

Abstract
Irreversible breakdown of cartilage extracellular matrix (ECM) by the collagenase matrix metalloproteinase 13 (MMP13) represents a key event in osteoarthritis (OA) progression. Although inflammation is most commonly associated with inflammatory joint diseases, it also occurs in OA and is thus relevant to the prevalent tissue destruction. Here, inflammation generates a cFOS AP-1 early response that indirectly affects MMP13 gene expression. To ascertain a more direct effect on prolonged MMP13 production we examined the potential molecular events occurring between the rapid, transient expression of cFOS and the subsequent MMP13 induction. Importantly, we show MMP13 mRNA expression is mirrored by nascent hnRNA transcription. Employing ChIP assays, cFOS recruitment to the MMP13 promoter occurs at an early stage prior to gene transcription and that recruitment of transcriptional initiation markers also correlated with MMP13 expression. Moreover, protein synthesis inhibition following early FOS expression resulted in a significant decrease in MMP13 expression thus indicating a role for different regulatory factors modulating expression of the gene. Subsequent mRNA transcriptome analyses highlighted several genes induced soon after FOS that could contribute to MMP13 expression. Specific small interfering RNA-mediated silencing highlighted that ATF3 was as highly selective for MMP13 as cFOS. Moreover, ATF3 expression was AP-1(cFOS/cJUN)-dependent and expression levels were maintained after the early transient cFOS response. Furthermore, ATF3 bound the proximal MMP13 AP-1 motif in stimulated chondrocytes at time points that no longer supported binding of FOS. Consequently, these findings support roles for both cFOS (indirect) and ATF3 (direct) in effecting MMP13 transcription in human chondrocytes.
Keywords: AP1 transcription factor (AP-1), arthritis, cell signaling, chondrocyte, inflammation, matrix metalloproteinase (MMP)
Introduction
Cartilage extracellular matrix (ECM)7 degradation within synovial joints (especially knee, hip, hands, and spine) is a pathological process synonymous with patient disability and pain in those suffering from either osteoarthritis (OA) or rheumatoid arthritis (RA). In reality, OA and RA are not single diseases but rather pathologies with various initiating factors that often include inflammation; both diseases have the common end point of irreversible destruction of the cartilage ECM. Although more marked in RA, inflammatory mediators released by immune cells (and joint tissue cells) orchestrate the aberrant production of catabolic factors that initiate destruction in both diseases. Numerous mediators including tumor necrosis factor α (TNFα), interleukin (IL)-17, and IL-1 have been reported to constitute this inflammatory response (1, 2). Our own findings have shown that such mediators, when in combination with the IL-6 family cytokines, oncostatin M (OSM) or IL-6 itself, promote marked expression of matrix metalloproteinases (MMPs) (3–7), the enzymes known to be responsible for cartilage damage. Potent expression of MMPs is therefore very likely within most inflammatory milieux, including OA.
Of the MMPs, it is specifically the collagenases MMP1 and MMP13 that are understood to cleave the main structural component of cartilage, type II collagen, thus effecting the irreversible loss of ECM architecture and function. Current dogma indicates MMP1 (produced by synovial fibroblasts) to be key in RA (predominantly a synovium-driven pathology), whereas MMP13 (synthesized by chondrocytes) is the major effector enzyme in OA, which is thought largely to be a cartilage-mediated disease. Although this representation is somewhat oversimplified, both enzymes remain key therapeutic targets for disease-modifying agents (8).
Under normal physiological conditions, chondrocytes maintain normal cartilage ECM homeostasis to enable smooth, frictionless joint articulation. In disease, a variety of stimuli such as Toll-like receptor ligands and inflammatory cytokines (3, 6, 9–11) elicit intracellular signaling cascades in chondrocytes that culminate in greatly increased MMP13 expression. Moreover, epigenetic studies have shown promoter alterations such as methylation status (12, 13) can further contribute to the elevated MMP13 expression, which is a hallmark of OA.
Regulation of eukaryotic gene expression represents a vital control mechanism that ensures appropriate cellular function. Transcriptional control can be supplemented at various levels, including alterations in ribosome translational efficiency and stability of mRNA transcripts and protein. Cartilage ECM proteolysis is subject to further regulation, because most MMPs are produced as inactive zymogens requiring proteolytic activation for activity (14), and a family of endogenous inhibitors (tissue inhibitors of metalloproteinases) provide additional control over the activities of these potent enzymes. Considerable research has focused on better understanding the molecular mechanisms by which transcription of MMPs is regulated, with the activator protein (AP)-1 transcription factor being considered a key factor for MMP expression (15–20). AP-1 is a protein dimer expressed ubiquitously, and is comprised of a FOS (cFOS, FOSB, FOSL1, FOS-like antigen (FRA)1 or FRA2) and JUN (cJUN, JUNB or JUND) heterodimer or a JUN homodimer. Variations in AP-1 composition dictate specificity and allow for differential gene regulation in specific tissues (21). Several studies including our own have confirmed AP-1 dependence for MMP13 induction by diverse stimuli including phorbol ester, parathyroid hormone, TNFα, IL-1β, and IL-17 (15–25), whereas we and others (17–19, 23, 24), have indicated cFOS·cJUN heterodimers are key regulators of MMP13 in chondrocytes. Moreover, we have shown pro-catabolic stimuli such as IL-1 + OSM markedly enhance MMP expression compared with IL-1 alone, both in vitro and in vivo (5–7, 19), with a concomitant increase in FOS induction (19). However, despite a functional role of FOS as an immediate early gene in regulating MMP13 expression via specific cell signaling pathways (18, 24–30), the complete mechanism underpinning transcriptional activation of this MMP remains to be defined.
In this context several studies suggest other transcriptional regulators may act, in concert with cFOS·cJUN, to effect MMP13 transcription (23, 25, 27–29). To further understand the regulation of MMP13 expression in the context of pathological ECM turnover, we have assessed MMP13 transcriptional regulation following a potent pro-catabolic stimulus known to promote cartilage destruction (5, 6, 31). Here, we demonstrate that cFOS·cJUN AP-1 is not the sole AP-1 contributor to the initiation of transcription of MMP13. We identify, for the first time, that cFOS·cJUN also induces activating transcription factor 3 (ATF3) to modulate MMP13 expression in chondrocytes. These data indicate a further mechanism in regulating this gene that may aid in defining more potential therapeutic targets to abrogate cartilage ECM degradation associated with OA.
