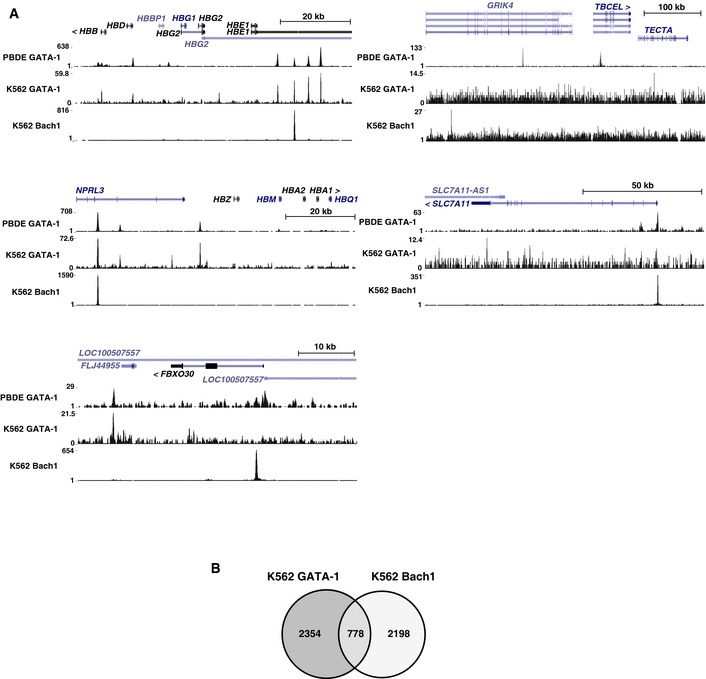
- Venn diagram depicting relationships between GATA‐1 and Bach1 target genes in K562 cells. The definition of a “target” gene was as follows: the gene had a peak in one of the promoter regions (2 kb upstream of transcription start site (TSS) to 200 bp downstream of TSS), a peak in one of the introns, or a peak in the region 10 kb upstream of TSS, which was not in an intron or promoter of another gene. All target genes were listed in Table EV3.
In principle, the distinct regulatory modes may reflect different requirements for chromatin modification at different target genes. Whereas ER‐GATA‐1 induced trimethylated histone H3 at lysine 4 (H3K4me3), a mark associated with active promoters, at the Hbb‐b1 promoter, H3K4me3 was not reduced in β‐estradiol‐treated double‐mutant as compared with WT1 cells (Fig EV3). Although H3K4me3 levels were slightly increased in β‐estradiol‐treated WT1 cells, the levels were quite high at promoters of Bach1‐sensitive (Fbxo30) and Bach1‐insensitive genes (Sqstm1 and Slc30a1) in WT1 and double‐mutant cells. ER‐GATA‐1 increased trimethylated histone H3 at lysine 36 (H3K36me3), a mark associated with active transcription, at open reading frames of Bach1‐sensitive or Bach1‐insensitive genes in WT1 cells; this mark decreased at Bach1‐sensitive and Bach1‐insensitive loci in double‐mutant cells, with the exception of Sqstm1. These results indicate that GATA‐1 target genes differing in Bach1 sensitivity are not discriminated by mechanisms establishing/maintaining H3K4me3 and H3K36me3. Furthermore, GATA‐1 target gene sensitivities to FOG‐1, TAL1, Lmo2, and Mi2β knockdowns do not predict the unique heme or Bach1 regulation described herein (Fig 8A), highlighting this new mode of GATA factor regulation.
Figure EV3. Quantitative ChIP analysis of H3K4me3 and H3K36me3 levels at promoters and open reading frames of Bach1‐sensitive and Bach1‐insensitive genes.
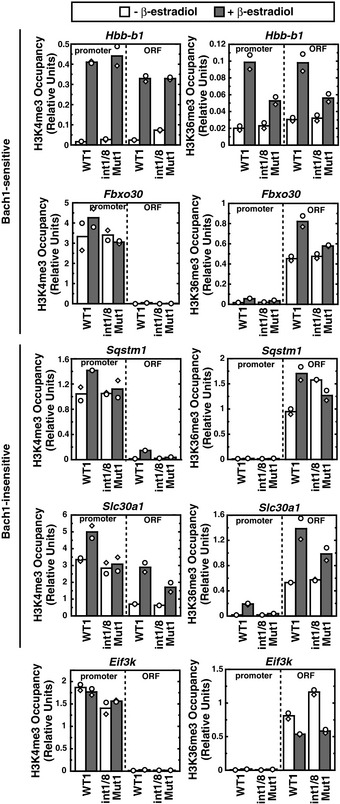
Quantitative ChIP analysis revealed H3K4me3 and H3K36me3 levels in untreated or β‐estradiol‐treated WT1 and double‐mutant clone 1 (n = 2; bars show the mean and individual data points are shown; each data point was based on two measurements).
Figure 8. GATA factor/heme circuit as an essential determinant of a cell type‐specific transcriptome that controls cellular differentiation.
