

Abstract
ABC transporters are polytopic membrane proteins that utilize ATP binding and hydrolysis to facilitate transport across biological membranes. Forty-eight human ABC transporters have been identified in the genome, and the majority of these are linked to heritable disease. Mutations in the ABCC6 (ATP binding cassette transporter C6) ABC transporter are associated with pseudoxanthoma elasticum, a disease of altered elastic properties in multiple tissues. Although ∼200 mutations have been identified in pseudoxanthoma elasticum patients, the underlying structural defects associated with the majority of these are poorly understood. To evaluate the structural consequences of these missense mutations, a combination of biophysical and cell biological approaches were applied to evaluate the local and global folding and assembly of the ABCC6 protein. Structural and bioinformatic analyses suggested that a cluster of mutations, representing roughly 20% of the patient population with identified missense mutations, are located in the interface between the transmembrane domain and the C-terminal nucleotide binding domain. Biochemical and cell biological analyses demonstrate these mutations influence multiple steps in the biosynthetic pathway, minimally altering local domain structure but adversely impacting ABCC6 assembly and trafficking. The differential impacts on local and global protein structure are consistent with hierarchical folding and assembly of ABCC6. Stabilization of specific domain-domain interactions via targeted amino acid substitution in the catalytic site of the C-terminal nucleotide binding domain restored proper protein trafficking and cell surface localization of multiple biosynthetic mutants. This rescue provides a specific mechanism by which chemical chaperones could be developed for the correction of ABCC6 biosynthetic defects.
Keywords: ABC transporter, biosynthesis, calcification, protein stability, protein structure, abcc6, pseudoxanthoma elasticum
Introduction
Pseudoxanthoma elasticum (PXE)2 is a multisystem autosomal recessive disease that results in the dystrophic mineralization of elastic tissues putatively due to the loss of one or more circulatory factors (1, 2). The mineralization and subsequent degradation of elastic fibers impacts multiple organs, including the vasculature, eyes, skin, and gastrointestinal tract (3). Patients suffer from premature arteriosclerosis, reduced peripheral circulation, loss of central vision, loss of skin tone, and bleeding in the digestive tract (4). Recent work suggests that a loss of circulating pyrophosphates, resulting from altered nucleotide secretion and conversion by nucleotide pyrophosphatases in the circulatory system, is associated with ectopic mineralization (5, 6). At present, only symptomatic treatments exist for PXE (4, 7).
PXE is caused by mutations in the ATP-binding cassette transporter C6 (ABCC6), a member of the multidrug resistant group of ABC proteins (1, 8–10). ABC transporters are active transporters that couple the energy of ATP hydrolysis to the directional transport of solutes (11). Although multiple putative substrates have been identified for ABCC6, the PXE-specific solute(s) and the mechanisms of ABCC6 functional regulation have not been fully elucidated (5, 9, 12). ABCC6 is composed of five domains: three transmembrane domains (TMD0/1/2) and two cytosolic nucleotide binding domains (NBD1/2) (13, 14). The core TMDs (TMD1/2) facilitate solute movement across the membrane, whereas the NBDs provide the energy for transport through nucleotide binding and hydrolysis. The NBDs heterodimerize in response to binding two ATP molecules within the conserved domain-domain interface. This dimerization induces conformational changes in the NBDs that propagate through the TMDs (15–17). ATP hydrolysis putatively reverses these conformational changes, thereby promoting transport cycling and multiple rounds of solute efflux. The NBDs are physically coupled to the TMDs through extensions of the TMD helices and intracellular loops, and these interactions are disrupted in multiple ABC transporter disease states (17–20). It is not known how the N-terminal TMD (TMD0) contributes to ABCC6 function or related physiology, although this domain is implicated in protein-protein interactions in other ABCC subfamily members (21, 22).
More than 200 PXE-related ABCC6 mutations have been identified by genetic screening in patients with varying disease severity (4, 23). Although mutations have been identified across all five ABCC6 domains, disease-associated mutations appear to be enriched within the NBD-NBD interface and the TMD-NBD interfaces (24). The disruption of the native domain-domain interactions across these interfaces potentially contributes to alterations in protein biosynthesis and/or function. Studies on CFTR, a closely related member of the C subfamily of human ABC proteins (ABCC7), has shown that disruption of the TMD-NBD interface is, in part, responsible for the biosynthetic defects associated with the most common cystic fibrosis mutation, F508del (20, 25, 26). Disruption of the NBD-NBD interface by the G551D mutation has been shown to adversely impact channel function (27). Orthologous mutations have been identified in multiple human ABC proteins and putatively give rise to similar biosynthetic and functional defects.
The most common clusters of mutations found in the PXE patient population lie within the conserved TMD-NBD interface (24). These mutations putatively disrupt native state structure and potentially impact the folding and assembly of the NBDs and TMDs. In addition, these mutations may also disrupt the energetic coupling between these domains, altering transport function. Several mutational “hotspots” within the TMD-NBD interface emerge from genetic testing of PXE patients. One such hotspot is located in NBD2. This cluster of mutations putatively interfaces with intracellular loops from TMD1 and TMD2 and includes the Arg-1314, Leu-1335, and Arg-1339 residues. Missense mutations at these positions are associated with moderate to severe PXE (28).
To elucidate the molecular defects associated with this cluster of mutations at the TMD-NBD interface in ABCC6, a combination of cell biological and biochemical experiments was utilized to evaluate changes in ABCC6 biosynthesis and structure. Both local domain folding and appropriate domain-domain assembly were required for proper ABCC6 maturation and were disrupted by disease-causing mutations. These biosynthetic defects could be rescued by an intragenic second-site suppressor mutation, and suppression was dependent on the formation of the NBD heterodimer. Together, these data suggest that ABCC6 folding is hierarchical, requiring local domain folding and global domain-domain assembly and that stabilization of these native domain-domain interactions facilitates biosynthetic rescue of multiple PXE-causing mutations. This data provide insight into the biosynthetic pathway of ABCC6 and suggest a single mechanism for the rescue of multiple disease-causing alleles.
Results
Disease-causing Mutations in NBD2 Alter ABCC6 Biosynthesis
The structures of multiple homologous ABC transporters have been solved by x-ray crystallography and provide templates to evaluate structure-function relationships in ABCC6 (17, 29, 30). The structure of Caenorhabditis elegans Pgp was used to model the location of mutations associated with PXE (Fig. 1, A and B). A putative cluster of PXE-causing missense mutations at positions Arg-1314, Leu-1335, and Arg-1339 was identified from the PXE-patient mutation database based on their frequency of occurrence in the patient population, their putative location in the structure of the ABCC6 model, and the frequency at which similar or identical mutations were reported in other ABC-transporters (see ClinVar; www.ncbi.nlm.nih.gov). Together, these mutations represent ∼20% of PXE patients identified with missense alleles. Patients with the R1314W, L1335P, or R1339C mutations showed strong cardiovascular, vision, gastrointestinal, and/or epidermal symptoms. Structural analyses of ABCC6 homology models suggested these three sites were located in, or immediately proximal to, the NBD2-TMD interface.
FIGURE 1.

ABCC6 biosynthesis and trafficking. The NBD2 mutations, R1314W, L1335P, and R1339C, were assessed for their impacts on ABCC6 structure and biosynthesis. A, a schematic representation of an ABCC6 homology model is shown. The core TMDs are colored cyan and magenta, and the NBDs are colored green and yellow. The Arg-1314, Leu-1335, and Arg-1339 side chains are shown as blue spheres. B, a schematic and surface representation of NBD2 and the contacting intracellular loops is shown. Intracellular loop four, from TMD1, is shown in cyan, and ICL5 (intracellular loop 5) from TMD2 is shown in magenta. The surface of NBD2 associated with the Arg-1314, Leu-1335, and Arg-1339 residues is shown in blue. C, a representative Western blot of HEK293 cells expressing the wild type, R1314W, L1335P, and R1339C mutants is shown. D, densitometric analysis of Western blots of steady state ABCC6 is shown. E, representative Western blotting after cell surface biotinylation is shown. F, densitometric analysis of cell surface resident ABCC6 is shown. G, representative confocal immunofluorescence images of HEK293 cells expressing the wild type and mutant ABCC6 proteins are shown. ABCC6 is stained in green, WGA is shown in red, and DAPI is shown in blue. H, a representative Western blot of HEK293 cells expressing ABCC6 in the presence and absence of lactacystin is shown. I, densitometric analysis of Western blots of ABCC6 after lactacystin treatment is shown normalized to the expression of wildtype ABCC6. Western blots and immunofluorescence images are representative of n ≥ 4 independent experiments. The identities of band B and C are indicated in C and H. PARP1 is shown as a loading control in C, E, and F. Data shown are summary or representative of n ≥ 3 independent experiments. Quantified data are mean ± S.D. *, p < 0.01 using ANOVA with Tukey's post hoc test.
