

Abstract
Glucocorticoids are a general class of steroids that possess renoprotective activity in glomeruli through their interaction with the glucocorticoid receptor. However, the mechanisms by which glucocorticoids ameliorate proteinuria and glomerular disease are not well understood. In this study, we demonstrated that α actinin 4 (ACTN4), an actin-cross-linking protein known to coordinate cytoskeletal organization, interacts with the glucocorticoid receptor (GR) in the nucleus of human podocytes (HPCs), a key cell type in the glomerulus critical for kidney filtration function. The GR-ACTN4 complex enhances glucocorticoid response element (GRE)-driven reporter activity. Stable knockdown of ACTN4 by shRNA in HPCs significantly reduces dexamethasone-mediated induction of GR target genes and GRE-driven reporter activity without disrupting dexamethasone-induced nuclear translocation of GR. Synonymous mutations or protein expression losses in ACTN4 are associated with kidney diseases, including focal segmental glomerulosclerosis, characterized by proteinuria and podocyte injury. We found that focal segmental glomerulosclerosis-linked ACTN4 mutants lose their ability to bind liganded GR and support GRE-mediated transcriptional activity. Mechanistically, GR and ACTN4 interact in the nucleus of HPCs. Furthermore, disruption of the LXXLL nuclear receptor-interacting motif present in ACTN4 results in reduced GR interaction and dexamethasone-mediated transactivation of a GRE reporter while still maintaining its actin-binding activity. In contrast, an ACTN4 isoform, ACTN4 (Iso), that loses its actin-binding domain is still capable of potentiating a GRE reporter. Dexamethasone induces the recruitment of ACTN4 and GR to putative GREs in dexamethasone-transactivated promoters, SERPINE1, ANGPLT4, CCL20, and SAA1 as well as the NF-κB (p65) binding sites on GR-transrepressed promoters such as IL-1β, IL-6, and IL-8. Taken together, our data establish ACTN4 as a transcriptional co-regulator that modulates both dexamethasone-transactivated and -transrepressed genes in podocytes.
Keywords: Glucocorticoid receptor, podocyte, transcription coactivator, transcription corepressor, translation control, α actinin 4, podocyte, transactivation, transrepression
Introduction
It has been a longstanding clinical practice to use ligands for nuclear hormone receptors (NRs),2 including steroids, to treat kidney diseases despite the lack of a clear understanding of their mechanisms of action in this tissue. Recent studies in multiple experimental models have begun to explore the direct and indirect effects of NRs in renal cells to better utilize NR ligands as therapeutic agents for glomerular diseases such as minimal-change disease (MCD) and focal segmental glomerulosclerosis (FSGS) (1, 2). Glucocorticoids (GCs) as a general class of steroids possess renoprotective activity in glomeruli (3–7); however, steroid resistance and systemic toxicity remain major issues for their long-term use (4). This is, in part, due to a lack of understanding of the mechanism underlying transcriptional regulation by the glucocorticoid receptor (GRα) in podocytes, a key cell type in the glomerulus that forms the filtration barrier.
Glucocorticoids regulate a wide variety of physiological processes (8, 9). Both natural and synthetic GCs have shown to exert anti-inflammatory actions mediated by the glucocorticoid receptor, a member of the NR superfamily that functions as transcription factor and regulates gene expression in a cell type-specific and context-dependent manner (10, 11). In the absence of GCs, GR is sequestered by heat shock protein chaperones in the cytoplasm (12). When GCs are present, they bind to GR, allowing GR to dissociate from its chaperone proteins and translocate to the nucleus. Ligand-bound GR binds glucocorticoid response elements (GREs) or negative GREs and positively or negatively regulate target gene expression (13). Additionally, ligand-bound GR can repress transcription through interactions with other transcriptional regulators, such as NF-κB, a mechanism termed transrepression. The synthetic GC dexamethasone binds to GR and regulates its target gene expression in human podocytes (HPCs). Recent evidence showed that dexamethasone target genes affect the morphological and cytoskeletal response of podocytes (14–16). Dexamethasone can also relieve the podocyte apoptosis induced by puromycin aminonucleoside treatment (17) and down-regulate cytokines and VEGF expression in podocytes (15). These transrepressive effects have been proposed to explain why GCs are effective for the treatment of nephrotic syndrome. However, more research needs to be done to elucidate the mechanism mediated by dexamethasone, especially the potential side effects of dexamethasone on podocytes.
The actinin (ACTN) family proteins contain four members that bind filamentous actin and maintain cytoskeletal architecture (18). Multiple mutations in ACTN4 have been linked to FSGS (19, 20). Additionally, ACTN4 deficiency is also found in multiple human primary glomerulopathies, including sporadic FSGS, MCD, and IgA nephropathy (21–24). However, the molecular mechanisms by which ACTN4 maintains podocytes homeostasis and its biochemical activities remain largely unknown.
We have shown previously that ACTN4 potentiates transcriptional activation by MEF2 (25, 26), NF-κB (27, 28), and NRs (29–31). The newly identified nuclear function of ACTN4, its link to FSGS and MCD, and the beneficial effects of GCs on FSGS and MCD prompted us to investigate the role of ACTN4 in GC-mediated transcriptional regulation in podocytes and the effects of FSGS-linked ACTN4 mutations on GR transactivation activity. Our data define the novel function of ACTN4 as a co-activator that regulates GR-mediated transactivation in human podocytes. Furthermore, our study reveals a unique feature of ACTN4 in GR-mediated transrepression.
Results
Expression of GRα in Immortalized HPCs and Primary Mouse Podocytes
We have demonstrated previously that ACTN4 can reside in both the nucleus and the cytoplasm (29). To begin to investigate the role of ACTN4 in GR-mediated transcriptional regulation in HPCs, we first examined the expression and subcellular distribution of GRα in immortalized HPCs by immunofluorescence microscopy. In untreated HPCs, a large fraction of GRα is primarily localized in the cytoplasm. However, dexamethasone, a potent GRα agonist, induced nuclear translocation of GRα (Fig. 1).
FIGURE 1.
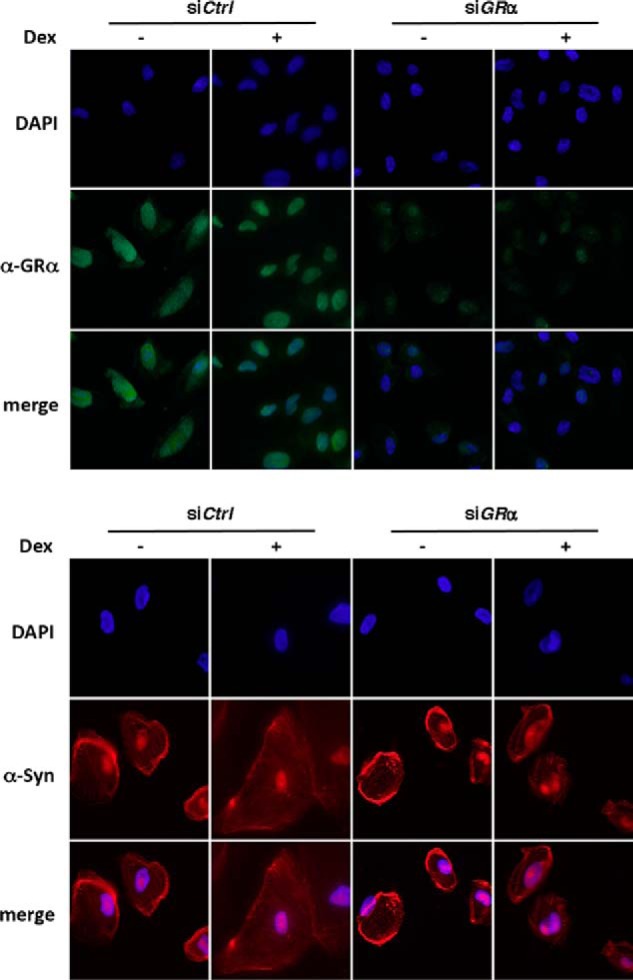
The effect of dexamethasone on nuclear translocation of GRα in podocytes. HPCs were transfected with either siCtrl or siGRα. Forty-eight hours post-transfection, cells were treated with or without 100 nm of dexamethasone (Dex) for 2 days. The cells were cultured for a total of 5 days, followed by immunostaining with anti-GRα or anti-synaptopodin antibodies and indirect fluorescence microscopy.
