

Abstract
Despite recent advances in therapeutics, diabetic retinopathy remains a leading cause of vision impairment. Improvement in the treatment of diabetic retinopathy requires a better understanding of the molecular mechanisms that cause neurovascular complications, particularly in type 2 diabetes. Recent studies demonstrate that rodents fed a high fat diet exhibit retinal dysfunction concomitant with attenuated Akt phosphorylation. The purpose of the present study was to evaluate the impact of a high fat/high sucrose diet on retinal insulin signaling and evaluate the mechanism(s) responsible for the changes. Mice fed a high fat/sucrose diet exhibited attenuated Akt phosphorylation in the retina as compared with mice fed normal chow. Retinas of mice fed a high fat/sucrose diet also exhibited elevated levels of activated JNK as well as enhanced p70S6K1 autoinhibitory domain phosphorylation. In cells, JNK activation enhanced p70S6K1 phosphorylation and mTORC1-dependent activation of the kinase, as evidenced by enhanced phosphorylation of key substrates. Rictor phosphorylation by p70S6K1 was specifically enhanced by the addition of phosphomimetic mutations in the autoinhibitory domain and was more sensitive to inhibition of the kinase as compared with rpS6. Notably, rictor and IRS-1 phosphorylation by p70S6K1 attenuate insulin action through a negative feedback pathway. Indeed, p70S6K1 inhibition prevented the repressive effect of JNK activation on insulin action in retinas. Overall, the results identify the JNK/S6K1 axis as a key molecular mechanism whereby a high fat/sucrose diet impairs insulin action in retina.
Keywords: c-Jun N-terminal kinase (JNK), insulin resistance, mammalian target of rapamycin (mTOR), retina, Type 2 diabetes, insulin action, retinopathy
Introduction
Diabetic retinopathy is the leading cause of vision loss in working age Americans. Almost everyone with type 1 diabetes and most people with type 2 diabetes develop retinopathy within two decades of disease onset (1). The incidence of type 2 diabetes is rapidly on the rise and accounts for up to 95% of diabetes prevalence (2). However, the overwhelming majority of experimental studies on diabetic retinopathy have been performed using animal models of type 1 diabetes. Development of type 2 diabetes is associated with consumption of a diet containing high fat and sucrose content, which has become extremely common in Western populations (3). Thus, in the present study we set out to investigate potential mechanisms whereby a Western diet contributes to diabetes-induced vision loss.
Diabetic retinopathy is caused by a combination of hyperglycemia and a reduction in insulin-mediated signaling, which results in neurovascular changes in the retina. Insulin provides a protective neurotrophic stimulus to retina by activating the protein kinase Akt through a signaling pathway involving the insulin receptor substrate 1/2 (IRS1/2)2 (4). In response to insulin, the activation loop of Akt is phosphorylated at Thr-308 by phosphoinositide-dependent kinase 1 (PDK1) (5), whereas the C-terminal hydrophobic motif is phosphorylated at Ser-473 by mTORC2 (mammalian target of rapamycin (mTOR) in complex 2) (6). Insulin-induced phosphorylation of Akt at Thr-308 occurs before Ser-473 (7), and phosphorylation of both residues produces higher in vitro kinase activity (8); thus, phosphorylation of Ser-473 is a key indicator of Akt activity.
In addition to mTORC2, insulin also promotes activation of mTORC1 and the subsequent phosphorylation and activation of the 70-kDa ribosomal protein S6 kinase 1 (p70S6K1). Chronic activation of p70S6K1 induces cellular insulin resistance by a mechanism known as the “negative-feedback loop” (9, 10), whereby p70S6K1 directly phosphorylates IRS1/2 (11) and the mTORC2 subunit rictor (rapamycin-insensitive companion of TOR) (12) on inhibitory residues to suppress insulin signaling to Akt. Like other AGC kinases, p70S6K1 has a bilobal fold structure with coordinating N- and C-terminal lobes (13). Regulation of p70S6K1 occurs through complex multisite phosphorylation whereby full activation of the kinase requires phosphorylation of both the hydrophobic motif at Thr-389 and the activation loop of catalytic domain at Thr-229 (14, 15). When dephosphorylated, a basic pseudo-substrate domain within the C-terminal lobe serves an autoinhibitory role by blocking the kinase domain (16). Phosphorylation of the pseudo-substrate domain relieves autoinhibition of the kinase by inducing a conformational change that allows for subsequent phosphorylation by mTORC1 and PDK1 at Thr-389 and Thr-229, respectively (17).
Our laboratory (19) and others (18) recently reported that the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) phosphorylates the pseudo-substrate domain of p70S6K1. The chronic inflammatory response associated with type 2 diabetes is characterized by abnormal cytokine production and activation of JNK (20, 21). Mice consuming a high fat diet exhibit early inflammasome activation within inner retinal layers and induction of JNK (22). Activation of JNK by proinflammatory cytokines inhibits insulin action at least in part by promoting inhibitory serine phosphorylation and degradation of IRS1/2 (23). Thus, the inhibitory effects of JNK activation on insulin action are potentially mediated through enhanced p70S6K1 activation. In this regard, both p70S6K1 (24) and JNK knock-out mice (20) are protected against diet-induced insulin resistance, suggesting that JNK-mediated activation of p70S6K1 potentially represents a key molecular mechanism whereby diet-induced inflammation attenuates insulin action in the retina leading to diabetes-induced vision loss.
Recent studies demonstrate high fat diet-induced retinal degeneration (25) and impaired retinal function (22, 26) in rodent models of type 2 diabetes. Moreover, these studies show attenuation of Akt phosphorylation in synaptic layers of the retina concomitant with high fat diet-induced retinal dysfunction (25, 26). The attenuation of Akt phosphorylation is most clearly observed in the outer segments of the outer plexiform layer, which is made up of synapses between photoreceptors, bipolar cells, and horizontal cells. The goal of the present study was to assess the development of insulin resistance in the retina of mice fed a Western diet (i.e. one containing high fat and sucrose content) using phosphorylation of Akt as a biomarker and to evaluate the mechanism(s) responsible for the observed changes. In support of the previous reports, we found that Akt phosphorylation was attenuated in the retina of mice after 2-weeks of consuming a Western diet. Of significance, the retina of mice fed a Western diet exhibited activated JNK as well as enhanced phosphorylation of p70S6K1 at Thr-421/Ser-424. We also present evidence that activation of the JNK signaling pathway represses insulin action through phosphorylation and activation of p70S6K1. Indeed, p70S6K1 inhibition was sufficient to prevent the repressive effect of JNK activation on insulin action in explant retinas.