To evaluate the impact of these mutations on the biosynthesis of full-length ABCC6, the wild type and mutant proteins were transiently expressed in HEK293 cells and analyzed by Western blotting. Previous studies have shown that ABCC6 is glycosylated, and the post-translational modification state of the protein can be used as an indicator of its movement through the secretory pathway (32). The core-glycosylated protein, which is associated with glycosylation in the endoplasmic reticulum, migrates at ∼155 kDa by gel electrophoresis (band B). The fully glycosylated protein, which is indicative of transit through the Golgi, migrates at ∼170 kDa by gel electrophoresis (band C). Immunoblot analysis of the wild type protein reveals both bands, consistent with steady state expression of the ABCC6 protein in the early and late compartments of the secretory pathway (Fig. 1C). No unglycosylated protein (band A) was detected under these conditions.
In contrast, the NBD2 mutants show reduced expression and a decrease in the production of the fully glycosylated, band C protein (Fig. 1, C and D). The L1335P mutant appears the least perturbed with respect to protein maturation. Both the core glycosylated and fully glycosylated proteins are reduced when compared with wild type. However, the relative quantity of fully glycosylated versus core glycosylated protein indicates a fraction of the expressed protein traffics through the Golgi. The R1314W mutant showed a similar decrease in the total quantity of expressed protein under steady state conditions. The quantity of fully glycosylated protein was reduced when compared with the core glycosylated species. The R1339C showed the most severe reduction in steady state protein levels of both the core and fully glycosylated species.
To further evaluate the biosynthetic effects of these mutants, the subcellular localization of the wild type and three mutants was assessed using cell surface biotinylation (Fig. 1, E and F). Consistent with the changes in steady state glycosylation seen between the wild type and mutant proteins, the cell surface resident ABCC6 was reduced with the introduction of the three NBD2 mutations. Fully glycosylated wild type protein was readily detected at the plasma membrane after modification with NHS-biotin and capture using streptavidin beads. In contrast, little to no mutant ABCC6 protein was detected at the cell surface using this method. The intracellular pools of ABCC6 and controls (PARP1) were seen in the cell lysates but not in the labeled cell surface fractions.
Subcellular localization was visualized using confocal imaging (Fig. 1G). The wild type protein appeared diffuse in the cytosol and at the plasma membrane. The intracellular fluorescence was consistent with protein transiting the biosynthetic and secretory pathways to the cell periphery. Co-staining with wheat germ agglutinin, a marker for the plasma membrane, demonstrated membrane localization of the wild type protein. Each of the mutants showed a reduction in membrane localization when compared with wild type ABCC6. The mutants showed strong intracellular staining, consistent with protein localized in the endoplasmic reticulum, early in the biosynthetic pathway. The apparent changes in total, plasma membrane, and intracellular staining were consistent with the biochemical analysis of ABCC6 trafficking by Western blotting and biotinylation.
To test whether the changes in steady state ABCC6 were the result of intracellular localization and an increase in the rate of protein degradation, cells were treated with lactacystin. The addition of lactacystin to cells expressing the wild type protein caused a reduction in the quantity of the fully glycosylated form of the protein (Fig. 1, H and I). Although the mechanisms linking proteasome inhibition to protein trafficking are not known, the loss of mature ABCC6 after lactacystin treatment is consistent with previous studies demonstrating altered trafficking of multiple classes of transmembrane proteins by proteasome inhibition (33–35). Lactacystin addition had minimal detectable effects on the core-glycosylated form of the wild type protein. Similarly, each of the three NBD2 mutants showed decreased efficiency of maturation in the presence of lactacystin. In contrast, the band B forms of each of the mutants increased when compared with untreated cells. The quantity of band B ABCC6 after lactacystin treatment was similar for the wild type and mutant proteins, suggesting that expression and biosynthesis of the four ABCC6 proteins was similar. These data are consistent with increased proteasome-mediated degradation of the mutant proteins underlying the changes in steady state expression seen by Western blotting.
Impact of R1314W, L1335P, and R1339C on NBD2
Models from other ABC transporters suggest that the biosynthesis of these multidomain proteins is a hierarchical process (25, 26). Mutations in the NBDs may directly affect local domain properties (stability, structure, function) or may disrupt the quaternary, native-state interactions between domains. To evaluate the specific molecular consequences of the NBD2 mutations on domain folding and structure, the wild type and mutant NBD2 proteins were expressed for analysis in isolation from other ABCC6 domains.
A β-galactosidase folding assay was used to evaluate the solubility of the NBD2 proteins transiently expressed in mammalian cells (36). This assay relies on the structural complementation of the α-fragment of β-galactosidase, fused to NBD2, co-expressed with the complementary ω-fragment. Enzymatic activity, produced from the α-fusion and its complementation with the ω-fragment, has previously been shown to report soluble protein, a surrogate for domain folding, and is measured using fluorescent substrates in kinetic experiments. When co-expressed with the ω-fragment, the wild type ABCC6 NBD2-α showed robust enzymatic activity (Fig. 2A). NBD2 expression in the absence of the ω-fragment or in the presence of the ω-fragment and the absence of the α-fusion resulted in β-galactosidase signals similar to those of control cells. The effect of the NBD2 mutations was similarly evaluated in HEK293 cells. In each case the mutant protein showed a decrease in β-galactosidase activity, consistent with a reduction in soluble protein (Fig. 2A). When compared with the wild type protein, the measured β-galactosidase activities were: R1314W, 56% ± 7%; L1335P, 76% ± 6%; R1339C, 59% ± 9%. In contrast, total protein levels in the whole cell lysate showed no significant difference between wild type and mutants as measured by Western blotting (Fig. 2A, inset). These data demonstrate that total steady state expression of the NBD2 proteins was similar and that the changes in β-galactosidase activity likely arose from changes in protein conformation, solubility, aggregation, and/or association with other cellular proteins.
FIGURE 2.

NBD2 folding and structure. The effects of the three NBD2 mutants were evaluated on the isolated NBD2 protein. A, NBD2 folding was assessed using the β-galactosidase structural complementation assay and normalized to the wild type NBD2 signal. Inset, a representative Western blot of total NBD2 protein expression is shown. B, Western blots of the whole cell lysate (WCL) and soluble fractions of NBD2 protein expressed in E. coli are shown. C, quantification of soluble NBD2 protein expression is shown. The open bars indicate the whole cell lysate, and the gray bars indicate the soluble fraction remaining after centrifugation. D, representative Coomassie Blue-stained gels are shown before and after boiling in the presence of nucleotide and reductant. E, chromatographs of the purified NBD2 proteins are shown after separation by analytical gel filtration. F, circular dichroism spectra of the purified NBD2 proteins are shown. G, SYPRO Orange thermal denaturation transitions are shown for the purified NBD2 proteins. Data shown are the summary or representative of n ≥ 3 independent experiments. Quantified data are the mean ± S.D. *, p < 0.01 using ANOVA with Tukey's post hoc test.
As a second, independent measure of the effects of the NBD2 mutants on domain folding and solubility, the proteins were expressed in Escherichia coli and assessed biochemically. The NBD2 proteins were expressed as fusion proteins with an N-terminal His6-Smt3 tag. Previous studies have shown that a similar construct reports on the soluble production of CFTR NBD proteins and can be used to assess the impact of disease-causing mutations on the isolated NBD (26).
When expressed in E. coli and after centrifugal separation, the soluble wild type protein represented a large fraction of the total protein expressed in bacterial cells, as assessed by Western blotting (Fig. 2B, SOL). Western blot analysis of each of the NBD2 mutants showed a modest decrease in NBD2 solubility (Fig. 2C, gray bars). Analysis of the whole cell lysate, which includes both the soluble, folded NBD2 protein and any insoluble material, demonstrated similar levels of expression for the wild type and mutant NBD2 proteins (Fig. 2B, WCL and Fig. 2C, open bars). The uniform total expression seen in the whole cell lysate suggested that the changes in soluble expression resulted from local alterations in the NBD properties and were not the result of decreased expression in the bacterial system. Densitometric analyses of the Western blots indicated the soluble production of each of the mutants in E. coli roughly mimicked the changes in solubility seen in mammalian cells when compared with the wild type protein (R1314W, 69% ± 17%; L1335P, 63% ± 14%; R1339C, 60% ± 13%). The solubility changes seen with the mutant NBD2 in both bacterial and mammalian expression systems are consistent with a local structural alteration in NBD2 and a change in one or more intrinsic properties.