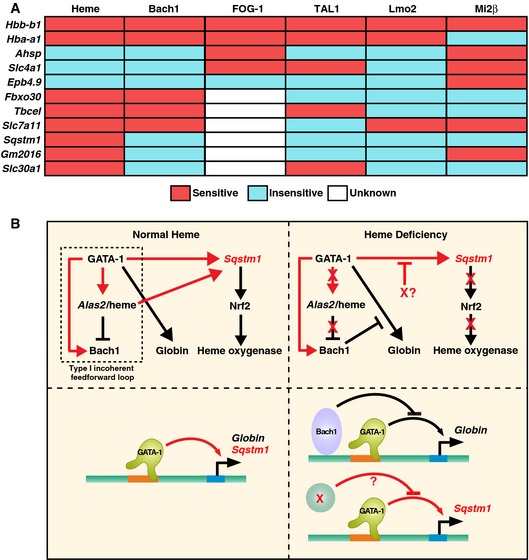
- The relationships between heme, Bach1, FOG‐1, TAL1, Lmo2, and Mi2β regulation of GATA‐1 target genes in G1E‐ER‐GATA‐1 cells. “Sensitive” implies significant deregulation upon heme deficiency, FOG‐1‐binding deficiency, or knockdown of the respective factor. TAL1, Lmo2, and Mi2β knockdowns were reported previously 76, 77. Expression of TAL1, Lmo2, and Mi2β decreased by 70%, 75%, and 50%, respectively, by the knockdown. FOG‐1 sensitivity was determined using G1E‐ER‐GATA‐1 (V205G) cells expressing mutated GATA‐1 defective in FOG‐1 binding 82.
- Cellular and molecular consequences of heme deficiency on GATA‐1 function. Type I incoherent feed‐forward loop 83 under normal heme conditions that controls heme biosynthesis and globin chain production. This loop also activates a p62‐dependent pathway that can induce Nrf2 and confer cytoprotection 70, and promote autophagy, a process vital for erythroid cell maturation 84, 85, 86, 87. In heme deficiency, Bach1‐sensitive and Bach1‐insensitive (X?) mechanisms disrupt multiple mechanistic steps, thus impairing hemoglobin biosynthesis, cytoprotection, and autophagy. As impaired autophagy and associated molecular defects would compromise cell survival and/or maturation, this underscores the critical implications of the heme mechanism to amplify GATA‐1 activity described herein. The red arrows indicate relationships derived from this study. The black arrows indicate relationships that were either known or predicted from existing knowledge.
Discussion
Elucidating cis regulatory mechanisms governing heme biosynthesis yielded the serendipitous discovery that heme is required for the master regulator GATA‐1 to establish/maintain the transcriptome of the erythroblast—the precursor to the red blood cell (Fig 8). When heme biosynthesis is normal, GATA‐1 establishes/maintains a rich target gene ensemble that promotes erythroid precursor survival and maturation 11, 12, 13, 14, 15, 16, and, in other contexts, promotes megakaryopoiesis or the generation and/or function of mast cells and eosinophils 44, 45, 46, 47, 48. We generated heme‐deficient erythroid cells by deleting two GATA‐1‐occupied cis elements that exhibited considerable differences in their activities. The quantitatively greater enhancer activity of the intron 1 element was not predictable from sequence features and chromatin attributes, thus highlighting the importance of analyzing cis element function at endogenous loci.
Using our heme‐deficient cell system with conditional heme rescue, we demonstrate that heme deficiency renders an important sector of GATA‐1 target genes GATA‐1 insensitive. The genes comprising this sector encode erythroid cell constituents vital for homeostasis and red blood cell genesis from progenitor cells. Heme export is required for erythropoiesis and protects erythroblasts from heme‐mediated toxicity 49, 50. As heme permits GATA‐1 to establish the erythroblast transcriptome, heme levels must suffice for GATA‐1 function, as well as its canonical role in hemoglobin assembly, while not being excessive, which is cytotoxic.
GATA factors utilize coregulators to confer activation or repression 27 and are controlled by signaling mechanisms that induce phosphorylation 51, 52, 53, acetylation 54, methylation 55, and sumoylation 56. How biochemical constituents, such as metal ions and metabolic intermediates, impact specific steps in GATA factor function is largely unexplored. In principle, such constituents might influence signaling mechanisms targeting GATA factors or protein interactors that control GATA factor activity. Analogous to the context‐dependent influence of heme on GATA‐1 function, GATA‐1 sumoylation 56 and GATA‐2 phosphorylation 53 influence only a subset of their respective target genes. Heme deficiency did not alter GATA‐1 sumoylation (Fig 7A, top) indicating that the heme activity cannot be explained by enhanced sumoylation. As heme only influenced a cohort of GATA‐1 target genes, it did not regulate GATA‐1 functions essential at all target loci. An intriguing mechanistic insight emerged from the finding that 5‐ALA did not rescue expression of all genes downregulated by heme deficiency (Fig 6). As the establishment and maintenance of GATA‐1 target gene transcription can be differentially regulated 57, a subset of the genes repressed upon heme deficiency might not be reactivated due to differential mechanisms mediating maintenance vs. establishment of transcription. While GATA‐1/heme suffices to maintain expression of certain genes repressed upon heme deficiency, these repressed genes might adopt chromatin attributes that are refractory to GATA‐1/heme‐mediated establishment of an active transcriptional state.