Results
Attenuated Akt Phosphorylation and Activation of JNK in the Retina of Mice Fed a Western Diet
Mice fed a Western diet for 2-weeks exhibited slightly elevated but not statistically different body weight (25.5 ± 1.1 versus 23.8 ± 0.6 g; p = 0.23) and blood glucose concentrations (189.5 ± 27.7 versus 163.5 ± 28.0 mg/dl; p = 0.53) as compared with mice receiving a control chow diet. Activation of Akt was assessed in the retina by examining its phosphorylation state. Phosphorylation of Akt on Ser-473 was attenuated in the retina of mice fed a Western diet for 2-weeks, as compared with mice receiving a control chow diet (Fig. 1A). One potential mechanism responsible for attenuated Akt phosphorylation is activation of JNK, which has been implicated in the development of complications associated with type 2 diabetes (20). Although the relative phosphorylation of JNK on Thr-183/Tyr-185 was similar (Fig. 1B), JNK expression was enhanced in the retina of mice fed a Western diet (Fig. 1C). Thus, phosphorylated JNK (Fig. 1D) as well as phosphorylation of its most well established substrate, c-Jun (Fig. 1E), were both enhanced in the retina of mice fed a Western diet. Phosphorylation of p70S6K1 on Thr-421/Ser-424 (Fig. 1F) and Thr-389 (Fig. 1G) was also elevated in the retina of mice fed a Western diet as compared with mice receiving a control diet. To assess the effect of a Western diet on p70S6K1 activity, we evaluated phosphorylation of rpS6 and rictor. As expected, phosphorylation of rpS6 on Ser-240/244 (Fig. 1H) and rictor on Thr-1135 (Fig. 1I) was enhanced in the retina of mice fed a Western diet, as compared with mice fed a control diet.
FIGURE 1.

Attenuated Akt phosphorylation and activation of JNK/p70S6K1 in the retina of mice fed a Western diet. Mice were fed either a Western (W) or control (C) diet for 2 weeks before retinal harvest as described under “Experimental Procedures.” Phosphorylation of Akt on Ser-473 (A), JNK phosphorylation on Ser-183/185 (B), JNK expression relative to GAPDH (C), JNK phosphorylation on Ser-183/185 relative to GAPDH (D.), c-Jun phosphorylation on Ser-63 (E.), p70S6K1 phosphorylation on Thr-421/Ser-424 (F) and Thr-389 (G), rpS6 phosphorylation on Ser-240/244 (H), and rictor phosphorylation on Thr-1135 (I) were assessed by Western blotting. Values are the means ± S.E. Statistical significance is denoted by: *, p < 0.05 versus control diet. Blots are representative of two experiments; within an experiment, two or three independent replicates were analyzed. Protein molecular mass in kDa is indicated at the left of the blots.
The JNK Agonist Anisomycin Promotes p70S6K1 Phosphorylation and Activation of the Kinase
To further investigate the effect of JNK activation on p70S6K1, HEK293E cells were chosen as a model system to directly manipulate these signaling pathways. After serum deprivation to reduce p70S6K1 phosphorylation, cells were treated with the JNK agonist anisomycin and/or insulin. Anisomycin enhanced phosphorylation of c-Jun on Ser-63 (Fig. 2A), whereas both insulin and anisomycin promoted p70S6K1 phosphorylation on Thr-421/Ser-424 (Fig. 2B). This is consistent with our report that JNK and mTORC1 coordinate phosphorylation of the p70S6K1 C-terminal domain (19). Similarly, administration of either insulin or anisomycin enhanced p70S6K1 phosphorylation on Thr-389 (Fig. 2C). Moreover, p70S6K1 phosphorylation at both Thr-421/424 and Thr-389 was increased to a greater extent in response to combined insulin and anisomycin treatment as compared with either one (Fig. 2, B and C). We assessed the effect of JNK activation on p70S6K1 activity by evaluating phosphorylation of rpS6, rictor, and eIF4B as well as expression of PDCD4. When cells were treated with either insulin or anisomycin, phosphorylation of rpS6 on Ser-240/244 was enhanced (Fig. 2D). Moreover, both insulin and anisomycin enhanced rictor phosphorylation on Thr-1135 (Fig. 2E) and eIF4B phosphorylation on Ser-422 (Fig. 2F); however, the effect of anisomycin on these sites was less than that of insulin alone, and no additive effect of insulin and anisomycin was observed. Phosphorylation of PDCD4 by p70S6K1 leads to its proteasome-dependent degradation (27). PDCD4 expression was reduced after administration of either insulin or anisomycin, and the effects were synergistic (Fig. 2G). Notably, anisomycin-induced phosphorylation of rpS6, rictor, eIF4B, and degradation of PDCD4 were absent in cells deficient for S6K1/2 (Fig. 2H). Overall, these findings demonstrate that JNK activation modulates phosphorylation of various p70S6K1 substrates.
FIGURE 2.

The JNK agonist anisomycin promotes p70S6K1 phosphorylation and activation of the kinase. HEK293E cells in culture were maintained in DMEM supplemented with 10% FBS and serum-deprived (SFM) for 2 h to repress phosphorylation of S6K1. During the last 30 min of serum deprivation, cells were treated with anisomycin (20 μm) and/or insulin (Ins; 100 nm) as indicated. Phosphorylation of c-Jun on Ser-63 (A), p70S6K1 on Thr-421/Ser-424 (B) and Thr-389 (C), rpS6 on Ser-240/244 (D), rictor on Thr-1135 (E), and eIF4B on Ser-422 (F) were assessed by Western blotting. G, expression of PDCD4 relative to GAPDH was also assessed by Western blotting. Protein loading was assessed by Ponceau S. Values are the means ± S.E. Statistical significance is denoted by the presence of different letters above each scatter plot on the graphs. Scatter plots with different letters are statistically different; p < 0.05. Results shown in A–G are representative of two independent experiments; within each experiment, three independent samples were analyzed. H, wild-type and S6K1/2 knock-out MEF were serum-deprived for 2 h and then treated with anisomycin (Aniso) as described above. Phosphorylation of rpS6 on Ser-235/236 and Ser-240/244, rictor on Thr-1135, and eIF4B on Ser-422 as well as expression of PDCD4, p70S6K1, and GAPDH were assessed by Western blotting. Blots shown in H are representative of three experiments; within each experiment, four independent samples were analyzed. Protein molecular mass in kDa is indicated at the left of the blots.
JNK Promotes p70S6K1 Phosphorylation and Facilitates mTORC1-dependent Activation of the Kinase
To further evaluate the role of JNK in activation of p70S6K1, we expressed a fusion protein composed of JNK1 and its activating kinase, MKK7, that allows for constitutive phosphorylation and activation of the expressed JNK1 (28). As a negative control, we used a variant of the fusion protein with mutations in the activating JNK1 phosphorylation sites (Thr-183/Tyr-185) that results in a JNK1 variant that cannot be activated (referred to hereafter as inactive JNK) (28). To ensure that mTORC1 signaling was active, cells were deprived of serum for 2 h and then treated with IGF1 to activate the kinase. As shown in Fig. 3A, IGF1 potently activated mTORC1, as assessed by phosphorylation of p70S6K1 and its downstream targets. Interestingly, IGF1 and anisomycin acted in an additive manner to promote phosphorylation of rictor and eIF4B but not rpS6 (Fig. 3A). Similarly, expression of active, but not inactive JNK enhanced p70S6K1 phosphorylation at Ser-421/Thr-424 as well as phosphorylation of rictor and eIF4B, but not rpS6, in IGF1-treated cells (Fig. 3B). On the other hand, knockdown of JNK with siRNA attenuated p70S6K1 phosphorylation on Thr-421/Ser-424 as well as phosphorylation of rpS6, rictor, and eIF4B (Fig. 3C). Similarly, anisomycin failed to promote phosphorylation of the p70S6K1 autoinhibitory domain or activation of the kinase in JNK1-deficient cells (Fig. 3D). Because both mTORC1 and JNK can phosphorylate the p70S6K1 autoinhibitory domain, we evaluated the role of mTORC1 in JNK-dependent activation of p70S6K1. Cells were treated with anisomycin in combination with either the mTORC1 inhibitor rapamycin or the mTORC1/2 inhibitor TORIN2. As expected, inhibition of mTORC1 attenuated but did not prevent the anisomycin-induced phosphorylation of p70S6K1 on Thr-421/Ser-424 (Fig. 3E). Notably, although treatment with rapamycin or TORIN2 had no effect on anisomycin-induced phosphorylation of JNK or c-Jun, both compounds dramatically attenuated phosphorylation of rpS6, rictor, and eIF4B (Fig. 3E). These findings suggest that JNK activation modulates p70S6K1 activity in an mTORC1-dependent manner.
