

Abstract
Familial hypercholesterolemia (FH) is characterized by severely elevated low density lipoprotein (LDL) cholesterol. Herein, we identified an FH patient presenting novel compound heterozygote mutations R410S and G592E of the LDL receptor (LDLR). The patient responded modestly to maximum rosuvastatin plus ezetimibe therapy, even in combination with a PCSK9 monoclonal antibody injection. Using cell biology and molecular dynamics simulations, we aimed to define the underlying mechanism(s) by which these LDLR mutations affect LDL metabolism and lead to hypercholesterolemia. Our data showed that the LDLR-G592E is a class 2b mutant, because it mostly failed to exit the endoplasmic reticulum and was degraded. Even though LDLR-R410S and LDLR-WT were similar in levels of cell surface and total receptor and bound equally well to LDL or extracellular PCSK9, the LDLR-R410S was resistant to exogenous PCSK9-mediated degradation in endosomes/lysosomes and showed reduced LDL internalization and degradation relative to LDLR-WT. Evidence is provided for a tighter association of LDL with LDLR-R410S at acidic pH, a reduced LDL delivery to late endosomes/lysosomes, and an increased release in the medium of the bound/internalized LDL, as compared with LDLR-WT. These data suggested that LDLR-R410S recycles loaded with its LDL-cargo. Our findings demonstrate that LDLR-R410S represents an LDLR loss-of-function through a novel class 8 FH-causing mechanism, thereby rationalizing the observed phenotype.
Keywords: cell biology, cholesterol metabolism, low density lipoprotein (LDL), lysosome, proprotein convertase subtilisin/kexin type 9 (PCSK9), familial hypercholesterolemia, human natural mutations, LDL receptor, subcellular localization, loss-of-function
Introduction
The levels of low density lipoprotein cholesterol (LDLc)5 are correlated with cardiovascular diseases, which are the main cause of death and morbidity worldwide (1). Among the best cholesterol-lowering agents are “statins” that inhibit HMG-CoA reductase and lower LDLc (by 40–60%) through up-regulation of the LDL receptor (LDLR) levels. Ezetimibe, which inhibits intestinal sterol absorption and reduces LDLc by ≤20%, is often administered with statins for a maximal decrease in LDLc levels (2).
Familial hypercholesterolemia (FH) is among the most common inherited metabolic disorders affecting 1:200 individuals worldwide and is characterized by severely elevated LDLc. Monogenic hypercholesterolemias are caused by mutations in genes involved in the uptake of low density lipoprotein (LDL) by the LDLR in hepatocytes (3). Autosomal recessive hypercholesterolemia is caused by mutations in the LDLR adaptor protein 1 (ARH) or its LDLRAP1 gene (4). Autosomal dominant familial hypercholesterolemia results from mutations in LDLR, apolipoprotein B (apoB), or proprotein convertase subtilisin/kexin type 9 (PCSK9). Loss-of-function (LOF) mutations in either LDLR (67%) or apoB (14%), the protein component of LDL that binds LDLR, result in FH and premature coronary heart disease (4). More than 1700 LDLR mutations were identified (5), and the wild-type (WT) LDLR structure was defined (Fig. 1A). Until recently, FH mutations in the LDLR were categorized into six different classes, depending on the receptor defect (Table 1) (6–12). In 2003, a third gene encoding the proprotein convertase subtilisin/kexin type 9 (PCSK9) (Fig. 1A) (13) was associated with FH (14), as it binds LDLR and enhances its degradation (15, 16) in endosomes/lysosomes (17). Indeed, PCSK9 gain-of-function (GOF) mutations were associated with hypercholesterolemia (14), especially D374Y, which enhances binding of PCSK9 to the LDLR by >10-fold (18), Conversely, LOF mutations result in hypocholesterolemia (19). The identification of the epidermal growth factor A (EGFA) domain of the LDLR as the binding site to PCSK9 catalytic subunit (20, 21) led to the discovery of a 7th class of LDLR mutations (H327Y) that enhances receptor binding to and degradation by PCSK9 (Table 1) (22).
FIGURE 1.

Domain organizations of LDLR and PCSK9 and structures of LDLR β-propeller domain and of PCSK9·LDLR complex. A, domain organizations of LDLR (top) and PCSK9 (bottom). LDLR is composed of the ligand binding domain (LBD), containing seven repeats (L1–L7) of ∼40 residues, the EGF precursor homology domain (EGFPH), consisting of EGFA, EGFB, six-bladed (1–6) β-propeller, and EGFC domains, the O-linked sugar regions, and the transmembrane domain (TMD). Mature PCSK9 is formed after auto-cleavage between the prodomain (Pro) and the catalytic domain (Catalytic). The hinge domain (H) and the C-terminal histidine-rich domain (CHRD) are indicated. B, LDLR β-propeller domain with its six blades and their corresponding residues. The positions of Arg410 and G592 in the β-propeller domain of the LDLR are displayed. C, overall structure of the PCSK9·LDLR complex (PDB code 3M0C) shows two interfaces of interaction between the two proteins as follows: a major interface between catalytic domain of PCSK9 (green) and the EGFA domain of LDLR (turquoise) and a minor interface (boxed) between the prodomain of PCSK9 (red) and the LDLR β-propeller domain (blue). Box, details of the prodomain/β-propeller interface; yellow dotted line shows the van der Waals interaction between Leu108 (PCSK9) and Leu647 (LDLR-WT), whereas the blue dotted line depicts the “putative” ionic interaction between the GOF mutation L108R (PCSK9) and Glu626 (LDLR-WT).
TABLE 1.
Functional classification of LDLR loss of function mutations
| Class | Phenotype | Example of mutant (Ref.) |
|---|---|---|
| 1 | No detectable LDLR protein | Stop 167 (7) |
| 2 | Either complete (2a) or partial (2b) block of transport of the LDLR from the ER | G565V (8) |
| 3 | Defective LDL binding | D227E (7) |
| 4 | Defective clustering in clathrin-coated pits | Y828C (9) |
| 5 | Recycling defective receptors | T454N (10) |
| 6 | Sorting defect in polarized epithelial cells | G844D (11) |
| 7 | Highly degraded by extracellular PCSK9 | H327Y (12) |
| 8a | Defective LDL delivery to lysosomes and resistance to extracellular PCSK9 | R410S |
a Proposed novel class is shown. LDLR is low density lipoprotein receptor; ER is endoplasmic reticulum; LDL is low density lipoprotein; PCSK9 is proprotein convertase subtilisin/kexin 9.
Complete loss of PCSK9 resulted in an unprecedented decrease in LDLc without apparent adverse effects, leading to the development of potent inhibitory PCSK9 monoclonal antibodies (mAbs). Large scale phase III clinical trials revealed that subcutaneous injection of these mAbs every 2 or 4 weeks results in ∼60% lowering of LDLc (23–25).
A suspected homozygote FH patient, referred to our Institut de Recherches Cliniques de Montréal (IRCM) lipid clinic in 2010, exhibited highly elevated LDLc despite maximal statin, ezetimibe, and PCSK9 inhibitor therapies. Genetic testing revealed the presence of two heterozygote mutations, R410S and G592E, one on each allele of the LDLR gene. Such mutations were previously reported individually and predicted to be damaging (7, 26). However, the R410S/G592E compound heterozygosity is novel. The underlying mechanisms of these two mutations are unknown, including the patient's resistance to PCSK9-mAb treatment. Therefore, our work sought to (i) identify the mechanism(s) by which the mutations R410S and G592E in the LDLR lead to hypercholesterolemia, as observed in our patient, and (ii) explain the patient's resistance to the PCSK9-mAb treatment, which would point to an alternative therapy for PCSK9-resistant patients. Herein, we provide evidence for a novel FH mechanism associated with LDLR-R410S, the latter representing a new class 8 LDLR mutation (Table 1), and we show that the LDLR-G592E does not effectively exit from the endoplasmic reticulum (ER), classifying it as a class 2b LDLR defect.
Results
Identification of a Compound Heterozygote FH Patient Resistant to Statin, Ezetimibe, and PCSK9-mAb Treatments
The prepositus, a 23-year-old man, was referred to the IRCM clinic for elevated LDLc and total cholesterol (Table 2). He had normal triglycerides and high density lipoprotein (HDL) levels, normal blood pressure, and no prior history of cardiovascular disease but presented bilateral xanthelasma of the eyelids without tendinous xanthoma. A diagnosis of homozygous FH was proposed based on high LDLc, a positive family history for hypercholesterolemia in both parents, and his poor response to statin therapy. Indeed, atorvastatin (10 mg) led to a modest 13% drop in LDLc compared with an expected ∼35% decrease, and 20 mg resulted in an additional 6% decrease (Fig. 2A). The patient was then switched to rosuvastatin (20 and 40 mg) and ezetimibe (10 mg) therapy, leading to a limited ∼37% reduction in LDLc compared with an expected ∼70% (27). Additional subcutaneous injections of 140 mg of the PCSK9-mAb Evolocumab every 2 weeks further lowered LDLc by only ∼15% (expected average 50–70%) (24, 28), suggestive of a resistance to PCSK9 action (Fig. 2A).
TABLE 2.
Average characteristics of the untreated subjects in each mutation group
TC is total cholesterol; HDLc is high density lipoprotein cholesterol; LDLc is low density lipoprotein cholesterol; TG is triglyceride. The numbers are listed as averages (± S.D. from the mean).
| Genotype | No. of subjects | Age | Sex, male/female | TC | HDLc | LDLc | TG | PCSK9 |
|---|---|---|---|---|---|---|---|---|
| years | mmol/liter | ng/ml | ||||||
| R410S/G592E | 1 | 23 | 1/0 | 11.5 | 0.8 | 10.2 | 1.0 | 82.0 |
| R410S/WT | 2 | 63.5 (± 16.3) | 1/1 | 7.8 (± 2.1) | 1.2 (± 0.1) | 5.9 (± 0.3) | 1.6 (± 0.3) | 90.5 (± 23.3) |
| WT/G592E | 2 | 63.5 (± 16.3) | 1/1 | 7.3 (± 1.2) | 1.3 (± 0.1) | 5.3 (± 0.3) | 1.5 (± 0.4) | 81.5 (± 2.1) |
| WT/WT | 8 | 38.2 (± 18.8) | 3/5 | 5.4 (± 1.3) | 1.3 (± 0.2) | 3.4 (± 1.4) | 1.4 (± 1.2) | 84.1 (± 24.5) |
FIGURE 2.

Evolution of LDLc concentration of patient III-3 following different therapies and family pedigree. A, LDLc concentrations following several lipid-lowering treatments. B, family pedigree. The prepositus is marked with an arrow. Round symbols, females. Square symbols, males. Black outlined symbols, family members for which data were available. Gray outlined symbols, family members for which no data were available. Symbols crossed through: deceased individuals. LDLR-R410S allele, dark gray shading. LDLR-G592E allele, light gray shading.