ACTN4 Potentiates and Interacts with GRα in a Ligand-dependent Manner
We have previously demonstrated that ACTN4 is a transcriptional coactivator for several transcription factors, including NRs (31). To test whether GR and ACTN4 interact in kidney cells, we carried out immunoprecipitation to examine whether endogenous GR and ACTN4 interact in HEK293T cells. In the absence of a GR agonist, α-ACTN4 and GRα did not interact; however, with the addition of dexamethasone, α-ACNT4 and GR co-immunoprecipitated (Fig. 2A). We also verified the association between ACTN4 and GR by GST pulldown assay (Fig. 2B). We then asked whether ACTN4 is also involved in GR-mediated transcriptional regulation. Overexpression of ACTN4 potentiates GRE reporter activity in a dose-dependent manner (Fig. 2C). In cultured human podocytes (Fig. 2D) and isolated mouse glomeruli (Fig. 2E), we similarly demonstrated that endogenous ACTN4 co-immunoprecipitated with GR. To further determine the role of ACTN4 in GR-mediated transcriptional activity, we carried out a transient transfection reporter assay in HPCs using a reporter construct harboring a GRE. We examined the role of ACTN4 in GR-mediated transcriptional regulation in a control HPC line and a previously established ACTN4 knockdown HPC cell line (shACTN4). We found that both the addition of dexamethasone and exogenously expressed GR increased reporter activity and that knockdown of ACTN4 significantly reduced GR- and dexamethasone-induced GRE activity (Fig. 2F). Based on these results, we conclude that ACTN4 potentiates dexamethasone-mediated GRE-driven reporter activity through its interaction with GR.
FIGURE 2.
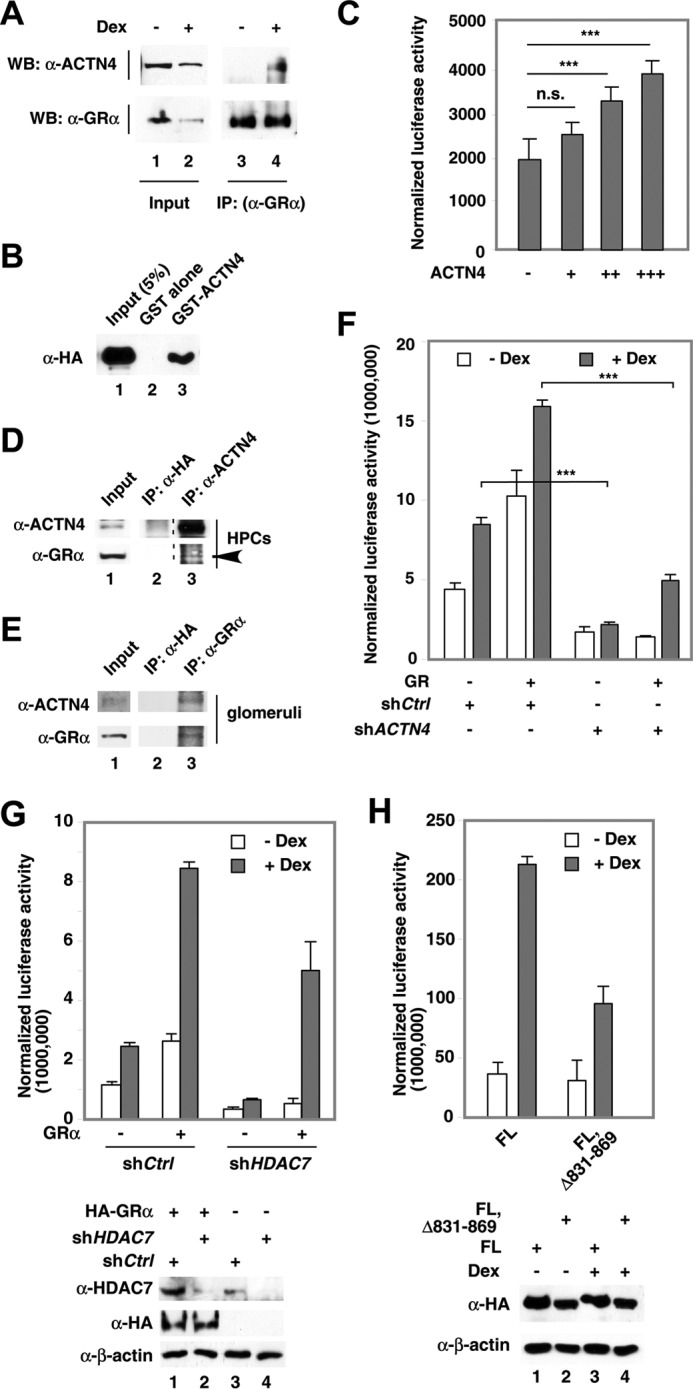
The effect of ACTN4 on GR-mediated transcriptional activation. A, HEK293T cells were treated with (+) or without (−) dexamethasone (Dex, 100 nm) for 8 h. Whole cell extracts were prepared and immunoprecipitated (IP) with anti-GR antibodies. Proteins were detected by Western blotting (WB) using anti-ACTN4 and anti-GRα antibodies as indicated. B, HeLa cells were transfected with an HA-GRα expression plasmid, and lysates were prepared and incubated with immobilized, bacterially expressed GST-ACTN4 fusion protein in the presence of dexamethasone. Pulldown fractions were subjected to Western blotting with HA antibodies. C, HEK293T cells were cotransfected with a GRE-luciferase reporter plasmid and HA-ACTN4 expression plasmid. The cells were then treated with dexamethasone for 12 h, and luciferase activity was measured. D, HPC whole cell extracts were prepared and immunoprecipitated with anti-ACTN4 antibodies and anti-HA antibodies as a control. Proteins were detected by Western blotting using anti-ACTN4 and anti-GR (arrowhead) antibodies as indicated. E, mouse glomerular extracts were prepared and immunoprecipitated with anti-GR or anti-HA antibodies, followed by Western blotting with anti-ACTN4 or GR antibodies. F, ACTN4 was stably knocked down by shRNAs in HPCs. shCtrl was used for comparison. A GRE-luciferase reporter plasmid and an HA-GR expression plasmid were transfected into shCtrl and shACTN4 HPCs, treated with vehicle (Veh) or dexamethasone for 12 h, and the reporter activity was analyzed. G, HDAC7 was stably knocked down by shRNAs in HPCs. shCtrl was used for comparison. A GRE-luciferase reporter plasmid and an HA-GR expression plasmid were transfected into shCtrl and shHDAC7 HPCs and treated with Veh or dexamethasone for 12 h, and the reporter activity was analyzed. H, HPCs were transiently transfected with ACTN4 (32), ACTN4 (FL, Δ831–869), ACTN4 (Iso), or ACTN4 (Iso, Δ441–479) and a GRE-Luciferase reporter plasmid and treated with or without dexamethasone, and reporter activity was measured. ***, p < 0.001; n.s., not significant.
ACTN4 was originally identified as an HDAC7-interacting protein (25). We therefore determined whether HDAC7 is required for ACTN4-mediated GR activity. Indeed, knockdown of HDAC7 decreased GRE-driven reporter activity with or without dexamethasone treatment (Fig. 2G). Furthermore, an ACTN4 mutant defective in interacting with HDAC7 (25) also lost its ability to potentiate GRE reporter activity (Fig. 2H). Based on these data, we conclude that HDAC7 regulates GR transactivation activity.