FIGURE 3.

JNK promotes p70S6K1 phosphorylation and facilitates activation of the kinase in an mTORC1-dependent manner. A and B, HEK293E cells in culture were maintained in DMEM supplemented with 10% FBS and serum-deprived (SFM) for 2 h to repress phosphorylation of p70S6K1. Cells were then treated with anisomycin (20 μm) and/or IGF1 (10 ng/ml) as indicated for 30 min. B, cells were transfected with FLAG-MKK7-JNK1 (Active JNK), FLAG-MKK7-JNK1 APF variant (Inactive JNK), or an empty vector control (EV) before serum deprivation and stimulation with IGF1. C, cells were transfected with siRNA directed against JNK1/2. D, wild-type and JNK1 knock-out MEF were serum-deprived for 2 h and then treated with anisomycin (Aniso) as described previously. E, cells were serum-deprived for 2 h to repress p70S6K1 phosphorylation. During the last 30 min of serum deprivation, cells were treated with anisomycin (20 μm), rapamycin (100 nm, Rapa), TORIN2 (10 nm, TORIN), or a combination as indicated. Phosphorylation of p70S6K1 on Thr-421/Ser-424, rpS6 on Ser-240/244, rictor on Thr-1135, eIF4B on Ser-422, c-Jun on Ser-63, and JNK on Thr-183/Tyr-185 as well as expression of wild-type and FLAG-tagged JNK and actin or GAPDH were assessed by Western blotting. Blots shown in A–C, and E and D are representative of three and two experiments, respectively; within each experiment, three independent samples were analyzed. Protein molecular mass in kDa is indicated at the left of the blots.
Acidic Mutations in the Autoinhibitory Domain of p70S6K1 Promote Rictor Phosphorylation
It was previously demonstrated that phosphorylation sites in the autoinhibitory domain of p70S6K1 promoted its phosphorylation at Thr-229 by PDK1 and enhanced activity of the kinase toward rpS6 C-terminal peptides in vitro (29). Importantly, mTORC1-dependent phosphorylation of the linker domain at Thr-389 is necessary for activation loop phosphorylation at Thr-229 and, thus, activity of the kinase (30). In agreement with these previous studies, mutation of Thr-389 to a phosphomimetic Glu residue was sufficient to promote p70S6K1 phosphorylation on Thr-229 even in cells deprived of serum and treated with rapamycin to prevent Thr-389 phosphorylation (Fig. 4A). Moreover, when combined with the T389E mutation, phosphomimetic substitutions in the autoinhibitory domain (i.e. S411D/S418D/T421E/S424D also known as D3E) enhanced Thr-229 phosphorylation to a greater extent compared with T389E alone. To determine the effect of these variant constructs on signaling downstream of p70S6K1, we assessed rpS6, rictor, and eIF4B phosphorylation. The overall level of rpS6 (Fig. 4B), rictor (Fig. 4C), and eIF4B (Fig. 4D) phosphorylation was greatest with expression of the T389E/D3E variant, whereas phosphorylation of these substrates by the T389A/D3E variant was not different from wild-type p70S6K1. However, in combination with T389E, the D3E mutation did not enhance phosphorylation of rpS6 or eIF4B as compared with T389E alone. Alternatively, rictor phosphorylation at Thr-1135 was enhanced by the D3E/T389E variant as compared with the T389E variant (Fig. 4C). Thus, phosphorylation of the p70S6K1 autoinhibitory domain may be particularly important with regards to phosphorylation of rictor.
FIGURE 4.

Effect of acidic mutations in the autoinhibitory domain of p70S6K1. HEK293E cells in culture were maintained in DMEM supplemented with 10% FBS. Cells were transfected with either empty vector (EV), HA-tagged p70S6K1 (WT), or a variant containing one of the following mutations: T389E, D3E, T389E/D3E, T389A/D3E. To reduce p70S6K1 phosphorylation and activation of mTORC1, cells were serum-deprived for 2 h and treated with rapamycin for the last 30 min. A, phosphorylation of p70S6K1 on Thr-229, Thr-389, and HA-tagged p70S6K1 expression was assessed by Western blotting. To assess the effects of p70S6K1 variant expression on downstream targets, phosphorylation of rpS6 on Ser-240/244 (B), rictor on Thr-1135 (C), and eIF4B on Ser-422 (D) as well as expression of GAPDH and HA-tagged p70S6K1 were assessed by Western blotting. Protein loading was assessed by Ponceau S. Values are the means ± S.E. Statistical significance is denoted by the presence of different letters above the scatter plots on the graphs. Scatter plots with different letters are statistically different, p < 0.05. Representative blots from five experiments are shown; within each experiment, two technical replicates were analyzed. Protein molecular mass in kDa is indicated at the left of the blots.
Variance in PF-47086 Sensitivity of p70S6K1 Substrate Phosphorylation Sites
Many multitarget protein kinases exhibit variability in their Michaelis-Menten constants (Km) for different substrates. An estimate of the Km of a kinase for its substrates can be obtained by treating cells with various concentrations of a selective kinase inhibitor and subsequently assessing the phosphorylation of downstream targets (e.g. Ref. (31). Therefore, in the present study cells were treated with increasing concentrations of the p70S6K1-specific inhibitor PF-4708671, and phosphorylation of rictor was compared with that of rpS6. In cells maintained in complete culture media, phosphorylation of rictor exhibited increased sensitivity toward PF-4708671 as compared with phosphorylation of rpS6 (Fig. 5A). Similarly, when cells were serum-deprived and stimulated with anisomycin, a much lower concentration of PF-4708671 was sufficient to attenuate phosphorylation of rictor as compared with rpS6 (Fig. 5B). Moreover, insulin and/or anisomycin promoted rpS6 phosphorylation on Ser-240/244 in cells treated with PF-470861; however, the effect was attenuated as compared with untreated cells (Fig. 5C). Alternatively, insulin and anisomycin failed to enhance rictor phosphorylation in cells treated with PF-470861. Overall, the results suggest that p70S6K1 has less affinity for rictor as compared with rpS6.
FIGURE 5.