Exome sequence analysis revealed no PCSK9 variation except for the presence of two missense mutations in the LDLR, namely R410S and G592E, one on each allele. Genetic analysis of the kindred revealed that the R410S and G592E mutations were inherited through the maternal and paternal lineage, respectively (Fig. 2B). Both Arg410 and Gly592 are fully conserved residues among species (data not shown) and are localized in the β-propeller domain of the LDLR, namely Arg410 in the β1 repeat and Gly592 in the β5 one (Fig. 1B).
Although the LDLR mutations R410S and G592E were previously reported, their compound heterozygosity, as in our FH patient, is new. For example, the combined heterozygote mutation, R410S on one allele and a deletion mutation on the other allele, was reported in 2013 in an 8-year-old FH patient (26). Although the early termination deletion mutant is expected to result in an LDLR-defective form, the consequence of the R410S mutation was not analyzed but was predicted to be pathogenic. The heterozygote G592E mutation was previously found either alone or in combination with other LOF mutations on the other allele (29). It resulted in 5–15% residual LDLR activity in fibroblasts and was predicted to be damaging (7).
Table 2 summarizes the characteristics of the subjects who participated in our study. Patients with the single LDLR mutation G592E (WT/G592E) displayed an elevated LDLc, similar to values reported in carriers of this mutation (7). Carriers of the single R410S mutation (R410S/WT) presented LDLc concentrations comparable with those with the single G592E mutation. The compound heterozygote patient (R410S/G592E) exhibited LDLc levels twice higher than subjects with a single mutation.
LDLR-R410S Is Well Expressed at the Cell Surface and the LDLR-G592E Is Degraded by the Proteasome
A segregation analysis revealed a genotype-to-phenotype correlation in the studied family pedigree, suggesting that the LDLR mutations R410S and G592E, individually or combined, are causative of FH, without affecting circulating PCSK9 levels that were in the normal range (Table 2) (30). Moreover, the underlying mechanisms of their effects on LDLR trafficking and their functional consequences in the development of FH are unknown.
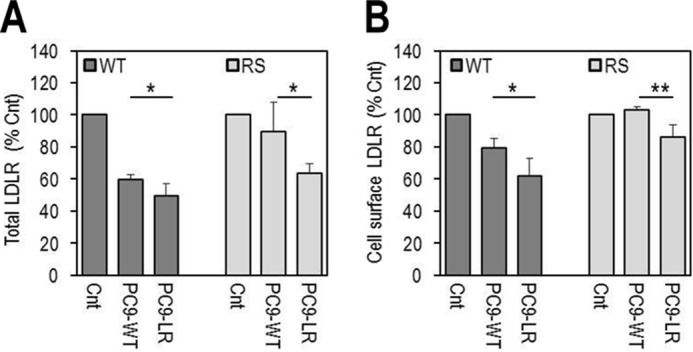
Accordingly, we first compared the cellular properties of the LDLR mutants with that of LDLR-WT (Fig. 3). Note that on Western blottings (WB) the LDLR appears as a doublet consisting of an LDLR that is not O-glycosylated (∼110 kDa) and one that is fully mature and O-glycosylated (∼140 kDa) (31). Thus, WB analyses of overexpressed LDLR-WT, -R410S, and -G592E and both mutants in HEK293 cells revealed that LDLR-WT and LDLR-R410S levels were similar, whereas those of LDLR-G592E were ∼30% of WT, and the equal combination R410S/G592E resulted in an intermediate ∼70% total LDLR protein level (Fig. 3A). This suggests that, different from the WT and R410S receptors, LDLR-G592E is less stable in these cells. We propose that the lower protein levels of this mutant are due to its retention in the ER, as evidenced by its endoglycosidase H sensitivity (Fig. 3B), and subsequent proteasomal degradation. Indeed, the LDLR-G592E levels were increased by ∼2-fold in the presence of the proteasome inhibitor MG132 (32), although this agent had little effect on LDLR-WT and LDLR-R410S (Fig. 3C). Whether the proteasomal degradation of the immature G592E receptor follows its ubiquitination or not is unknown. Finally, fluorescence-activated cell sorting (FACS) analysis of the LDLR confirmed that WT and R410S cell surface levels were similar, whereas those of G592E and R410S/G592E were ∼40 and ∼70% of WT (Fig. 3D).
FIGURE 3.

Total and cell surface expression of wild-type LDLR and its two mutants, R410S and G592E. HEK293 cells were transfected with V5-tagged LDLR-WT (WT), LDLR-R410S (RS), LDLR-G592E (GE), or both mutants (RS/GE). A, WB analysis of LDLR expression. B, LDLR sensitivity to endoglycosidase H (Endo H). C, LDLR levels after 18 h in absence (−) or presence (+) of 2.5 μmol/liter MG132, a proteasome inhibitor. D, FACS analyses of cell surface LDLR expression. E, immunofluorescence microscopy of total (PERM) and cell surface (NON PERM) LDLR in HepG2 cells overexpressing WT, RS, GE, or RS/GE. Quantification of cell surface LDLR is shown, using 12 transfected cells (EGFP-positive)/condition/experiment. Non-transfected cells expressed ∼100-fold less LDLR than transfected ones. Scale bar, 15 μm. Data are representative of at least two independent experiments. Quantifications are averages ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (t test).
Similar observations were found in liver-derived HepG2 cells using immunocytochemistry of the LDLR and its mutants (Fig. 3E) under permeabilized (total LDLR; upper panel) and non-permeabilized (cell surface LDLR; lower panel) conditions. The data showed that the cellular LDLR distribution/localization of LDLR-WT and LDLR-R410S was similar in both conditions. Conversely, G592E and the double mutant resulted in decreased cell surface LDLR levels, with an estimated ∼30% (G592E-LDLR) and ∼60% (R410S/G592E-LDLR) reaching the plasma membrane, as compared with WT-LDLR (Fig. 3E).
In conclusion, the cell surface expression of LDLR-G592E was drastically reduced compared with WT, mainly because of its retention in ER and subsequent degradation. This should result in increased circulating LDLc levels, as observed in all family members that carried the G592E mutation (Table 2). The cell surface expression of LDLR-WT and LDLR-R410S was similar and much higher than that of LDLR-G592E. However, individuals carrying a single R410S or G592E mutation presented equally increased LDLc levels (5.3–5.9 versus normal 3.4 mmol/liter). This raises the question of the functional activity of the LDLR-R410S and its regulation by PCSK9.
PCSK9-WT Binds Cell Surface LDLR-R410S but Does Not Lead to Its Degradation: Importance of LDLR-Arg410 for PCSK9 Function
It is a rare event to find hypercholesterolemic individuals resistant to the LDLc-lowering effect of a PCSK9-mAbs. In the present FH patient the circulating levels of PCSK9 were within normal range (∼82 ng/ml; Table 2). This eliminated the likelihood that the patient's resistance to PCSK9-mAbs is due to abnormally elevated levels of circulating PCSK9. We thus investigated the possibility that the LDLR-R410S is inadequately responding to PCSK9-enhanced LDLR degradation.
We reported that in cell lines PCSK9 enhances the degradation of the LDLR both by an intracellular pathway (Golgi to lysosomes, observed upon co-expression of PCSK9 and LDLR), and an extracellular one (early endosomes to lysosomes, observed upon incubation of cells with exogenous PCSK9) (33). Accordingly, co-expression of PCSK9-WT or its GOF mutant D374Y with LDLR-WT or LDLR-R410S in HEK293 cells followed by WB analyses revealed that LDLR-WT and LDLR-R410S are ≥90% degraded (Fig. 4A). Similar results were obtained by FACS analysis of cell surface LDLR levels (Fig. 4B). Thus, we conclude that the LDLR mutation R410S does not prevent PCSK9 from dragging the receptor toward lysosomal degradation in the intracellular pathway (33).
FIGURE 4.

Effects of PCSK9 on LDLR degradation. A and B, HEK293 cells were co-transfected with V5-tagged LDLR-WT (WT) or LDLR-R410S (RS) and PCSK9-WT (PC9-WT), PCSK9-DY (PC9-DY) or empty vector (v), as control, and analyzed by WB using anti-human LDLR (A), or by FACS for cell surface LDLR expression (B). C and D, HEK293 cells overexpressing V5-tagged WT or RS were incubated for 7 h with conditioned media from HEK293 cells (input): no PCSK9 control media (Cnt) or PCSK9 media (∼1.8 μg/ml), PC9-WT or GOF PC9-DY, and analyzed by WB with anti-human LDLR (C) or by FACS for cell surface LDLR expression (D). E, mouse primary hepatocytes lacking both PCSK9 and LDLR were transfected with V5-tagged WT or RS and incubated for 18 h with conditioned media from HEK293 as described in C. Cell lysates were immunoblotted with anti-V5-HRP for LDLR. Data are representative of at least three independent experiments. Quantifications are averages ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (t test).
However, because the latter pathway is not predominant in liver, where PCSK9 acts mostly extracellularly on hepatocytes, we next examined the effect of exogenously added PCSK9 on cellular LDLR. Unexpectedly, although extracellular PCSK9-WT or PCSK9-D374Y could enhance the degradation of LDLR-WT (by 50 or 60%, respectively), LDLR-R410S was resistant to the activity of either PCSK9 isoform, as observed by WB (Fig. 4C), or FACS (Fig. 4D) analyses.
Finally, we confirmed the above observations with exogenous PCSK9 in mouse primary hepatocytes isolated from mice lacking both PCSK9 and LDLR (34) and overexpressing LDLR-WT or LDLR-R410S (Fig. 4E). Collectively, our results suggest that the underlying mechanism of the FH phenotype observed in the proband (III-3) and the average 1.7-fold higher LDLc found in R410S heterozygote family members (I-2 and II-3) (Fig. 1B; Table 1) exclude a contribution by extracellular PCSK9.
In a next logical step, we evaluated the ability of extracellular PCSK9, non-tagged or a C-terminally tagged PCSK9-mCherry (35), to bind cell surface LDLR in HEK293 cells overexpressing the LDLR-WT or LDLR-R410S. FACS analysis (Fig. 5A) and direct fluorescence measurements (Fig. 5B) of surface-associated PCSK9 clearly demonstrated that both PCSK9 forms bind similarly the WT and R410S receptors at the plasma membrane. Furthermore, a dose-response curve at the cell surface of HEK293 cells measuring the interaction of each receptor with exogenous PCSK9 at 4 °C gave similar binding parameters (Kd ∼2 μm; Fig. 5C).
FIGURE 5.

PCSK9 displays similar binding to the cell surface LDLR-WT and LDLR-RS. A and B, HEK293 cells transfected with LDLR-WT (WT) or LDLR-R410S (RS) were incubated for 4 h at 4 °C with conditioned media from HEK293 cells: no PCSK9 control media (Cnt) or media containing PCSK9-WT. PCSK9 was non-tagged (PC9-WT) ∼1.3 μg/ml (A), or mCherry-tagged (PC9-mCherry), ∼1 μg/ml (B). PCSK9 binding to cell surface receptor was quantified by FACS (A) or fluorescence spectroscopy (B). Bars represent averages ± S.D. from three independent experiments. C, HEK293 cells overexpressing LDLR-WT (WT) or LDLR-RS (RS) were incubated with purified recombinant human PCSK9 for 4 h at 4 °C. The curve through the data is the fit to a rectangular hyperbola and is representative of two independent experiments. The average PCSK9 binding constants determined from the two experiments are 1.8 ± 0.8 μm (WT) and 1.3 ± 0.1 μm (RS).