Mapping of the Interaction Domains between ACTN4 and GR
To understand the molecular basis of the interaction between ACTN4 and GR, we mapped the interaction domains by GST pulldown assays with several ACTN4 constructs. ACTN4 (Iso) is a spliced isoform that is missing amino acids 89–478 of the full-length protein (25). The C-terminal truncation mutant ACTN4 (Iso, 2–102) contains an intact LXXLL nuclear receptor-interacting domain and part of the calponin homology 1 domain, whereas ACTN4 (95–521) contains the remaining C terminus of the ACTN4 isoform that includes part of Spectrin repeat (SR) 2, SR3, SR4, and the calmodulin-like domain (Fig. 3A). Immobilized, purified GST-ACTN4 full-length (32), ACTN4 (Iso), and truncated ACTN4 (Iso) mutants were incubated with whole cell lysate expressing HA-GR, followed by Western blotting with anti-HA antibodies. We found that amino acids 2–102 are sufficient to bind GR. However, the C terminus did not interact with GR. We further mapped the ACTN4 interaction domain in GR (Fig. 3B). The N terminus (1–419) of GR contains the transcription activation function 1 domain and interacts with ACTN4 in a dexamethasone-independent manner. The C terminus (488–777) contains the ligand-binding domain and interacts poorly with ACTN4. However, this interaction is enhanced by dexamethasone.
FIGURE 3.
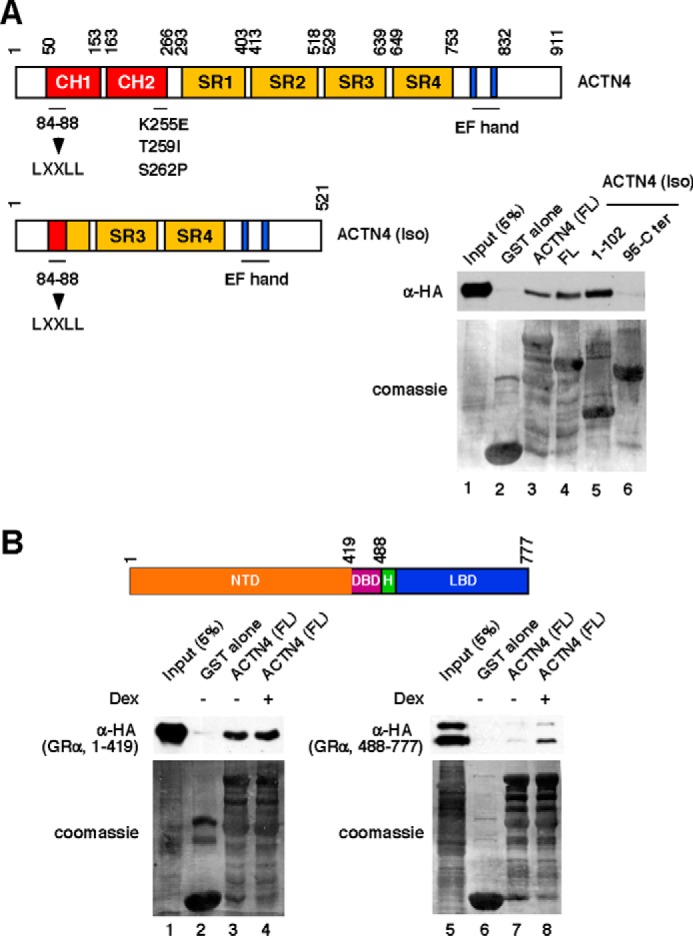
Mapping of ACTN4- and GR-interacting domains. A, top panel, schematic of ACTN4 functional domains (CH, calponin homology domain; CaM, calmodulin), the nuclear receptor-interacting motif, LXXLL (amino acids 84–88), and the C-terminal EF-hand calcium binding domains. The FSGS-linked K255E, T259I, and S262P mutants are indicated. Bottom panel, HeLa cells were transfected with an HA-GR expression plasmid. Whole cell lysates were incubated with immobilized GST and GST-ACTN4 and its variants. GST pulldown assays were performed as described for Fig. 2B. Pulldown fractions were analyzed by Western blotting with anti-HA antibodies. The expressions of GST fusion proteins were visualized by Coomassie Blue staining. B, top panel, schematic of GRα functional domains (NTD, N-terminal domain; DBD, DNA-binding domain; H, hinge domain; LBD, ligand-binding domain). Bottom panel, HeLa cells were transfected with HA-GR truncated mutants. Whole cell lysates were incubated with immobilized GST and GST-ACTN4. GST pulldown assays were performed as described. Dex, dexamethasone.
The Role of Actin-binding in ACTN4-mediated Transcriptional Regulation of GRE Reporter
We have previously identified an ACTN4 spliced isoform (called Iso) that is missing most of the actin-binding domain (31). We found that this isoform is still capable of potentiating GRE reporter activity (Fig. 4A) and capable of binding GR in vitro (Fig. 3A). The ability of NR co-activators to potentiate NR transcriptional activity depends on the NR-interacting motif LXXLL, which is commonly present in NR co-activators that include ACTN4, the histone acetyltransferases CREB-binding protein/p300, p300/CBP-associated factor, and the p160 family coactivator members (29). An ACTN4 mutant (LXXAA) failed to potentiate GRα reporter activity (Fig. 4B) and was unable to interact with GR (Fig. 4C). However, this mutant was still capable of interacting and potentiating NF-κB transcriptional activity (27). Furthermore, this mutant binds F-actin filaments comparably to the wild-type protein (Fig. 4D). Collectively, these data indicate that the inability of ACTN4 (LXXAA) to potentiate NR transcriptional activity is independent of its actin binding activity and that the actin-binding domain is dispensable for potentiating GRE reporter activity
FIGURE 4.
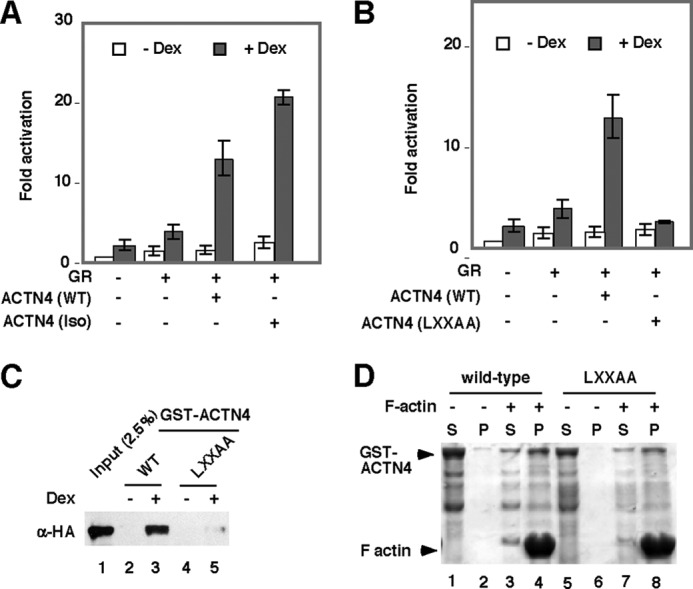
The effect of the ACTN4 LXXAA mutation on potentiating GR-mediated transcriptional activity. A and B, expression plasmids for ACTN4 (WT), ACTN4 (Iso) (A), or an ACTN4 mutant (LXXAA) (B) were co-transfected with a GR expression plasmid and GRE-luciferase reporter plasmid in HEK293T cells, followed by treatment with Veh or dexamethasone (Dex) for 12 h, and the reporter activity was analyzed. C, HeLa cells were transfected with an HA-GR expression plasmid, and lysates were prepared and incubated with bacterially expressed GST-ACTN4 (WT) or GST-ACTN4 (LXXAA) fusion proteins in the absence or presence of dexamethasone (100 nm). Pulldown fractions were subjected to Western blotting with HA antibodies. D, the LXXAA mutation does not affect ACTN4 F-actin filament binding activity. Recombinant GST, GST-ACTN4 (WT), and GST-ACTN4 (LXXAA) were purified and subjected to F-actin co-sedimentation assays. S, soluble fraction; P, pellet fraction. Note that the differences of GST-ACTN4 in the pellet fractions between lanes 2 and 4 and lanes 6 and 8 are similar.