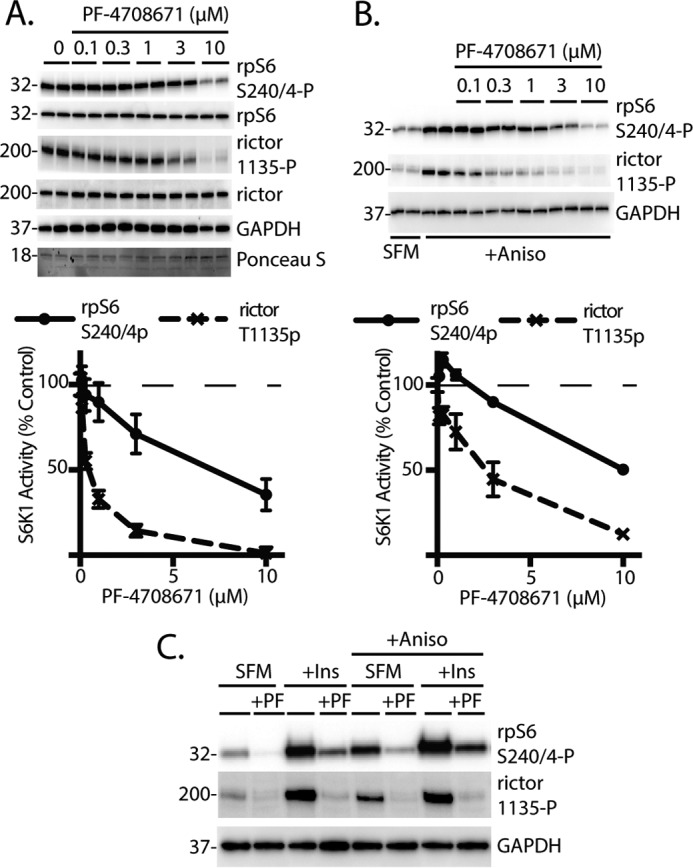
Effect of the p70S6K1 inhibition on phosphorylation of rpS6 and rictor. HEK293E cells in culture were maintained in DMEM supplemented with 10% FBS. A, HEK293E cells were treated with PF-4708671 (0.1–10 μm) as indicated. Protein loading was assessed by Ponceau S. B, cells were serum-deprived (SFM) for 2 h and treated with 0.1–10 μm PF-4708671 for the last 30 min. Cells were then treated with anisomycin (20 μm) for 30 min. Values are the means ± S.E. In some cases, bars representing S.E. are obscured. C, cells were serum-deprived and treated with 10 μm PF-4708671. Cells were then treated with anisomycin (20 μm) and/or insulin (10 nm) as indicated for 30 min. Phosphorylation rpS6 on Ser-240/244 and rictor on Thr-1135 as well as expression of GAPDH were assessed by Western blotting. Blots shown are representative of two experiments; within each experiment, three independent samples were analyzed. Protein molecular mass in kDa is indicated at the left of the blots.
Activation of JNK Represses Insulin Action through a p70S6K1-dependent Mechanism
It has been previously demonstrated that p70S6K1-mediated phosphorylation of rictor at Thr-1135 suppresses mTORC2-dependent phosphorylation and activation of Akt (12). Based on the finding that rictor phosphorylation was more sensitive to modulation of the kinase activity as compared with rpS6, we investigated the hypothesis that JNK attenuates insulin action by enhancing p70S6K1 activity. Expression of active JNK repressed insulin-stimulated phosphorylation of Akt on Ser-473 (Fig. 6A). Similarly, when cells were treated with anisomycin, insulin-stimulated phosphorylation of Akt on Ser-473 was also attenuated (Fig. 6B).
FIGURE 6.

JNK activation represses insulin action through an S6K1/2-dependent mechanism. A, HEK293E cells in culture were maintained in DMEM supplemented with 10% FBS. Cells were transfected with active JNK, inactive JNK, or an empty vector control (EV) as described in Fig. 3B. Cells were serum-deprived for 2 h to repress phosphorylation of S6K1 followed by treatment with insulin (Ins, 100 nm) as indicated. Blots shown in A are representative of four experiments performed in triplicate. Protein loading was assessed by Ponceau S. Phosphorylation of Akt on Ser-473 (B) and IRS-1 on Ser-636/639 (C) were evaluated by Western blotting in cells after 2 h of serum deprivation followed by treatment with insulin (Ins) and/or anisomycin (20 μm) for 30 min. Values are the means ± S.E. Statistical significance is denoted by the presence of different letters above the scatter plots on the graphs. Scatter plots with different letters are statistically different, p < 0.05. D, mice were fed either a Western (W) or control (C) diet as described in Fig. 1. Phosphorylation of IRS-1 on Ser-636/639 was assessed in retinal lysates by Western blotting. Values are the means ± S.E. Statistical significance is denoted by *; p < 0.05 versus control diet. Blots are representative of two experiments; within an experiment, three independent replicates were analyzed. E, wild-type and S6K1/2 knock-out MEF were serum-deprived for 2 h and then treated with anisomycin and/or insulin as described above. F, HEK293E cells were serum-deprived 2 h and then treated with anisomycin, rapamycin (100 nm, Rapa), TORIN2 (10 nm, TORIN), or a combination as indicated for 30 min. G, HEK293E cells in culture were maintained in DMEM supplemented with 10% FBS and serum-deprived for 2 h to repress phosphorylation of p70S6K1. Cells were then treated with anisomycin and/or IGF1 (10 ng/ml) as indicated for 30 min. H, HEK293E cells in culture were maintained in DMEM supplemented with 10% FBS or serum-deprived for 2 h followed by treatment with insulin or a combination of insulin and anisomycin for 0.5–60 min as indicated. Results shown in A, B–D, and E–H are representative of four, two, and three independent experiments, respectively; within each experiment two or three independent samples were analyzed. Protein molecular mass in kDa is indicated at the left of the blots.
In addition to rictor phosphorylation, p70S6K1 also represses insulin action and thus Akt phosphorylation through inhibitory serine phosphorylation of IRS-1 (9). When cells were treated with anisomycin, phosphorylation of IRS-1 on Ser-636/639 was enhanced (Fig. 6C). Similarly, phosphorylation of IRS-1 at Ser-636/639 was elevated in the retina of mice fed a Western diet (Fig. 6D), consistent with the idea that activation of the JNK/S6K1 axis promotes negative feedback. Importantly, the effect of anisomycin on IRS-1 phosphorylation was absent in cells deficient for S6K1/2, showing that JNK1-induced IRS-1 phosphorylation requires these proteins (Fig. 6E). Furthermore, the effect of anisomycin on IRS-1 phosphorylation was attenuated by either rapamycin or TORIN2 (Fig. 6F) in a manner that was consistent with p70S6K1 substrate phosphorylation observed in Fig. 3D. This supports the previous report that mTOR and JNK play coordinating roles in IRS-1 serine phosphorylation (32). The repressive effect of JNK activation on Akt phosphorylation is further supported by the finding that in combination with IGF1, anisomycin enhanced IRS-1 phosphorylation at Ser-636/639 and prevented Akt phosphorylation at Ser-473 (Fig. 6G). To further explore how p70S6K1 phosphorylation is enhanced over time, leading to repressed Akt phosphorylation, we performed a time-course where cells were treated with insulin in the presence or absence of anisomycin. Whereas insulin caused a progressive enhancement in p70S6K1 phosphorylation at Thr-421/Ser-424 over 60 min, the combination of insulin and anisomycin maximally enhanced p70S6K1 phosphorylation at Thr-421/Ser-424 after just 30 s and was associated with attenuated phosphorylation of Akt at Ser-473 (Fig. 6H). Thus, anisomycin-induced p70S6K1 autoinhibitory domain phosphorylation is associated with attenuated insulin-stimulated Akt phosphorylation.