Thus, the resistance of the LDLR-R410S to extracellular PCSK9-induced degradation in cell lines and mouse primary hepatocytes is not due to the impairment of PCSK9 binding to this receptor, but it is likely a consequence of the R410S mutation.
We next addressed the possibility of rescuing the LDLR-R410S phenotype. X-ray crystallography of the LDLR·PCSK9 complex revealed that Leu108 in the PCSK9-prodomain makes hydrophobic contacts with Leu647 in the β6-propeller domain of the LDLR (Fig. 1C) (21). This was supported by the identification of L108R as a GOF mutation of PCSK9, likely by enhancing such interaction via an ionic contact between the PCSK9 mutant Arg108 and Glu626 in the β6-propeller domain of the LDLR (36). Because the LDLR Arg410 (β1-propeller domain) is spatially adjacent to Glu626, we compared the effects of exogenous PCSK9-WT and PCSK9-L108R GOF mutant on LDLR-R410S (Fig. 6). This GOF mutant partially restored the ability of PCSK9 to enhance LDLR-R410S degradation following an 18-h incubation, as evidenced by a more sensitive ELISA (compared with WB) for total LDLR (from 11% with PCSK9-WT to 37% with PCSK9-L108R; Fig. 6A), and by cell surface FACS analysis (from 0% with PCSK9-WT to 14% with PCSK9-L108R; Fig. 6B). Generally, a higher decrease in total LDLR measured by ELISA compared with cell surface LDLR assayed by FACS has already been reported (37), and it likely emphasizes that only a small fraction of total LDLR remains at the cell surface (38). Thus, PCSK9-L108R GOF mutation can functionally but partially enhance/rescue the ability of PCSK9 to degrade the LDLR-R410S, likely reflecting an increased affinity of PCSK9 for the LDLR (36).
FIGURE 6.
GOF PCSK9-L108R in the prodomain rescues PCSK9-mediated degradation of LDLR-R410S via the extracellular pathway. HEK293 cells overexpressing LDLR-WT (WT) or LDLR-R410S (RS) were incubated for 18 h with conditioned media from HEK293 cells: no PCSK9 control-media (Cnt) or PCSK9-media (∼1.1 μg/ml), PCSK9-WT (PC9-WT), or GOF PCSK9-L108R (PC9-LR). A, total LDLR quantification by ELISA. B, FACS analyses of cell surface LDLR. Data are representative of four independent experiments. Bars are averages ± S.D. *, p < 0.05; **, p < 0.01 (t test).
To investigate the importance of Arg410, we generated three other LDLR mutants, namely R410K (positive charge), R410E (negative charge), and R410A (neutral amino acid). The level of expression of these mutants was tested in HEK293 cells by ELISA for total LDLR (Fig. 7A), FACS for cell surface LDLR (Fig. 7B), and in HepG2 cells by immunocytochemistry (Fig. 7E) and their functionality by DiI-LDL uptake in HepG2 cells (Fig. 7F). The DiI-LDL was chosen because it was previously shown to undergo the same endocytic degradative pathway as the unlabeled LDL (39). The data showed that only the expression of LDLR-R410E was lower than LDLR-WT and correlated with its lower functional activity (Fig. 7F). In contrast, LDLR-WT, -R410S, -R410K, and -R410A reached the cell surface to a similar extent in both cell lines (Fig. 7, B and E). The LDLR-R410E is less stable as its levels were lower, and in contrast to the other mutants, it was sensitive to endoglycosidase H digestion, suggesting that it was mostly retained in the ER (Fig. 7C). Similar to the LOF mutant LDLR-G592E (Fig. 3C), the LDLR-R410E levels were increased in the presence of MG132, in support of its degradation by the proteasome (data not shown). This suggests that a negative charge (glutamic acid) in place of either Gly592 or Arg410 is damaging and likely prevents the correct folding of the protein in the ER.
FIGURE 7.

Effect of loss of Arg410 on LDLR expression, functionality, and sensitivity to extracellular PCSK9-mediated degradation. A–D, HEK293 cells transfected with LDLR-WT (WT) or LDLR-R410 mutants (RS, RK, RE, RA) were analyzed for total LDLR expression by ELISA (A), cell surface LDLR expression by FACS (B), sensitivity of LDLR to endoglycosidase H, by WB (C), and cell surface LDLR expression, by FACS, following an 18-h incubation with conditioned media from HEK293 cells: no PCSK9 control media (Cnt), or PCSK9 media (∼1.1 μg/ml), PCSK9-WT (PC9-WT), or GOF PCSK9-DY (PC9-DY) (D). E and F, immunofluorescence microscopy in HepG2 cells transfected with WT or RS, RK, RE, RA mutants. E, total LDLR, under permeabilized conditions (PERM), and cell surface LDLR, under non-permeabilized conditions (NON PERM). F, DiI-LDL internalization after 4-h incubation with 5 μg/ml DiI-LDL at 37 °C. Quantifications in E and F were derived from analyses of 12 transfected cells (EGFP-positive)/condition/experiment. Scale bar, 15 μm. Bars in A, B, and F are averages ± S.D. of five independent experiments, and bars in C and D are averages ± S.D. of two independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (t test).
Overnight incubations of HEK293 cells overexpressing LDLR-WT, -R410S, -R410K, or -R410A with exogenous PCSK9-WT or PCSK9-D374Y revealed that these LDLR mutants were still partially resistant to PCSK9-WT based on FACS analyses (Fig. 7D). However, under these longer incubation conditions, the mutant receptors became sensitive to the GOF PCSK9-D374Y. We conclude that the integrity of LDLR-Arg410 is critical for its sensitivity to PCSK9-WT but not for the degradation by PCSK9-D374Y, especially after extended incubation times.
Thus, the resistance of the patient to the PCSK9-mAb therapy is likely due to the inability of exogenous PCSK9 to efficiently enhance the degradation of LDLR-R410S. However, this would not explain the FH phenotype, unless the LDLR-R410S has also lost its functionality.
LDLR-R410S Binds LDL but Fails to Efficiently Internalize and Degrade It
To test the hypothesis that the LDLR-R410S has a lower activity toward LDL, binding of 125I-LDL to the cell surface of HEK293 cells overexpressing LDLR-WT or LDLR-R410S was assessed at 4 °C, and its time-dependent internalization and degradation were evaluated at 37 °C (Fig. 8, A–C). Both overexpressed cell surface receptors bound 125I-LDL similarly, with estimated dissociation constants of 0.42 ± 0.05 nm for LDLR-WT and 0.30 ± 0.08 nm for LDLR-R410S (Fig. 8A), assuming a molecular mass of 5.5 × 105 g/mol for apoB-100, the sole LDLR ligand in LDL. The rate of cellular internalization of 125I-LDL by LDLR-R410S over 120 min was found to be ∼40% lower than that observed with LDLR-WT (Fig. 8B), and it was associated with an ∼50% decrease in 125I-LDL intracellular degradation over 8 h (Fig. 8C).
FIGURE 8.

Functionality of LDLR-WT and LDLR-R410S. A–C, HEK293 cells transfected with LDLR-WT (WT) or LDLR-R410S (RS) were evaluated. A, 125I-LDL binding to LDLR after a 1-h incubation at 4 °C with increasing concentrations of 125I-LDL (200 cpm/pg). B, time-dependent 125I-LDL internalization after continuous incubation with 2 ng of 125I-LDL at 37 °C. C, time-dependent 125I-LDL degradation after continuous incubation with 2 ng of 125I-LDL (4 ng/ml) at 37 °C. At each time point, the medium was precipitated with ice-cold TCA, and the TCA-soluble material was used as a measure of the degraded 125I-LDL. Data are representative of three independent experiments each performed in four biological replicates. Points are the averages ± S.D. within one experiment. D and E, HepG2 cells overexpressing WT or RS were incubated with DiI-LDL (5 μg/ml) and analyzed by confocal microscopy as follows: DiI-LDL binding to the cell surface after 4-h incubation at 4 °C (D) or DiI-LDL internalization after 4 h at 37 °C (E). Quantifications in D and E were derived from analyses of 12 transfected cells (EGFP-positive)/condition/experiment. Scale bar, 15 μm. Data are representative of at least two independent experiments. Bars are averages ± S.D. **, p < 0.01 (t test).
We recapitulated these results by immunofluorescence in HepG2 cells overexpressing either receptor and incubated with fluorescent DiI-LDL for 4 h at 4 °C (Fig. 8D) or 37 °C (Figs. 7F and 8E). DiI-LDL binding was similar to both receptors, but its internalization was decreased by ∼45% with LDLR-R410S (Fig. 8E).
Hence, the LDLR-R410S exhibits a defect in LDL internalization/degradation, suggesting a decreased functionality compared with LDLR-WT. Although this conclusion agrees with the FH phenotype, the underlying mechanism of the observed LOF still remains unknown.
LDLR-R410S Exhibits Lower LDL Release in Acidic Endosomes and Reduced LDL Delivery to Late Endosomes/Lysosomes
We analyzed the intracellular localization of DiI-LDL in HepG2 cells by quantifying its co-localization with specific markers of early endosomes (EEA1) and late endosomes/lysosomes (Lamp1) (Fig. 9A). Thus, after 4-h incubations at 37 °C, the fluorescence associated with early endosomes was similar for both receptors, whereas that in late endosomes/lysosomes was ∼50% lower for LDLR-R410S versus LDLR-WT. Hence, the R410S mutation does not affect the early steps of DiI-LDL internalization but results in a less efficient delivery of the cargo to late endosomes/lysosomes compared with the WT receptor.
FIGURE 9.

Residue Arg410 of the LDLR is important for LDL release in acidic early endosomes and for its delivery to late endosomes. A, HepG2 cells overexpressing LDLR-WT (WT) or LDLR-R410S (RS) were incubated with DiI-LDL (5 μg/ml) for 4 h at 37 °C and analyzed by immunofluorescence under permeabilized conditions. Upper panels, co-localization of DiI-LDL with markers for early endosomal compartment (EEA1) and quantification. Lower panels, co-localization of DiI-LDL with markers for late endosomal/lysosomal compartment (Lamp1) and quantification. Quantifications were derived from analyses of 12 transfected cells (EGFP-positive)/condition/experiment. Scale bar, 15 μm. B and C, HEK293 cells overexpressing WT or RS were incubated for 1 h at 4 °C with 15 μg/ml DiI-LDL at pH 7.4, washed, and switched to pH 5.3 for an additional hour at 4 °C. At each pH, the amount of fluorescence from DiI-LDL bound to each receptor was measured by FACS. B, flow cytometry plots for WT (left) and RS (right). C, quantification of DiI-LDL bound at each pH relative to pH 7.4. D, HEK293 cells overexpressing WT or RS were incubated with DiI-LDL (6 μg/ml) for 1 h at 4 °C, washed, and then switched to fresh serum-free DMEM at 37 °C for the indicated times. DiI-LDL released into media was measured and reported relative to DiI-LDL bound at time 0. Data are averages ± S.D. of at least three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (t test).