To assess the structural effects of these mutations, the NBD2 proteins were purified from E. coli for in vitro analyses. The NBD2 proteins all expressed robustly in E. coli, and the soluble fraction of each protein was purified using affinity and size exclusion chromatography (Fig. 2D). The NBD2 proteins appeared monomeric after purification with the exception of R1339C, which spontaneously formed disulfide bonds that were resistant to reduction in the absence of heat.
Hydrodynamic properties were assessed using analytical gel filtration chromatography to evaluate the solution properties of each of the purified NBDs. The wild type protein eluted as a single symmetrical peak at ∼12.5 ml (Fig. 2E). The peak showed no significant signs of tailing or asymmetry, and no protein was found in the column void volume. Based on known gel filtration standards, this elution volume was consistent with a monomeric NBD with an apparent molecular mass of ∼25–30 kDa. Similarly, the elution chromatographs of L1335P and R1314W showed no significant differences from wild type in either the column void or outside of the monomer peak. In contrast, the elution chromatographs of R1339C showed two predominant peaks. A peak corresponding to the monomer and consistent with the wild type NBD was observed. In addition, a peak corresponding to a dimer eluted with an apparent molecular mass of ∼50–55 kDa, as determined by gel filtration standards. The monomer and dimer peaks measured by gel filtration in solution were consistent with the disulfide-conjugated dimers seen by SDS-PAGE for the R1339C protein. The R1339C monomer could be isolated from the dimer by gel filtration and, in the presence of reductant, remained monomeric in vitro (Fig. 2, E–G; mR1339C).
The secondary structures of the purified NBD2 proteins were characterized by circular dichroism (CD) spectroscopy. The CD spectrum of the wild type NBD protein was consistent with a mixed α/β-protein and spectra reported from similar NBD proteins (32, 37). The spectra showed an absorbance minimum near 205–208 nm, corresponding to α-structure, with a second minimum and shoulder between 215 and 225 nm, corresponding to both α- and β-structures in the NBD2 protein (Fig. 2F). The purified R1314W and L1335P proteins showed CD spectra similar to the wild type NBD2 protein, consistent with acquisition of a wild type-like native state. The R1339C showed differences from both the wild type and the other mutant NBD2 proteins in the dimeric form. In comparison to the wild type protein, the minimum near 205–208 nm was reduced, whereas the second minimum and shoulder between 215–222 nm was increased. These data suggest that the R1339C mutant had an influence on the secondary structure of the NBD2 protein and correlated with the spontaneous disulfide formation seen in the R1339C mutant. The CD spectrum of the monomeric R1339C was similar to the other NBD2 proteins. It is not clear if these changes in secondary structure led to, or are the result of, spontaneous disulfide formation by the R1339C substitution but suggested domain structural properties are adversely impacted by this substitution.
Finally, to assess the impact of the NBD2 mutations on domain stability, thermal denaturation experiments were performed using SYPRO Orange binding as a fluorescent monitor of protein unfolding (38). The stabilities of each of the purified NBD2 proteins were assessed in the presence of 2 mm ATP. The wild type protein showed a sigmoidal increase in dye fluorescence with a midpoint of 48.5 ± 0.3 °C (Fig. 2G). The mutant proteins showed qualitatively similar sigmoidal unfolding transitions with only minor variations in thermal unfolding parameters. The R1314W mutant unfolded with a transition midpoint at 48.5 ± 0.4 °C. Similarly, the L1335P protein unfolded with a midpoint of 48.2 ± 0.3 °C. The R1339C protein showed a slight reduction in thermal stability with a transition midpoint of 47.4 ± 0.5 °C. The similarity in thermal stability is consistent with the high degree of surface exposure of the NBD2 substitutions and suggests that surface properties, but not core structure or stability, is more strongly perturbed by the three mutations.
NBD Dimerization Rescues ABCC6 Trafficking Defects
The folding and assembly of NBD2 occurs late in the biosynthesis of the ABCC6 transporter. As such, it is possible that stabilization of the native NBD2 interaction with other domains of the protein may serve as a means to rescue ABCC6 NBD2 biosynthetic defects. Previous studies have shown that the NBD-NBD dimer can be stabilized by multiple mutations to the catalytic residues associated with ATP hydrolysis (15, 16). Substitution of the Walker B glutamate to glutamine results in the stabilization of multiple NBD dimers in both bacterial and mammalian systems (Fig. 3A) (15, 39). To test the effects of these catalytic mutations, Asn and Gln were substituted for the canonical Asp and Glu in the NBDs (D778N and E1427Q).
FIGURE 3.

Rescued ABCC6 folding by domain-domain stabilization. The catalytic E1427Q mutation was used to stabilize the ATP-bound NBD dimer and to assess the rescue of ABCC6 folding. A, a schematic showing the putative domain-domain stabilization by the E1427Q mutant is shown. The catalytic mutation, E1427Q, putatively blocks ATP hydrolysis at the NBD2 Walker A/B composite site. The TMDs are shown in red, the NBDs are shown in blue, and the ATP is shown as orange spheres. The ATP molecules are numbered based on their association with the NBD1 or NBD2 Walker A/B sequences. B, representative Western blots of the NBD1 and NBD2 catalytic mutants in the otherwise wild type ABCC6 background are shown. C, representative Western blots of the E1427Q double mutants are shown. D, densitometric analysis of Western blots of steady state ABCC6 is shown. E, representative Western blots of cell surface biotinylated ABCC6 are shown. F, densitometric analysis of Western blots of steady state ABCC6 is shown. G, confocal immunofluorescence images of the E1427Q mutants are shown. ABCC6 is shown in green, WGA is shown in red, and DAPI is shown in blue. Yellow indicates colocalization of the ABCC6 and agglutinin staining. H, representative Western blots of Walker A ATP-binding site mutations are shown with Glu-1427 (E) or E1427Q (Q). Western blots and immunofluorescence images are representative of n ≥ 4 independent experiments. The identities of band B and C are indicated in C. PARP1 is shown as a loading control in B, C, E, and H. Data shown are summary or representative of n ≥ 3 independent experiments. Quantified data are mean ± S.D. *, p < 0.01 using ANOVA with Tukey's post hoc test.
The biosynthesis of the D778N and E1427Q proteins was first assessed by Western blotting after expression in HEK293 cells (Fig. 3B). The introduction of the D778N mutant dramatically reduced total expression of ABCC6. The D778N mutant was excluded from further evaluation due to its negative impact on ABCC6 expression. In contrast, the E1427Q marginally increased total ABCC6 expression and had no negative effect on the apparent levels of band C protein. The E1427Q mutation was then introduced into the NBD2 mutant backgrounds, and maturation was assessed using Western blotting. The inclusion of the E1427Q mutant on the R1314W, L1335P, and R1339C backgrounds dramatically increased the quantity of fully glycosylated protein under steady state conditions (Fig. 3, C and D). In each case, the quantity of band C protein increased 2.5–3-fold.
The subcellular localization of the E1427Q NBD2 mutants was then assessed to evaluate changes in ABCC6 trafficking. The redistribution of ABCC6 within the cell was first assessed biochemically using cell surface biotinylation. The E1427Q double mutants showed dramatically increased amounts of cell surface ABCC6 as compared with the single NBD2 mutations alone (Fig. 3, E and F). These data were consistent with stabilization of the ABCC6 protein and redistribution from intracellular compartments to the plasma membrane. To further confirm the trafficking of the E1427Q-rescued ABCC6 NBD2 mutants from intracellular compartments to the plasma membrane, immunofluorescence was used (Fig. 3G). Cells expressing E1427Q showed ABCC6 plasma membrane localization consistent with the biochemical analyses. Cells expressing the E1427Q-rescued R1314W, R1335P, and R1339C proteins showed a significant increase in the plasma membrane localization, as evaluated by colocalization with agglutinin when compared with the NBD2 mutations in the wild type background (Figs. 1G and 3G). In addition, both the number of cells and total fluorescence associated with the NBD2 mutants increased with the E1427Q suppressor.
To further evaluate the E1427Q mechanism of rescue, PXE-causing mutations in the NBD2 Walker A sequence were introduced into the full-length protein: G1299S and S1307P. The Gly-1299 and Ser-1307 sites are located proximal to the nucleotide in the NBD2 ATP composite site. The G1299S and S1307P mutants both showed decreased steady state protein levels and failed to mature, as assessed by Western blotting (Fig. 3H). The introduction of the E1427Q mutant had no discernible effects on either the G1299S or S1307P mutants. This indicated that the E1427Q-mediated rescue was not universal for all ABCC6 mutants.