FSGS-linked ACTN4 Mutants Failed to Activate Transcription Mediated by GR
We next determined whether the known FSGS-linked ACTN4 mutations K255E, S258P, and T262I affect the ability of ACTN4 to potentiate GR-mediated transactivation (Fig. 5A). We found that FSGS-linked ACTN4 mutants significantly reduced GRE reporter activity (Fig. 5A, lanes 3–5), and the reduction of GRE activities was also observed in GR-overexpressing cells (Fig. 5A, lanes 8–10). This would suggest that the FSGS-linked mutations are functional. Additionally, FSGS-linked ACTN4 failed to bind GR, as demonstrated by GST pulldown assays (Fig. 5B, lanes 3–5). In sum, the above data would suggest that FSGS-linked ACTN4 mutants have functionally lost their ability to potentiate dexamethasone-mediated transcriptional activation.
FIGURE 5.
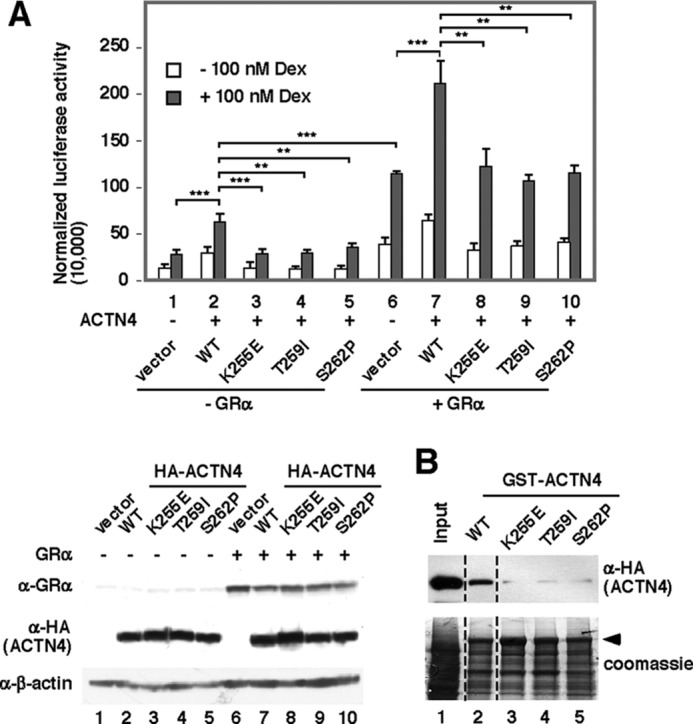
The effect of FSGS-linked ACTN4 on GR-mediated transcriptional activation. A, a GRE-containing reporter construct was co-transfected with or without plasmids expressing GR and ACTN4 (WT) or FSGS-linked ACTN4 mutants along with β-gal as indicated. The cells were treated with or without 100 nm dexamethasone (Dex) overnight. Luciferase activity was measured and normalized to β-gal activity. The expression of GR, HA-ACTN4 wild-type, and variants is shown. B, lysates were incubated with immobilized bacterially expressed GST-ACTN4 (WT) and GST-ACTN4 (FSGS-linked mutants) fusion proteins in the presence of the 100 nm dexamethasone. Pulldown fractions were subjected to immunoblotting with anti-HA antibodies. **, p < 0.01; ***, p < 0.001.
Knockdown of ACTN4 Has Little or No Effect on Dexamethasone-mediated Nuclear Translocation of GR
Because both ACTN4 and GR localize in the nucleus and cytoplasm, our observation that loss of ACTN4 reduces GRE-mediated reporter activity raises the possibility that ACTN4 plays a role in dexamethasone-induced nuclear translocation of GR. To test this possibility, we determined whether knockdown of ACTN4 affects dexamethasone-induced nuclear translocation of GR. Control and ACTN4 knockdown HPCs were treated with or without dexamethasone, followed by immunofluorescence microscopy and subcellular fractionation. Dexamethasone induced endogenous GR nuclear translocation in HPCs, which was confirmed by cell fractionation (Fig. 6A). Subcellular fractionation of HPCs further indicated that knockdown of ACTN4 had little or no effect on dexamethasone-induced nuclear translocation of GR (Fig. 6B). Additionally, immunofluorescence microscopy demonstrated that GR was capable of translocating to the nucleus in ACTN4 knockdown HPCs (Fig. 6C). Taken together, our data suggest that the loss of ACTN4 impairs dexamethasone-mediated transactivation by GR without affecting dexamethasone-induced GR nuclear translocation.
FIGURE 6.

The effect of ACTN4 knockdown on dexamethasone-induced GR nuclear translocation in HPCs. A, HPCs were treated with DMSO or dexamethasone (Dex, 100 nm) for 4 h, followed by subcellular fractionation and Western blotting with the indicated antibodies. B, shCtrl and shACTN4 HPCs were treated and analyzed as in A. Only the nuclear fraction is shown. C, shCtrl and shACTN4 HPCs were treated with or without dexamethasone (100 nm) for 1 h, followed by immunostaining with the indicated antibodies, and visualized by fluorescence microscopy. N, nuclear; C, cytoplasmic.
ACTN4 and GR Interact Exclusively in the Nucleus of HPCs
To further investigate the mechanism underlying ACTN4-mediated activation of GR, we determined in which cellular compartment the GR:ACTN4 interaction takes place. We addressed this issue by performing coimmunoprecipitation with nuclear or cytoplasmic extracts prepared from HPCs (Fig. 7A). We demonstrated that endogenous ACTN4 and GR interact in the nucleus but not the cytoplasm. We further demonstrated that immobilized, purified GST-ACTN4 was capable of pulling down nuclear HA-GR in vitro (Fig. 7B). In contrast, no interaction was observed when cytoplasmic extracts were used for a pulldown assay. Thus, we conclude that endogenous ACTN4 and GR interact in the nucleus but not in the cytoplasm. The current model for the mechanism of GR activation proposes that cytoplasmic GR is sequestered by HSP70/HSP90 (33). To investigate the role of HSP90 on GR translocation in HPCs, we carried out cell fractionation to examine the endogenous GR localization following knockdown of HSP90. Interestingly, the abundance of cytoplasmic GR in HSP90 knockdown HPCs was also reduced (Fig. 8A). We also carried out endogenous immunoprecipitation to examine the interaction between GR and ACTN4. We found that knockdown of HSP90 enhanced the association between cytoplasmic GR and ACTN4 (Fig. 8B).
FIGURE 7.
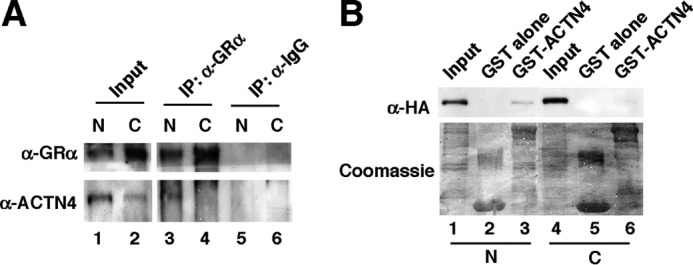
The interaction between ACTN4 and GR only occurred in the nucleus in HPCs. A, HPCs were treated with 100 nm dexamethasone for 4 h, and nuclear (N) and cytoplasmic (C) fractions were prepared. Coimmunoprecipitation (IP) was carried out using anti-GR antibodies, followed by Western blotting with the indicated antibodies. B, HeLa cells were transiently transfected with an HA-GR expression plasmid. Nuclear (4) and cytoplasmic (4) lysates were incubated with immobilized GST or GST-ACTN4. Pulldown assays were performed with nuclear extracts expressing HA-GR (lanes 1–3) and with cytoplasmic extracts (lanes 4–6). Pulldown fractions were analyzed by Western blotting with anti-HA antibodies. The expressions of GST fusion proteins were visualized by Coomassie Blue staining.