Results
MMP13 Gene Expression Is Associated with an Early Induction of cFOS
We have previously reported that the potent cytokine combination of IL-1 + OSM induces time-dependent collagenolytic MMP13 expression, typically reaching maximal induction at 24 h (31–33). We now confirm a detailed time course for MMP13 induction in primary human articular chondrocytes, exhibiting detectable stimulation of mRNA expression from around 6 h post-stimulation (Fig. 1A). The expression of MMP13 was associated with an early, rapid but transient induction of FOS mRNA (Fig. 1B) as we have previously reported (33). This was also reflected in detectable cFOS protein in the nuclear fraction at 1 h, which persisted up to 3–6 h (Fig. 1, C and D), and was accompanied by cJUN phosphorylation (Fig. 1, E and F). These data are consistent with the prevailing paradigm of cFOS·phospho-cJUN dependence for proinflammatory cytokine induction of MMP13 (18, 19, 21–25, 34). Importantly, we verified that nascent hnRNA mirrored the mRNA expression indicative of transcription of the gene independent of mRNA synthesis/stability (Fig. 1G).
FIGURE 1.

IL-1 + OSM-stimulated MMP13 mRNA expression is preceded by nascent hnRNA formation and involves early induction of cFOS in human chondrocytes. Chondrocytes were stimulated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for the indicated durations. Total RNA was isolated, reverse transcribed, and subjected to real-time RT-PCR for MMP13 mRNA (A), cFOS mRNA (B), or MMP13 nascent hnRNA (G) as described under “Experimental Procedures.” Data are expressed relative to 18S rRNA and presented as fold-increase compared with basal expression. PCR data (n = 4) are representative of at least three separate chondrocyte populations. Cells were subjected to subcellular fractionation and protein from the cytosolic (a), membrane-bound (b), soluble nuclear (c), chromatin-bound, (d) and cytoskeletal (e) fractions resolved using SDS-PAGE and immunoblotted with antibodies to cFOS (C) or phosphorylated cJUN (E). Combined densitometric scans of five separate blots for each protein in the soluble nuclear fraction, relative to t = 0 h, are shown in D and F, respectively, and were obtained from separate chondrocyte populations. All data are presented as mean (± S.D.), where ***, p < 0.001; **, p < 0.01; *, p < 0.05 IL-1 + OSM-treated compared with control; ANOVA.
Enrichment of cFOS at the MMP13 Promoter Is an Early, Transient Response Event
Initially, ChIP assays revealed that cFOS enrichment was only detectable 1 h post-stimulation (Fig. 2A, inset), further suggesting an early, rapid but transient recruitment of cFOS to this promoter region. Further assays also confirmed enrichment of phospho-Ser5 RNA Pol II (pRNA Pol II) at the proximal MMP13 promoter 24 h post-stimulation (Fig. 2B, inset), in line with mRNA/hnRNA expression at this time point (see Fig. 1, A and B). In this context it is worthy to note that Ser5 phosphorylation of RNA Pol II has been shown to be concentrated near the promoter of genes, whereas Ser2 phosphorylation of RNA Pol II is observed throughout the gene (35). Consequently, no enrichment in the 3′-UTR of MMP13 was observed in the assays presented herein (see Fig. 2B). Assays employing an anti-acetyl(Lys9/14)-histone H3 antibody indicated histone H3 was acetylated at the MMP13 promoter at all time points (Fig. 2C) thus allowing appropriate regulation of transcription of the gene.
FIGURE 2.

ChIP analyses of the MMP13 proximal promoter. Human chondrocytes were treated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for the indicated durations. Cells were then subject to DNA-protein cross-linking, lysis, and DNA shearing. Immunoprecipitation for cFOS (A), pRNA Pol II (B), or AcH3 (C) was followed by isolation of complexed genomic DNA and subsequent real-time RT-PCR for the proximal (closed bars) and 3′-UTR (used for normalization; open bars) regions of MMP13 as indicated. Data (mean ± S.D.) are pooled from three (five for inset data) separate chondrocyte populations. Statistical comparisons are: *, p < 0.05 (24 h IL-1 + OSM stimulation versus basal); #, p < 0.05 (1 h IL-1 + OSM stimulation versus basal). D, chondrocytes were treated with IL-1 (0.05 ng/ml) and OSM (10 ng/ml) for 24 h. Emetine (10 μm final concentration) was added at the indicated times after IL-1 + OSM stimulation and real-time RT-PCR performed on extracted RNA relative expression levels of MMP13 mRNA were normalized to 18S rRNA, where ***, p < 0.001 (IL-1 + OSM + emetine versus basal); ###, p < 0.001 (IL-1+OSM versus basal). Data (mean ± S.D., n = 6) are representative of three separate experiments each using chondrocyte cultures from different donors.
Although there was a requirement for MMP13 induction by IL-1 + OSM via cFOS, the delay in transcription of hnRNA/mRNA combined with the rapid turnover of cFOS (see Fig. 1B–D) (36) indicated that other AP-1 regulators were synthesized following FOS induction, and thus contribute to transcription of the gene. A role for de novo protein formation was confirmed, after the early induction of cFOS, employing the protein synthesis inhibitor emetine. Here, 24 h IL-1 + OSM-stimulated cells were incubated with the inhibitor and MMP13 mRNA expression subsequently monitored. Emetine was observed to significantly reduce MMP13 expression, which persisted up to 6–8 h post-stimulation (see Fig. 2D). Consequently, IL-1 + OSM stimulation and induction of cFOS·p-cJUN heterodimers promote the rapid formation of transcriptional regulator(s), which further drive MMP13 expression. To determine these novel regulators, genome-wide microarray analyses were performed on primary chondrocyte populations stimulated at specific time points with IL-1 + OSM and where MMP13 induction occurred at 24 h. Analysis of the data confirmed that the 24-h time point had the highest number of changes in gene expression relative to basal (Fig. 3, A and B) as previously reported for a chondrocyte cell line (33). Furthermore, ingenuity pathway analysis of the 24-h dataset highlighted multiple proinflammatory signaling pathways, including IL-1β, IL-17, and TNFα (see supplemental Table S1), as well as multiple regulators and upstream regulators (supplemental Tables S2 and S3), indicating that IL-1 + OSM is indeed a potent model proinflammatory stimulus. Although heat maps (Fig. 3A) indicated relatively few changes in gene expression at the earlier time points; marked induction of several transcriptional regulators, including FOS, JUN, and ATF3, was observed at 1 (Fig. 3C) and 1.25 h (Fig. 3D). Strikingly, FOS was one of the most highly induced genes at 1 h (Fig. 3C), which almost halved by 1.25 h (Fig. 3D) and was one of the most highly down-regulated genes at 24 h (Fig. 3E) further illustrating the transient nature of FOS expression. MMP13 expression was detectable at 24 h but barely detectable at the early time points (Fig. 3, F and G), correlating with the transient expression profile of FOS and the more sustained expression of ATF3 (see Figs. 3F and 4). Furthermore, JUN expression was elevated and sustained at both 1 and 1.25 h (Fig. 3, C and D), whereas data from the arrays also indicated increased expression from 1 to 1.25 h of FOSL1, FOSB, JUNB, RELA (p65), immediate early responsive gene 3 (IER3), and interferon regulatory factor 1 (IRF1), which were all confirmed by real-time RT-PCR in multiple chondrocyte populations (Fig. 4). Subsequent siRNA assays were employed to determine the effect of knockdown of these and cFOS·cJUN transcriptional regulators on MMP13 expression in chondrocytes stimulated with IL-1 + OSM. Here, knockdowns of not only FOS/FOSL1 but also ATF3 resulted in substantial inhibition of MMP13 expression (p < 0.0001 compared with siRNA controls (see Table 1)).