NBD Dimerization Underlies the E1427Q-mediated Rescue of ABCC6
To characterize the mechanisms by which the E1427Q mutant rescues the trafficking of the NBD2 mutants, the effect of E1427Q was assessed in the isolated NBD2 protein. If the mechanism underlying NBD2 correction was associated with ligand-induced stabilization of NBD2, the E1427Q mutant would be predicted to enhance the local domain properties. The introduction of the E1427Q mutation into the otherwise wild type NBD2 background resulted in a decrease in NBD2 solubility, measured in the β-galactosidase assay (Fig. 4A). Similarly, the inclusion of the E1427Q mutation in the R1314W, L1335P, and R1339C NBD2 backgrounds decreased NBD solubility. The relative decrease in solubility between the wild type and mutant NBDs was similar in magnitude and suggested that the E1427Q mutant had negative effects in each of these isolated proteins. These data demonstrate that E1427Q did not directly stabilize isolated NBD2.
FIGURE 4.

NBD structure, dimerization, and biosynthetic correction of ABCC6. The role of NBD dimerization in the E1427Q-mediated ABCC6 rescue was assessed. A, soluble NBD2, assessed using the β-galactosidase assay is shown normalized to the wild type NBD2 signal. B, a schematic and surface representation of the ABCC6 NBD heterodimer is shown with the G755R substitution shown in blue. C, soluble NBD1 protein, measured using the β-galactosidase assay is shown normalized to wild type NBD1 signal. D, representative Western blots of the wild type, E1427Q, and G755R mutant ABCC6 proteins are shown with and without the NBD2 mutations. E, densitometric analysis of Western blots is shown for the NBD2, G755R and E1427Q mutants. F, a schematic showing the trans complementation of NBD2 co-expressed with full-length or truncated forms of ABCC6 is shown. G, a representative blot of the biosynthesis of the ΔNBD2 protein is shown. The core and glycosylated forms are indicated with B and C, respectively, with B′ and C′ indicated the core and fully glycosylated species of the truncated ABCC6 protein. H, co-expression of NBD2 with the ΔNBD2 protein is assessed by the β-galactosidase assay. Data shown are summary or representative of n ≥ 3 independent experiments. Quantified data are mean ± S.D. *, p < 0.01 using ANOVA with Tukey's post hoc test.
The observation that the E1427Q mutant had no positive effects on NBD2 folding in isolation but was able to rescue ABCC6 maturation is consistent with the stabilization of the ATP-bound NBD heterodimer. Structural analyses of these NBD homo- and heterodimers indicate that the Walker A and B sequence from one NBD are juxtaposed with the LSGGQ signature sequence of the opposing protomer. Analysis of the ABC transporter mutation database revealed a disease-causing mutation, G755R, which is located within the LSGGQ signature sequence of NBD1 and is identified in multiple ABC transporter-related diseases (40). Structural modeling using ATP-bound NBD dimers as a template for the ABCC6 NBDs demonstrates the G755R mutation would putatively block NBD dimerization by introducing steric and electrostatic clashes with the ATP bound at the NBD2 composite site (Fig. 4B). This modeling further suggests that the G755R mutant would likely not affect protein biosynthesis but would impact the ATP-dependent function of the native protein, consistent with the effects of similar mutations in CFTR (27).
The G755R substitution was introduced into the NBD1 protein in isolation and in the full-length ABCC6 protein to probe its effect on ABCC6 biosynthesis and the potential role of NBD dimerization in NBD2 mutant suppression. Minimal changes in NBD1 solubility were observed using the β-galactosidase assay described above when the G775R mutation was expressed in mammalian cells (Fig. 4C). The G755R mutant protein showed 110% ± 8% β-galactosidase activity when compared with the wild type NBD1 protein. As with NBD2, the ω- and α-fusion controls showed background β-gal activity to be similar to that of the HEK-293 cells alone. Steady state analysis of the G755R mutant in the full-length ABCC6 protein revealed no discernible changes in protein maturation, as assessed by Western blotting (Fig. 4C). Total ABCC6 expression was unchanged by the G755R mutant as was the relative quantities of band B and band C protein. These data are consistent with a putative functional defect associated with the G755R mutant and are consistent with findings of other substitutions within the LSGGQ signature sequence for CFTR.
The G755R mutant was then introduced into the background of the E1427Q wild type and NBD2 mutant proteins and assessed by Western blotting. In the background of the wild type and E1427Q mutant, the G755R mutant had no discernible effect on full-length ABCCC6 maturation (Fig. 4, D and E). Both the wild type and E1427Q protein showed similar glycosylation patterns with and without the G755R mutation. Similarly, the R1314W, L1335P, and R1339C mutants appeared to be largely unaffected by the G775R mutation. However, the G755R mutation blocked the suppressive effects of the E1427Q mutation in the R1314W, L1335P, and R1339C proteins and reduced steady state protein and band C production in the triple mutants (Fig. 4, D and E). The loss of suppression by the G755R/E1427Q combination suggested that the NBD heterodimerization was blocked by the G755R mutant and that NBD dimerization was mechanistically important for E1427Q biosynthetic rescue.
To further probe domain association as a mechanism underlying E1427Q-mediated rescue, a domain-domain interaction assay was developed to probe the interactions of NBD2 with the N-terminal domains of ABCC6 in trans (Fig. 4F). A truncated ABCC6 protein was generated by introducing a stop codon upstream of the NBD2 sequence (ΔNBD2) and for co-expression studies with the NBD2-α fusion protein. Similar truncations in CFTR have been shown to traffic through the secretory pathway and have ATP-independent functional properties at the plasma membrane (26, 41, 42). Stabilization of the free NBD2 reporter by native interactions with the truncated ABCC6 was then assessed using the β-galactosidase assay. The ΔNBD2 protein expressed at low levels in HEK293 cells, as determined by Western blotting and compared with the full-length protein (Fig. 4G). The protein showed two distinct glycosylation species, consistent with both core (band B′) and fully (band C′) glycosylated protein and trafficking of the protein through the secretory pathway. Thus, the steady state expression and apparent stability of ABCC6 are decreased by the ΔNBD2 truncation, but NBD2 is not strictly obligatory for transit through the secretory pathway.
The NBD2-α fusion proteins were then co-expressed with the full-length or ΔNBD2 ABCC6 proteins, and NBD2 levels were evaluated by the β-galactosidase assay. Co-expression of the wild type and E1427Q NBD proteins with the full-length ABCC6 protein showed minimal differences to that seen when NBD2 was expressed alone (Figs. 4, A and H). The E1427Q mutant reduced NBD2 solubility when expressed alone or co-expressed with full-length ABCC6. Co-expression of the NBD2-α fusion with the ΔNBD2 ABCC6, in contrast, showed an increase in soluble wild type NBD2 production as compared with that co-expressed with the full-length protein. Co-expression of the E1427Q NBD2 protein with ΔNBD2 ABCC6 resulted in a further increase in NBD2 solubility when compared with that expressed in the full-length ABCC6 background. Wild type NBD2 solubility increased to 142% ± 5% when co-expressed with the ΔNBD2 ABCC6, as compared with its co-expression with the full-length protein. E1427Q NBD2 increased from 75% ± 8% to 205% ± 15%, as compared with wild type NBD2, when co-expressed with the full-length and ΔNBD2 protein, respectively.
The dramatic increase in E1427Q NBD2 solubility that was seen in the ΔNBD2 co-expression was reversed when the G755R mutant was introduced into the ΔNBD2 background. The G755R mutation had minimal effect on the wild type NBD2 solubility in the ΔNBD2 background (142% ± 5% versus 148% ± 4%). In contrast, the G755R mutant reversed the effect of the E1427Q mutation co-expressed with ΔNBD2 (205% ± 15% to 110% ± 6%). These data are consistent with ΔNBD2 complementation of the NBD2-α fusion in this co-expression system. Furthermore, these data suggest that the E1427Q mutant can act by stabilizing the NBD heterodimer, which is disrupted by the G755R mutation.
Domain Folding and Assembly Steps in ABCC6 Biosynthesis
To further understand how changes in NBD properties are correlated with those of the full-length protein, a direct comparison between domain folding and ABCC6 maturation was performed. Densitometric analyses of the fully glycosylated ABCC6 were plotted against the β-galactosidase activity measured for the isolated NBD proteins, normalized to the wild type proteins (Fig. 5, green circle). When plotted using this approach, the three NBD2 mutants appeared to show a stronger influence on the maturation of the full-length protein than on the domain in isolation (Fig. 5, blue circles).
FIGURE 5.