To better understand the above observations, we next tested the hypothesis that LDL release from LDLR at the acidic pH of endosomes/lysosomes (6) is less effective with the mutant receptor. Consequently, we FACS-analyzed in HEK293 cells the amount of DiI-LDL bound to either LDLR-WT or LDLR-R410S both before and after lowering the pH from 7.4 to 5.3. The data showed that ∼40% of DiI-LDL remained bound to the LDLR-WT at acidic versus neutral pH, as reported previously (40). In addition, ∼50% more DiI-LDL remained bound to LDLR-R410S at pH 5.3 compared with LDLR-WT (Fig. 9, B and C). Our cautious interpretation is that at acidic pH, LDLR-R410S binds DiI-LDL stronger than the LDLR-WT, thereby delivering less LDL to lysosomes. Moreover, because LDL binding was equal for both receptors, but the overall cell-associated LDL and its degradation were lower in cells expressing LDLR-R410S versus LDLR-WT (Fig. 8), it is possible that some of the internalized LDL by LDLR-R410S may be “regurgitated” outside of the cell (41). Because a previous report showed that fluorescent DiI does not leak out of the cells upon DiI-LDL degradation (39), we also measured the time-dependent increase of DiI-LDL fluorescence into fresh medium at 37 °C after a 1-h DiI-LDL binding at 4 °C followed by extensive washes. The data revealed that cells overexpressing LDLR-R410S released ∼50% higher levels of DiI-LDL compared with LDLR-WT (Fig. 9D). Also, time-dependent FACS analyses of LDLR cell surface recovery in the presence of LDL, showed that both receptors recycle back to the cell surface with similar rates (data not shown).
Therefore, entry of LDLR-bound LDL in early endosomes and cell surface recycling of the LDLR are similar for WT and R410S receptors. A notable difference between the two receptors is the tighter association of LDL with LDLR-R410S at acidic pH. Altogether, our results led us to conclude that this mutation resulted in impaired delivery of LDL to lysosomes and in recycling of the LDLR-R410S loaded with its cargo, which is released back into the medium. We estimate an ∼50% split between degradation and regurgitation or “retro-endocytosis” (42) of the LDLR-R410S·LDL complex. The consequence would be a LOF of the LDLR and a lower LDL clearance from the circulation, in agreement with the patient's FH phenotype.
Molecular Dynamics Simulation of LDLR-WT and LDLR-R410S Suggests Flexibility Differences between the β-Propeller Domains of the Two Receptors
It has been documented (6, 43) that long range intramolecular movements at the interface of the ligand binding repeats L4 and L5 and the top of the β-propeller (including blades β4 and β5) of LDLR are important for the release of receptor-bound LDL at the acidic pH of endosomes. Hence, the flexibility of some β-blades might be essential in this process.
Our experimental observations showed that the LDLR-R410S exhibits a defective LDL delivery to late endosomes/lysosomes, likely due to a tighter LDL receptor association at acidic pH. Because the R410S mutation occurs in the β1-blade of the LDLR β-propeller domain, we aimed to test potential differences in the flexibility of the β-propeller of WT and mutant receptors at neutral and acidic pH. This was achieved by molecular dynamics (MD) simulation of the entire β-propeller domain of the two receptors at pH 7 and 5.
Indeed, LDLR-WT and LDLR-R410S displayed differences in the predicted structural flexibilities of the β-propeller domains at neutral and acidic pH (Fig. 10 and supplemental Movie 1). At pH 7, although the β-propeller domain of LDLR-R410S is slightly less flexible than that of LDLR-WT, both structures exhibit a similar high flexibility of the segment comprising amino acids (aa) 458–465 of the β2-blade. However, at pH 5 there is a predicted substantial difference in the mobility of the two receptors. Thus, except for the highly mobile β2-loop (aa 458–465) common to both receptors and the modestly flexible β6-region (aa 635–636) in LDLR-R410S, the remainder of the β-propeller structure of LDLR-R410S is predicted to be rigid, whereas in LDLR-WT, blades β3 (aa 503–507) and β4 (aa 546–550) are flexible. Although the MD simulations did not account for LDL binding, the predicted major loss in flexibility of blades β3 and β4 in the β-propeller of LDLR-R410S at pH 5 supports the herein proposed model where, at acidic pH, LDL binds stronger to and is retained longer by the LDLR-R410S as compared with LDLR-WT (Fig. 9, B and C).
FIGURE 10.

At pH 5, the β-propeller mobility of LDLR-R410S is reduced compared with LDLR-WT, and for both receptors the structures at pH 5 are more mobile than the ones at pH 7. A, β-propeller configurations of LDLR-WT (WT) and LDLR-R410S (RS) at pH 7 (left) and pH 5 (right) were generated by molecular dynamics simulations starting with the LDLR-WT structure from PDB code 1N7D. The six blades (β1–β6) and flexible amino acid regions are indicated. The B-factor putty scale is shown as a measure of region flexibility (blue-to-red translated into rigid-to-flexible). B, plots of root mean squared fluctuations values (Å) for each β-propeller residue of LDLR-WT (blue trace) and LDLR-R410S (red trace) at pH 7 (top) and pH 5 (bottom) that were used to calculate the B-factors and generate the configurations in A.
Discussion
In the 1980s, the advent of statins, which inhibit the synthesis of cholesterol, has significantly reduced mortality and events associated with cardiovascular complications. FH is a dominant genetic disease mostly caused by LOF variants in the LDLR gene (6). Pathogenic changes in LDLR result in lower levels of active hepatic LDLR protein and hence in impaired clearance and increased levels of circulating LDLc. Although >1700 LDLR mutations have been identified, <10% have been mechanistically validated in cells (5). The discovery of PCSK9 (13) and its implication in LDLR metabolism (14, 15) have revolutionized the field of regulation of LDLc. Circulating PCSK9, which mainly originates from liver hepatocytes (44), enhances the degradation of the LDLR in endosomes/lysosomes and hence reduces the clearance rate of plasma LDLc (45).
Clearance of circulating LDLc is mostly afforded by the hepatic LDLR. LDL particles bind LDLR at the cell surface, and the LDLR·LDLc complex is endocytosed into clathrin-coated vesicles. Fusion of these vesicles with early endosomes (EEA1-positive) is followed by acidification to pH ∼6, which leads to a change in LDLR conformation from “open to closed” and triggers the dissociation of LDL from the LDLR (46). Subsequently, LDL is delivered to lysosomes where it is hydrolyzed, and cholesterol is redistributed within the cell. In turn, the LDLR is either recycled back to the cell surface for re-use (∼100–200 times each day), or it is sorted to late endosomes/lysosomes for degradation (47). The latter pathway is favored when PCSK9 binds the cell surface LDLR (18) and is more efficacious with PCSK9 GOF mutants such as D374Y and L108R (36, 45). The main binding interface (∼1000 Å2) occurs between the catalytic domain of PCSK9 and the EGFA domain of the LDLR (Fig. 1C) (12, 20, 21). Additionally, in the prodomain of PCSK9, Leu108 makes a hydrophobic contact with Leu647 in the β-propeller domain of the LDLR, thereby creating a second minor interface (∼70 Å2) (21).
The underlying mechanisms favoring the LDLR recycling pathway as opposed to its sorting to endosomes/lysosomes for degradation in the presence of PCSK9 are unknown. It has been proposed that, in the absence of PCSK9, specific protein binders to the cytosolic tail of the LDLR or an intermediate protein may regulate the LDLR recycling pathway (48). It has also been established that the C-terminal domain of PCSK9 (CHRD; Fig. 1A) is critical for the sorting of the LDLR·PCSK9 complex to endosomes/lysosomes (49–52). Possibly, a protein “X” would bind the LDLR·PCSK9 complex at the cell surface and sort it to degradation compartments (45, 49, 53, 54), likely in competition with the recycling pathway. The involvement of a similar or different protein X may also be applicable to the intracellular pathway that occurs following exit of the complex from the trans-Golgi network, and where the recycling pathway is absent. This hypothesis would predict that specific LDLR mutations may affect differently the intracellular versus the extracellular pathway.
Herein, we undertook an extensive characterization of two LDLR mutations, R410S and G592E, found in an FH patient of Italian origin. Based on our studies, neither of these mutations resulted in a complete loss of LDLR function. Therefore, it was not surprising that the 10.2 mmol/liter LDLc levels of our FH patient (Table 2) were lower than the ∼16.2 mmol/liter value reported for an 8-year-old FH patient presenting two LDLR mutations, R410S on one allele and a complete LOF on the other allele (26). The novel combination of R410S and G592E in our patient resulted in a doubling of the LDLc levels observed in single mutants (Table 2), suggesting the mutations are additive, with each providing similar contributions to the FH phenotype. In addition, the retention in the ER of LDLR-G592E (Fig. 3B) and of other LDLR mutants at this position, such as G592D, G592R, and G592A (data not shown), reflects the importance of the conserved Gly592 for the correct LDLR folding. Finally, in line with the lower levels of LDLR-G592E reaching the cell surface, DiI-LDL internalization was found to be 5-fold lower in HepG2 cells overexpressing this mutant versus LDLR-WT (data not shown).
Notably internalization of 125I-LDL in HEK293 cells expressing either the LDLR-WT or -R410S is similar at early time points (up to 40 min; Fig. 8B), when LDL internalization is mostly a function of the endocytic rate of the LDLR·LDL complex, as we also observed by immunocytochemistry on DiI-LDL accumulated in EEA1-positive endosomes (Fig. 9A, upper panel). In contrast, at later times the LDLR-R410S-expressing cells accumulate ∼40% less 125I-LDL than those expressing LDLR-WT (Fig. 8B), as also observed in Lamp1-positive organelles (Fig. 9A, lower panel). At later times (>40 min) the amount of cellular accumulation of LDL is a balance between endocytosis, retro-endocytosis, and degradation.