FIGURE 3.

IL-1 + OSM stimulation induces expression of several transcriptional regulators prior to MMP13 expression. Human chondrocytes were treated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for the indicated time points and total RNA isolated. A–F, RNA from three separate populations (numbers 1, 2, and 3) was profiled using the Human HT-12v4 Expression Beadchip. A heat map of all significantly regulated genes is presented. It should be noted that the modest differences in gene expression between 1 and 1.25 h are reflected in these time points being grouped by chondrocyte population (A). Volcano plots are presented depicting genes significantly regulated >±2-fold, as indicated by the vertical dotted lines, at 24 (B), 1 (C), and 1.25 h (D), as well as a plot of change in fold-expression from 24 versus 1 h (E): genes of note are highlighted in red. F, expression profiles for MMP13, FOS, and ATF3 from the microarray dataset for each chondrocyte population are shown. G, RNA from six separate chondrocyte populations was subjected to real-time RT-PCR for MMP13, and relative expression levels (normalized to 18S rRNA) at the indicated times determined. The plots show mean ± S.D. (n = 4). Data from the populations used in the microarray analyses are highlighted by the solid symbols (#1, ▴; #2 ■; #3, ●).
FIGURE 4.

Expression of potential transcriptional MMP13 regulators following IL-1 + OSM stimulation. Six separate human chondrocyte populations were treated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for the indicated durations. Total RNA was isolated, reverse transcribed, and subjected to real-time RT-PCR for the indicated genes as described under “Experimental Procedures.” Data are expressed relative to 18S rRNA and presented as fold-increase compared with basal expression (mean ± S.D., n = 4). Note: the same symbols have been used as in Fig. 3E to denote individual populations.
TABLE 1.
MMP13 induction is primarily dependent on FOS family members and ATF3 following IL-1 + OSM stimulation of chondrocytes
Prior to stimulation with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml), human chondrocytes were transfected with siRNA specific for the indicated genes, or a non-targeting control (siCon; all 100 nm), and mRNA expression levels (mean ± S.D., n = 6) of MMP13 were measured at 24 h post-stimulation, relative to siCon-treated cells, normalized to 18S rRNA. The percentage inhibition of MMP13 expression relative to IL-1 + OSM + siCon was then calculated. Statistical comparisons are versus IL-1 + OSM + siCon (Student's two-tailed unpaired t test), where ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05. The data are pooled from three separate experiments, each using chondrocyte cultures from different donors.
| Gene (siRNA) | MMP13 |
|
|---|---|---|
| % Inhibition | p value | |
| ± S.E. | ||
| ATF3 | 42.4 (15.2) | **** |
| cFOS | 42.2 (9.5) | **** |
| FOSB | 29.8 (19.9) | ** |
| FOSL1 | 39.1 (8.9) | **** |
| IER3 | 18.4 (11.7) | * |
| IRF1 | 19.5 (14.8) | * |
| JUN | 23.9 (13.3) | ** |
| RELA | 17.7 (10.9) | * |
Prolonged Expression of ATF3 Modulates MMP13 Transcription
The above siRNA data thus implicated a potent role for ATF3 in regulating MMP13 expression. Temporal expression of ATF3 in IL-1 + OSM-treated cells was thus determined to observe any overlap with FOS expression. Here, ATF3 mRNA and nuclear protein was significantly increased at 1 h but maintained up to 24 h (see Fig. 5, A-C) in contrast to FOS expression (Fig. 1, B-D) where levels decreased substantially after 1 h. siRNAs knockdowns of FOS/JUN in IL-1 + OSM-stimulated cells were subsequently employed to determine any association with ATF3 expression. Knockdown of both FOS and JUN mRNA significantly reduced ATF3 mRNA levels (Fig. 5D). Control Western blots on nuclear extracts confirmed siRNA knockdowns of cFOS and phospho-cJUN was also mirrored at the protein level, where the latter also reflected total cJUN levels (see Fig. 5E). DNA affinity precipitation assays (DAPAs) were subsequently performed on IL-1 + OSM-stimulated cells to ensure that the temporal binding of cFOS and ATF3 to the MMP13 proximal promoter AP-1 element was reflective of their respective transient and sustained levels of expression. Here, specific AP-1 oligonucleotide-protein binding was initially determined in the presence of 50× excess non-biotinylated AP-1 oligonucleotide (data not shown). Further DAPAs confirmed the ability of both of these factors to bind at 1 h; whereas only ATF3 was still bound to this element 6 h post-stimulation (Fig. 6A). This was associated with the maintained expression of ATF3 at 6 h in contrast to the observed decreased expression of cFOS (see Fig. 6B, Western blot input).
FIGURE 5.