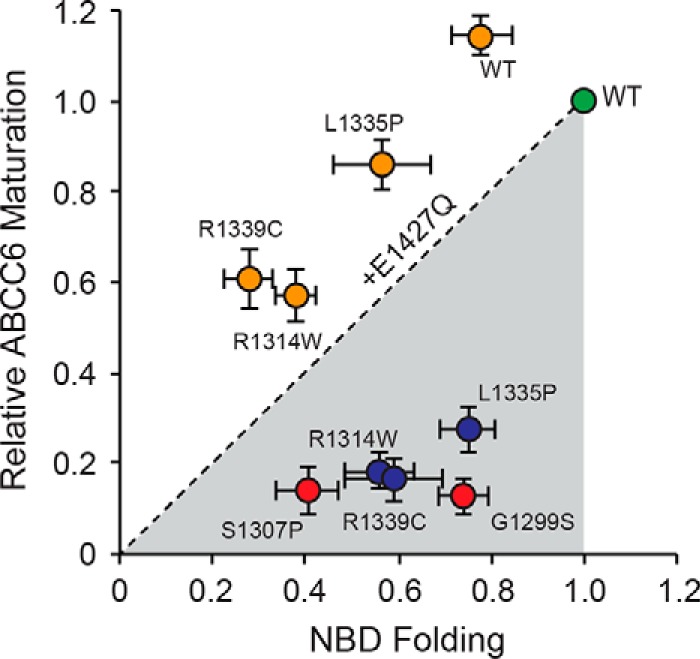
Relationship between NBD and full-length ABCC6 folding. The impact of the NBD2 mutations on the production of full-length and soluble NBD2 proteins is shown. NBD2 folding, measured as a function of soluble NBD2 production is plotted along the x axis. Full-length ABCC6 protein production, measured as the relative quantity of band C, fully glycosylated protein, is plotted on the y axis. Values for both are normalized against the wild type signal, green. The R1314W, L1335P, and R1339C single mutants are shown in red; the E1427Q variant proteins are shown in orange. The dashed line represents the unity relationship between NBD folding and ABCC6 maturation.
A strict correlation between NBD folding and full-length maturation would result in clustering of the data points along the unity axis between the origin and the wild type data point. The NBD2 mutants appeared to diverge from this axis, suggesting an uncoupling of ABCC6 biosynthetic events. These data and structural models suggest that the R1314W, L1335P, and R1339C mutations likely contribute to modest changes in local structural properties of the NBD but significantly alter TMD-NBD interactions in the full-length protein. Introduction of the E1427Q mutation decreased NBD2 solubility in all mutants tested but increased full-length biosynthesis dramatically for the R1314W, L1335P, and R1339C proteins (Fig. 5, orange circles). Each of the E1427Q double mutants shifted from below the unity line to above, despite the decrease in NBD2 solubility. This shift suggests that the mechanisms underlying rescue are likely associated with the stabilization of native domain-domain interactions and not the effect of the E1427Q mutant on the NBD2 alone.
Discussion
The folding of polytopic membrane proteins is inherently complicated, requiring the translation, folding, assembly, and membrane insertion of individual domains. For ABC transporters, as with many membrane proteins, mutations are often associated with the disruption of these processes. Here we show that a cluster of mutations associated with PXE in NBD2 is associated with altered ABCC6 biosynthesis and provide a mechanism by which these altered biosynthetic events can be reversed.
At least two different effects of the tested NBD2 mutations can be seen in these experiments. First, each of the mutants had modest effects on the biosynthesis and folding of NBD2 in the absence of other ABCC6 domains in mammalian and bacterial cells (Figs. 2 and 5). The observation that a decrease in NBD2 solubility occurred in both systems suggested that the intrinsic properties of the NBD itself had changed and these effects were not cell-type specific. Biochemical and biophysical measurements of the purified NBDs suggested that the variant NBD2 protein could adopt structures similar to those of the wild type, although changes in protein yield were observed for each of the variants (Fig. 2). With the exception of R1339C, the NBD2 variants showed similar hydrodynamic, structural, and thermodynamic properties. The R1339C protein was unique in its propensity to form disulfide bonds. However, when the monomeric R1339C NBD2 protein was isolated, it showed similar structural properties and a modest decrease in thermodynamic stability compared with the other NBD2 proteins. These data suggest that all the NBD2 variants can adopt similar structures.
Second, these mutations had severe impacts on the biosynthesis of full-length ABCC6. Given the location of the mutations and their proximity to the conserved TMD-NBD interface, the data suggest that the mutations likely altered domain-domain association in the full-length protein (Figs. 1, 3, and 5). Structural analyses of multiple ABC proteins suggest that the three mutations are located in or proximal to the TMD-NBD2 interface and likely contribute to domain-domain assembly. Thus, the change in physical and chemical character for each of these mutations would potentially alter the energetics of the domain-domain interaction in this interface, uncoupling NBD2 from the TMDs. Changes in local domain dynamics, solubility, and/or domain-domain association events all may contribute to the recognition of aberrant ABCC6 by cellular quality control systems and are implicated in the identification of other mutant ABC proteins (43). Further studies are needed to resolve differences in domain dynamics and other fine-grain structural details associated with the NBD2 variants and their handling in the cell.
Similar alterations in biosynthesis have been shown with missense mutations at the Phe-508 locus of NBD1 in CFTR and Pgp (18, 19). Missense mutations, located within the homologous TMD-NBD1 interface, have been shown to have minimal to moderate effects on NBD1 folding and moderate to severe effects on CFTR maturation. Suppression of the F508del and Phe-508 missense mutations, both genetically or pharmacologically, underlies hierarchical CFTR folding models and is consistent with the effects of the NBD2 mutations reported here (25, 42, 44). In CFTR, stabilization of the NBD itself is sufficient to partially restore its biosynthesis (26, 37, 44). Similarly, stabilization of the TMD-NBD interaction is also sufficient to partially restore biosynthesis (26). Together, the biosynthetic rescue associated with the stabilization of intradomain structure and interdomain interactions is additive and facilitates increased CFTR maturation (25).
Given the location of NBD2 within the primary sequence of ABCC6, its translation occurs after that of the TMDs and NBD1, providing a potential scaffold onto which the NBD can fold. As such, mutations that putatively stabilize native domain-domain interactions were evaluated for their ability to rescue mutant ABCC6 folding. The catalytic residues in the Walker B sequences of the NBDs were selected based on prior work showing their role in NBD dimerization. The D778N substitution in NBD1 decreased ABCC6 maturation, similar to results with CFTR trafficking (26, 45). In contrast, the E1427Q mutation in NBD2 appeared to traffic normally and rescued the NBD2 R1314W, L1335P, and R1339C variants (Figs. 3–5). This is consistent with functional and biochemical studies of CFTR wherein dimer stabilizing mutations in NBD2 increase the biochemical stability of the full-length protein and channel open potential (39, 46).
In contrast to the other NBD2 mutations, neither the G1299S nor the S1307P mutants, both proximal to the ATP binding site in NBD2, could be rescued by E1427Q. Given the mild effects of G1299S on NBD2 folding, this could be due to the alterations within the ATP binding site and a subsequent loss of ATP binding and efficacy of the E1427Q mutant. In contrast, the more severe impacts of the S1307P mutation on NBD2 folding suggest that the core structure of the NBD2 protein may be perturbed. Further investigation of the structural consequences of these mutations is required to identify the exact mechanisms by which they alter ABCC6 biosynthesis. However, the inability of the E1427Q mutation to rescue these mutations demonstrates that the mechanism is not global for all NBD2 PXE-causing mutations.
The observed rescue could be the result of at least two different effects of the E1427Q substitution; that is, intradomain stabilization resulting from ligand-induced stabilization via the non-hydrolyzed ATP molecule or stabilization of the NBD heterodimer via decreased ATPase activity. The inclusion of the E1427Q mutant had negative effects on the isolated NBD2 but strong positive effects on the folding of the full-length mutant proteins. This suggested that intradomain stability was not increased with the E1427Q mutant and pointed to stabilization of the NBD heterodimer as a mechanism of rescue in the full-length protein (Figs. 3 and 5). Consistent with this, disruption of the putative NBD heterodimer by the G755R mutant reversed the effects of the E1427Q suppressor in full-length ABCC6.
Given the conservation of the ATPase reaction cycle and mechano-chemistry and the location of the G755R substitution in the LSGGQ signature sequence, it is likely that this mutant disrupts NBD dimerization and this counteracts the effects of the E1427Q mutant (Fig. 4). As G755R equivalent mutations are associated with multiple ABC-transporter diseases, these data suggest that this substitution may generally result in altered protein function and not biosynthesis, consistent with measurements of multiple LSGGQ mutants in CFTR (40, 47). This result, along with those of G551D and G1349D in CFTR also argue against a requirement for the formation of the NBD heterodimer in normal protein biosynthesis. These functional mutations show normal protein biosynthesis despite the putative disruption of the NBD heterodimer (48).