Our collective data revealed that the R410S mutation has a dramatic impact on the LDLR activity (Figs. 8 and 9) and on the structural dynamics of the β-propeller domain at both pH 7 and 5 (Fig. 10 and supplemental Movie 1), namely relative to LDLR-WT, LDLR-R410S bound LDL tighter at acidic pH, displayed a reduced LDL delivery to late endosomes/lysosomes, and an increased release in the medium of the bound/internalized LDL (Fig. 9D). This suggests that some (estimated at ∼50%) of the LDLR-R410S recycles together with its LDL-cargo. Regurgitation or retro-endocytosis of up to 10% of the internalized LDL has been reported for the native LDL receptor (41). Although the R410S mutation did not affect the PCSK9 binding to the cell surface LDLR (Fig. 5), it prevented the extracellular PCSK9-induced LDLR degradation in endosomes/lysosomes (Fig. 4, C–E). This result explains the resistance of the FH patient to the PCSK9-mAb treatment (Fig. 2A), leading us to probe ways to bypass the resistance of LDLR-R410S to extracellular PCSK9. Thus, we hypothesized that the R410S mutation could reduce the stabilizing secondary interaction between Leu108 in the prodomain of PCSK9 and Leu647 in the β6-blade of the LDLR β-propeller domain (Fig. 1C) (21). Indeed, upon overnight incubations, the PCSK9 GOF mutation L108R partially reversed the resistance to PCSK9-induced degradation of the LDLR-R410S (Fig. 6), as did the GOF PCSK9-D374Y that strengthens the PCSK9 interaction with the LDLR-EGFA domain (Fig. 7D) (18). Because Arg410 is highly conserved in most mammalian LDLRs, we mutated this residue to Lys, Ala, and Glu. The R410A and R410K mutants behaved similarly to R410S in all our tests (Fig. 7). However, LDLR-R410E was largely retained in the ER and consequently displayed a reduced DiI-LDL uptake (Fig. 7, E and F), likely emphasizing that a negative charge is not tolerated at this position.
Unexpectedly, the LDLR-R410S and LDLR-WT were equally sensitive to PCSK9-induced degradation when co-expressed in HEK293 cells (Fig. 4, A and B) and in mouse primary hepatocytes (data not shown), in contrast to cells incubated with extracellular PCSK9 (see above). This clearly emphasizes a major difference in the trafficking regulation of the LDLR·PCSK9 complex in the intracellular versus the extracellular pathway (33). R410S is the first example of an LDLR natural mutation that can differentiate between the two pathways. Presently, the only known trafficking difference between the intracellular versus extracellular pathway is the implication of clathrin light chain versus clathrin heavy chain in vesicular budding from the trans-Golgi network or cell surface, respectively (33). Therefore, the LDLR-R410S could provide a unique tool for the discovery of specific regulators of the LDLR·PCSK9 complex in each pathway.
Presently, LDLR LOF mutations are grouped in seven different classes depending on the pathway affected and the observed receptor defect (Table 1). We propose that the G592E mutation best belongs to class 2b, as it only partially reaches the cell surface, rather than its original classification as a class 5 mutation, which pertains to recycling defective alleles (7). Based on its functional phenotype, the R410S mutation failed to fit in any of the proposed LDLR mutant classes. We therefore suggest a novel 8th class defined as an LDLR that is “defective in LDL delivery to lysosomes and resistant to extracellular PCSK9-induced degradation” (Table 1). It is a matter of speculation whether some of the other natural FH-causing mutations (>50) reported around Arg410 in the β1-blade, but not characterized functionally, would also fit in this classification. Interestingly, the β1-blade mutant LDLR-R416W was suggested to be a recycling-defective class 5 receptor (10).
It is interesting that the levels of circulating PCSK9 in our double heterozygote FH patient were similar to those of his family members and are in the normal range (Table 2). It should be noted that the average circulating levels of PCSK9 in 20 untreated heterozygote FH patients was reported to be ∼1.5-fold higher than in non-FH controls, with three of them exhibiting values close to normal (55).
This raises the question of the presence in some FH patients of clearance receptor(s) of circulating PCSK9 other than the LDLR. Indeed, studies in LDLR KO mice revealed a slower clearance rate of plasma PCSK9 (56), but the lack of LDLR also up-regulates the levels of other PCSK9 targets in liver, e.g. LRP1 (54) and VLDL receptor (57, 58). Thus, in our patient the partial contribution of LDLR-G592E and LDLR-R410S to the clearance of PCSK9 and the possible presence of compensatory mechanisms may be responsible for the observed relatively normal levels of circulating PCSK9.
The limited response of our FH patient to a maximal dose of rosuvastatin and ezetimibe (Fig. 2A) can now be rationalized by a major LOF of R410S and G592E. Although statins, as inhibitors of HMG-CoA reductase, should increase the mRNA, and hence protein levels of these mutants, their limited functionality would not be enough to effectively reduce LDLc. A treatment option would be the drug lomitapide recently approved for the treatment of homozygous FH patients (59). This inhibitor of microsomal triglyceride transport protein reduces the levels of apoB in circulation by inhibiting its lipidation in the ER. Alternatively, the patient could be treated with mipomersen, a modified single-strand antisense oligonucleotide designed to degrade apoB-100 by specifically binding to its mRNA (59). Nevertheless, increased hepatic fat and long term hepatic safety are the major issues for both lomitapide and mipomersen, and therefore patients treated with these drugs should be closely monitored (59).
In conclusion, our study defined the mechanism underlying the FH phenotype observed in a patient with high LDLc despite maximal treatments with lipid-lowering drugs and provided evidence for the existence of a new class 8 LDLR LOF mutation. The approach used combined clinical profiling data, genetics, cell biology, and MD simulations to unravel the molecular details of disease-causing mutations. Application of these approaches to other LDLR mutations can help the advancement toward more effective personalized treatments aimed at lowering LDLc and the incidence of cardiovascular complications.
Experimental Procedures
Case Study at the IRCM Clinic
Clinical studies were conducted at the IRCM lipid clinic and were approved by the Institutional Review Board. All subjects provided written informed consent.
DNA Analysis from Human Subjects
DNA was extracted from white blood cells with the QIAmp blood maxi kit (Qiagen). The LDLR exon was sequenced using the VariantSeqr approach (Thermo Fisher Scientific), a system involving PCR sequencing of exons (and the 5′ upstream region). Briefly, PCRs were carried out with 10 ng/μl genomic DNA. Sanger sequencing was performed, and fragments were subjected to capillary electrophoresis on a 3130xl genetic analyzer with POP-7. Results were analyzed with the ViiA 7 software version 1.2.2 (AB Applied Biosystems, Life Technologies, Inc.).
Lipid and Lipoprotein Determination
Plasma was collected in EDTA tubes following an overnight 12-h fast. Total cholesterol, triglyceride, and HDLc concentrations were assayed on an automated analyzer (Abbott Biochromatic Analyzer model 100). LDLc was calculated using the Friedwald formula. Plasma PCSK9 concentrations were measured using an in-house ELISA as described previously (60).
cDNAs, Cell Culture, and Transfections
The cDNAs encoding for human LDLR and its mutants were cloned in pIRES-EGFP (where IRES is internal ribosome entry site and EGFP is enhanced green fluorescent protein) vector containing a C-terminal V5 tag. Point mutations of the LDL receptor were created by site-directed mutagenesis, and the identity of each mutant was confirmed by DNA sequencing. Full-length human PCSK9 was cloned in pmCherry-N1, a mammalian expression vector (Clontech) designed to express a protein of interest fused to the N terminus of mCherry (mCherry is a mutant fluorescent protein derived from the tetrameric Discosoma sp. red fluorescent protein). The constructions containing the human PCSK9, WT or D374Y, cloned in pIRES-EGFP were previously described (16). HEK293 (human embryonic kidney-derived epithelial) and HepG2 (human hepatocellular carcinoma) cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) (HEK293 cells) or in Eagle's minimal essential medium (HepG2 cells) supplemented with 10% (v/v) fetal bovine serum (Invitrogen) and were maintained at 37 °C under 5% CO2. HEK293 cells were seeded in poly-l-lysine (50 μg/ml)-coated 12-well plates (3 × 105 cells/well) or 24-well plates (1.5 × 105 cells/well) and the following day were transfected using jetPRIME (PolyPlus) and a total of 0.5 μg of cDNA or 0.25 μg of cDNA, respectively. 24 h post-transfection, the culturing medium was changed to serum-free, and the cells were treated according to each experiment. Alternatively, for immunofluorescence experiments, HepG2 cells (0.5 × 105 cells/well) were plated on poly-l-lysine-coated round microscope coverslips that were placed in a 24-well cell culture plate and 24 h later were transfected with 0.25 μg of cDNA using FuGENE® HD (Promega). Transfected HepG2 cells were maintained in culture during 48 h, including last 24 h in serum-free medium, to achieve maximal expression of the receptor.
Sources of Human PCSK9
For all experiments that required addition of extracellular PCSK9 at a single fixed concentration, PCSK9 was obtained from 24-h serum-free conditioned media of HEK293 cells overexpressing stably, non-tagged, or V5-tagged PCSK9 (WT or D374Y) or transiently PCSK9-mCherry. The PCSK9 concentrations in the conditioned media were measured using an in-house ELISA as described previously (60). For the experiment that required PCSK9 titration to the HEK293 cells transiently expressing LDLR, purified recombinant human PCSK9-His was obtained from ACRO Biosystems (PC9-H5223).
Isolation, Culture, and Transfection of Mouse Primary Hepatocytes
Mouse primary hepatocytes were isolated from livers of 8–10-week-old male mice lacking both PCSK9 and LDLR (34) by a two-step collagen perfusion method, as described previously (61). Cells were seeded in 12-well plates (CellBind, 3337; Corning) in Williams' medium E supplemented with 10% (v/v) FBS at a density of 2.5 × 105 cells/well. After 2 h, the medium was replaced with hepatozyme medium (Invitrogen) for 12 h prior to transfection. Transfections were performed with Effectene (Qiagen) using 3 μg of cDNA/well, following the manufacturer's instructions (transfection efficiencies 10–15%). 24 h post-transfection, the cells were washed once and incubated for an additional 18 h with conditioned media from naive HEK293 cells (control) or overexpressing either WT or D374Y human PCSK9 (1.8 μg/ml).
Enzymatic Digestion of Glycosyl Moieties
Proteins (30 μg) from cell lysates were incubated with endoglycosidase H (New England Biolabs) for 90 min at 37 °C. Products were separated on 8% Tris glycine SDS-polyacrylamide gels and transferred to a polyvinylidene difluoride (PVDF) membrane (0.45 μm; PerkinElmer Life Sciences), and LDLR protein was revealed by WB (see below).
Western Blotting
Following the incubation times and treatments specific to each experiment, cultured cells were washed twice with ice-cold PBS and lysed for 40 min on ice in radioimmunoprecipitation assay (RIPA) buffer in the presence of a mixture of protease inhibitors (Roche Applied Science). Cell lysates were cleared by centrifuging for 12 min at 15,000 × g at 4 °C, and the total protein content was measured using the DC Protein assay (Bio-Rad). Thirty to 50 μg of protein were separated on 8% Tris glycine SDS-polyacrylamide gels and transferred to a PVDF membrane. Western blotting was performed for human LDLR-V5 (goat anti-human LDLR, 1:1000, AF2148, R&D Systems or anti-V5-HRP, 1:5000, R96125, Invitrogen), β-actin (rabbit anti-β-actin, 1:5000, A2066, Sigma), or GAPDH (rabbit anti-GAPDH, 1:2500; ab9485; Abcam), and human PCSK9 (rabbit anti-human PCSK9, 1:2500; in-house antibody). After incubation with the appropriate secondary antibodies, if required, the membranes were revealed using Clarity Western ECL Substrate (Bio-Rad) and imaged with a GelDoc XR+ instrument (Bio-Rad), and the bands of interest were quantified using ImageLab 5.2.1 software (Bio-Rad).