GATA‐1 induction of globin chains, ALAS‐2/heme biosynthesis, and Bach1, with heme repressing Bach1, constitutes a type I incoherent feed‐forward loop 58. This loop is a core of a complex network that establishes/maintains the erythroid cell transcriptome. One component of the mechanism by which heme amplifies GATA‐1 activity involves downregulating the heme‐sensing repressor Bach1. In heme deficiency, Bach1 accumulated and repressed a subset of GATA‐1/heme target genes. Disruption of heme biosynthesis and 5‐ALA rescue in cells with or without GATA‐1 and Bach1 indicates a balance between GATA‐1 Bach1 and dictates transcription at select GATA‐1 target genes.
This relationship between GATA‐1 and Bach1 could not have been predicted from existing knowledge of Bach1 function. Whereas Bach1 occupancy at the HS2 DNase I hypersensitive site of the β‐globin locus control region decreased significantly during the initial 8 h of hemin treatment of MEL cells, Hbb‐b1 expression was not activated 17. Sun et al 17 suggested that this reflects lack of recruitment of the activator p45/NF‐E2 to HS2. P45/NF‐E2 regulates β‐like globin expression in MEL cell systems 59, 60, 61, but p45/NF‐E2 knockout mice do not exhibit defective β‐like globin gene expression 62, 63. In our study, Hbb‐b1 expression was considerably higher in WT cells in comparison with heme‐deficient double‐mutant cells (Figs 3 and 5A and C). Although it is formally possible that NF‐E2/p45 levels/activity are altered in the double‐mutant cells, thus contributing to decreased Hbb‐b1 expression, we demonstrated that Bach1 accumulated in heme‐deficient, double‐mutant G1E‐ER‐GATA‐1 cells. Importantly, Bach1 knockdown in this context almost completely restored Hbb‐b1 expression (Fig 7E). Our results indicate that Bach1 represses Hbb‐b1 expression in a heme‐dependent manner.
MafK is a heterodimeric partner of Bach1 and p45/NF‐E2, which occupies HS2 when Hbb‐b1 expression is both repressed and activated 64. Upon hemin‐induced MEL cell maturation, p45/NF‐E2 replaces Bach1 at HS2, which correlates with Hbb‐b1 activation. After maturation, MafK occupancy was unaffected. As expected, heme downregulated Bach1 chromatin occupancy 17, since heme was known to stimulate Bach1 proteolysis via the ubiquitin–proteasome system 18. While these mechanistic studies implicate Bach1 in repressing β‐like globin gene expression in MEL cells, how GATA‐1 might interface with the respective mechanisms was unclear.
At a cohort of GATA‐1/heme‐regulated genes, heme amplification of GATA‐1 function was Bach1 insensitive. Whereas heme deficiency reduced GATA‐1‐mediated activation of these genes, and Bach1 accumulated, a major reduction in Bach1 did not abrogate the inhibitory effect on GATA‐1 function. These genes included Sqstm1, Gm2016, and Slc30a1. Sqstm1 encodes p62/sequestosome 1, a selective autophagy receptor that binds autophagic cargo, linking it to LC3/GABARAP autophagy machinery 65, 66. By binding ubiquitin, p62/sequestosome 1 promotes protein degradation via the proteasome 67, 68. While GATA‐1 activates genes encoding components of the autophagy machinery, thus instigating autophagy upon erythroid maturation 43, Sqstm1 was not known to be GATA‐1‐regulated.
p62/sequestosome 1 is also a signaling adapter 69 and can activate the nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2) transcription factor 70, a mediator of stress responses 71. GATA‐1/heme induction of p62/sequestosome 1 provides additional mechanistic insights into how GATA‐1 instigates autophagy and links GATA‐1 and Nrf2 mechanisms. Nrf2 induces heme oxygenase‐1 in response to cell stress, and heme oxygenase‐1 catalyzes heme degradation 72. Building upon the type I incoherent feed‐forward loop (Fig 8B), (i) GATA‐1 induces the heme biosynthetic enzyme Alas2; (ii) GATA‐1/heme induces Bach1; (iii) Bach1 represses heme oxygenase‐1; (iv) GATA‐1/heme induces p62/sequestosome; (v) p62/sequestosome induces Nrf2; and (vi) Nrf2 induces heme oxygenase‐1. This iterative cycle has important consequences for autophagy and cytoprotection in erythroid cells and likely for a broad cadre of systems. The other Bach‐1‐insensitive GATA‐1/heme‐activated genes encode a zinc transporter (Slc30a1 73) and a gene of unknown function (GM2016).
Analogous to the heme‐sensitive or heme‐insensitive GATA‐1 target genes and the Bach1‐sensitive or Bach1‐insensitive GATA‐1/heme target genes, the coregulators FOG‐1 74, 75, SetD8 76, Mi2β 75, and LMO2 77, and transcription factors FoxO3 43 and TAL1 77 mediate certain, but not all, GATA‐1 actions. While the FOG‐1‐dependent genes Hbb‐b1 and Hba‐a1 were heme‐activated, other FOG‐1‐dependent genes, Ahsp, Slc4a1, Hebp1, Ptdss2, and Abcb10, as well as FOG‐1‐independent genes, Epb4.9, Klf1, and Tac2, were not heme‐regulated. With the exception of Hbb‐b1 and Hba‐a1, GATA‐1‐K137‐sumoylation‐dependent genes (Ahsp, Slc4a1, Abcb10, Hebp1, Ptdss2, Tac2, Kit, and Lyl1) were not heme‐regulated. The GATA‐1/heme‐activated gene Hbb‐bh1 and GATA‐1‐repressed/heme‐activated genes, Ephx2, Apoc1, P2rx1, and Rab44, were SetD8 sensitive. Mi2β‐sensitive genes, Kit, Clec10a, Rgs19, and Clec4d, were not heme‐regulated. Bach1‐sensitive (Fbxo30, Tbcel, and Slc7a11) and Bach1‐insensitive genes, Sqstm1, Gm2016, and Slc30a1, were SetD8 insensitive. Thus, heme and Bach1 regulation of GATA‐1 function represents new modes of transcriptional control not predicted by established mechanisms (Fig 8A).