The trans-complementation studies performed using ABCC6 and ABCC6 truncations co-expressed with NBD2 further suggest the mechanism of rescue is associated with stabilization of the NBD heterodimer. Similar, hemichannel complementation studies have been performed with CFTR and demonstrated that functional channels could be reconstituted from the assembly of multiple channel fragments in trans (49, 50). Co-expression of NBD2 with ΔNBD2 ABCC6, which putatively provided a native docking site for the NBD2 protein expressed in trans, led to increased NBD2 signal in the β-galactosidase assay (Fig. 4). The increase in soluble NBD2 protein was consistent with structural complementation between the ΔNBD2 ABCC6 and NBD2 proteins, resulting from interactions with the TMDs, NBD1, or both.
Co-expression of the wild type NBD2 with ΔNBD2 ABCC6 putatively facilitates assembly of the TMD-NBD interaction with a catalytically active and transient NBD heterodimer and would be predicted to provide some stabilization of the NBD2 protein. Co-expression of E1427Q NBD2 with the ΔNBD2 protein resulted in a significant and further increase in the NBD2 signal. As the TMD-NBD interface is putatively unaffected by the E1427Q substitution, this increase in NBD2 signal likely arises from the formation of the TMD-NBD interaction and a stable NBD heterodimer. In contrast, the G755R substitution, which would putatively disrupt this stable NBD heterodimer, reversed the effects of the ΔNBD2 co-expressed with E1427Q NBD2. This further suggested that the stable NBD heterodimer formation was necessary for the E1427Q-mediated rescue of NBD2 variants. Importantly, none of these effects was seen when the NBD2 protein was co-expressed with the full-length ABCC6 protein, demonstrating an available NBD2 docking site in ABCC6 was required for these stabilization of NBD2. This was consistent with the trans complementation of NBD2 with the various ΔNBD2 ABCC6 variants.
These data suggest that stabilization of NBD domain-domain interactions may provide a mechanism for biosynthetic correction of multiple disease-causing alleles. It is not yet clear that this biosynthetic rescue would be sufficient to rescue PXE pathophysiology, as the exact function of ABCC6 in normal and disease pathology is incompletely understood. Moreover, stabilization of the NBD heterodimer decreases specific transport activity in other ABC proteins (51). As such, the effects of the stable NBD heterodimer and protein trafficking would need to be balanced with changes in protein activity. It is possible, however, that sufficient increases in transporter numbers at the plasma membrane would overcome kinetic defects in the transport cycle and restore near normal function. This dimer-mediated biosynthetic mechanism suggests that small molecule therapeutics could potentially be identified that stabilize the NBD dimer interface and serve as folding correctors. Although previous studies suggest the NBD dimer interface might be targeted to potentiate transporter function, these studies suggest that targeting this interface may also be useful in correcting biosynthetic defects, a therapeutic strategy currently being explored for both ABCC6 and CFTR (28, 39, 46, 47, 52).
The relative rarity of individual PXE-causing alleles in ABCC6 and the distribution of these alleles are problematic for allele-specific drug screening, however. As with other personalized medicine development efforts, rare diseases and rare alleles remain a frustration both economically and practically. Phenotypic drug development screens are often focused on targeting single alleles. In contrast, the identification of mechanisms that facilitate rescue of multiple alleles provides a potential strategy for therapeutic development efforts addressing a broader population of patients. The demonstration that the E1427Q-mediated rescue is more general to a set of mutations establishes this mechanistically. The tested ABCC6 NBD2 mutations, with the exception of those associated with the Walker A ATP binding sequence, were at least partially rescued by the E1427Q mechanism. It is possible that a small molecule that mimics this mechanism of dimer stabilization would rescue multiple alleles. Such a mechanism would potentially provide specificity for ABCC6, unlike general small molecule treatments (28, 53, 54). By extension, similar approaches could be utilized to correct trafficking in other ABC proteins.
Recent work with CFTR correctors and potentiators, which are used clinically, suggests that common theratypes (classes of mutations that respond similarly to specific therapeutics) are likely to emerge as drug therapies are extended throughout the patient population (55). By characterizing the mechanisms of biosynthetic disruption and correction, rational drug discovery efforts could leverage common mechanisms of rescue for multiple alleles. Establishing mechanisms by which multiple alleles can be corrected reduces the practical limitations of screening for multiple rare genotypes, thereby simplifying therapeutic development for patients suffering from rare diseases.
Experimental Procedures
Homology Modeling of ABCC6 and the Isolated NBDs
The structures of C. elegans Pgp (PDB 4F4C) and Staphylococcus aureus Sav1866 were used as starting structures for homology models of human ABCC6 (17, 29). The coding sequences were aligned using T-Coffee and manually curated. The aligned sequences were used to generate an initial template using the Swiss Model server (56, 57). Models were further refined using the Chiron server (58). Similar modeling was performed on the NBD sequences in isolation using the human MRP1 NBD1 (PDB 4C3Z) and the human CFTR NBD2 (PDB 3GD7) as templates (59). Structural images were rendered in PyMOL.
Cloning of Full-length ABCC6 and NBDs
Full-length ABCC6 cDNA was PCR-amplified and inserted into pcDNA3.1 for CMV-driven transient expression in mammalian cells (32). The ΔNBD2 ABCC6 protein spanned residues 1–1253 and was similarly cloned and expressed using pcDNA3.1. The NBD1 cDNA, coding residues 623–870, and NBD2 cDNA, coding residues 1254–1503, was PCR-amplified and inserted into pcDNA3.1 containing an in-frame fusion of the HA tag and the α- fragment of β-galactosidase for structural complementation (pcDNA3.1-NBD2-HA-α), as previously described (26, 32, 36). The NBD coding sequences were similarly cloned into the pSmt3 expression vector as an in-frame fusion to N-terminal His6 and SUMO tags for expression in E. coli, as previously described (26, 31, 32). PCR-based site-directed mutagenesis was used to introduce sequence changes in all plasmids and was confirmed by automated DNA sequencing.
Cell Culture, Biotinylation, and Western Blotting
HEK293 cells (ATCC) were routinely maintained in DMEM (Gibco) with 10% FBS (Hyclone) and 100 units/ml penicillin and streptomycin (Hyclone) at 37 °C with 5% CO2. Plasmids were transfected into HEK293 cells using XtremeGENE (Roche Applied Science) following the manufacturer's protocols and expressed for 48 h before analysis. Cells were lysed with radioimmune precipitation assay buffer (50 mm Tris, 150 mm NaCl, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1% IGEPAL CA-630, Roche Complete Protease Inhibitors), and the cell lysates were cleared by centrifugation (21,000 RCF, 4 °C, 15 min). For cell surface biotinylation experiments, EZ-link NHS-SS-biotin was used to label cell surface proteins following the manufacturer's protocols (Thermo Scientific). Biotinylated proteins were captured by incubation with streptavidin-agarose beads at 4 °C (Thermo Scientific). Biotinylated proteins were eluted using DTT and assessed by Western blotting. ABCC6 was detected using the M6II-31 rat monoclonal antibody (Santa Cruz). The anti-PARP-1 P116/P25 antibody (EMD Millipore) or anti-tubulin antibody (EMD Millipore) was used for loading controls. Western blots were performed on at least three independent transfections. Densitometric analyses were performed on at least three independent experiments.
Immunofluorescence Staining
HEK293 cells were plated on poly-lysine-treated glass coverslips and transfected with ABCC6 for immunofluorescence staining (32). Briefly, cells were washed 3 times in PBS and fixed using 2% paraformaldehyde in PBS for 10 min at 4 °C. Cells were blocked with BSA and stained using the ABCC6 antibodies described above. AlexaFluor secondary antibodies were used to visualize ABCC6 using AlexaFluor-labeled secondary antibodies (Life Technologies). DAPI (Sigma) and wheat germ agglutinin (WGA, Sigma) were used to stain the nuclei and plasma membranes, respectively. Immunofluorescence was visualized on a Fluoview 1000 confocal microscope (Olympus). Immunofluorescence images were collected from multiple fields from at least four independent experiments.
Structural Complementation Assay
pcDNA3.1-NBD-HA-α and pcDNA3.1-ω-plasmids were co-transfected into HEK293 cells using XtremeGENE (Roche Applied Science), as previously described for CFTR (26). Forty-eight hours post-transfection, cells were washed twice in PBS and lysed in 1× reporter lysis buffer (Promega). The lysates were mixed with fluorescein-di-β-galactoside (FDG) at a final concentration of 5 μm FDG. Fluorescence was monitored using a BioTek Synergy 4 microplate reader in kinetic mode at 37 °C. Enzymatic activity was fit using first order kinetics under steady state conditions, and mean reaction velocity was used to quantify soluble proteins yields. The data presented represent at least four independent experiments for each protein variant.