To evaluate the effect of p70S6K1 activation on insulin-stimulated Akt phosphorylation, we performed chemical inhibition of p70S6K1 with PF-4708671. PF-4708671 promoted insulin-stimulated phosphorylation of Akt on Ser-473 in a dose-dependent manner (Fig. 7A). Whereas anisomycin attenuated insulin-stimulated Akt phosphorylation, cells treated with PF-470861 in combination with insulin and anisomycin exhibited similar Akt Ser-473 phosphorylation as cells treated with insulin alone (Fig. 7B). Despite the presence of the p70S6K1 inhibitor, anisomycin still modestly reduced insulin-stimulated phosphorylation of Akt at Ser-473 as compared with cells treated with insulin and PF-4708671 alone. Thus, JNK activation may also work through p70S6K1-independent mechanisms. Notably, anisomycin-induced phosphorylation of IRS-1, rictor, and rpS6 was absent in cells deficient for S6K1/2 but could be rescued with expression of S6K1 (Fig. 7C). Alternatively, expression of S6K2 was sufficient for anisomycin-induced rpS6 phosphorylation and modestly facilitated rictor phosphorylation. However, S6K2 was insufficient for anisomycin-induced IRS-1 phosphorylation. Thus, some of the effects of JNK activation on negative feedback pathways downstream of S6K1/2 may be facilitated by S6K2, but these effects were largely S6K1-dependent.
FIGURE 7.

Inhibition of p70S6K1 maintains insulin action in retinas upon JNK activation. A, HEK293E cells were serum-deprived for 2 h and treated with 0.1–10 μm PF-47086 for the last 30 min to inhibit p70S6K1. Cells were then treated with insulin (100 nm) for 30 min as indicated. B, cells were serum-deprived and treated with PF-47086 (10 μm) to inhibit p70S6K1. Cells were then treated with anisomycin and/or insulin as indicated for 30 min. C, S6K1/2 knock-out MEF were transfected with HA-tagged S6K1, HA-tagged S6K2, or an EV control followed by serum deprivation (SFM) and treatment with insulin and anisomycin as described above. Phosphorylation of rpS6 on Ser-240/244, rictor on Thr-1135, and eIF4B on Ser-422 as well as expression of Akt, IRS-1, GAPDH, and HA-tagged S6K1/2 were assessed by Western blotting. Blots shown in A–C are representative of three independent experiments; within each experiment three independent samples were analyzed. D, explant tissue culture was used to study insulin action in retina without the influence of the blood-retinal barrier. Mouse retinas were removed and incubated in tissue culture medium in either the presence or absence of PF-4708671 (10 μm) for 15 min. Retinas were treated with anisomycin (20 μm) and/or insulin (100 nm) as indicated for 30 min. Phosphorylation of Akt on Ser-473, p70S6K1 on Thr-421/Ser-424, and c-Jun on Ser-63 were evaluated by Western blotting. Values are the means ± S.E. Statistical significance is denoted by the presence of different letters above the scatter plots on the graphs. Scatter plots with different letters are statistically different; p < 0.05. Results in D are representative of two experiments; within each experiment, two or three independent samples were analyzed. Protein molecular mass in kDa is indicated at the left of the blots. E, working model for the mechanism whereby a Western diet attenuates insulin action in the retina.
Inhibition of p70S6K1 Maintains Insulin Action in Retinal Tissue upon JNK Activation
A retina explant system was used to evaluate the effect of JNK activation and p70S6K1 inhibition on insulin action in retinal tissue. In support of our findings in cells in culture, anisomycin-induced JNK activation enhanced phosphorylation of p70S6K1 at Thr-421/Ser-424 and c-Jun at Ser-63 in retinal explants (Fig. 7D). Moreover, insulin enhanced phosphorylation of Akt on Ser-473, whereas JNK activation with anisomycin prevented the effect. Alternatively, when retinas were treated with the p70S6K1 inhibitor PF-4708671, insulin-stimulated phosphorylation of Akt at Ser-473 was similar in the presence and absence of JNK activation. Thus, p70S6K1 inhibition blocked the repressive effect of JNK activation on insulin action in retina.
Discussion
In the present study we set out to investigate the early impact of a high fat/sucrose diet on insulin signaling in retina and explore the mechanisms responsible for the observed changes. Overall, the findings support a model where diet-induced JNK activation represses insulin action through p70S6K1-dependent negative feedback (Fig. 7B). A link between inflammation and insulin resistance in the retina has been previously proposed (33), yet direct evidence for the molecular mechanism has remained limited. High fat diet-induced retinal inflammation was recently reported in rodent models of type 2 diabetes (22, 34). Notably, feeding a high fat diet induces inflammatory markers and JNK phosphorylation in the inner retina before the onset of electroretinogram abnormalities or detectable vascular disease (22), suggesting that JNK activation is an early event in disease progression. Herein we extend the previous studies by demonstrating that JNK acts to modulate phosphorylation of substrates downstream of S6K1/2 by enhancing the phosphorylation of residues on rictor and IRS1 that are associated with attenuated insulin action.
In the retina of mice fed a Western diet for 2 weeks, Akt phosphorylation was attenuated as compared with control mice fed a control chow diet. Rodent models of type 1 diabetes exhibit progressive impairment of retinal insulin receptor signaling through Akt and suggest that attenuation of this pathway contributes to early stages of disease progression (35). In models of type 1 diabetes, attenuated Akt activity in the retina is typically reported in the absence of a change in the phosphorylation state of the protein (e.g. Ref. 35). Alternatively, attenuated retinal Akt phosphorylation has been observed in models of type 2 diabetes and was associated with high fat diet-induced retinal dysfunction (25, 26). Thus, the mechanisms whereby Akt signaling is impaired in type 1 and 2 diabetes are potentially different. However, enhanced JNK activity has been reported both in the retina of rats after streptozotocin administration (36) as wll as in the retina of mice consuming a high fat diet (22).
In addition to being regulated by phosphorylation in response to cell stress, JNK is also regulated by gene expression (37). In the present study retinas of mice fed a Western diet for 2 weeks exhibited elevated levels of activated JNK. However, with longer periods of high fat feeding this may not be the case. In the retina of mice fed a high fat diet for 3–12 months, no change in JNK expression was observed; however, JNK phosphorylation was elevated relative to expression of the protein (22). In the present study, JNK activation enhanced p70S6K1 phosphorylation and mTORC1-dependent activation of the kinase, as evidenced by enhanced phosphorylation of rpS6, eIF4B, rictor, and serine residues on IRS-1. Because rictor and IRS-1 phosphorylation by p70S6K1 attenuate insulin action upstream of Akt, JNK-mediated activation of p70S6K1 must be sufficient to promote kinase activity despite attenuated activation of Akt. Notably, anisomycin promoted phosphorylation of the p70S6K1 hydrophobic motif at Thr-389, a site most closely linked to mTORC1 kinase activity. Previous reports demonstrate that in response to osmotic stress, JNK promotes p70S6K1 phosphorylation at Thr-389 by directly phosphorylating raptor, a component of mTORC1 (38). This is in contrast to a more recent report that JNK contributes to disassociation of mTORC1 via direct phosphorylation of raptor and mTOR in response to proteotoxic stress (39). One possible explanation to reconcile these findings is that JNK-mediated phosphorylation of the p70S6K1 autoinhibitory domain is sufficient to promote mTORC1-dependent phosphorylation of Thr-389 despite attenuated mTORC1 activity (i.e. JNK phosphorylation makes p70S6K1 a better substrate for a less active mTORC1). This is consistent with the idea that phosphorylation of the C-terminal domain induces a conformational change in p70S6K1 that facilitates subsequent phosphorylation and ultimately full activation (17).