IL-1 + OSM induces sustained expression of ATF3, which is cFOS/cJUN-dependent. A, human chondrocytes were treated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for the indicated durations. Total RNA was isolated, reverse transcribed, and subjected to real-time RT-PCR for ATF3 as described under “Experimental Procedures.” Data are expressed relative to 18S rRNA and presented as fold-increase compared with basal expression (mean ± S.D., n = 4). All data are representative of three separate experiments each using chondrocyte cultures from different donors. B, cells were subjected to subcellular fractionation and proteins from the cytosolic (a), membrane-bound (b), soluble nuclear (c), chromatin-bound (d), and cytoskeletal (e) fractions resolved using SDS-PAGE and immunoblotted with an antibody to ATF3. C, relative density (compared with t = 0 h) data of combined densitometric scans for the soluble nuclear fractions of three separate blots (mean ± S.D.) are presented. All statistical comparisons are IL-1 + OSM-treated compared with basal (ANOVA), where ***, p < 0.001; **, p < 0.01. D, human chondrocytes were treated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for 1.25 h. Prior to stimulation, chondrocytes were transfected with siRNA specific for FOS, JUN, or a non-targeting control (siCon; all 100 nm). Following stimulation, total RNA was isolated, reverse transcribed, and subjected to real-time RT-PCR for ATF3. Data are expressed relative to 18S rRNA and plotted relative to the IL-1 + OSM + siCon stimulation (set to 100%; mean ± S.D., n = 8). Statistical comparisons are specific siRNA versus siCon (ANOVA), where ***, p < 0.001; **, p < 0.01. E, nuclear fractions were also isolated following stimulation and immunoblotted for cFOS and p-cJUN*S63 or the soluble nuclear marker protein lamin A/C to assess protein expression following siRNA knockdown.
FIGURE 6.
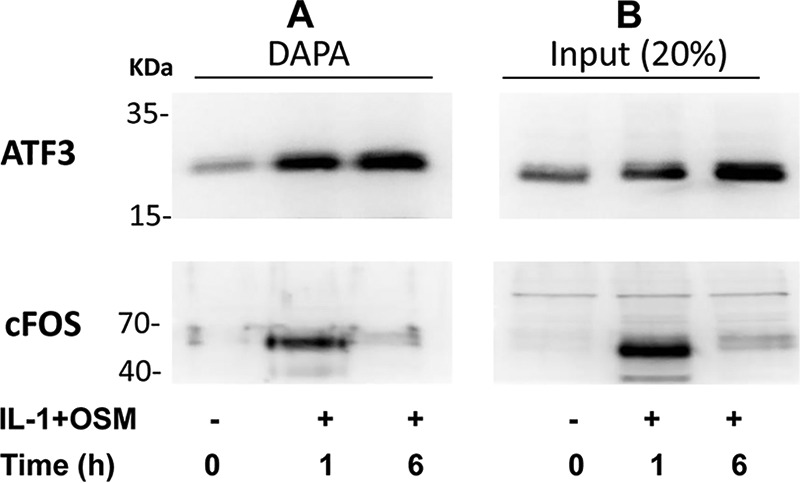
ATF3, but not cFOS, binds the proximal MMP13 AP-1 motif at a transcriptionally active time point in IL-1 + OSM-stimulated chondrocytes. Human chondrocytes were treated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for the durations indicated. Nuclear lysates were prepared and subjected to either DAPA with the ATF3 or cFOS antibodies (A) or Western blotting alone (input) (B), as described under “Experimental Procedures.” All data are representative of three separate experiments each using chondrocyte cultures from different donors.
Discussion
Proinflammatory cytokines that are associated with cartilage destruction and arthritis have been extensively reported by several groups including our own (6, 7, 9, 37, 38). In this context, cytokine combinations such as IL-1 + OSM represent highly useful model stimuli to study complex inflammatory mechanisms in arthritis, promoting cartilage breakdown (9, 31, 38–41) that correlates with significantly enhanced collagenolytic MMP expression (4–7, 18, 19, 30–33). Indeed, ingenuity pathway analysis of the 24-h time point indicated that the stimulus of IL-1 + OSM up-regulated numerous genes, many regulators of gene expression, associated with various other proinflammatory stimuli such as IL-1, IL-17, and TNFα, indicative of IL-1 + OSM being a potent model procatabolic stimulus.
Such cytokine-mediated inflammation is an important contributory factor for arthritis initiation and progression. This is the case not only in RA, where anti-cytokine biologics are now a key part of therapeutic strategies (41), but is also increasingly recognized in OA (2). Pathological inflammatory milieux are unlikely to comprise only a single mediator and so induction via several cytokines may be common; this may explain, in part, how relatively low level joint inflammation can nevertheless perpetuate cartilage destruction. Thus, the cytokine-stimulated molecular pathways and regulators that drive the expression of collagenolytic MMPs are likely to be common to many proinflammatory stimuli and therefore potential therapeutic targets.