In Vivo Protein Solubility
The pSmt3-NBD2 plasmid was used for protein expression in E. coli. Soluble protein yields were assessed after centrifugation as previously described (26). Briefly, culture densities were assessed using A600 and adjusted to normalize for differences in growth. Cultures were then sonicated in lysis buffer (50 mm Tris, 150 mm NaCl, 2 mm ATP, 1 mm DTT, 10% glycerol), and an aliquot of sonicated sample was taken for the total expression controls. The samples were then clarified by centrifugation (40,000 RCF, 30 min, 4 °C), and aliquots of the supernatant were taken as the soluble fraction. Samples were diluted with SDS-PAGE sample buffer and assessed by electrophoresis and Western blotting. Solubility measurements were taken on at least three independent expression experiments for each protein assessed.
Protein Expression and Purification
NBD2 protein was expressed and purified as previously described (26, 31). Expression cultures of BL21(DE3) E. coli were grown at 37 °C until A600 = 0.7–0.8. The cultures were then shifted to 15 °C, and protein was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Protein was expressed at 15 °C overnight (18–20 h). Cells were collected by centrifugation and resuspended in lysis buffer (50 mm Tris, 150 mm NaCl, 10% glycerol, 2 mm ATP, and 1 mm β-mercaptoethanol and 5 mm imidazole). Cells were lysed by sonication, and the soluble material was used for purification after centrifugation (30000 RCF, 30 min). The His6-SUMO-NBD protein was captured using nickel-nitrilotriacetic acid FastFlow resin (GE Healthcare). The column was washed (lysis buffer with 60 mm imidazole), and the protein was eluted (lysis buffer with 400 mm imidazole). Fractions containing the protein were then concentrated using Amicon Ultra Concentrators (Millipore), and the protein was further purified using a Sephacryl S-300 HR gel filtration column (GE Healthcare) in lysis buffer. The His-SUMO tags were cleaved by incubating with His-tagged Ulp1, which specifically cleaves the Smt3 moiety from the C-terminal fusion partner. The cleaved His-Smt3 and His-Ulp1 proteins were removed with a nickel-nitrilotriacetic acid column. The unbound NBD2 protein was collected and concentrated to 8–10 mg/ml using Amicon Concentrators (Millipore) and flash-frozen in liquid nitrogen. Protein was stored at −80 °C before use and cleared by centrifugation after thawing and before analysis. Protein characterization was performed on at least two independent protein preparations.
Circular Dichroism Spectroscopy
CD spectra of the NBDs were collected using a Jasco 810 circular dichroism spectrometer and a 0.1-cm quartz cuvette. Baseline spectra (buffers) were collected and subtracted from the spectra of the NBD containing samples. Data were converted to molar ellipticity per residue (degree cm2 dmol−1).
Analytical Gel Filtration Chromatography
Purified NBD proteins were injected on a Superdex S75 10/300 GL (GE Healthcare) pre-equilibrated in lysis buffer using a Shimadzu Prominence HPLC and monitored by UV absorbance at 220 and 280 nm. Gel filtration standards (Bio-Rad) were used to calibrate the elution volumes by apparent molecular mass.
Thermal Denaturation Experiments
Purified NBD protein was diluted to 1–5 μm concentration in lysis buffer including 2 mm ATP, 1 mm β-mercaptoethanol and supplemented with 5× SYPRO Orange (Life Technologies) in a final volume of 50 μl, essentially as described (38). Reactions were transferred to 96-well plates, sealed with optical plastic tape (Corning), and read on an ABI 7900HT Q-PCR machine between 25 and 95 °C at multiple ramp rates between 1 and 2.5 °C/min using the ROX dye parameters.
Curve-fitting, Densitometry, and Statistical Analysis
Densitometry, curve-fitting, and statistical analysis were performed using ImageJ, SigmaPlot and Prism software. ANOVA analysis and Tukey's post hoc test were performed in Prism.
Author Contributions
Y. R. and P. H. T. conceived the project. Y. R. and P. H. T. designed and performed the experiments, analyzed the data, and wrote and edited the manuscript. P. H. T. supervised the project.
Acknowledgments
We thank members of the CFTR 3D Structure and Folding Consortia for constructive criticism.
This work was supported in part by NIDDK, National Institutes of Health Grant DK083284 (to P. H. T.). This work was also supported by Cystic Fibrosis Foundation Therapeutics Grant THIBOD13XX0 (to P. H. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PXE
- pseudoxanthoma elasticum
- ABCC6
- ATP binding cassette transporter C6
- TMD
- transmembrane domain
- NBD
- nucleotide binding domain
- CFTR
- cystic fibrosis transmembrane conductance regulator
- WGA
- wheat germ agglutinin
- RCF
- relative centrifugal force
- ANOVA
- analysis of variance.
References
- 1.Le Saux O., Urban Z., Tschuch C., Csiszar K., Bacchelli B., Quaglino D., Pasquali-Ronchetti I., Pope F. M., Richards A., Terry S., Bercovitch L., de Paepe A., and Boyd C. D. (2000) Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 25, 223–227 [DOI] [PubMed] [Google Scholar]
- 2.Jiang Q., Endo M., Dibra F., Wang K., and Uitto J. (2009) pseudoxanthoma elasticum is a metabolic disease. J. Invest. Dermatol. 129, 348–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodman R. M., Smith E. W., Paton D., Bergman R. A., Siegel C. L., Ottesen O. E., Shelley W. M., Pusch A. L., and McKusick V. A. (1963) Pseudoxanthoma elasticum: a clinical and histopathological study. Medicine 42, 297–334 [PubMed] [Google Scholar]
- 4.Marconi B., Bobyr I., Campanati A., Molinelli E., Consales V., Brisigotti V., Scarpelli M., Racchini S., and Offidani A. (2015) Pseudoxanthoma elasticum and skin: clinical manifestations, histopathology, pathomechanism, perspectives of treatment. Intractable Rare Dis. Res. 4, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jansen R. S., Duijst S., Mahakena S., Sommer D., Szeri F., Váradi A., Plomp A., Bergen A. A., Oude Elferink R. P., Borst P., and van de Wetering K. (2014) ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 34, 1985–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen R. S., Küçükosmanoglu A., de Haas M., Sapthu S., Otero J. A., Hegman I. E., Bergen A. A., Gorgels T. G., Borst P., and van de Wetering K. (2013) ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. U.S.A. 110, 20206–20211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uitto J., Jiang Q., Váradi A., Bercovitch L. G., and Terry S. F. (2014) Pseudoxanthoma elasticum: diagnostic features, classification, and treatment options. Expert Opin. Orphan Drugs 2, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorgels T. G., Hu X., Scheffer G. L., van der Wal A. C., Toonstra J., de Jong P. T., van Kuppevelt T. H., Levelt C. N., de Wolf A., Loves W. J., Scheper R. J., Peek R., and Bergen A. A. (2005) Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 14, 1763–1773 [DOI] [PubMed] [Google Scholar]
- 9.Iliás A., Urbán Z., Seidl T. L., Le Saux O., Sinkó E., Boyd C. D., Sarkadi B., and Váradi A. (2002) Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J. Biol. Chem. 277, 16860–16867 [DOI] [PubMed] [Google Scholar]
- 10.Klement J. F., Matsuzaki Y., Jiang Q. J., Terlizzi J., Choi H. Y., Fujimoto N., Li K., Pulkkinen L., Birk D. E., Sundberg J. P., and Uitto J. (2005) Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol. Cell. Biol. 25, 8299–8310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dean M., Rzhetsky A., and Allikmets R. (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166 [DOI] [PubMed] [Google Scholar]
- 12.Hosen M. J., Zubaer A., Thapa S., Khadka B., De Paepe A., and Vanakker O. M. (2014) Molecular docking simulations provide insights in the substrate binding sites and possible substrates of the ABCC6 transporter. PloS ONE 9, e102779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai J., Daoud R., Alqawi O., Georges E., Pelletier J., and Gros P. (2002) Nucleotide binding and nucleotide hydrolysis properties of the ABC transporter MRP6 (ABCC6). Biochemistry 41, 8058–8067 [DOI] [PubMed] [Google Scholar]
- 14.Slot A. J., Molinski S. V., and Cole S. P. (2011) Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 50, 179–207 [DOI] [PubMed] [Google Scholar]
- 15.Moody J. E., Millen L., Binns D., Hunt J. F., and Thomas P. J. (2002) Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 277, 21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaitseva J., Jenewein S., Jumpertz T., Holland I. B., and Schmitt L. (2005) H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 24, 1901–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson R. J., and Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 18.Thibodeau P. H., Brautigam C. A., Machius M., and Thomas P. J. (2005) Side chain and backbone contributions of Phe508 to CFTR folding. Nat. Struct. Mol. Biol. 12, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du K., Sharma M., and Lukacs G. L. (2005) The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 12, 17–25 [DOI] [PubMed] [Google Scholar]