In summary, heme amplifies GATA‐1 activity to establish a cell type‐specific transcriptome. As a component of a complex network, a GATA‐1/heme‐dependent incoherent feed‐forward loop ensures the identity and physiological function of the developing red blood cell and constitutes a new paradigm for GATA factor control of cellular differentiation and phenotypes. A dual mechanism involving Bach1‐sensitive and Bach1‐insensitive components underlies the heme activity to sculpt a genetic network that drives cellular differentiation. From systems biological/pathophysiological perspectives, it will be instructive to determine how parameters influence the heme/GATA factor circuit in homeostasis and disease, thereby dictating red blood cell development and function, and perhaps impacting a broader repertoire of GATA factor‐expressing cell types.
Materials and Methods
Cell culture
G1E‐ER‐GATA‐1 cells were cultured in Iscove's modified Dulbecco's medium (GIBCO) containing 15% FBS (Gemini), 1% penicillin/streptomycin (Gemini), 2 U/ml erythropoietin, 120 nM monothioglycerol (Sigma), and 0.6% conditioned medium from a Kit ligand‐producing CHO cell line, and 1 μg/ml puromycin (Gemini). ER‐GATA‐1 activity was induced by addition of 1 μM β‐estradiol (Steraloids) to the media. To rescue heme biosynthesis, 1 mM 5‐aminolevulinic acid hydrochloride (5‐ALA; Sigma) was added to the media.
Generation of genomic deletions in cells using CRISPR/Cas9 system
Guide sequences for gene targeting were designed with online tools (http://crispr.mit.edu/). A U6 promoter‐driven sgRNA expression cassette was amplified by hemi‐nested PCR and cloned into SmaI‐cut pBluescript (Addgene). 10 μg sgRNA‐expressing plasmids were co‐nucleofected into 3 × 106 G1E‐ER‐GATA‐1 cells with 10 μg Cas9‐expressing plasmid 25 using Amaxa Nucleofector Kit R (Lonza). 72 h after transfection, genomic DNA of the cell population was extracted and examined to detect mutations using T7 endonuclease I that cleaves heteroduplexes consisting of wild‐type and mutated DNA sequences. Genomic DNA flanking the target site was amplified by PCR and amplicons were denatured, reannealed, and incubated with T7 endonuclease I (New England BioLabs) for 10 min at 37°C. Clonal cell lines were isolated by dilution in a 48‐well plate. Genomic DNA flanking the mutated site was amplified by PCR and mutations were detected by direct sequencing of the PCR products.
Western blotting
1 × 106 cells were boiled in SDS lysis buffer (50 mM Tris, pH 6.8, 2% β‐mercaptoethanol, 2% SDS, 0.04% bromophenol blue, 10% glycerol) for 15 min. Samples were resolved by SDS–PAGE and detected with Pierce ECL 2 (Life Technologies). Following antibodies were used: rat anti‐GATA‐1 (Santa Cruz Biotechnology; sc‐265), rabbit anti‐Bach1, mouse anti‐β‐actin (Cell Signaling; 8H10D10), and mouse anti‐α‐tubulin (Calbiochem; CP06). Following secondary antibodies were used: goat anti‐mouse‐IgG‐HRP, goat anti‐rat‐IgG‐HRP (Santa Cruz Biotechnology; sc‐2005, sc‐2006), and protein A‐HRP (Sigma; P8651).
RT–qPCR
Total RNA was purified from 2 × 106 cells with TRIzol (Life Technologies). 0.75 μg RNA was treated with DNase I (Life Technologies) for 15 min at room temperature. DNase I was inactivated by addition of EDTA and by heating at 65°C for 10 min. To synthesize cDNA, DNase I‐treated RNA was incubated with 125 ng of a 5:1 mixture of oligo(dT) primers and random hexamer at 68°C for 10 min. RNA/primers were incubated with Moloney MLV reverse transcriptase (Life Technologies), 10 mM DTT, RNAsin (Promega), and 0.5 mM deoxynucleoside triphosphates at 42°C for 1 h, and then heat inactivated at 98°C for 5 min. Real‐time PCRs were conducted with Power SYBR Green Master Mix (Applied Biosystems) using ViiA 7 Real‐Time PCR system (Applied Biosystems).
Quantitative ChIP
1 × 107 cells were cross‐linked in 1% (v/v) formaldehyde for 10 min at room temperature, sonicated, and immunoprecipitated with rabbit anti‐GATA‐1 antiserum, rabbit anti‐H3K4me3 (Active motif; 39159), rabbit anti‐H3K36me3 (Active motif; 61101), or preimmune serum. ChIP samples were quantitated using ViiA 7 Real‐Time PCR system with Power SYBR Green Master Mix.