MMP13 is thought to be the key collagenase in OA (8). Despite a confirmed role for AP-1 in the transcriptional activation of many MMP genes, including MMP13 (13, 15–19), there is a temporal discord between the expression of the rapid FOS response (typically 30–60 min) and the subsequent expression of collagenolytic MMPs (typically from 6 to 24 h). We have previously confirmed a role for cFOS·cJUN AP-1 regulators in MMP13 transcription (18, 19), and here further demonstrate the rapid, early but transient induction of FOS in human chondrocytes. ChIP assays, performed at a time when MMP13 mRNA was maximally detectable (24 h) confirmed no enrichment of cFOS to the proximal promoter region of the gene, although recruitment of phospho-Ser5 RNA Pol was observed at 24 h. This confirms there is indeed a delay between the time of FOS expression and when the MMP13 gene is transcribed. Although cFOS was recruited transiently to the promoter at early time points, no pRNA Pol II was detected thus suggesting the gene remained transcriptionally silent but potentially poised as acetylation of H3-histones was observed at all time points studied. This indicates that at early time points, recruitment of a cFOS·cJUN AP-1 complex may not be sufficient to effect MMP13 gene transcription. Moreover, this suggests that further transcriptional regulators are required for the marked MMP13 expression observed following IL-1 + OSM stimulation. Inhibition of protein synthesis after IL-1 + OSM stimulation and, importantly, following expression of cFOS protein (as well as cJUN phosphorylation) confirmed a role for the subsequent expression of different regulatory proteins in the transcriptional activation of MMP13. To determine these effectors, genome-wide microarray analyses of stimulated human chondrocytes was employed: this confirmed that FOS was one of the most highly induced genes following IL-1 + OSM stimulation at 1 h, and that this expression was very transient. Although heat maps indicated relatively few changes in gene expression between 1 and 1.25 h, probably reflecting in part the heterogeneity of primary chondrocyte populations, several genes (encoding known modulators of gene transcription) were nevertheless expressed at 1 h and appeared to have a more sustained gene expression profile at 1.25 h. These included ATF3, FOSL1, FOSB, JUNB, RELA, IER3, and IRF1. However, siRNA knockdowns of these factors indicated that only FOS family members and ATF3 had the most potent inhibitory effect on MMP13 expression. It is most probable that both FOSB and FOSL1 could also contribute to AP-1-dependent ATF3 expression, but herein we have focused on cFOS as this is considered a prime AP-1 effector protein for MMP expression (15–25) and silencing of this AP-1 member had the most marked effect on MMP13 induction. Our data thus implicate an important role for ATF3 in affecting MMP13 expression subsequent to the transcription of FOS. Expression of ATF3 at both the mRNA and protein levels indicated increased expression at the early 1-h time point as for cFOS but in contrast this was sustained over 24 h where maximum MMP13 levels were observed. Importantly, siRNA knockdowns of both FOS and JUN significantly reduced ATF3 expression, further confirming a transcriptional relationship between these factors and the continued expression of ATF3 in regulating MMP13 transcription. ATF3, like other bZIP transcriptional regulators, can form both homo- or heterodimers to either activate or repress gene expression, and in many instances is dependent on cell type and the promoter sequence of the target gene (42, 43). Indeed, the proximal AP-1 motifs for MMP1 and MMP13 are different, and whereas silencing ATF3 reduced MMP13 expression suggesting a transcriptional activator role, MMP1 expression was unaffected.8 Futhermore, binding to the proximal AP-1 promoter element of MMP13 was observed for both cFOS and ATF3 at the early 1-h time point with only ATF3 bound at the much longer time point of 6 h. Consequently, these studies provide strong evidence that ATF3 is transcriptionally dependent upon the initial inflammation-induced expression of cFOS·cJUN so as to mediate prolonged MMP13 expression. In this context, ATF3 is a well established stress-inducible gene (44), considered a hub for cellular adaptive responses (44, 45), which has previously been shown to be involved in chondrocyte differentiation (46). Moreover, very recent studies have shown that ATF3 regulates AP-1-dependent gene expression in murine osteoclast precursors (47), and that increased expression of ATF3 in mouse and human chondrocytes is a prime mediator of OA development (48). Importantly, deletion of ATF3 was observed to abrogate the onset of OA (48). In conclusion, data presented herein indicate that cytokine-induced expression of MMP13 is dependent on the initial transient formation of cFOS·cJUN dimers, which subsequently promote sustained formation of ATF3 to modulate expression of the gene.
Experimental Procedures
Materials
All chemicals were obtained from Sigma (Poole, UK) unless otherwise stated and of the highest purity available. All cytokines were recombinant human. IL-1α was a generous gift from Dr. Keith Ray (GlaxoSmithKline, Stevenage, UK). OSM was prepared in-house as described (49). siRNA reagents were screened for toxicity using the Toxilight assay of adenylate kinase release (Lonza, Wokingham, UK).
Chondrocytes
Human chondrocytes were isolated by enzymatic digestion of intact articular cartilage from OA patients undergoing joint replacement surgery as described (50). All subjects gave informed consent and the study was approved by the Newcastle and North Tyneside Joint Ethics Committee. Chondrocytes were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 IU of penicillin, 100 μg/ml of streptomycin, 40 units/ml of nystatin.
Cell Fractionation and Immunoblotting
Chondrocyte lysates were prepared as described previously (30, 50). In some experiments chondrocytes were subjected to subcellular fractionation using the NE-PER Nuclear and Cytoplasmic Protein Extraction Kit or Subcellular Protein Fractionation Kit (both from ThermoFisher Scientific, Loughborough, UK). Lysates or fractions were resolved by SDS-PAGE, transferred to PVDF membranes, and subsequently probed with the following antibodies: lamin A/C (number 4777) was purchased from Cell Signaling Technology (Danvers, MA); cFos (sc7202), cJun*S63 (sc7980), and ATF3 (sc188) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Transfection and RNA Interference
Primary human chondrocytes were prepared and cultured as above, and transfected as described previously (30). As an initial screen for modulation of MMP13 expression, Qiagen FlexiPlate small interfering RNA (siRNA) reagents were used. Dharmacon ON-TARGETplusTM SMARTpools® (ThermoFisher Scientific) of 4 specific siRNA duplexes (total of 100 nm siRNA; see supplemental Table S1) were used for follow-up studies where appropriate; silencing efficacy was ≥90% as assessed by Western blotting (data not shown). After transfection, cells were stimulated for 1.25 h to measure expression of transcriptional regulators, or 24 h for MMP genes. Changes in mRNA expression were calculated by comparison with cells transfected with 100 nm siCONTROL (siCon: non-targeting siRNA #2, catalog number 001210-02; Dharmacon).
Chromatin Immunoprecipitation (ChIP)
ChIP experiments were performed according to the standard protocol detailed in the EZ-ChIP kit (Merck-Millipore). Briefly, human chondrocytes were cultured until 70–80% confluent, at which point they were treated with IL-1 (0.05 ng/ml) in combination with OSM (10 ng/ml) for various durations. Cellular components were then cross-linked using formaldehyde for 5 min with agitation, and the reaction quenched with 0.125 m glycine for 5 min (with agitation). Cells were then washed twice and scraped into PBS supplemented with protease inhibitors. Cells were pelleted, resuspended in lysis buffer, and lysates were sonicated in a Bioruptor Plus (Diagenode) sonicating water bath (15 cycles of 30 s on, 30 s off; full power) to shear chromatin. Samples were then pre-cleared with the appropriate agarose beads for 1 h and subjected to overnight immunoprecipitation with relevant antibodies (10 μg) or corresponding isotype controls: anti-phospho-RNA polymerase II CTD repeats YSPTSPS (Ser5 pRNA Pol II; ab5131: Abcam, Cambridge, UK), anti-acetyl(Lys9/14)-histone H3 (AcH3, number 06-599; Millipore, Dundee, UK), and anti-cFos (Santa Cruz). Antibody·protein·DNA complexes were extracted with agarose beads and washed sequentially with kit-supplied buffers. DNA was then eluted and cross-links reversed (4 h incubation at 65 °C with 5 m NaCl, and then a 1-h incubation at 37 °C with RNase and proteinase K to degrade RNA and protein, respectively). DNA was purified by spin column and then used as input for SYBR Green real-time PCR, using the following MMP13 primers: proximal promoter/transcription start site: For, 5′-GAAAAAGTCGCCACGTAAGC-3′ and Rev, 5′-CGACAATGAGTCCAGCTCAA-3′; 3′-untranslated region (3′-UTR): For, 5′-TCGGCACAAAATACAGGTCA-3′ and Rev, 5′-GCCTCCCCTTTTTAGACCAC-3′. Data were normalized to background using isotype-control antibodies and variations in amount of DNA were normalized using the percent input method and shown as fold-change over basal in unstimulated cells at t = 0 h.