Fluorescence-activated Cell Sorting (FACS) Analysis of LDL Receptor Expression, LDL Binding and Release, and PCSK9 Binding
Flow cytometry was performed using a CyAn ADP flow cytometer (DAKO; Beckman Coulter), and collected data were analyzed using Summit software (Summit Software Inc.).
LDLR Cell Surface Expression by FACS
Transfected HEK293 cells grown for 24 h and treated according to each experimental design were placed on ice, detached by gentle up and down pipetting, washed with PBS, 0.5% BSA, 1 g/liter glucose (wash buffer), incubated for 10 min at 4 °C with a mouse anti-human LDLR antibody (1:250; MAB2148; R&D Systems), washed again, and finally incubated for 5 min at 4 °C with secondary antibody Alexa Fluor 647-conjugated goat anti-mouse IgG (1:500; A21235; Molecular Probes). After staining, cells were washed once more and resuspended in wash buffer containing 1.67 μg/ml propidium iodide (P4864; Sigma). For each sample, Alexa Fluor 647 fluorescence of 5000 high EGFP and live events (propidium iodide negative) was acquired for data analysis. All measurements were performed at least in triplicate.
LDL Binding and Release
HEK293 cells overexpressing LDLR-WT or LDLR-R410S and grown for 24 h were placed on ice, detached, washed as above, and incubated with 15 μg/ml DiI-LDL (J65330; Alfa Aesar) for 1 h at 4 °C in pH 7.4 buffer (RPMI 1640 + l-glutamine serum-free medium, 1.5 mm CaCl2, where RPMI is Roswell Park Memorial Institute medium). To test for low pH-induced LDL ligand release, following two washes the cells were shifted to pH 5.3 by replacing the pH 7.4 buffer with RPMI 1640 + l-glutamine serum-free medium, 25 mm succinate, 1.5 mm CaCl2, pH 5.3, for 1 additional hour at 4 °C. For each sample, DiI-LDL fluorescence of 5000 high EGFP events was acquired for data analysis.
PCSK9 Binding to the Cell Surface LDLR
HEK293 cells transfected with LDLR-WT or LDLR-R410S and grown for 24 h were placed on ice, washed once with ice-cold PBS, and incubated for 4 h at 4 °C with conditioned media from naive HEK293 cells (control) or HEK293 cells overexpressing either WT or D374Y human PCSK9 (1.3 μg/ml). Following one wash, cells were processed for FACS as described above, except that the staining was performed with a rabbit anti-human PCSK9 antibody (1:250; in-house antibody) and a secondary antibody Alexa Fluor 647-conjugated goat anti-rabbit IgG (1:500; A21245; Molecular Probes). For each sample, Alexa Fluor 647 fluorescence of 2000 live events was acquired for analysis.
Confocal Laser Scanning Microscopy Analysis of LDL Receptor Expression and Localization and DiI-LDL Binding and Internalization in HepG2 Cells
Samples were visualized using a Plan-Apochromat 63 × 1.4 oil objective of an LSM-710 confocal laser-scanning microscope (Carl Zeiss) with sequential excitation and capture image acquisition with a digital camera. Images were processed with ZEN software. Image analysis to quantify the fluorescence intensities and co-localization was accomplished using Imaris ×64 8.2.0.
Cell Surface LDLR Expression Analysis
48 h post-transfection, the cells were washed twice with PBS and fixed with PBS, 4% paraformaldehyde (10 min). After blocking with PBS, 2% BSA (1 h), samples were incubated at 4 °C overnight with goat anti-human LDLR polyclonal antibody (1:200; AF2148; R&D Systems), washed with PBS, and incubated with the appropriate fluorescent secondary antibody for 1 h at room temperature. Nuclei were stained by incubating the samples with PBS, 1 μg/ml Hoechst for 2 min. Coverslips were mounted on a glass slide with Mowiol (Sigma). To analyze LDLR total expression, the same protocol was followed, except after the blocking step and prior to incubation with primary antibody, cells were permeabilized with PBS, 0.2% Triton X-100 for 5 min.
LDL Binding
Transfected HepG2 cells grown for 48 h were washed twice with cold PBS and then incubated with DiI-LDL (5 μg/ml) for 4 h at 4 °C. Next, the samples were fixed and nuclei were stained as described above. To analyze LDL internalization, 48 h post-transfection the cells were washed twice with PBS and incubated with DiI-LDL (5 μg/ml in fresh serum-free media) for 4 h at 37 °C. The samples were then processed as described above.
DiI-LDL Co-localization with Early Endosomal and Late Endosomal/Lysosomal Compartments
Cells were labeled under permeabilizing conditions with antibodies against EEA1 (goat anti-EEA1, 1:200, sc-6415, Santa Cruz Biotechnology) and Lamp1 (mouse anti-Lamp1, 1:200, BD555798, PharmingenTM), respectively.
Fluorescence Spectroscopy Analysis of PCSK9-mCherry Binding
HEK293 cells overexpressing WT or mutant LDLR and grown for 24 h in 24-well plates were placed on ice, washed with ice-cold PBS, and incubated with ice-cold PCSK9-mCherry conditioned media (∼1 μg/ml) for 4 h at 4 °C. After three washes with PBS, the cells were fixed with PBS, 4% formaldehyde,1 μg/ml Hoechst 33258 (300 μl/well) for 20 min at room temperature, followed by three final PBS washes and fluorescence scanning (bottom read) on a SpectraMax i3 plate reader (Molecular Devices). Bound PCSK9-mCherry was measured in each well as the average fluorescence intensity (587 nm excitation/612 nm emission) of 32 points equally distributed in a fill pattern.
PCSK9 Binding to HEK293 Cells Overexpressing WT and R410S LDL Receptor
HEK293 cells overexpressing LDLR-WT or LDLR-R410S and grown for 24 h in 6-well plates were placed on ice, washed with ice-cold PBS, and then incubated for 4 h at 4 °C with increasing concentrations of pre-cooled purified recombinant human PCSK9 (ACRO Biosystems), in DMEM serum-free media (0.9 ml/well). Subsequently, the cells were washed four times with ice-cold PBS (+Ca, +Mg) and lysed on ice in 0.5 ml/well of pre-cooled, non-denaturing cell lysis buffer (20 mm Tris-HCl, pH 8, 137 mm NaCl, 2 mm Na2EDTA, 1% Nonidet P-40, 10% glycerol) in the presence 4% protease inhibitor mixture without EDTA (Roche Applied Science) for 40 min with gentle rotation. Cell lysates were cleared by centrifuging for 12 min at 15,000 × g at 4 °C. The supernatants corresponding to the non-denatured cell lysates were subjected to measurement of cell-bound PCSK9 levels (in-house ELISA) (60), total human LDLR protein levels (human LDLR DuoSet ELISA development kit, DY218; R&D Systems; see below), and total protein (Bio-Rad DC Protein assay). PCSK9 binding to cells overexpressing the two receptors was estimated from the plot of nanograms of PCSK9 bound/ng of LDLR versus PCSK9 ligand concentration (μg/ml) and by fitting the data to a one-site binding equation (rectangular hyperbola) in SigmaPlot version 11 (Systat Software Inc.).
125I-LDL Binding, Uptake and Degradation Assays
LDL was prepared from fresh human plasma by sequential ultracentrifugation at a density range of 1.006–1.063 g/ml and was dialyzed and diluted to 3 mg/ml with saline (0.9% sodium chloride). Radioiodination of LDL was performed as described (62). The specific activity of the 125I-LDL was between 200 and 800 cpm/pg of protein, and more than 97% of the radioactivity was bound to protein as demonstrated by tricarboxylic acid (TCA) precipitation.
125I-LDL Binding Assay
Prior to the binding assay, HEK293 cells, seeded in 24-well plates and transfected as described above, were incubated for 24 h in DMEM serum-free media, washed once with 1 ml/well sample medium (DMEM, 25 mm HEPES, 2 mg/ml BSA, pH 7.45), and then incubated in 1 ml/well of the same medium for 10 min at 37 °C. Next, cells were placed on ice, pre-chilled for 15 min, and incubated for 1 h with increasing concentrations of precooled 125I-LDL (in 0.5-ml sample medium/well). Nonspecific binding was determined in the presence of a 50-fold excess of unlabeled LDL. Subsequently, the cells were washed four times with ice-cold PBS, 2 mg/ml BSA (1 ml/well), solubilized in 0.1 n NaOH (0.5 ml/well), and placed on a shaker for 1 h. In each well, the amount of ligand bound was determined by measuring the radioactivity of 0.250 ml of the solution in a Gamma counter (PerkinElmer Life Sciences catalog no. 2470). Specific 125I-LDL binding was calculated as total binding minus nonspecific binding. Protein content of each well was determined by the bicinchoninic acid protein assay (Pierce). Bound 125I-LDL was reported as kcpm/mg protein.
Internalization Assay
Cells were incubated continuously with 2 ng of 125I-LDL (in 0.5 ml of sample medium/well) at 37 °C for different times as follows: 15, 30, 45, 60, and 120 min. Next, cells were processed as for the binding assay. For each time point, internalized 125I-LDL was expressed as kcpm/mg protein.
Degradation Assay
The experiment was set up as described for the internalization assay, except with different time points as follows: 1–4, 6, and 8 h. At the end of each time point, the amount of degraded 125I-LDL was measured as the soluble 125I in the medium after protein precipitation with equal volumes of TCA. The TCA-precipitated proteins were centrifuged at 13,000 rpm for 5 min, and the radioactivity of 250 μl of the supernatant (TCA-soluble) was measured. The cells were processed as for the binding assay, and protein content of each well was determined by the bicinchoninic acid protein assay (Pierce). For each time point, degraded 125I-LDL was reported as kcpm/mg protein.
Fluorescence Spectroscopy Analysis of DiI-LDL Release into Media
HEK293 cells overexpressing LDLR-WT or LDLR-R410S and grown for 24 h in 24-well plates were placed on ice, washed with ice-cold PBS, and incubated with ice-cold DiI-LDL (6 μg/ml in 0.3 ml/well of fresh serum-free media) for 1 h at 4 °C. For each receptor, a control was incubated with a serum-free media void of DiI-LDL and used for background correction of each sample. After two washes with cold PBS, the DiI-LDL fluorescence in the last wash was similar to that of the background control, demonstrating that that all excess labeled LDL was washed off. Fresh pre-warmed serum-free medium was then added (0.3 ml/well), and the cells were incubated at 37 °C for different times as follows: 0, 10, 60, 120, and 240 min. For each time point, the media were collected and centrifuged at 2000 × g to remove any cell debris, and the DiI-LDL fluorescence intensity (520 nm excitation/575 nm emission) of the supernatant was measured on a SpectraMax i3 plate reader (Molecular Devices). DiI-LDL bound to the cells overexpressing either receptor was calculated from the difference between the fluorescence intensity of the DiI-LDL media before and after 1 h of incubation at 4 °C. DiI-LDL release into media was expressed relative to DiI-LDL bound at time 0.