FIGURE 8.
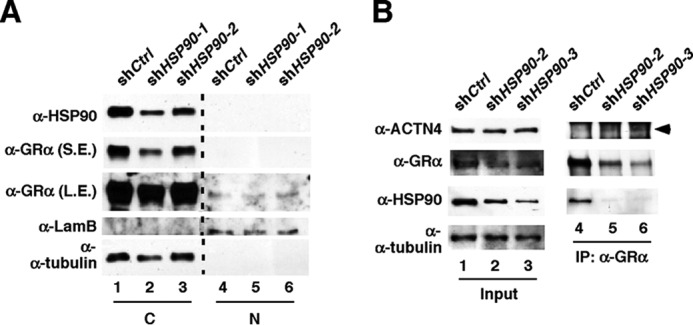
The effect of knocking down HSP90 on the association between ACTN4 and GRα. Knockdown of HSP90 enhances the association of cytoplasmic GR and ACTN4. A, HSP90 was stably knocked down by shRNAs in HPCs. Nuclear (N) and cytoplasmic (C) GR and HSP90 were detected by Western blotting with the indicated antibodies. S.E., shorter exposure; L.S., longer exposure. B, shCtrl and HSP90 knockdown (shHSP90) HPCs were grown and harvested, and nuclear and cytoplasmic extracts were prepared. Cytoplasmic extracts were used for immunoprecipitation (IP) with anti-GR antibodies, and the immunoprecipitates were subjected to Western blotting with the indicated antibodies. Arrowhead, ACTN4 band.
ACTN4 Is Recruited to the Promoters of GR-transactivated Genes
We have previously carried out microarray gene expression studies in HPCs and identified several dexamethasone target genes (14). We further validated several dexamethasone target genes by quantitative RT-PCR. Taking advantage of the ENCODE database, we found that the dexamethasone-inducible gene promoters Serpin family E member 1 (SERPINE1), angiopoietin-like 4 (ANGPTL4), chemokine (C-C motif) ligand 20 (CCL20), and serum amyloid A1 (SAA1) promoters contain one putative GR binding site (Fig. 9A). These GR binding sites are next to a histone 3 lysine 27 acetyl mark that is often found in active regulatory elements. We next asked whether dexamethasone promotes the recruitment of GR to these putative GR target sites using ChIP assays. Our data indicated that, in response to dexamethasone, both GR and ACTN4 recruitment was increased on the putative GR binding sites in the promoters of SERPINE1 (Fig. 9B), ANGPTL4 (Fig. 9C), CCL20 (Fig. 9D), and SAA1 (Fig. 9E).
FIGURE 9.
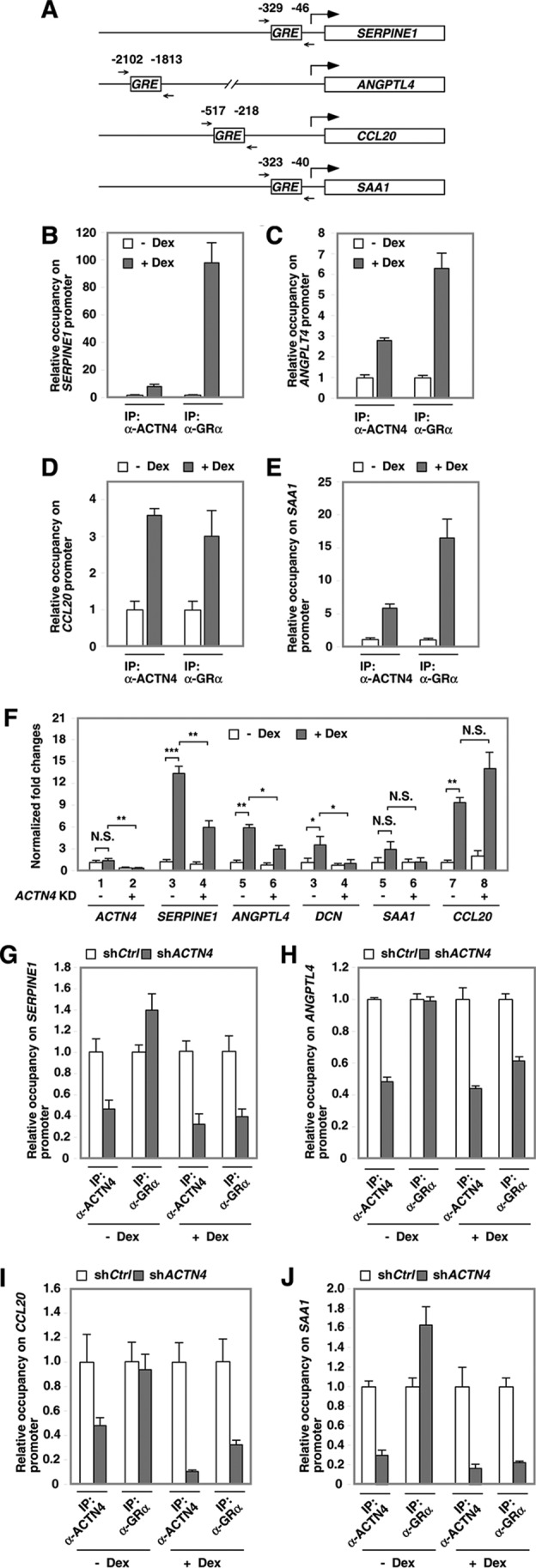
The recruitment of ACTN4 and GR with GR transactivated promoters in HPCs. A, diagrams of mapped GREs and PCR primer sets in GR-transactivated genes. B–E, HPCs were treated with vehicle or dexamethasone (Dex) for 4 h, followed by ChIP assays with anti-ACTN4 and anti-GR antibodies. Immunoprecipitated (IP) chromatin was analyzed by qPCR using the primers flanking the mapped putative GR-binding sites (ENCODE) on the indicated promoters. F, control and ACTN4 knockdown HPCs were treated with vehicle or dexamethasone for 8 h prior to harvest. Total RNA was prepared, and quantitative RT-PCR analysis was conducted using gene-specific primers. G–J, HPCs stably expressing shCtrl or shACTN4 were treated with vehicle or dexamethasone for 4 h prior to harvest. The relative recruitment of ACTN4 and GR to SERPINE1 (4), ANGPTL4 (4), CCL20 (4), and SAA1 (17) promoters was determined by normalizing the PCR products from shACTN4 cells to that of shCtrl cells. *, p < 0.05; **, p < 0.01; ***, p < 0.001; N.S., not significant.
To determine the role of ACTN4 in mediating transactivation of these GR target genes, control and ACTN4 knockdown HPCs were treated with or without dexamethasone, and their mRNA expression was assessed by RT-PCR. We found that knocking down ACTN4 significantly decreased mRNA levels of SERPINE1, ANGPTL4, and DCN (Fig. 9F). However, SAA1 and CCL20 expression was slightly increased in ACTN4 knockdown HPCs. These results indicate that GR-transactivated genes are not all co-activated by ACTN4. In the absence of dexamethasone, loss of ACTN4 reduced the association of ACTN4 with the promoters of SERPINE1, ANGPTL4, SAA1, and CCL20 without significantly affecting GR occupancy on these promoters (Fig. 9, G–J). Interestingly, in the presence of dexamethasone, loss of ACTN4 reduced both the recruitment of ACTN4 and GR to GR binding sites. This observation suggests that GR binding to these promoters may partly depend on ACTN4 in dexamethasone-treated HPCs.