Heme quantitation
Cells (7–8 × 107 WT or double‐mutant cells) were resuspended in cold hypotonic buffer (8.1 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4, 10 mM NaCl) and lysed using three successive rounds of freezing at −80°C, followed by thawing on ice. Cell lysates were centrifuged at 17,000 g for 30 min at 4°C. The supernatants were centrifuged at 100,000 g for 60 min at 4°C. Electronic absorption spectra of the cleared lysates were recorded on a double‐beam Varian Cary 4 Bio spectrophotometer from 300 to 650 nm. The oxyhemoglobin concentration in the cleared lysates was calculated using extinction coefficients for human oxyhemoglobin monomers 78. The total heme concentration in the cleared lysates was quantified using the pyridine hemochrome assay. Briefly, 200 μl of a stock solution comprised of 0.1 N NaOH and 33% pyridine (v/v) was combined with 100 μl of the cleared lysate to convert all heme into a complex with pyridine. After adding 2–5 mg of sodium dithionite, the absorption spectrum was recorded between 500 and 600 nm. Known extinction coefficients of the reduced pyridine hemochromogen of protoheme were used to calculate the heme concentrations 79.
Retroviral infection
Two distinct shRNAs targeting Bach1 transcripts were designed and cloned into MSCV‐PIG (IRES‐GFP) vector provided by Mitchell Weiss. Transfecting 293T cells with 15 μg of MSCV‐PIG vector and pCL‐Eco packaging vector produced retrovirus expressing shRNA targeting luciferase or Bach1. G1E‐ER‐GATA‐1 cells (2 × 105) were added to 100 μl viral supernatant, polybrene (8 μg/ml), and HEPES buffer, and then spinoculated at 1,200 g for 90 min at 30°C. Three days post‐infection, cells were treated with β‐estradiol, and after 48 h, cells were harvested for RT–qPCR and Western blotting analyses.
Sequences of oligonucleotides for construction of shRNA plasmids
shBach1‐1: TGCTGTTGACAGTGAGCGCGCTTCCAGTTTCTCAAGTTTATAGTGAAGCCACAGATGTATAAACTTGAGAAACTGGAAGCATGCCTACTGCCTCGGA.
shBach1‐2: TGCTGTTGACAGTGAGCGCGCCCGTATGCTTGTGTGATTATAGTGAAGCCACAGATGTATAATCACACAAGCATACGGGCATGCCTACTGCCTCGGA.
RNA sequencing
RNA samples from three biological replicates were used for the analysis. Total RNA was purified in TRIzol (Life Technologies). Samples were prepared using TruSeq RNA Sample Prep Kit (Illumina) and sequenced on an Illumina HiSeq 2000. Transcript quantification and differential expression were conducted using the software packages RSEM 80 and DESeq2 81, respectively. RSEM v1.2.20 was provided with a reference transcript set consisting of all protein coding and long intergenic noncoding transcripts from the Ensembl release 79 annotation of the NCBI Build 38 mouse genome assembly. Default parameters and Bowtie v1.1.1 were used for transcript quantification with RSEM. Gene‐level read counts were then given as input to DESeq2 v1.6.3 for differential expression analysis between pairs of conditions. DESeq2 was run with default parameters except for betaPrior = F, cooksCutoff = F, and alpha = 0.05. Genes with Benjamini–Hochberg FDR values < 0.05 were deemed to be differentially expressed between a given pair of conditions. The accession number for the RNA‐seq data is GEO:GSE74371. Ensembl Gene IDs for various cohorts of differentially expressed genes were given as input to The Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/) for Gene Ontology (GO) analysis. Enriched GO terms were clustered and all significant GO terms (P < 0.05) were listed. The GO term categories, GOTERM_BP_FAT, GOTERM_MF_FAT, and GOTERM_CC_FAT, were used.
Author contributions
NT, EM, JNB, and EHB conceived and designed the experiments. NT and EM performed the experiments. NT, EM, JNB, CND, and EHB analyzed the data. KI and DY contributed reagents/materials/analysis tools. NT, JNB, CND, and EHB wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File
Acknowledgements
This work was supported by NIH grants DK50107 (to E.H.B.) and HG007019 (to E.H.B. and C.N.D.) and Cancer Center Support Grant P30 CA014520. We thank Jin‐Soo Kim and Mitchell Weiss for Cas9 and MSCV vectors, respectively.