Notably, cells treated with insulin and anisomycin exhibited no further activation of p70S6K1 (based on phosphorylation of rpS6, rictor, and eIF4B) than cells treated with insulin alone. However, anisomycin enhanced IRS-1 phosphorylation, whereas insulin had no effect. This finding would seem to suggest that JNK is responsible for IRS-1 phosphorylation. However, anisomycin-induced IRS-1 phosphorylation at Ser-636/639, both, required S6K1/2 and was rapamycin-sensitive. One of the simplest explanations for these findings is that p70S6K1-dependent phosphorylation of IRS-1 at Ser-636/639 requires prior phosphorylation by JNK. Indeed, JNK co-immunoprecipitates with IRS-1 and phosphorylates the protein in a manner that is dependent on Ser-307 (40). Together these findings are consistent with a model wherein IRS-1 phosphorylation at Ser-307 by JNK is permissive of phosphorylation at Ser-636/639 by p70S6K1 (Fig. 7E).
The JNK inhibitor SP600125 was recently found to have protective effects on hyperglycemia-induced renal damage in a mouse model of type 1 diabetes (41). Although cells treated with SP600125 exhibit attenuated phosphorylation of the p70S6K1 autoinhibitory domain (18, 19), the use of this chemical inhibitor fails to provide meaningful insight into the role of JNK or the JNK/S6K1 axis per se, as SP600125 is equally effective at direct inhibition of a number of other kinases including p70S6K1 (42). In the present study we made use of a highly selective inhibitor of p70S6K1, PF-4708671. This compound does not inhibit the activity of S6K2 or a panel of 87 other protein and lipid kinases, with the exception of MSK1 (43).
PF-4708671 inhibited insulin- and anisomycin-induced phosphorylation of rpS6, rictor, and eIF4B. However, rictor phosphorylation was more sensitive PF-4708671, as compared with rpS6, suggesting that it is an inferior substrate of the kinase and principally phosphorylated when p70S6K1 is most active. The vast majority of studies evaluating the effect of autoinhibitory domain phosphorylation on p70S6K1 activity have assessed immune complex kinase activity toward an rpS6 C-terminal peptide (e.g. Refs. 30 and 44). Thus, little is known with regard to the impact of this domain on the phosphorylation of other p70S6K1 targets. To evaluate the role of JNK-mediated autoinhibitory domain phosphorylation on p70S6K1 activity, we expressed variants of the kinase containing phosphomimetic substitutions in the C terminus in combination with the T389E or T389A mutation. Notably, rictor phosphorylation was specifically enhanced by the D3E/T389E variant as compared with the T389E variant. Thus, JNK-mediated phosphorylation of the p70S6K1 autoinhibitory domain may shift substrate specificity of the kinase in favor of rictor by modulating access to the kinase domain.
Although diabetic retinopathy is more traditionally associated with the vascular lesions that accompany the later stages of this disease, numerous reports have conclusively demonstrated that neuro-retinal function is impaired before the onset of microvascular complications (for review, see Ref. 45). Accordingly, therapeutic approaches that prevent neuro-retinal dysfunction are of particular interest. One such approach would be to maintain insulin action in the retina, as impaired insulin signaling in the retina contributes to diabetes-induced neurodegeneration and blindness (25, 35, 46). This study identifies the JNK/S6K1 axis as a potential molecular target to maintain insulin action in retina and prevent complications associated with diabetic retinopathy.
Experimental Procedures
Animals
10-Week-old C57BL/6 male mice were obtained from Charles River. They were maintained on a 12:12-h reverse light dark cycle. Mice were fed ad libitum for 2 weeks, 4 h per day either a control (TD 08485) or a Western (TD 88137) diet containing 42% kcal from fat and 34% kcal from sucrose (Envigo). Mice were anesthetized by isoflurane inhalation 16 h after the previous feeding to control for acute effects of diet. Retinas were flash-frozen in liquid nitrogen, and lysates were prepared as previously described (47). Protein concentrations were assessed by the Bradford assay, and supernatants were combined with a 2× Laemmli buffer, boiled for 5 min, and analyzed via Western blotting.
Ex Vivo Retina Tissue Culture
For retinal explants, mice were fed the control diet ad libitum for 2 weeks before retinal extraction. Retinas were harvested and placed directly into Dulbecco's modified Eagle's medium (DMEM) containing 5.6 mmol/liter glucose, 10% FBS, and 1% penicillin-streptomycin. Retinas were rocked continuously for 15 min in a humidified atmosphere consisting of 5% CO2 at 37 °C and 5% CO2. Where indicated, retinas were treated with 10 μm PF-4708671 for 15 min before exposure to 100 nm insulin and/or 20 μm anisomycin for 30 min and homogenized as described above.
Cell Culture
HEK293E, S6K1/2 wild-type (WT), S6K1/2 double knock-out (DKO), JNK1−/−, and JNK1+/+ mouse embryonic fibroblasts (MEF) were maintained in DMEM supplemented with 5.6 mmol/liter glucose, 10% FBS, and 1% penicillin-streptomycin. S6K1/2 WT and DKO MEF were kind gifts from Dr. Sara Kozma (University of Cincinnati). JNK1−/− and JNK1+/+ MEF were kindly provided by Dr. Michael Karin (University of California, San Diego). For S6K1/2 and JNK1 overexpression studies, cells were transfected with Lipofectamine 2000 as previously described (48). Plasmids for expression of FLAG-MKK7-JNK1 (active JNK) and the FLAG-MKK7-JNK1 APF variant (inactive JNK) were obtained from Dr. Rodger Davis (University of Massachusetts Medical School) via Addgene. Plasmids for expression of HA-tagged p70S6K1 as well as T389E, D3E, T389E/D3E, and T389A/D3E variants were generously provided by Dr. John Blenis (Weill Cornell Medicine). The plasmid for expression of HA-tagged S6K2 was also obtained from Dr. Blenis via Addgene. Where indicated, cells were serum-deprived for 2 h, after which the following were added to the medium for 30 min: 10 nm TORIN2 (TOCRIS Bioscience), 100 nm rapamycin (Sigma), 0.1–10 μm PF-4708671 (Sigma), 20 μm anisomycin (Sigma), 100 nm insulin (Lilly), and 10 ng/ml IGF-1 (Sigma). For cell culture experiments, all inhibitors were prepared in the vehicle dimethyl sulfoxide (DMSO). As a vehicle control, cells were treated with DMSO in the absence of inhibitors. The cells were washed with sterile PBS, harvested in 200 μl of 1× Laemmli buffer, and boiled for 5 min before analysis by Western blotting.
Western Blotting Analysis
Lysates were fractionated using Criterion Pre-cast 4–20% gels (Bio-Rad). Proteins were transferred to PVDF, and the membrane was blocked in 5% milk, washed in TBST, and incubated with the appropriate primary antibody and secondary antibodies (see Table 1). The antigen-antibody interaction was visualized by enhanced chemiluminescence using a ProteinSimple Fluorochem E imaging system (Santa Clara, CA). Blots were quantified using Image J Software (NIH, Bethesda, MD).
TABLE 1.