- 20.He L., Aleksandrov L. A., Cui L., Jensen T. J., Nesbitt K. L., and Riordan J. R. (2010) Restoration of domain folding and interdomain assembly by second-site suppressors of the deltaF508 mutation in CFTR. FASEB J. 24, 3103–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babenko A. P., and Bryan J. (2003) Sur domains that associate with and gate KATP pores define a novel gatekeeper. J. Biol. Chem. 278, 41577–41580 [DOI] [PubMed] [Google Scholar]
- 22.Pratt E. B., Tewson P., Bruederle C. E., Skach W. R., and Shyng S. L. (2011) N-terminal transmembrane domain of SUR1 controls gating of Kir6.2 by modulating channel sensitivity to PIP2. J. Gen. Physiol. 137, 299–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plomp A. S., Florijn R. J., Ten Brink J., Castle B., Kingston H., Martín-Santiago A., Gorgels T. G., de Jong P. T., and Bergen A. A. (2008) ABCC6 mutations in pseudoxanthoma elasticum: an update including eight novel ones. Mol. Vis. 14, 118–124 [PMC free article] [PubMed] [Google Scholar]
- 24.Fülöp K., Barna L., Symmons O., Závodszky P., and Váradi A. (2009) Clustering of disease-causing mutations on the domain-domain interfaces of ABCC6. Biochem. Biophys. Res. Commun. 379, 706–709 [DOI] [PubMed] [Google Scholar]
- 25.Mendoza J. L., Schmidt A., Li Q., Nuvaga E., Barrett T., Bridges R. J., Feranchak A. P., Brautigam C. A., and Thomas P. J. (2012) Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 148, 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thibodeau P. H., Richardson J. M. 3rd, Wang W., Millen L., Watson J., Mendoza J. L., Du K., Fischman S., Senderowitz H., Lukacs G. L., Kirk K., and Thomas P. J. (2010) The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 285, 35825–35835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y., Devor D. C., Engelhardt J. F., Ernst S. A., Strong T. V., Collins F. S., Cohn J. A., Frizzell R. A., and Wilson J. M. (1993) Molecular basis of defective anion transport in L cells expressing recombinant forms of CFTR. Hum. Mol. Genet. 2, 1253–1261 [DOI] [PubMed] [Google Scholar]
- 28.Pomozi V., Brampton C., Fülöp K., Chen L. H., Apana A., Li Q., Uitto J., Le Saux O., and Váradi A. (2014) Analysis of pseudoxanthoma elasticum-causing missense mutants of ABCC6 in vivo; pharmacological correction of the mislocalized proteins. J. Invest. Dermatol. 134, 946–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin M. S., Oldham M. L., Zhang Q., and Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shintre C. A., Pike A. C., Li Q., Kim J. I., Barr A. J., Goubin S., Shrestha L., Yang J., Berridge G., Ross J., Stansfeld P. J., Sansom M. S., Edwards A. M., Bountra C., Marsden B. D., et al. (2013) Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. U.S.A. 110, 9710–9715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mossessova E., and Lima C. D. (2000) Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 5, 865–876 [DOI] [PubMed] [Google Scholar]
- 32.Xue P., Crum C. M., and Thibodeau P. H. (2014) Regulation of ABCC6 trafficking and stability by a conserved C-terminal PDZ-like sequence. PloS ONE 9, e97360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., and Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 34.Gong Q., Keeney D. R., Molinari M., and Zhou Z. (2005) Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J. Biol. Chem. 280, 19419–19425 [DOI] [PubMed] [Google Scholar]
- 35.Hirai M., Kadowaki N., Kitawaki T., Fujita H., Takaori-Kondo A., Fukui R., Miyake K., Maeda T., Kamihira S., Miyachi Y., and Uchiyama T. (2011) Bortezomib suppresses function and survival of plasmacytoid dendritic cells by targeting intracellular trafficking of Toll-like receptors and endoplasmic reticulum homeostasis. Blood 117, 500–509 [DOI] [PubMed] [Google Scholar]
- 36.Wigley W. C., Stidham R. D., Smith N. M., Hunt J. F., and Thomas P. J. (2001) Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nat. Biotechnol. 19, 131–136 [DOI] [PubMed] [Google Scholar]
- 37.Protasevich I., Yang Z., Wang C., Atwell S., Zhao X., Emtage S., Wetmore D., Hunt J. F., and Brouillette C. G. (2010) Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 19, 1917–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 [DOI] [PubMed] [Google Scholar]
- 39.Vergani P., Lockless S. W., Nairn A. C., and Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gyimesi G., Borsodi D., Sarankó H., Tordai H., Sarkadi B., and Hegeds T. (2012) ABCMdb: a database for the comparative analysis of protein mutations in ABC transporters, and a potential framework for a general application. Hum. Mutat. 33, 1547–1556 [DOI] [PubMed] [Google Scholar]
- 41.Pollet J. F., Van Geffel J., Van Stevens E., Van Geffel R., Beauwens R., Bollen A., and Jacobs P. (2000) Expression and intracellular processing of chimeric and mutant CFTR molecules. Biochim. Biophys. Acta 1500, 59–69 [DOI] [PubMed] [Google Scholar]
- 42.Cui L., Aleksandrov L., Chang X. B., Hou Y. X., He L., Hegedus T., Gentzsch M., Aleksandrov A., Balch W. E., and Riordan J. R. (2007) Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 365, 981–994 [DOI] [PubMed] [Google Scholar]
- 43.Farinha C. M., Matos P., and Amaral M. D. (2013) Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: not just from the endoplasmic reticulum to the Golgi. FEBS J. 280, 4396–4406 [DOI] [PubMed] [Google Scholar]
- 44.Pissarra L. S., Farinha C. M., Xu Z., Schmidt A., Thibodeau P. H., Cai Z., Thomas P. J., Sheppard D. N., and Amaral M. D. (2008) Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem. Biol. 15, 62–69 [DOI] [PubMed] [Google Scholar]
- 45.Lewis H. A., Buchanan S. G., Burley S. K., Conners K., Dickey M., Dorwart M., Fowler R., Gao X., Guggino W. B., Hendrickson W. A., Hunt J. F., Kearins M. C., Lorimer D., Maloney P. C., Post K. W., et al. (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23, 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aleksandrov L. A., Jensen T. J., Cui L., Kousouros J. N., He L., Aleksandrov A. A., and Riordan J. R. (2015) Thermal stability of purified and reconstituted CFTR in a locked open channel conformation. Protein Expr. Purif. 116, 159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeCarvalho A. C., Gansheroff L. J., and Teem J. L. (2002) Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator delta f508. J. Biol. Chem. 277, 35896–35905 [DOI] [PubMed] [Google Scholar]
- 48.Bompadre S. G., Sohma Y., Li M., and Hwang T. C. (2007) G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J. Gen. Physiol. 129, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan K. W., Csanády L., Seto-Young D., Nairn A. C., and Gadsby D. C. (2000) Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH2-terminal nucleotide binding domain. J. Gen. Physiol. 116, 163–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Csanády L., Chan K. W., Seto-Young D., Kopsco D. C., Nairn A. C., and Gadsby D. C. (2000) Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J. Gen. Physiol. 116, 477–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tombline G., Bartholomew L. A., Tyndall G. A., Gimi K., Urbatsch I. L., and Senior A. E. (2004) Properties of P-glycoprotein with mutations in the “catalytic carboxylate” glutamate residues. J. Biol. Chem. 279, 46518–46526 [DOI] [PubMed] [Google Scholar]
- 52.Bompadre S. G., Li M., and Hwang T. C. (2008) Mechanism of G551D-CFTR (cystic fibrosis transmembrane conductance regulator) potentiation by a high affinity ATP analog. J. Biol. Chem. 283, 5364–5369 [DOI] [PubMed] [Google Scholar]
- 53.Rubenstein R. C., and Zeitlin P. L. (2000) Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am. J. Physiol. Cell Physiol. 278, C259–C267 [DOI] [PubMed] [Google Scholar]
- 54.Perlmutter D. H. (2002) Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 52, 832–836 [DOI] [PubMed] [Google Scholar]
- 55.Cutting G. R. (2015) Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet. 16, 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 57.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramachandran S., Kota P., Ding F., and Dokholyan N. V. (2011) Automated minimization of steric clashes in protein structures. Proteins 79, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaptal V., Magnard S., Gueguen-Chaignon V., Falson P., Di Pietro A., and Baubichon-Cortay H. (2014) Nucleotide-free crystal structure of nucleotide-binding domain 1 from human ABCC1 supports a “general-base catalysis” mechanism for ATP hydrolysis. Biochemistry & Pharmacology: Open Access 3, 150 [Google Scholar]