EMBO Reports (2016) 17: 249–265
References
- 1. Gunther V, Lindert U, Schaffner W (2012) The taste of heavy metals: gene regulation by MTF‐1. Biochim Biophys Acta 1823: 1416–1425 [DOI] [PubMed] [Google Scholar]
- 2. Fleischhacker AS, Kiley PJ (2011) Iron‐containing transcription factors and their roles as sensors. Curr Opin Chem Biol 15: 335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953 [DOI] [PubMed] [Google Scholar]
- 4. Tsukada Y, Fang J, Erdjument‐Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain‐containing proteins. Nature 439: 811–816 [DOI] [PubMed] [Google Scholar]
- 5. Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, Hayashi N, Yamamoto M, Shibahara S, Fujita H et al (2001) Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J 20: 2835–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Igarashi K, Watanabe‐Matsui M (2014) Wearing red for signaling: the heme‐bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J Exp Med 232: 229–253 [DOI] [PubMed] [Google Scholar]
- 7. Chen JJ (2014) Translational control by heme‐regulated eIF2alpha kinase during erythropoiesis. Curr Opin Hematol 21: 172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of Mammalian iron metabolism. Cell 142: 24–38 [DOI] [PubMed] [Google Scholar]
- 9. Evans T, Felsenfeld G (1989) The erythroid‐specific transcription factor Eryf1: a new finger protein. Cell 58: 877–885 [DOI] [PubMed] [Google Scholar]
- 10. Tsai SF, Martin DI, Zon LI, D'Andrea AD, Wong GG, Orkin SH (1989) Cloning of cDNA for the major DNA‐binding protein of the erythroid lineage through expression in mammalian cells. Nature 339: 446–451 [DOI] [PubMed] [Google Scholar]
- 11. Pevny L, Simon MC, Robertson E, Klein WH, Tsai SF, D'Agati V, Orkin SH, Costantini F (1991) Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA‐1. Nature 349: 257–260 [DOI] [PubMed] [Google Scholar]
- 12. Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH (1996) Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA‐1. Proc Natl Acad Sci USA 93: 12355–12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujiwara T, O'Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, Choi K, Farnham PJ, Bresnick EH (2009) Discovering hematopoietic mechanisms through genome‐wide analysis of GATA factor chromatin occupancy. Mol Cell 36: 667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, Yu D, Hardison R, Weiss MJ, Orkin SH et al (2009) Insights into GATA‐1‐mediated gene activation versus repression via genome‐wide chromatin occupancy analysis. Mol Cell 36: 682–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng Y, Wu W, Kumar SA, Yu D, Deng W, Tripic T, King DC, Chen K‐B, Zhang Y, Drautz D et al (2009) Erythroid GATA1 function revealed by genome‐wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res 19: 2172–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, Weiss MJ (2004) Global regulation of erythroid gene expression by transcription factor GATA‐1. Blood 104: 3136–3147 [DOI] [PubMed] [Google Scholar]
- 17. Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K (2004) Heme regulates the dynamic exchange of Bach1 and NF‐E2‐related factors in the Maf transcription factor network. Proc Natl Acad Sci USA 101: 1461–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zenke‐Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, Igarashi K (2007) Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol 27: 6962–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grass JA, Jing H, Kim S‐I, Martowicz ML, Pal S, Blobel GA, Bresnick EH (2006) Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol Cell Biol 26: 7056–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wozniak RJ, Boyer ME, Grass JA, Lee Y, Bresnick EH (2007) Context‐dependent GATA factor function: combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J Biol Chem 282: 14665–14674 [DOI] [PubMed] [Google Scholar]
- 21. Khandekar M, Brandt W, Zhou Y, Dagenais S, Glover TW, Suzuki N, Shimizu R, Yamamoto M, Lim KC, Engel JD (2007) A Gata2 intronic enhancer confers its pan‐endothelia‐specific regulation. Development 134: 1703–1712 [DOI] [PubMed] [Google Scholar]
- 22. Johnson KD, Hsu AP, Ryu MJ, Wang J, Gao X, Boyer ME, Liu Y, Lee Y, Calvo KR, Keles S et al (2012) Cis‐element mutated in GATA2‐dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest 122: 3692–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao X, Johnson KD, Chang YI, Boyer ME, Dewey CN, Zhang J, Bresnick EH (2013) Gata2 cis‐element is required for hematopoietic stem cell generation in the mammalian embryo. J Exp Med 210: 2833–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lim KC, Hosoya T, Brandt W, Ku CJ, Hosoya‐Ohmura S, Camper SA, Yamamoto M, Engel JD (2012) Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. J Clin Invest 122: 3705–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hewitt KJ, Kim DH, Devadas P, Prathibha R, Zuo C, Sanalkumar R, Johnson KD, Kang YA, Kim JS, Dewey CN et al (2015) Hematopoietic signaling mechanism revealed from a stem/progenitor cell cistrome. Mol Cell 59: 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S (2010) GATA switches as developmental drivers. J Biol Chem 285: 31087–31093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bresnick EH, Katsumura KR, Lee HY, Johnson KD, Perkins AS (2012) Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res 40: 5819–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeVilbiss AW, Sanalkumar R, Johnson KD, Keles S, Bresnick EH (2014) Hematopoietic transcriptional mechanisms: from locus‐specific to genome‐wide vantage points. Exp Hematol 42: 618–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pal S, Cantor AB, Johnson KD, Moran T, Boyer ME, Orkin SH, Bresnick EH (2004) Coregulator‐dependent facilitation of chromatin occupancy by