ACTN4 Plays a Role in GR-transrepressed Genes in a Promoter Context-dependent Manner
It has been established experimentally that transrepression accounts for the majority of the anti-inflammatory properties of GCs. We have demonstrated previously that dexamethasone treatment represses the expression of NF-κB target genes such as IL-1β, IL-6, and IL-8 in HPCs. The current transrepression model suggests that dexamethasone induces the association of GR with NF-κB and histone deacetylases (HDACs) to transrepress NF-κB target genes (27). The ENCODE database indicates that there is one NF-κB binding site (NRE) is present in the promoters of IL-1β and IL-6 but none in the IL-8 promoter (Fig. 10A, light gray squares), although we have previously mapped NF-κB sites in IL-1β, IL-6, and IL-8 promoters, according to a published database (34) (Fig. 10A, dark gray squares). Additionally, a putative GR site (GRE) was mapped. With this information, we carried out ChIP assays and examined the association of ACTN4, GR, and NF-κB on these NF-κB-inducible promoters in response to dexamethasone treatment. Our data indicate that dexamethasone promotes the recruitment of GR to both putative NF-κB binding sites in the promoter of IL-1β (Fig. 10B). Similarly, dexamethasone promotes the recruitment of GR to both NF-κB binding sites on the IL-6 promoter (Fig. 10C), although to a lesser extent. Furthermore, dexamethasone promotes the recruitment of GR to the GRE on the IL-8 promoter, as mapped by the ENCODE project (Fig. 10D). However, dexamethasone had no or very little effect on GR recruitment to the previously mapped NRE in the IL-8 promoter (Fig. 10D, right). Increased association of ACTN4 with the NF-κB sites was also detected in all but this site. Indeed, dexamethasone treatment reduced the association of ACTN4 with the GRE in the IL-8 promoter (∼ 50%) (Fig. 10D, left). Last, dexamethasone treatment did not affect the association of p65 with the ENCODE-mapped NRE or GR binding sites (GRE). In fact, dexamethasone treatment resulted in reduced association of p65 with previously mapped NREs in IL-1β, IL-6, and IL-8 promoters.
FIGURE 10.
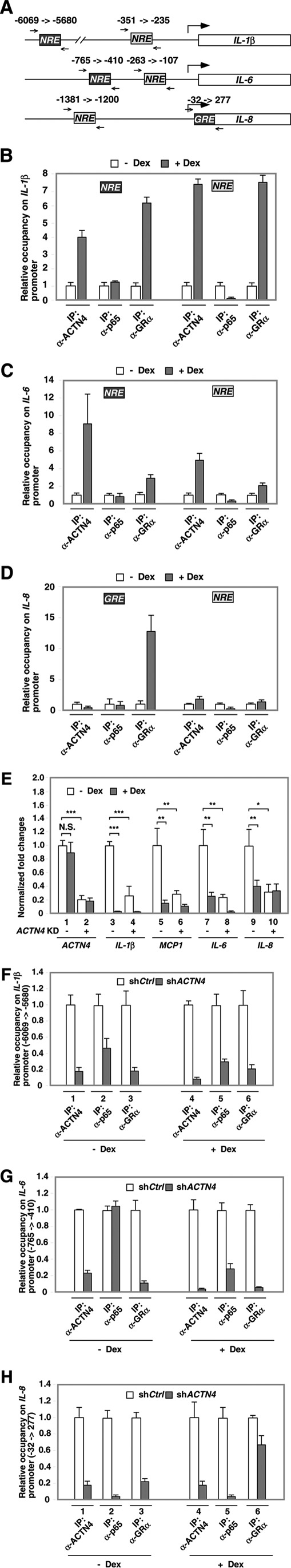
The effects of dexamethasone on the recruitment of GR, ACTN4, and p65 to NF-κB targeted promoters. A, diagrams of mapped NREs or putative GRE and PCR primer sets. Dark gray squares indicate p65 or GR binding sites on NF-κB-inducible genes. Light gray squares indicate NF-κB sites described in our previous paper (27). B–D, HPCs were treated with vehicle or dexamethasone (Dex) for 4 h, followed by ChIP assays with anti-ACTN4, anti-GR, and anti-p65 antibodies. Immunoprecipitated (IP) chromatin was analyzed by qPCR using the primers (A, small arrows) flanking the putative p65-binding sites in the promoters of IL-1β, IL-6, and IL-8. E, control and ACTN4 knockdown HPCs were treated with vehicle or dexamethasone for 8 h prior to harvest. Total RNA was extracted, and quantitative RT-PCR analysis was conducted using gene-specific primers. F–H, HPCs stably expressing shCtrl or shACTN4 were treated with vehicle or dexamethasone for 4 h prior to harvest. The relative recruitment of ACTN4, GR, and p65 to the promoters of IL-1β, IL-6, and IL-8 was determined by normalizing the PCR products from shACTN4 cells to that of shCtrl cells. *, p < 0.05; **, p < 0.01; ***, p < 0.001; N.S., not significant.
Knockdown of ACTN4 reduced expression of IL-1β, IL-6, IL-8, and MCP-1 in HPCs in the absence of dexamethasone (Fig. 10E). ACTN4 knockdown further reduced dexamethasone-mediated repression of IL-6 and MCP-1 but had no effect on IL-8 expression (Fig. 10E, lanes 9 and 10). Interestingly, ACTN4 knockdown HPCs resulted in reduced association of GR and p65 with the NF-κB sites (NRE, dark gray square, Fig. 10A) on the IL-1β, IL-6, and IL-8 promoters (Fig. 10, F and G). In the absence of dexamethasone, knockdown of ACTN4 reduced the expression of NF-κB target genes, although to varying degrees. This is consistent with our previous findings that ACTN4 is a NF-κB co-activator (27). Furthermore, loss of ACTN4 also reduced the association of GR with the NF-κB sites on IL-1β, IL-6, and IL-8 promoters (Fig. 10, F, lane 3, and G) and significantly blocked the recruitment of p65 (>90%) to the IL-8 promoter (Fig. 10H, lane 2). Thus, the reduced expression of IL-8 in ACTN4 knockdown HPCs could potentially be due to the loss of p65 binding on this promoter. However, although knockdown of ACTN4 reduced the recruitment of p65 (>90%) to the putative GRE site of the IL-8 promoter (Fig. 10H, lane 5), only a 60% decrease in IL-8 mRNA levels was observed. These results suggest that other transcription factors also play a role in controlling IL-8 expression. Unlike the IL-8 promoter, much more potent dexamethasone-mediated transrepression activity was observed in IL-1β expression, and this correlated with significant recruitment of GR to both NREs in the IL-1β promoter (Fig 10B).
Discussion
Our observations suggest a regulatory mechanism of ligand-induced nuclear translocation of GR and dexamethasone-mediated transcriptional regulation that is similarly to that described previously in other tissues. First, dexamethasone induces dissociation of cytoplasmic GR from HSP90, thus promoting nuclear translocation of GR in HPCs. Furthermore, dexamethasone possesses both transactivation and transrepression activity of GR target genes in HPCs (Fig. 11).
FIGURE 11.