Human LDLR ELISA in Cell Lysates
Following the treatments specific to each experiment, HEK293 cells were washed twice with ice-cold PBS and lysed on ice with ice-cold non-denaturing cell lysis buffer (20 mm Tris-HCl, pH 8, 137 mm NaCl, 2 mm Na2EDTA, 1% Nonidet P-40, 10% glycerol, 4% protease inhibitor mixture without EDTA) for 40 min with gentle rotation. Cell lysates were cleared by centrifuging for 12 min at 15,000 × g at 4 °C. The supernatants corresponding to the non-denatured cell lysates were saved and subjected to measurement of total human LDLR protein levels (human LDLR DuoSet ELISA development kit, DY218; R&D Systems) and of total protein (Bio-Rad DC Protein assay), following the manufacturers' protocol. The optical densities of the colored products were determined using a SpectraMax i3 plate reader (Molecular Devices). All measured LDLR concentrations (picograms/ml) were corrected for total protein concentration (micrograms/ml).
Statistical Analysis
Quantifications are defined as averages ± S.D. The statistical significance was evaluated by Student's t test, and probability values (p) of <0.05 were considered significant. Data were analyzed, and graphics were generated with SigmaPlot version 11 (Systat Software Inc.).
In Silico MD Simulation
MD simulation was run under the GROMACS package (63). All atoms were considered, making hydrogen atoms explicit. To characterize interactions between atoms in these systems, it has been shown that the force field AMBER99SB-ILDN (64) used in the simulations is perfectly appropriate. The initial configurations for the WT β-propeller and its mutant were extracted from the Protein Data Bank (PDB). The six blades of the β-propeller are interlocked via through-space interactions such that the whole system looks like a stable cylinder. Several PCSK9-LDLR crystals (PDB codes 3P5B and 3M0C) give incomplete coordinates for the β-propeller, particularly for an external loop that protrudes from the blade β2 surface (residues 457–464). In contrast, a crystal of the LDLR alone grown at acidic pH (PDB code 1N7D) could be used to prepare the input files as follows. The sequence 398–660 was numerically extracted from the PDB file, and it was N-capped as an acetamide. The C end was treated as a carboxylate. From that first WT input geometry, the mutant R410S was built with non-clashing. Finally, coordinates corresponding to the two pH 5 input files were generated by protonating the His residues (His to Hip). The polypeptides were then put in a box with periodic boundary conditions. Explicit water molecules following the TIP3P model (65) have been added. Moreover, Na+ or Cl− ions were also inserted to keep the charge of the whole system neutral. A typical simulation box contains 48,000 atoms and shows a volume of 474,552 (783) Å3.
The resulting systems were minimized using the robust steep descent algorithm with a 1000.0 kJ/mol/nm maximum force tolerance. Energies as a function of minimization steps were plotted to ensure that a minimal energy configuration was reached. Concerning the subsequent steps, the leap-frog integrator was employed to integrate Newton's equation of motion with a time step of 2 fs. The cutoff for the non-bond interactions was set to 1.0 nm. Long range electrostatics interactions were computed with the Particle Mesh Ewald (PME) algorithm (66). A first 100-ps MD run was carried out at 22 °C in the statistical microcanonical (NVT) ensemble, in which the number of particles, volume, and temperature are kept constant. It was verified that such duration is sufficient to reach a thermal equilibrium by plotting the temperature versus time. Berendsen thermostat was employed to tune the atom velocities in agreement with the appropriate Maxwell distribution (67). A 1-ns MD in the NPT ensemble was then run by keeping the number of particles, the pressure, and the temperature constant. The pressure was considered equilibrated at 1.0 bar. The isotropic Parrinello-Rahman barostat implemented into the GROMACS package was employed to ensure the specified pressure with the isothermal compressibility of water (68). Temperature and pressure as function of time were plotted to certify the equilibration of those properties. The system was then considered to be in equilibrium making possible data acquisition. MDs were run in the NPT for 30 ns at a temperature of 300 K (27 °C). The acid environment has also been considered. For this, the above procedure was repeated with the protonated histidine. Analysis of the backbone motion was achieved through the computation of the root mean squared fluctuations (RMSF) (standard deviation), defined by Equation 1,
| (Eq. 1) |
The positions' fluctuations for the α-carbons were calculated with respect to the average configuration. Mean values were computed for each residue. A higher RMSF indicates a greater displacement of the atoms with time, and thus a higher mobility of the segments (69). Therefore, mobility of all the residues can be isolated, unveiling the contribution of each segment to the whole mobility of the molecule. To ease visualization, B-factors were calculated from the RMSF values using Equation 2.
| (Eq. 2) |
The relationship between the RMSF and B-factors is thoroughly described in the literature (70).Once the RMSF values were converted to temperature factors, all atom coordinates were written in a PDB file with their respective B-factor for future visualization. All this procedure was done using the RMSF code within GROMACS package (71).
Author Contributions
D. S.-R. and E. G. contributed equally to this work, conducted most of the experiments, and analyzed the results. R. E. prepared the primary hepatocytes. J. H. did most of the cloning work. M. C. A. was responsible for all cell cultures and FACS acquisition data. Some Western blottings were performed by C. B. Y. L. D., A. F., and A. S. performed all the in silico molecular dynamics simulations. A. B. identified the FH patient and followed the family history, clinical parameters, and treatments, sometimes with the help of Z. A. R. S. K. performed all the time-dependent 125I-LDL uptake and degradation experiments in HEK293 cells. N. G. S. analyzed the data and wrote the manuscript with D. S.-R. and E. G. as well as conceived and followed the project.
Supplementary Material
Acknowledgments
We are grateful to all members of the Seidah laboratory for technical support and discussions. We thank the following for technical assistance: Valérie Menier for cell culture, Éric Massicotte for FACS analyses, and Dominic Filion for microscopy and image analyses. We also thank Brigitte Mary for editorial assistance.
This work was supported in part by Canadian Institutes of Health Research Foundation Grant 148363 and Canada Research Chair 216684 and in part by Pfizer Aspire CV IIR Grant W1207162 and Fondation Leducq Grant 13CVD03 (to N. G. S.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movie S1.
- LDLc
- low density lipoprotein cholesterol
- FH
- familial hypercholesterolemia
- LDLR
- low density lipoprotein receptor
- apoB
- apolipoprotein B
- LOF
- loss-of-function
- GOF
- gain-of-function
- EGFA
- epidermal growth factor A
- ER
- endoplasmic reticulum
- WB
- Western blotting
- MD
- molecular dynamics
- aa
- amino acid
- RMSF
- root mean squared fluctuation
- TCA
- tricarboxylic acid
- PDB
- Protein Data Bank
- IRCM
- Institut de Recherches Cliniques de Montréal
- IRES
- internal ribosome entry site
- EGFP
- enhanced green fluorescent protein
- DiI-LDL
- 3,3′-dioctadecylindocarbocyanine-low density lipoprotein.
References
- 1.LaRosa J. C., Grundy S. M., Waters D. D., Shear C., Barter P., Fruchart J. C., Gotto A. M., Greten H., Kastelein J. J., Shepherd J., Wenger N. K., and Treating to New Targets (TNT) Investigators (2005) Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N. Engl. J. Med. 352, 1425–1435 [DOI] [PubMed] [Google Scholar]
- 2.Cannon C. P., Blazing M. A., Giugliano R. P., McCagg A., White J. A., Theroux P., Darius H., Lewis B. S., Ophuis T. O., Jukema J. W., De Ferrari G. M., Ruzyllo W., De Lucca P., Im K., Bohula E. A., et al. (2015) Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 372, 2387–2397 [DOI] [PubMed] [Google Scholar]
- 3.Brown M. S., and Goldstein J. L. (1986) A receptor-mediated pathway for cholesterol homeostasis. Science 232, 34–47 [DOI] [PubMed] [Google Scholar]
- 4.Goldstein J. L., Hobbs H. H., and Brown M. S. (2001) in The Metabolic and Molecular Bases of Inherited Disease (Valle D., Scrive C. R., Beaudet A. L., Sly W. S., Childs B., and Volgestein B., eds) pp. 2863–2913, McGraw-Hill, New York [Google Scholar]
- 5.Fokkema I. F., Taschner P. E., Schaafsma G. C., Celli J., Laros J. F., and den Dunnen J. T. (2011) LOVD Version 2.0: the next generation in gene variant databases. Hum. Mutat. 32, 557–563 [DOI] [PubMed] [Google Scholar]
- 6.Jeon H., and Blacklow S. C. (2005) Structure and physiologic function of the low density lipoprotein receptor. Annu. Rev. Biochem. 74, 535–562 [DOI] [PubMed] [Google Scholar]
- 7.Hobbs H. H., Brown M. S., and Goldstein J. L. (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1, 445–466 [DOI] [PubMed] [Google Scholar]
- 8.Sørensen S., Ranheim T., Bakken K. S., Leren T. P., and Kulseth M. A. (2006) Retention of mutant low density lipoprotein receptor in endoplasmic reticulum (ER) leads to ER stress. J. Biol. Chem. 281, 468–476 [DOI] [PubMed] [Google Scholar]
- 9.Ranheim T., Kulseth M. A., Berge K. E., and Leren T. P. (2006) Model system for phenotypic characterization of sequence variations in the LDL receptor gene. Clin. Chem. 52, 1469–1479 [DOI] [PubMed] [Google Scholar]
- 10.Etxebarria A., Benito-Vicente A., Palacios L., Stef M., Cenarro A., Civeira F., Ostolaza H., and Martin C. (2015) Functional characterization and classification of frequent low density lipoprotein receptor variants. Hum. Mutat. 36, 129–141 [DOI] [PubMed] [Google Scholar]
- 11.Koivisto U. M., Hubbard A. L., and Mellman I. (2001) A novel cellular phenotype for familial hypercholesterolemia due to a defect in polarized targeting of LDL receptor. Cell 105, 575–585 [DOI] [PubMed] [Google Scholar]
- 12.Bottomley M. J., Cirillo A., Orsatti L., Ruggeri L., Fisher T. S., Santoro J. C., Cummings R. T., Cubbon R. M., Lo Surdo P., Calzetta A., Noto A., Baysarowich J., Mattu M., Talamo F., De Francesco R., et al. (2009) Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 284, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 13.Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., and Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., et al. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 15.Maxwell K. N., and Breslow J. L. (2004) Adenoviral-mediated expression of Pcsk9 in mice results in a low density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. U.S.A. 101, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., et al. (2004) NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 17.Nassoury N., Blasiole D. A., Tebon Oler A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., and Seidah N. G. (2007) The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 18.Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., et al. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 19.Cohen J., Pertsemlidis A., Kotowski I. K., Graham R., Garcia C. K., and Hobbs H. H. (2005) Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 [DOI] [PubMed] [Google Scholar]