GATA‐1. Proc Natl Acad Sci USA 101: 980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnson KD, Kim S‐I, Bresnick EH (2006) Differential sensitivities of transcription factor target genes underlie cell type‐specific gene expression patterns. Proc Natl Acad Sci USA 103: 15939–15944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Campagna DR, de Bie CI, Schmitz‐Abe K, Sweeney M, Sendamarai AK, Schmidt PJ, Heeney MM, Yntema HG, Kannengiesser C, Grandchamp B et al (2014) X‐linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA‐binding site mutations. Am J Hematol 89: 315–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaneko K, Furuyama K, Fujiwara T, Kobayashi R, Ishida H, Harigae H, Shibahara S (2014) Identification of a novel erythroid‐specific enhancer for the ALAS2 gene and its loss‐of‐function mutation which is associated with congenital sideroblastic anemia. Haematologica 99: 252–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weiss MJ, Yu C, Orkin SH (1997) Erythroid‐cell‐specific properties of transcription factor GATA‐1 revealed by phenotypic rescue of a gene‐targeted cell line. Mol Cell Biol 17: 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grass JA, Boyer ME, Pal S, Wu J, Weiss MJ, Bresnick EH (2003) GATA‐1‐dependent transcriptional repression of GATA‐2 via disruption of positive autoregulation and domain‐wide chromatin remodeling. Proc Natl Acad Sci USA 100: 8811–8816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, Weiss MJ (1999) GATA‐1 and erythropoietin cooperate to promoter erythroid cell survival by regulating bcl‐xl expression. Blood 94: 87–96 [PubMed] [Google Scholar]
- 36. Katsumura KR, DeVilbiss AW, Pope NJ, Johnson KD, Bresnick EH (2013) Transcriptional mechanisms underlying hemoglobin synthesis. Cold Spring Harb Perspect Med 3: a015412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bank A (1968) Hemoglobin synthesis in beta‐thalassemia: the properties of the free alpha‐chains. J Clin Invest 47: 860–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rana AP, Ruff P, Maalouf GJ, Speicher DW, Chishti AH (1993) Cloning of human erythroid dematin reveals another member of the villin family. Proc Natl Acad Sci USA 90: 6651–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu X, Kong Y, Dore LC, Abdulmalik O, Katein AM, Zhou S, Choi JK, Gell D, Mackay JP, Gow AJ et al (2007) An erythroid chaperone that facilitates folding of alpha‐globin subunits for hemoglobin synthesis. J Clin Invest 117: 1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dailey HA, Meissner PN (2013) Erythroid heme biosynthesis and its disorders. Cold Spring Harb Perspect Med 3: a011676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bloomer JR, Brenner DA, Mahoney MJ (1977) Study of factors causing excess protoporphyrin accumulation in cultured skin fibroblasts from patients with protoporphyria. J Clin Invest 60: 1354–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tahara T, Sun J, Igarashi K, Taketani S (2004) Heme‐dependent up‐regulation of the alpha‐globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun 324: 77–85 [DOI] [PubMed] [Google Scholar]
- 43. Kang YA, Sanalkumar R, O'Geen H, Linnemann AK, Chang CJ, Bouhassira EE, Farnham PJ, Keles S, Bresnick EH (2012) Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol 32: 226–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH (1997) A lineage‐selective knockout establishes the critical role of transcription factor GATA‐1 in mekagaryocyte growth and platelet development. EMBO J 16: 3965–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Migliaccio AR, Rana RA, Sanchez M, Lorenzini R, Centurione L, Bianchi L, Vannucchi AM, Migliaccio G, Orkin SH (2003) GATA‐1 as a regulator of mast cell differentiation revealed by the phenotype of the GATA‐1low mouse mutant. J Exp Med 197: 281–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cantor AB, Iwasaki H, Arinobu Y, Moran TB, Shigematsu H, Sullivan MR, Akashi K, Orkin SH (2008) Antagonism of FOG‐1 and GATA factors in fate choice for the mast cell lineage. J Exp Med 205: 611–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu C, Cantor AB, Yang H, Browne C, Wells RA, Fujiwara Y, Orkin SH (2002) Targeted deletion of a high‐affinity GATA‐binding site in the GATA‐1 promoter leads to selective loss of the eosinophil lineage in vivo. J Exp Med 195: 1387–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ohneda K, Moriguchi T, Ohmori S, Ishijima Y, Satoh H, Philipsen S, Yamamoto M (2014) Transcription factor GATA1 is dispensable for mast cell differentiation in adult mice. Mol Cell Biol 34: 1812–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J et al (2008) A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 319: 825–828 [DOI] [PubMed] [Google Scholar]
- 50. Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL (2004) Identification of a human heme exporter that is essential for erythropoiesis. Cell 118: 757–766 [DOI] [PubMed] [Google Scholar]
- 51. Crossley M, Orkin SH (1994) Phosphorylation of the erythroid transcription factor GATA‐1. J Biol Chem 269: 16589–16596 [PubMed] [Google Scholar]