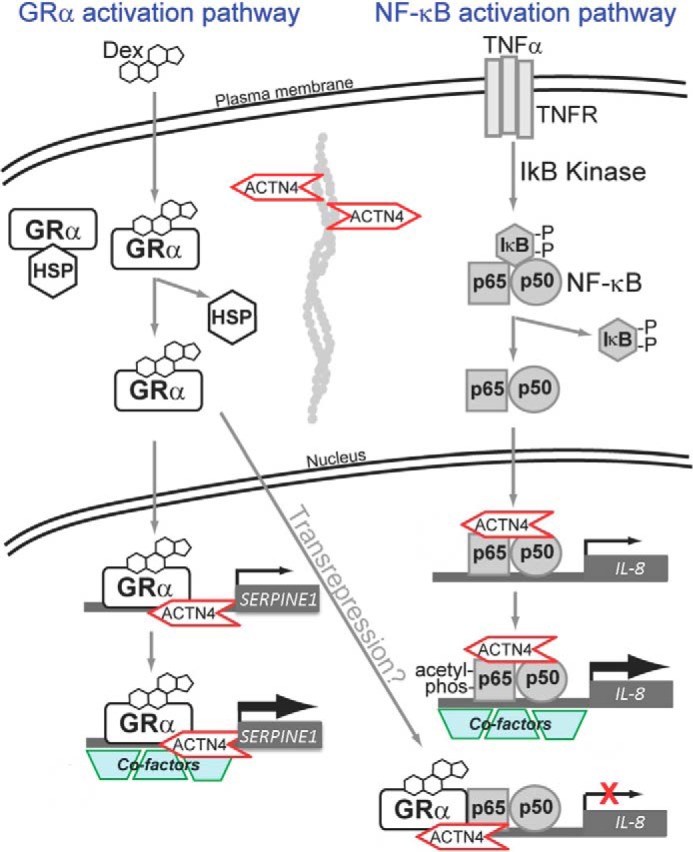
A model depicting the mechanism by which ACTN4 regulates dexamethasone- and TNFα-mediated transcriptional regulation in HPCs.
The observations that both GR and ACTN4 localize in both the nucleus and cytoplasm raised several intriguing questions: in which subcellular compartments do GR and ACTN4 interact, does ACTN4 modulate ligand-dependent nuclear translocation of GR, and does the actin-binding activity of ACTN4 play a role in regulating GR transcription activity? Our results indicated that GR and ACTN4 only interact in the nucleus. This is in part due to sequestering of GR by HSP90 and dexamethasone-inducible nuclear translocation of GR. Thus, loss of ACTN4 had little effect on dexamethasone-induced nuclear translocation of GR. Furthermore, we found that the actin binding-deficient ACTN4 isoform, ACTN4 (Iso), still potently activates GR transcriptional activity and that the ACTN4 mutant, LXXLL, although it is capable of binding actin filaments, failed to activate a GRE-driven reporter. Based on these observations, we conclude that the ability of ACTN4 to co-activate GR transcription activity is independent of its actin-binding activity.
Dexamethasone induces the expression of GR target genes through positive GREs or composite binding motifs (13). We have demonstrated previously that dexamethasone induces the expression of SERPINE1, ANGPLT4, CCL20, and SAA1 in HPCs (14). In this report, we showed that dexamethasone promotes the recruitment of GR to the promoters of these dexamethasone-transactivated genes. Furthermore, knockdown of ACTN4 blocked dexamethasone-induced expression ofSERPINE1, ANGPTL4, and SAA1 and slightly increased CCL20 mRNA (Fig. 10). ChIP assays validated the enhanced recruitment of ACTN4 to these GR-targeted regions in response to dexamethasone, consistent with the observation that dexamethasone enhances the association of ACTN4 with GR. However, loss of ACTN4 did not significantly reduce the association of GR with its targeted promoters, indicating that the recruitment of GR to these promoters does not rely on the association of ACTN4 with these promoters. Together, these data support a model in which dexamethasone-bound GRα recruits ACTN4 to GRα-targeted promoters to potentiate transcription.
Although commonly used for its ability to suppress inflammation, dexamethasone also solicits unwanted side effects (35). SERPINE1 encodes plasminogen activator inhibitor 1 (PAI-1), which is present in trace amounts in healthy kidneys but increased in a wide variety of both acute and chronically diseased kidneys. Reduced PAI-1 activity has been shown to be protective for the development of albuminuria and glomerulosclerosis in experimental diabetes (36). Moreover, CCL20 is up-regulated in patients with progressive IgA nephropathy (32). Podocyte-specific overexpression of ANGPTL4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome in animal models (37). Thus, it is possible that the ability of dexamethasone to induce SERPINE1, ANGPTL4, and CCL20 expression accounts for some of its adverse effects. SAA1 is induced by dexamethasone in other tissues (38), and its expression is associated with kidney diseases (39). However, although SAA1-containing amyloid fibers interfere with kidney filtration, leading to kidney failure, the exact function of SAA1 in the kidney remains largely unknown.
It has been well established that the beneficial anti-inflammatory effect of glucocorticoids is through the suppression of transcription factors, including the NF-κB family of transcription factors. The observation that ACTN4 potentiates both NF-κB (27) and GR transactivation activity prompted us to investigate the role of ACTN4 in dexamethasone-mediated transrepression activity. Indeed, dexamethasone enhanced the recruitment of GR to NF-κB binding sites on the promoters for IL-1β and IL-6. Only the IL-8 promoter was an exception (Fig. 9). Interestingly, dexamethasone also promoted the recruitment of ACTN4 to NF-κB target sites on the IL-1β and IL-6 promoters but reduced the recruitment of GR (50%) to the GRE on the IL-8 promoter. Neither dexamethasone-mediated transrepression of IL-1β nor IL-6 required ACTN4, as knockdown of ACTN4 had little effect on the ability of dexamethasone to repress IL-1β or IL-6. In contrast, loss of ACTN4 dramatically reduces dexamethasone-mediated transrepression of IL-8 expression. These observations suggest that dexamethasone-mediated transrepression is promoter-dependent.
In the absence of dexamethasone, knockdown of ACTN4 reduced the expression of NF-κB target genes, although to varying degrees. This is reminiscent of our previous findings that ACTN4 is a NF-κB co-activator (27). Furthermore, loss of ACTN4 also reduced the association of GR with the NF-κB sites on the IL-1β, IL-6, and IL-8 promoters and significantly blocked the recruitment of p65 (>90%) to the IL-8 promoter. Thus, the reduced expression of IL-8 in ACTN4 knockdown HPCs could be due to loss of p65 binding to its promoter. However, although knockdown of ACTN4 reduced the recruitment of p65 (>90%) to −32 → 277 of the IL-8 promoter, only a 60% decrease in the IL-8 mRNA level was observed. These data suggest that other transcription factors also play an important role to control IL-8 expression. Unlike the IL-8 promoter, a much more potent dexamethasone-mediated transrepression activity was observed in IL-1β expression, and this correlates with significant recruitment of GR to both NREs in the IL-1β promoter.
The mechanism underlying the pathogenesis of ACTN4-linked FSGS is not fully understood but is likely complex. Work by others has described cytoplasmic roles of ACTN4 mutants and cytoskeletal anomalies, and we have characterized an additional role involving transcriptional disturbances from its interaction with NRs. Thus, the FSGS-linked ACTN4 mutations may have both gain-of-function (enhanced actin-binding activity) and loss-of-function mutations (decreased transcription activity). Our continued work to characterize the NR-associated transcriptional loss-of-function activity is important considering the first-line treatment for FSGS are glucocorticoids. Future investigations to understand the pathogenesis of ACTN4-linked glomerulopathies and the possible renoprotective functions of glucocorticoid treatment could lead to better treatment options for FSGS patients.
Experimental Procedures
Plasmid Construction
CMX-HA-ACTN4, GST-ACTN4 (WT), different ACTN4 mutation expression plasmids, as well as GST-ACTN4 deletion constructs were described previously (25). The expression plasmids CMX-HA-GR (full-length, 1–419 and 488-C terminus) were generated by PCR and subcloned into the CMX-1H vector (27). The GRE-Luc reporter plasmid was a gift from Dr. Ron Evans (The Salk Institute).