- 20.Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., and Deisenhofer J. (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lo Surdo P., Bottomley M. J., Calzetta A., Settembre E. C., Cirillo A., Pandit S., Ni Y. G., Hubbard B., Sitlani A., and Carfí A. (2011) Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNutt M. C., Kwon H. J., Chen C., Chen J. R., Horton J. D., and Lagace T. A. (2009) Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HEPG2 cells. J. Biol. Chem. 284, 10561–10570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein E. A., and Raal F. (2014) Reduction of low density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu. Rev. Med. 65, 417–431 [DOI] [PubMed] [Google Scholar]
- 24.Seidah N. G., Awan Z., Chrétien M., and Mbikay M. (2014) PCSK9: a key modulator of cardiovascular health. Circ. Res. 114, 1022–1036 [DOI] [PubMed] [Google Scholar]
- 25.Sabatine M. S., Giugliano R. P., Wiviott S. D., Raal F. J., Blom D. J., Robinson J., Ballantyne C. M., Somaratne R., Legg J., Wasserman S. M., Scott R., Koren M. J., Stein E. A., and Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators (2015) Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1500–1509 [DOI] [PubMed] [Google Scholar]
- 26.Bertolini S., Pisciotta L., Rabacchi C., Cefalù A. B., Noto D., Fasano T., Signori A., Fresa R., Averna M., and Calandra S. (2013) Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis 227, 342–348 [DOI] [PubMed] [Google Scholar]
- 27.Lind S., Olsson A. G., Eriksson M., Rudling M., Eggertsen G., and Angelin B. (2004) Autosomal recessive hypercholesterolaemia: normalization of plasma LDL cholesterol by ezetimibe in combination with statin treatment. J. Intern. Med. 256, 406–412 [DOI] [PubMed] [Google Scholar]
- 28.Langslet G., Emery M., and Wasserman S. M. (2015) Evolocumab (AMG 145) for primary hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 13, 477–488 [DOI] [PubMed] [Google Scholar]
- 29.Huijgen R., Kindt I., Defesche J. C., and Kastelein J. J. (2012) Cardiovascular risk in relation to functionality of sequence variants in the gene coding for the low density lipoprotein receptor: a study among 29,365 individuals tested for 64 specific low density lipoprotein-receptor sequence variants. Eur. Heart J. 33, 2325–2330 [DOI] [PubMed] [Google Scholar]
- 30.Awan Z., Seidah N. G., MacFadyen J. G., Benjannet S., Chasman D. I., Ridker P. M., and Genest J. (2012) Rosuvastatin, proprotein convertase subtilisin/kexin type 9 concentrations, and LDL cholesterol response: the JUPITER trial. Clin. Chem. 58, 183–189 [DOI] [PubMed] [Google Scholar]
- 31.Pedersen N. B., Wang S., Narimatsu Y., Yang Z., Halim A., Schjoldager K. T., Madsen T. D., Seidah N. G., Bennett E. P., Levery S. B., and Clausen H. (2014) Low density lipoprotein receptor class A repeats are O-glycosylated in linker regions. J. Biol. Chem. 289, 17312–17324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee D. H., and Goldberg A. L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 [DOI] [PubMed] [Google Scholar]
- 33.Poirier S., Mayer G., Poupon V., McPherson P. S., Desjardins R., Ly K., Asselin M. C., Day R., Duclos F. J., Witmer M., Parker R., Prat A., and Seidah N. G. (2009) Dissection of the endogenous cellular pathways of PCSK9-induced LDLR degradation: evidence for an intracellular route. J. Biol. Chem. 284, 28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denis M., Marcinkiewicz J., Zaid A., Gauthier D., Poirier S., Lazure C., Seidah N. G., and Prat A. (2012) Gene inactivation of PCSK9 reduces atherosclerosis in mice. Circulation 125, 894–901 [DOI] [PubMed] [Google Scholar]
- 35.Poirier S., Hamouda H. A., Villeneuve L., Demers A., and Mayer G. (2016) Trafficking dynamics of PCSK9-induced LDLR degradation: focus on human PCSK9 mutations and C-terminal domain. PLoS ONE 11, e0157230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abifadel M., Guerin M., Benjannet S., Rabès J. P., Le Goff W., Julia Z., Hamelin J., Carreau V., Varret M., Bruckert E., Tosolini L., Meilhac O., Couvert P., Bonnefont-Rousselot D., Chapman J., Carrié A., Michel J. B., Prat A., Seidah N. G., and Boileau C. (2012) Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis 223, 394–400 [DOI] [PubMed] [Google Scholar]
- 37.Weider E., Susan-Resiga D., Essalmani R., Hamelin J., Asselin M. C., Nimesh S., Ashraf Y., Wycoff K. L., Zhang J., Prat A., and Seidah N. G. (2016) Proprotein convertase subtilisin/kexin type 9 (PCSK9) single domain antibodies are potent inhibitors of low density lipoprotein receptor degradation. J. Biol. Chem. 291, 16659–16671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roubtsova A., Chamberland A., Marcinkiewicz J., Essalmani R., Fazel A., Bergeron J. J., Seidah N. G., and Prat A. (2015) PCSK9 deficiency unmasks a sex/tissue-specific subcellular distribution of the LDL and VLDL receptors in mice. J. Lipid Res. 56, 2133–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pitas R. E., Innerarity T. L., Weinstein J. N., and Mahley R. W. (1981) Acetoacetylated lipoproteins used to distinguish fibroblasts from macrophages in vitro by fluorescence microscopy. Arteriosclerosis 1, 177–185 [DOI] [PubMed] [Google Scholar]
- 40.Beglova N., Jeon H., Fisher C., and Blacklow S. C. (2004) Cooperation between fixed and low pH-inducible interfaces controls lipoprotein release by the LDL receptor. Mol. Cell 16, 281–292 [DOI] [PubMed] [Google Scholar]
- 41.Brown M. S., Anderson R. G., and Goldstein J. L. (1983) Recycling receptors: the round-trip itinerary of migrant membrane proteins. Cell 32, 663–667 [DOI] [PubMed] [Google Scholar]
- 42.Aulinskas T. H., Oram J. F., Bierman E. L., Coetzee G. A., Gevers W., and van der Westhuyzen D. R. (1985) Retro-endocytosis of low density lipoprotein by cultured human skin fibroblasts. Arteriosclerosis 5, 45–54 [DOI] [PubMed] [Google Scholar]
- 43.Beglova N., and Blacklow S. C. (2005) The LDL receptor: how acid pulls the trigger. Trends Biochem. Sci. 30, 309–317 [DOI] [PubMed] [Google Scholar]
- 44.Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., and Prat A. (2008) Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 45.Awan Z., Dubuc G., Faraj M., Dufour R., Seidah N. G., Davignon J., Rabasa-Lhoret R., and Baass A. (2014) The effect of insulin on circulating PCSK9 in postmenopausal obese women. Clin. Biochem. 47, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 46.Rudenko G., Henry L., Henderson K., Ichtchenko K., Brown M. S., Goldstein J. L., and Deisenhofer J. (2002) Structure of the LDL receptor extracellular domain at endosomal pH. Science 298, 2353–2358 [DOI] [PubMed] [Google Scholar]
- 47.Brown M. S., Herz J., and Goldstein J. L. (1997) LDL-receptor structure. Calcium cages, acid baths and recycling receptors. Nature 388, 629–630 [DOI] [PubMed] [Google Scholar]
- 48.Denis N., Palmer-Smith H., Elisma F., Busuttil A., Wright T. G., Bou Khalil M., Prat A., Seidah N. G., Chrétien M., Mayne J., and Figeys D. (2011) Quantitative proteomic analysis of PCSK9 gain of function in human hepatic HuH7 cells. J. Proteome Res. 10, 2011–2026 [DOI] [PubMed] [Google Scholar]
- 49.Zhang D. W., Garuti R., Tang W. J., Cohen J. C., and Hobbs H. H. (2008) Structural requirements for PCSK9-mediated degradation of the low density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benjannet S., Rhainds D., Hamelin J., Nassoury N., and Seidah N. G. (2006) The proprotein convertase PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 281, 30561–30572 [DOI] [PubMed] [Google Scholar]
- 51.Holla Ø. L., Cameron J., Tveten K., Strøm T. B., Berge K. E., Laerdahl J. K., and Leren T. P. (2011) Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J. Lipid Res. 52, 1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saavedra Y. G., Day R., and Seidah N. G. (2012) The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 is needed for the extracellular low density lipoprotein receptor (LDLR) degradation pathway. J. Biol. Chem. 287, 43492–43501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Butkinaree C., Canuel M., Essalmani R., Poirier S., Benjannet S., Asselin M.-C., Roubtsova A., Hamelin J., Marcinkiewicz J., Chamberland A., Guillemot J., Mayer G., Sisodia S. S., Jacob Y., Prat A., and Seidah N. G. (2015) Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9-mediated low density lipoprotein receptor degradation but interact with each other. J. Biol. Chem. 290, 18609–18620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canuel M., Sun X., Asselin M.-C., Paramithiotis E., Prat A., and Seidah N. G. (2013) Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS ONE 8, e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raal F., Panz V., Immelman A., and Pilcher G. (2013) Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high dose statin therapy. J. Am. Heart Assoc. 2, e000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tavori H., Fan D., Blakemore J. L., Yancey P. G., Ding L., Linton M. F., and Fazio S. (2013) Serum proprotein convertase subtilisin/kexin type 9 and cell surface low density lipoprotein receptor: evidence for a reciprocal regulation. Circulation 127, 2403–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poirier S., Mayer G., Benjannet S., Bergeron E., Marcinkiewicz J., Nassoury N., Mayer H., Nimpf J., Prat A., and Seidah N. G. (2008) The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283, 2363–2372 [DOI] [PubMed] [Google Scholar]
- 58.Topchiy E., Cirstea M., Kong H. J., Boyd J. H., Wang Y., Russell J. A., and Walley K. R. (2016) Lipopolysaccharide is cleared from the circulation by hepatocytes via the low density lipoprotein receptor. PLoS ONE 11, e0155030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rader D. J., and Kastelein J. J. (2014) Lomitapide and mipomersen: two first-in-class drugs for reducing low density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 129, 1022–1032 [DOI] [PubMed] [Google Scholar]
- 60.Dubuc G., Tremblay M., Paré G., Jacques H., Hamelin J., Benjannet S., Boulet L., Genest J., Bernier L., Seidah N. G., and Davignon J. (2010) A new method for measurement of total plasma PCSK9: clinical applications. J. Lipid Res. 51, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., and Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vassiliou G., and Stanley K. K. (1994) Exogenous receptor-associated protein binds to two distinct sites on human fibroblasts but does not bind to the glycosaminoglycan residues of heparan sulfate proteoglycans. J. Biol. Chem. 269, 15172–15178 [PubMed] [Google Scholar]
- 63.Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., and Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 66.Darden T., York D., and Edersen L. (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 67.Berendsen H. J., Postma J. P., van Gunsteren W. F., DiNola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 68.Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 [Google Scholar]
- 69.Vendome J., Posy S., Jin X., Bahna F., Ahlsen G., Shapiro L., and Honig B. (2011) Molecular design principles underlying β-strand swapping in the adhesive dimerization of cadherins. Nat. Struct. Mol. Biol. 18, 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuzmanic A., and Zagrovic B. (2010) Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 98, 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.