- 52. Zhao W, Kitidis C, Fleming MD, Lodish HF, Ghaffari S (2006) Erythropoietin stimulates phosphorylation and activation of GATA‐1 via the PI3‐kinase/AKT signaling pathway. Blood 107: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Katsumura KR, Yang C, Boyer ME, Li L, Bresnick EH (2014) Molecular basis of crosstalk between oncogenic Ras and the master regulator of hematopoiesis GATA‐2. EMBO Rep 15: 938–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lamonica JM, Vakoc CR, Blobel GA (2006) Acetylation of GATA‐1 is required for chromatin occupancy. Blood 108: 3736–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hosokawa H, Kato M, Tohyama H, Tamaki Y, Endo Y, Kimura MY, Tumes DJ, Motohashi S, Matsumoto M, Nakayama KI et al (2015) Methylation of Gata3 protein at Arg‐261 regulates transactivation of the Il5 gene in T helper 2 cells. J Biol Chem 290: 13095–13103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee H‐Y, Johnson KD, Fujiwara T, Boyer ME, Kim S‐I, Bresnick EH (2009) Controlling hematopoiesis through sumoylation‐dependent regulation of a GATA factor. Mol Cell 36: 984–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee HY, Johnson KD, Boyer ME, Bresnick EH (2011) Relocalizing genetic loci into specific subnuclear neighborhoods. J Biol Chem 286: 18834–18844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Milo R, Shen‐Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U (2002) Network motifs: simple building blocks of complex networks. Science 298: 824–827 [DOI] [PubMed] [Google Scholar]
- 59. Lu SJ, Rowan S, Bani MR, Ben‐David Y (1994) Retroviral integration within the Fli‐2 locus results in inactivation of the erythroid transcription factor NF‐E2 in Friend erythroleukemias: evidence that NF‐E2 is essential for globin expression. Proc Natl Acad Sci USA 91: 8398–8402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kotkow KJ, Orkin SH (1995) Dependence of globin gene expression in mouse erythroleukemia cells on the NF‐E2 heterodimer. Mol Cell Biol 15: 4640–4647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mosser EA, Kasanov JD, Forsberg EC, Kay BK, Ney PA, Bresnick EH (1998) Physical and functional interactions between the transactivation domain of the hematopoietic transcription factor NF‐E2 and WW domains. Biochemistry 37: 13686–13695 [DOI] [PubMed] [Google Scholar]
- 62. Shivdasani RA, Orkin SH (1995) Erythropoiesis and globin gene expression in mice lacking the transcription factor NF‐E2. Proc Natl Acad Sci USA 92: 8690–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shivdasani RA, Rosenblatt MF, Zucker‐Franklin D, Jackson CW, Hunt P, Saris CJ, Orkin SH (1995) Transcription factor NF‐E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell 81: 695–704 [DOI] [PubMed] [Google Scholar]
- 64. Brand M, Ranish JA, Kummer NT, Hamilton J, Igarashi K, Francastel C, Chi TH, Crabtree GR, Aebersold R, Groudine M (2003) Dynamic changes in transcription factor complexes during erythroid differentiation revealed by quantitative proteomics. Nat Struct Mol Biol 11: 73–80 [DOI] [PubMed] [Google Scholar]
- 65. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- 67. Vadlamudi RK, Joung I, Strominger JL, Shin J (1996) p62, a phosphotyrosine‐independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin‐binding proteins. J Biol Chem 271: 20235–20237 [DOI] [PubMed] [Google Scholar]
- 68. Kirkin V, McEwan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34: 259–269 [DOI] [PubMed] [Google Scholar]
- 69. Moscat J, Diaz‐Meco MT, Wooten MW (2007) Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci 32: 95–100 [DOI] [PubMed] [Google Scholar]
- 70. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12: 213–223 [DOI] [PubMed] [Google Scholar]
- 71. Motohashi H, Yamamoto M (2004) Nrf2‐Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 10: 549–557 [DOI] [PubMed] [Google Scholar]
- 72. Igarashi K, Sun J (2006) The heme‐Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal 8: 107–118 [DOI] [PubMed] [Google Scholar]
- 73. Palmiter RD, Huang L (2004) Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch 447: 744–751 [DOI] [PubMed] [Google Scholar]
- 74. Crispino JD, Lodish MB, MacKay JP, Orkin SH (1999) Use of altered specificity mutants to probe a specific protein‐protein interaction in differentiation: the GATA‐1:FOG complex. Mol Cell 3: 219–228 [DOI] [PubMed] [Google Scholar]
- 75. Johnson KD, Boyer ME, Kang JA, Wickrema A, Cantor AB, Bresnick EH (2007) Friend of GATA‐1‐independent transcriptional repression: a novel mode of GATA‐1 function. Blood 109: 5230–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. DeVilbiss AW, Boyer ME, Bresnick EH (2013) Establishing a hematopoietic genetic network through locus‐specific integration of chromatin regulators. Proc Natl Acad Sci USA 110: E3398–E3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fujiwara T, Lee HY, Sanalkumar R, Bresnick EH (2010) Building multifunctionality into a complex containing master regulators of hematopoiesis. Proc Natl Acad Sci USA 107: 20429–20434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Waterman MR (1978) Spectral characterization of human hemoglobin and its derivatives. Methods Enzymol 52: 456–463 [DOI] [PubMed] [Google Scholar]
- 79. Winterhalter KH, Ioppolo C, Antonini E (1971) Distribution of heme in systems containing heme‐free and heme‐bound hemoglobin chains. Biochemistry 10: 3790–3795 [DOI] [PubMed] [Google Scholar]
- 80. Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kim S‐I, Bultman SJ, Jing H, Blobel GA, Bresnick EB (2007) Dissecting molecular steps in chromatin domain activation during hematopoietic differentiation. Mol Cell Biol 27: 4551–4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shoval O, Alon U (2010) SnapShot: network motifs. Cell 143: 326.e1 [DOI] [PubMed] [Google Scholar]
- 84. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A et al (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK (2010) Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA 107: 832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li‐Harms X, Milasta S, Lynch J, Wright C, Joshi A, Iyengar R, Neale G, Wang X, Wang YD, Prolla TA et al (2014) Mito‐protective autophagy is impaired in erythroid cells of aged mtDNA‐mutator mice. Blood 125: 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB (2008) Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112: 1493–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File