DAPA
Chondrocyte nuclear lysates were generated as described above (ThermoFisher Scientific). Nuclear protein (80 μg) was incubated with double-stranded biotinylated oligonucleotide (35 pmol) containing the proximal MMP13 AP-1 binding site (5′-[biotin]-TAAGTGATGACTCACCATTGC-3′; 5′-GCAATGGTGAGTCATCACTTA-3′) in 500 μl of binding buffer (12 mm HEPES, pH 7.9, 4 mm Tris-HCl, 60 mm KCl, 5% (w/v) glycerol, 0.5 mm EDTA, 1 mm DTT, and 1 mini-protease inhibitor mixture tablet (Roche)/10 ml of buffer) for 45 min at 4 °C. Streptavidin-coated agarose beads, pre-cleared with BSA, were then added and incubated for 2 h with rotation at 4 °C. Beads were pelleted by brief centrifugation, supernatants were discarded, and pellets washed three times with 1 ml of binding buffer for 5 min at 4 °C with rotation. Beads were finally resuspended in 50 μl of sample buffer, incubated at 100 °C for 5 min, pelleted, and supernatants removed for SDS-PAGE (as above).
Gene Expression Analyses
RNA was typically stabilized in cell lysates in a 96-well format and cDNA synthesized using the Cells-to-SignalTM kit (Life Technologies) as directed. Real-time PCR assays were conducted using primers (30) and conditions described previously (18, 34). TaqMan assays used Universal Probe Library probes (Roche Applied Sciences) as directed (see supplemental Table S4). Nascent hnRNA transcript expression of MMP13 was determined via reverse transcription and TaqMan (37) employing 5′-AGCACCCTTCTCATGACCTC-3′ (For) and 5′-TCCCCTGGTCTTGTGTGAG-3′ (Rev) primers spanning the Exon7/Intron7 boundary with probe number 82.
For genome-wide analyses, RNA was isolated from chondrocytes using RNeasy Mini Kit (Qiagen, Crawley, UK) according to the manufacturer's protocol. RNA from three individual patient samples was stimulated with IL-1 + OSM for three durations. Changes in whole genome expression were then profiled using the Human HT-12v4 Expression Beadchip (Illumina, Saffron Walden, UK). Amplification, labeling, hybridization, and detection were all performed by Cambridge Genomic Services (Cambridge University, UK). Raw expression data (GEO accession GSE86578) were analyzed using Agilent GeneSpring GX11 (Agilent Technologies, Santa Clara, CA). Raw data were normalized with a quantile algorithm and the baseline transformed to the median of all samples. Relative gene expression of the probe set was determined by normalizing raw expression values for each probe set to the unstimulated control from each chondrocyte population. The selection of genes for further investigation was based on detailed functional analysis of statistically significant gene expression performed with Ingenuity Pathways Analysis (Ingenuity Systems). Analysis of RNA expression data in the context of known biological response and regulatory networks allowed for the identification of genes with relevant biological function.
Statistical Analyses
Statistical differences between sample groups were assessed using one-way analysis of variance (ANOVA) with a post hoc multiple comparison test or Student's two-tailed unpaired t test, where ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05. For clarity, only selected comparisons are presented in some figures.
Author Contributions
A. D. R., G. J. L., and G. N. E.-F. designed the research; C. M. C., C. D. M., and D. J. W. performed the research; C. M. C., C. D. M., G. J. L., A. S., G. N. E.-F., and A. D. R. analyzed the data; G. N. E.-F. and A. D. R. wrote the paper.
Supplementary Material
Acknowledgments
We thank all the collaborators mentioned for generous provision of reagents. We are also grateful to the orthopaedic surgeons and research nurses at the Freeman and RVI hospitals in Newcastle for the generous supply of joint tissue. Clinical and translational research in the Musculoskeletal Research Group is supported by the Northumberland, Tyne and Wear Comprehensive Local Research Network.
This work was supported by the Nuffield Foundation (Oliver Bird Rheumatism Programme), the JGW Patterson Foundation, Arthritis Research Campaign Grants 18726, 19322, and 19485, and a United Kingdom National Institute for Health Research Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Hospitals National Health Service Foundation Trust. The authors declare that they have no conflicts of interest with the contents of this article. The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research, or the Dept. of Health.

This article contains supplemental Tables S1–S4.
C. D. Macdonald, G. J. Litherland, G. N. Europe-Finner, and A. D. Rowan, unpublished data.
- ECM
- extracellular matrix
- AP-1
- activator protein-1
- ATF3
- activating transcription factor-3
- DAPA
- DNA affinity precipitation assay
- IER3
- immediate early responsive gene 3
- IRF1
- interferon regulatory factor 1
- MMP13
- matrix metalloproteinase 13
- OA
- osteoarthritis
- OSM
- oncostatin M
- RA
- rheumatoid arthritis
- ANOVA
- analysis of variance.