Antibodies used for Western blotting
Primary and secondary antibodies used for Western blotting are listed by manufacturer, catalog number, and lot number. Species of origin as well as antibody specificity to human (H), mouse (M), and rat (R) are also indicated.
| Antibodies | Catalog # | Lot # | Species of origin | Specificity |
|---|---|---|---|---|
| Cell Signaling Technology antibodies | ||||
| AKT-p (Ser-473) | 4060 | 19 | Rabbit | H, M, R |
| AKT | 4691 | 27 | Rabbit | H, M, R |
| JNK-p (Thr-183/Tyr-185) | 9251 | 15 | Rabbit | H, M, R |
| SAPK/JNK | 9252 | 15 | Rabbit | H, M, R |
| c-Jun-p (Ser-63) | 9261 | 14 | Rabbit | H, M, R |
| c-Jun | 9165 | 14 | Rabbit | H, M, R |
| p70S6K1-p (Thr-421/Ser-424) | 9204 | 11 | Rabbit | H, M, R |
| p70S6K1-p (Thr-389) | 9234 | 11 | Rabbit | H, M, R |
| p70S6K1-T | 2708 | 11 | Rabbit | H, M, R |
| rpS6-p(Ser-240/Ser-244) | 5364 | 6 | Rabbit | H, M, R |
| rpS6-p(Ser-235/Ser-236) | 2211 | 11 | Rabbit | H, M, R |
| rpS6 | 2217 | 4 | Rabbit | H, M, R |
| Rictor-p (Tyr-1135) | 3806 | 5 | Rabbit | H, M |
| rictor | 9476 | 5 | Rabbit | H, M |
| eIF4B-p (Ser-422) | 3591 | 6 | Rabbit | H, M, R |
| eIF4B | 3592 | 3 | Rabbit | H, M, R |
| PDCD4 | 9535 | 4 | Rabbit | H, M, R |
| IRS-1-p (Ser-636/639) | 2388 | 14 | Rabbit | H, M, R |
| IRS-1 | 2382 | 4 | Rabbit | H, M, R |
| actin | 3700 | 14 | Mouse | H, M, R |
| Abcam antibodies | ||||
| p70S6K1-p (Ser-229) | ab59208 | GR145615-6 | Rabbit | H, M, R |
| Santa Cruz antibodies | ||||
| GAPDH | 6C5 | K0315 | Mouse | H, M, R |
| α-Tubulin | DM1A | C0112 | Mouse | H, M, R |
| p70S6K1-p (Ser-411) | sc-7983 | K1715 | Rabbit | H, M, R |
Statistical Analysis
Data are expressed as the mean ± S.E. Student's t test or one-way analysis of variance was used to analyze signaling data. Fischer's LSD test was used to identify specific differences if a significant overall F-value was observed after one-way analysis of variance. Relationships were determined by Pearson product moment correlation analysis. Significance was set at p < 0.05 for all analyses.
Author Contributions
W. P. M. researched the data and wrote the manuscript. S. R. and T. D. M researched the data. S. R. K. designed the experiments and reviewed/edited the manuscript. M. D. D. designed the experiments, researched the data, and wrote/reviewed/edited the manuscript.
Acknowledgments
We gratefully acknowledge Drs. Leonard Jefferson and Alistair Barber for critically evaluating this manuscript. We also thank Dr. Sara Kozma for providing S6K1/2 DKO MEF and Dr. Michael Karin for providing JNK knock-out MEF. Finally, we thank Allyson Torro and Joshua Moore for technical assistance in performance of the studies described herein.
This work was supported, in whole or in part, by National Institutes of Health Grants EY023612 (to M. D. D.) and DK13499 (to S. R. K.). This work was also supported by The American Diabetes Association Pathway to Stop Diabetes Grant 1-14-INI-04. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IRS1/2
- insulin receptor substrate 1/2
- PDK1
- phosphoinositide-dependent kinase 1
- mTORC2
- mammalian target of rapamycin (mTOR) in complex 2
- p70S6K1
- 70-kDa ribosomal protein S6 kinase 1
- D3E
- S411D/S418D/T421E/S424D
- DKO
- double knock-out
- MEF
- mouse embryonic fibroblasts.
References
- 1.Fong D. S., Aiello L. P., Ferris F. L. 3rd, and Klein R. (2004) Diabetic retinopathy. Diabetes Care 27, 2540–2553 [DOI] [PubMed] [Google Scholar]
- 2.U. S. Department of Health and Human Service (2014) National Diabetes Statistics Report, 2014: Estimates of Diabetes and Its Burden in the United States, p. 9, National Center for Chronic Disease Prevention and Health Promotion, Division of Diabetes Translation, CDC (Centers for Disease Control and Prevention) [Google Scholar]
- 3.Parillo M., and Riccardi G. (2004) Diet composition and the risk of type 2 diabetes: epidemiological and clinical evidence. Br. J. Nutr. 92, 7–19 [DOI] [PubMed] [Google Scholar]
- 4.Barber A. J., Nakamura M., Wolpert E. B., Reiter C. E., Seigel G. M., Antonetti D. A., and Gardner T. W. (2001) Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J. Biol. Chem. 276, 32814–32821 [DOI] [PubMed] [Google Scholar]
- 5.Alessi D. R., James S. R., Downes C. P., Holmes A. B., Gaffney P. R., Reese C. B., and Cohen P. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7, 261–269 [DOI] [PubMed] [Google Scholar]
- 6.Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 7.Humphrey S. J., Yang G., Yang P., Fazakerley D. J., Stöckli J., Yang J. Y., and James D. E. (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., and Hemmings B. A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 9.Shah O. J., Wang Z., and Hunter T. (2004) Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14, 1650–1656 [DOI] [PubMed] [Google Scholar]
- 10.Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R., Gout I., Downes C. P., and Lamb R. F. (2004) The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Gao Z., Yin J., Quon M. J., and Ye J. (2008) S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 283, 35375–35382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dibble C. C., Asara J. M., and Manning B. D. (2009) Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 29, 5657–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearce L. R., Komander D., and Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell. Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 14.Han J. W., Pearson R. B., Dennis P. B., and Thomas G. (1995) Rapamycin, wortmannin, and the methylxanthine SQ20006 inactivate p70s6k by inducing dephosphorylation of the same subset of sites. J. Biol. Chem. 270, 21396–21403 [DOI] [PubMed] [Google Scholar]
- 15.Magnuson B., Ekim B., and Fingar D. C. (2012) Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441, 1–21 [DOI] [PubMed] [Google Scholar]
- 16.Banerjee P., Ahmad M. F., Grove J. R., Kozlosky C., Price D. J., and Avruch J. (1990) Molecular structure of a major insulin/mitogen-activated 70-kDa S6 protein kinase. Proc. Natl. Acad. Sci. U.S.A. 87, 8550–8554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price D. J., Mukhopadhyay N. K., and Avruch J. (1991) Insulin-activated protein kinases phosphorylate a pseudosubstrate synthetic peptide inhibitor of the p70 S6 kinase. J. Biol. Chem. 266, 16281–16284 [PubMed] [Google Scholar]