References
- 1.Koenders M. I., Joosten L. A., and van den Berg W. B. (2006) Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Ann. Rheum. Dis. 65, iii29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldring M. B., and Goldring S. R. (2007) Osteoarthritis. Osteoarthritis. J. Cell. Physiol. 213, 626–634 [DOI] [PubMed] [Google Scholar]
- 3.Hui W., Cawston T., and Rowan A. D. (2003) Transforming growth factor β1 and insulin-like growth factor 1 block collagen degradation induced by oncostatin M in combination with tumour necrosis factor α from bovine cartilage. Ann. Rheum. Dis. 62, 172–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hui W., Rowan A. D., Richards C. D., and Cawston T. E. (2003) Oncostatin M in combination with tumor necrosis factor α induces cartilage damage and matrix metalloproteinase expression in vitro and in vivo. Arthritis. Rheum. 48, 3404–3418 [DOI] [PubMed] [Google Scholar]
- 5.Koshy P. J., Henderson N., Logan C., Life P. F., Cawston T. E., and Rowan A. D. (2002) Interleukin 17 induces cartilage collagen breakdown: novel synergistic effects in combination with proinflammatory cytokines. Ann. Rheum. Dis. 61, 704–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowan A. D., Hui W., Cawston T. E., and Richards C. D. (2003) Adenoviral gene transfer of interleukin-1 in combination with oncostatin M induces significant joint damage in a murine model. Am. J. Pathol. 162, 1975–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowan A. D., Koshy P. J., Shingleton W. D., Degnan B. A., Heath J. K., Vernallis A. B., Spaull J. R., Life P. F., Hudson K., and Cawston T. E. (2001) Synergistic effects of glycoprotein 130 binding cytokines in combination with interleukin-1 on cartilage collagen breakdown. Arthritis Rheum. 44, 1620–1632 [DOI] [PubMed] [Google Scholar]
- 8.Rowan A. D., Litherland G. J., Hui W., and Milner J. M. (2008) Metalloproteases as potential therapeutic targets in arthritis treatment. Expert Opin. Ther. Targets 12, 1–18 [DOI] [PubMed] [Google Scholar]
- 9.Zhang Q., Hui W., Litherland G. J., Barter M. J., Davidson R., Darrah C., Donell S. T., Clark I. M., Cawston T. E., Robinson J. H., Rowan A. D., and Young D. A. (2008) Differential Toll-like receptor-dependent collagenase expression in chondrocytes. Ann. Rheum. Dis. 67, 1633–1641 [DOI] [PubMed] [Google Scholar]
- 10.Radwan M., Gavriilidis C., Robinson J. H., Davidson R., Clark I. M., Rowan A. D., and Young D. A. (2013) Matrix metalloproteinase-13 expression in response to double-stranded RNA in human chondrocytes. Arthritis. Rheum. 65, 1290–1301 [DOI] [PubMed] [Google Scholar]
- 11.Bui C., Barter M. J., Scott J. L., Xu Y., Galler M., Reynard L. N., Rowan A. D., and Young D. A. (2012) cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 26, 3000–3011 [DOI] [PubMed] [Google Scholar]
- 12.Roach H. I., Yamada N., Cheung K. S., Tilley S., Clarke N. M., Oreffo R. O., Kokubun S., and Bronner F. (2005) Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 52, 3110–3124 [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto K., Otero M., Imagawa K., de Andrés M. C., Coico J. M., Roach H. I., Oreffo R. O., Marcu K. B., and Goldring M. B. (2013) Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J. Biol. Chem. 288, 10061–10072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Page-McCaw A., Ewald A. J., and Werb Z. (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auble D. T., and Brinckerhoff C. E. (1991) The AP-1 sequence is necessary but not sufficient for phorbol induction of collagenase in fibroblasts. Biochemistry 30, 4629–4635 [DOI] [PubMed] [Google Scholar]
- 16.Uchida M., Shima M., Chikazu D., Fujieda A., Obara K., Suzuki H., Nagai Y., Yamato H., and Kawaguchi H. (2001) Transcriptional induction of matrix metalloproteinase-13 (collagenase-3) by 1α,25-dihydroxyvitamin D3 in mouse osteoblastic MC3T3-E1 cells. J. Bone Miner. Res. 16, 221–230 [DOI] [PubMed] [Google Scholar]
- 17.Benbow U., and Brinckerhoff C. E. (1997) The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol. 15, 519–526 [DOI] [PubMed] [Google Scholar]
- 18.Litherland G. J., Elias M. S., Hui W., Macdonald C. D., Catterall J. B., Barter M. J., Farren M. J., Jefferson M., and Rowan A. D. (2010) Protein kinase C isoforms ζ and ι mediate collagenase expression and cartilage destruction via STAT- and ERK-dependent c-fos induction. J. Biol. Chem. 285, 22414–22425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Catterall J. B., Carrère S., Koshy P. J., Degnan B. A., Shingleton W. D., Brinckerhoff C. E., Rutter J., Cawston T. E., and Rowan A. D. (2001) Synergistic induction of matrix metalloproteinase 1 by interleukin-1α and oncostatin M in human chondrocytes involves signal transducer and activator of transcription and activator protein 1 transcription factors via a novel mechanism. Arthritis Rheum. 44, 2296–2310 [DOI] [PubMed] [Google Scholar]
- 20.Benderdour M., Tardif G., Pelletier J. P., Di Battista J. A., Reboul P., Ranger P., and Martel-Pelletier J. (2002) Interleukin 17 (IL-17) induces collagenase-3 production in human osteoarthritic chondrocytes via AP-1 dependent activation: differential activation of AP-1 members by IL-17 and IL-1β. J. Rheumatol. 29, 1262–1272 [PubMed] [Google Scholar]
- 21.Chinenov Y., and Kerppola T. K. (2001) Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20, 2438–2452 [DOI] [PubMed] [Google Scholar]
- 22.Hess J., Porte D., Munz C., and Angel P. (2001) AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J. Biol. Chem. 276, 20029–20038 [DOI] [PubMed] [Google Scholar]
- 23.Liacini A., Sylvester J., Li W. Q., Huang W., Dehnade F., Ahmad M., and Zafarullah M. (2003) Induction of matrix metalloproteinase-13 gene expression by TNF-α is mediated by MAP kinases, AP-1, and NF-κB transcription factors in articular chondrocytes. Exp. Cell Res. 288, 208–217 [DOI] [PubMed] [Google Scholar]
- 24.Schmucker A. C., Wright J. B., Cole M. D., and Brinckerhoff C. E. (2012) Distal interleukin-1β (IL-1β) response element of human matrix metalloproteinase-13 (MMP-13) binds activator protein 1 (AP-1) transcription factors and regulates gene expression. J. Biol. Chem. 287, 1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selvamurugan N., Chou W. Y., Pearman A. T., Pulumati M. R., and Partridge N. C. (1998) Parathyroid hormone regulates the rat collagenase-3 promoter in osteoblastic cells through the cooperative interaction of the activator protein-1 site and the runt domain binding sequence. J. Biol. Chem. 273, 10647–10657 [DOI] [PubMed] [Google Scholar]
- 26.Mengshol J. A., Vincenti M. P., and Brinckerhoff C. E. (2001) IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 29, 4361–4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vincenti M. P., Coon C. I., and Brinckerhoff C. E. (1998) Nuclear factor κB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1β-stimulated synovial fibroblasts. Arthritis Rheum. 41, 1987–1994 [DOI] [PubMed] [Google Scholar]
- 28.Westermarck J., Seth A., and Kähäri V. M. (1997) Differential regulation of interstitial collagenase (MMP-1) gene expression by ETS transcription factors. Oncogene 14, 2651–2660 [DOI] [PubMed] [Google Scholar]
- 29.Porte D., Tuckermann J., Becker M., Baumann B., Teurich S., Higgins T., Owen M. J., Schorpp-Kistner M., and Angel P. (1999) Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene 18, 667–678 [DOI] [PubMed] [Google Scholar]
- 30.Litherland G. J., Dixon C., Lakey R. L., Robson T., Jones D., Young D. A., Cawston T. E., and Rowan A. D. (2008) Synergistic collagenase expression and cartilage collagenolysis are phosphatidylinositol 3-kinase/Akt signaling-dependent. J. Biol. Chem. 283, 14221–14229 [DOI] [PubMed] [Google Scholar]
- 31.Cawston T. E., Curry V. A., Summers C. A., Clark I. M., Riley G. P., Life P. F., Spaull J. R., Goldring M. B., Koshy P. J., Rowan A. D., and Shingleton W. D. (1998) The role of oncostatin M in animal and human connective tissue collagen turnover and its localization within the rheumatoid joint. Arthritis Rheum. 41, 1760–1771 [DOI] [PubMed] [Google Scholar]
- 32.Koshy P. J., Lundy C. J., Rowan A. D., Porter S., Edwards D. R., Hogan A., Clark I. M., and Cawston T. E. (2002) The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: a time-course study using real-time quantitative reverse transcription–polymerase chain reaction. Arthritis Rheum. 46, 961–967 [DOI] [PubMed] [Google Scholar]
- 33.Barksby H. E., Hui W., Wappler I., Peters H. H., Milner J. M., Richards C. D., Cawston T. E., and Rowan A. D. (2006) Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: implications for cartilage destruction and repair. Arthritis Rheum. 54, 540–550 [DOI] [PubMed] [Google Scholar]
- 34.Vincenti M. P., and Brinckerhoff C. E. (2002) Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 4, 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komarnitsky P., Cho E. J., and Buratowski S. (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Healy S., Khan P., and Davie J. R. (2013) Immediate early response genes and cell transformation. Pharmacol. Ther. 137, 64–77 [DOI] [PubMed] [Google Scholar]
- 37.Yue C., Ponzio T. A., Fields R. L., and Gainer H. (2008) Oxytocin and vasopressin gene expression and RNA splicing patterns in the rat supraoptic nucleus. Physiol. Genomics 35, 231–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karsdal M. A., Madsen S. H., Christiansen C., Henriksen K., Fosang A. J., and Sondergaard B. C. (2008) Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity. Arthritis Res. Ther. 10, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durigova M., Troeberg L., Nagase H., Roughley P. J., and Mort J. S. (2011) Involvement of ADAMTS5 and hyaluronidase in aggrecan degradation and release from OSM-stimulated cartilage. Eur. Cell Mater. 21, 31–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Little C. B., Flannery C. R., Hughes C. E., Goodship A., and Caterson B. (2005) Cytokine induced metalloproteinase expression and activity does not correlate with focal susceptibility of articular cartilage to degeneration. Osteoarthritis Cartilage 13, 162–170 [DOI] [PubMed] [Google Scholar]
- 41.Thalayasingam N., and Isaacs J. D. (2011) Anti-TNF therapy. Best Pract. Res. Clin. Rheumatol. 25, 549–567 [DOI] [PubMed] [Google Scholar]
- 42.Gellersen B., Kempf R., and Telgmann R. (1997) Human endometrial stromal cells express novel isoforms of the transcriptional modulator CREM and up-regulate ICER in the course of decidualization. Mol. Endocrinol. 11, 97–113 [DOI] [PubMed] [Google Scholar]
- 43.Bailey J., Phillips R. J., Pollard A. J., Gilmore K., Robson S. C., and Europe-Finner G. N. (2002) Characterization and functional analysis of cAMP response element modulator protein and activating transcription factor 2 (ATF2) isoforms in the human myometrium during pregnancy and labor: identification of a novel ATF2 species with potent transactivation properties. J. Clin. Endocrinol. Metab. 87, 1717–1728 [DOI] [PubMed] [Google Scholar]
- 44.Hai T., Wolfgang C. D., Marsee D. K., Allen A. E., and Sivaprasad U. (1999) ATF3 and stress responses. Gene Expr. 7, 321–335 [PMC free article] [PubMed] [Google Scholar]
- 45.Nilsson M., Ford J., Bohm S., and Toftgård R. (1997) Characterization of a nuclear factor that binds juxtaposed with ATF3/Jun on a composite response element specifically mediating induced transcription in response to an epidermal growth factor/Ras/Raf signaling pathway. Cell Growth Differ. 8, 913–920 [PubMed] [Google Scholar]
- 46.James C. G., Woods A., Underhill T. M., and Beier F. (2006) The transcription factor ATF3 is upregulated during chondrocyte differentiation and represses cyclin D1 and A gene transcription. BMC Mol. Biol. 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukasawa K., Park G., Iezaki T., Horie T., Kanayama T., Ozaki K., Onishi Y., Takahata Y., Yoneda Y., Takarada T., Kitajima S., Vacher J., and Hinoi E. (2016) ATF3 controls proliferation of osteoclast precursor and bone remodeling. Sci. Rep. 6, 30918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iezaki T., Ozaki K., Fukasawa K., Inoue M., Kitajima S., Muneta T., Takeda S., Fujita H., Onishi Y., Horie T., Yoneda Y., Takarada T., and Hinoi E. (2016) ATF3 deficiency in chondrocytes alleviates osteoarthritis development. J. Pathol. 239, 426–437 [DOI] [PubMed] [Google Scholar]
- 49.Staunton D., Hudson K. R., and Heath J. K. (1998) The interactions of the cytokine-binding homology region and immunoglobulin-like domains of gp130 with oncostatin M: implications for receptor complex formation. Protein Eng. 11, 1093–1102 [DOI] [PubMed] [Google Scholar]
- 50.Cleaver C. S., Rowan A. D., and Cawston T. E. (2001) Interleukin 13 blocks the release of collagen from bovine nasal cartilage treated with proinflammatory cytokines. Ann. Rheum. Dis. 60, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.