- 18.Zhang J., Gao Z., and Ye J. (2013) Phosphorylation and degradation of S6K1 (p70S6K1) in response to persistent JNK1 Activation. Biochim. Biophys. Acta 1832, 1980–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin T. D., Dennis M. D., Gordon B. S., Kimball S. R., and Jefferson L. S. (2014) mTORC1 and JNK coordinate phosphorylation of the p70S6K1 autoinhibitory domain in skeletal muscle following functional overloading. Am. J. Physiol. Endocrinol. Metab. 306, E1397–E1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., and Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 21.Tang J., and Kern T. S. (2011) Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 30, 343–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajagopal R., Bligard G. W., Zhang S., Yin L., Lukasiewicz P., and Semenkovich C. F. (2016) Functional deficits precede structural lesions in mice with high fat diet-induced diabetic retinopathy. Diabetes 65, 1072–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguirre V., Uchida T., Yenush L., Davis R., and White M. F. (2000) The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser-307. J. Biol. Chem. 275, 9047–9054 [DOI] [PubMed] [Google Scholar]
- 24.Tremblay F., Brûlé S., Hee Um S., Li Y., Masuda K., Roden M., Sun X. J., Krebs M., Polakiewicz R. D., Thomas G., and Marette A. (2007) Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 104, 14056–14061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marçal A. C., Leonelli M., Fiamoncini J., Deschamps F. C., Rodrigues M. A., Curi R., Carpinelli A. R., Britto L. R., and Carvalho C. R. (2013) Diet-induced obesity impairs AKT signalling in the retina and causes retinal degeneration. Cell Biochem. Funct. 31, 65–74 [DOI] [PubMed] [Google Scholar]
- 26.Chang R. C., Shi L., Huang C. C., Kim A. J., Ko M. L., Zhou B., and Ko G. Y. (2015) High-fat diet-induced retinal dysfunction. Invest. Ophthalmol. Vis. Sci. 56, 2367–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorrello N. V., Peschiaroli A., Guardavaccaro D., Colburn N. H., Sherman N. E., and Pagano M. (2006) S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471 [DOI] [PubMed] [Google Scholar]
- 28.Lei K., Nimnual A., Zong W. X., Kennedy N. J., Flavell R. A., Thompson C. B., Bar-Sagi D., and Davis R. J. (2002) The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH2-terminal kinase. Mol. Cell. Biol. 22, 4929–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pullen N., Dennis P. B., Andjelkovic M., Dufner A., Kozma S. C., Hemmings B. A., and Thomas G. (1998) Phosphorylation and activation of p70s6k by PDK1. Science 279, 707–710 [DOI] [PubMed] [Google Scholar]
- 30.Dennis P. B., Pullen N., Pearson R. B., Kozma S. C., and Thomas G. (1998) Phosphorylation sites in the autoinhibitory domain participate in p70(s6k) activation loop phosphorylation. J. Biol. Chem. 273, 14845–14852 [DOI] [PubMed] [Google Scholar]
- 31.Kang S. A., Pacold M. E., Cervantes C. L., Lim D., Lou H. J., Ottina K., Gray N. S., Turk B. E., Yaffe M. B., and Sabatini D. M. (2013) mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiratani K., Haruta T., Tani A., Kawahara J., Usui I., and Kobayashi M. (2005) Roles of mTOR and JNK in serine phosphorylation, translocation, and degradation of IRS-1. Biochem. Biophys. Res. Commun. 335, 836–842 [DOI] [PubMed] [Google Scholar]
- 33.Liu X., Mameza M. G., Lee Y. S., Eseonu C. I., Yu C. R., Kang Derwent J. J., and Egwuagu C. E. (2008) Suppressors of cytokine-signaling proteins induce insulin resistance in the retina and promote survival of retinal cells. Diabetes 57, 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mancini J. E., Ortiz G., Croxatto J. O., and Gallo J. E. (2013) Retinal upregulation of inflammatory and proangiogenic markers in a model of neonatal diabetic rats fed on a high-fat-diet. BMC Ophthalmol. 13, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reiter C. E., Wu X., Sandirasegarane L., Nakamura M., Gilbert K. A., Singh R. S., Fort P. E., Antonetti D. A., and Gardner T. W. (2006) Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin. Diabetes 55, 1148–1156 [DOI] [PubMed] [Google Scholar]
- 36.Poulaki V., Joussen A. M., Mitsiades N., Mitsiades C. S., Iliaki E. F., and Adamis A. P. (2004) Insulin-like growth factor-I plays a pathogenetic role in diabetic retinopathy. Am. J. Pathol. 165, 457–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss L., Whitmarsh A. J., Yang D. D., Rincón M., Davis R. J., and Flavell R. A. (2000) Regulation of c-Jun NH2-terminal kinase (Jnk) gene expression during T cell activation. J. Exp. Med. 191, 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwak D., Choi S., Jeong H., Jang J. H., Lee Y., Jeon H., Lee M. N., Noh J., Cho K., Yoo J. S., Hwang D., Suh P. G., and Ryu S. H. (2012) Osmotic stress regulates mammalian target of rapamycin (mTOR) complex 1 via c-Jun N-terminal kinase (JNK)-mediated Raptor protein phosphorylation. J. Biol. Chem. 287, 18398–18407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su K. H., Cao J., Tang Z., Dai S., He Y., Sampson S. B., Benjamin I. J., and Dai C. (2016) HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat. Cell Biol. 18, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee Y. H., Giraud J., Davis R. J., and White M. F. (2003) c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J. Biol. Chem. 278, 2896–2902 [DOI] [PubMed] [Google Scholar]
- 41.Hong Z., Hong Z., Wu D., and Nie H. (2016) Specific MAPK inhibitors prevent hyperglycemia-induced renal diseases in type 1 diabetic mouse model. Mol. Cell Biochem. 419, 1–9 [DOI] [PubMed] [Google Scholar]
- 42.Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pearce L. R., Alton G. R., Richter D. T., Kath J. C., Lingardo L., Chapman J., Hwang C., and Alessi D. R. (2010) Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 431, 245–255 [DOI] [PubMed] [Google Scholar]
- 44.Hannan K. M., Thomas G., and Pearson R. B. (2003) Activation of S6K1 (p70 ribosomal protein S6 kinase 1) requires an initial calcium-dependent priming event involving formation of a high-molecular-mass signalling complex. Biochem. J. 370, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antonetti D. A., Barber A. J., Bronson S. K., Freeman W. M., Gardner T. W., Jefferson L. S., Kester M., Kimball S. R., Krady J. K., LaNoue K. F., Norbury C. C., Quinn P. G., Sandirasegarane L., Simpson I. A., and JDRF Diabetic Retinopathy Center Group (2006) Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 55, 2401–2411 [DOI] [PubMed] [Google Scholar]
- 46.Reiter C. E., and Gardner T. W. (2003) Functions of insulin and insulin receptor signaling in retina: possible implications for diabetic retinopathy. Prog. Retin. Eye Res. 22, 545–562 [DOI] [PubMed] [Google Scholar]
- 47.Dennis M. D., Kimball S. R., Fort P. E., and Jefferson L. S. (2015) Regulated in development and DNA Damage 1 is necessary for hyperglycemia-induced vascular endothelial growth factor expression in the retina of diabetic rodents. J. Biol. Chem. 290, 3865–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller W. P., Mihailescu M. L., Yang C., Barber A. J., Kimball S. R., Jefferson L. S., and Dennis M. D. (2016) The translational repressor 4E-BP1 contributes to diabetes-induced visual dysfunction. Invest. Ophthalmol. Vis. Sci. 57, 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]