Antibodies and Chemicals
The anti-ACTN4 and anti-HDAC7 antibodies have been described previously (27). For immunoprecipitation assays, anti-HA antibodies (Santa Cruz Biotechnology, sc-805), anti-GR antibodies (sc-8992), anti-p65 antibodies (sc-372), anti-HSP90 (sc-69703), anti-Lamin B (sc-6216), and anti-α-tubulin (T-5168, Sigma-Aldrich) were used. For immunostaining, anti-GR antibodies and anti-synaptopodin antibodies were used, and rhodamine phalloidin (Cytoskeleton, Inc., PHDR1) was used to detect actin. The secondary antibodies for immunofluorescence were from Life Technologies (anti-mouse or anti-rabbit Alexa Fluor 488 or Alexa Fluor 594). Dexamethasone (D4902) was purchased from Sigma-Aldrich.
siRNA Transfection
Human podocytes were subjected to siRNA transfection with either a non-targeting siRNA (siCtrl) or with siRNA against GR (GR-1(J003424-07)) or (GR-1(J003424-09)) according to the protocol of the manufacturer (Dharmacon). Twenty-four hours after transfection, the medium was replaced with RPMI 1640 supplemented with 10% charcoal-stripped fetal bovine serum, 50 units/ml penicillin G, and 50 μg/ml streptomycin sulfate, and insulin, transferrin, and sodium selenite mixture (ITS) (Sigma). The next day, the cells were treated with or without 100 nm dexamethasone.
Cell Culture and Differentiation
HEK293T and HeLa cells were grown in DMEM supplemented with 10% FBS and penicillin-streptomycin (50 units/ml) at 37 °C in 5% CO2. HPCs were cultured in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum, penicillin-streptomycin (50 units/ml), and insulin-transferrin-selenite (Sigma). HPCs were grown at a permissive temperature of 33 °C and transferred to 37 °C to induce differentiation for 3 days prior to treatment. Cells were serum-starved in RPMI 1640 supplemented with 2% fetal bovine serum, penicillin-streptomycin (50 units/ml), and insulin-transferrin-selenite.
Immunofluorescence Microscopy
HPCs were fixed in 3.7% paraformaldehyde in PBS for 30 min at room temperature and permeabilized in PBS with addition of 0.1% Triton X-100 and 10% goat serum for 10 min. The cells were washed three times with PBS and incubated in a PBS-goat serum (10%) plus 0.1% Tween 20 solution (ABB) for 60 min. Incubation with primary antibodies was carried out for 120 min in ABB. The cells were washed three times in 1× PBS, and the secondary antibodies were added for another hour in the dark at room temperature in ABB. DAPI was applied to the samples after the final wash to visualize cell nuclei. Images were visualized using a Leica epifluorescence microscope.
Generation of Stable Knockdown Cells
shACTN4 and shHDAC7 stable cells were described previously (27). Lentiviral vectors harboring shRNAs against human HSP90α (TRCN0000008490, TRCN0000008493, and TRCN0000008494) were purchased from Sigma-Aldrich. The packaging plasmids used were pMD2.G and psPAX2, which were kindly provided by Dr. Barbara Bedogni. The establishment of stable ACTN4 and HSP90α knockdown cells was performed as described previously (21). After cotransfection into the HEK293T packaging cell line with shRNA and packaging plasmids, the supernatants containing lentiviral particles were collected and used to infect the HEK293T and HPC cell lines. Cells were stably infected with lentivirus and selected in puromycin-containing medium (1.5 μg/ml) for 2 days. Knockdown of ACTN4 and HSP90α was confirmed by Western blotting.
Preparation of Mouse Glomeruli
Glomeruli were isolated using the standard sieving protocol, and protein extracts were prepared as described previously (27). Glomeruli from 20-week-old male C57BL/6 mice (n = 5) were pooled.
Subcellular Fractionation of HPCs
Isolation of nuclei and cytoplasm from HPCs and extraction of nuclear and cytosolic proteins were performed according to a published protocol (27). Nuclear and cytoplasmic fractions were separated on SDS-polyacrylamide gels, followed by Western blotting with the indicated antibodies. Lamin B and α-tubulin served as nuclear and cytoplasmic markers, respectively.
In Vitro Protein-Protein Interaction Assays
GST fusion proteins were expressed in the Escherichia coli DH5α strain, affinity-purified, and immobilized on glutathione-Sepharose 4B beads. Immobilized, purified GST and GST-ACTN4 fusion proteins were incubated with whole cell extracts expressing HA-GR, followed by extensive washes as described previously (29). The pulldown fractions were subjected to Western blotting with HA antibodies. The immobilized GST and GST fusion proteins were visualized by Coomassie Brilliant Blue staining.
Coimmunoprecipitation
HPCs were treated with DMSO or dexamethasone (100 nm) and harvested, and whole cell lysates or nuclear extracts were prepared. Glomerular extracts were precleared with Protein A beads (RepliGen). Immunoprecipitation and Western blotting assays were carried out following our published protocol (29).
Transient Transfection Reporter Assays
HEK293T or HPCs cells stably expressing shRNA against human ACTN4 (shACTN4) or control shRNA (shCtrl) were cultured in 24-well plates. Cells were cotransfected with a GRE-driven luciferase reporter and a Renilla luciferase control reporter construct using Lipofectamine 2000. To determine the effects of wild-type or mutant ACTN4 on GRE reporter activity, HEK293 cells were transiently transfected with the indicated ACTN4 expression plasmids. The cells were lysed in reporter lysis 5× buffer 12 h after dexamethasone treatment and 48 h after transfection (Promega). The luciferases activity was determined using a Dual-Luciferase assay kit (Promega). Luciferase activity was normalized with that of Renilla luciferase. Each transfection was performed in triplicate.
RNA Isolation and qPCR
Total RNA was extracted using the RNeasy isolation kit (Qiagen) and reverse-transcribed using a reverse transcription kit (Bio-Rad) according to the instructions of the manufacturer. iQ SYBR Green PCR Supermix (Bio-Rad and Qiagen) and the CFX96 real-time PCR detection system (Bio-Rad) were used to quantify cDNAs according to the instructions of the manufacturer, and 18S rRNA was used for normalization. The relative mRNA expression was calculated by the 2−(ΔΔCt) method. The PCR primers used in this study and their sequences were described previously (27).
Actin-binding Assays
Actin binding protein spin-down assay biochem kits were obtained from Cytoskeleton and used following the instructions of the manufacturer. Both wild-type and LXXAA mutant GST-ACTN4 proteins were purified from E. coli. F-actin was polymerized and incubated with GST-ACTN4 proteins at room temperature. The mixture was incubated on ice and then centrifuged at 150,000 × g for 1.5 h at 24 °C. Both the supernatants and the pellets were subjected to 8% SDS-PAGE, followed by transfer to PVDF membranes. Proteins were visualized by Coomassie Blue staining or Western blotting.
ChIP Assays
ChIP assays were carried out as described previously (27). Immunoprecipitation was performed using anti-ACTN4 and anti-p65 antibodies. The primer sequences for ChIP assays for GRE were as follows: SERPIN1, CTAGGCTTTTTGGGTCACCCGGCATGG (forward) and CTAGGCTTTTTGGGTCACCCGGCATGG (reverse); CCL20, GTATGCTCAACATCCTGT (forward) and AGAGCTGCTTTACTCATT (reverse); ANGPTL4, AGCTGTCTGCCCTGCAATGTACAAGTT (forward) and AGGATTCCTGGAGCAGGGGTCCTAAGA (reverse); and SAA1, ACTTCACACCTTCCAGCAGCCCAGGTG (forward) and CTGCTGGTTTCTTGGGAGCGAAGAAAA (reverse). The primer sequences for the ChIP assays for p65 and GR were as follows: IL-1β (−6069 → −5680), GTGCTGTGTGAATTT (forward) and TCCAGCAACAAAGCT (reverse); IL-1β (−351→ −235), CTGTGTGTCTTCCACTTTGTCCC (forward) and TGCATTGTTTTCCTGACAATCG (reverse); IL-8 (−32 → 277), CTTGTTTACCACAAG (forward) and ACATGTTACTAGTAT (reverse); IL-8 (−1381 → −1200), CTTGTTTACCACAAG (forward) and ACATGTTACTAGTAT (reverse); IL-6 (−765 →−410), ATGGAGATGAATCAC (forward) and GTTTACTCTTAACTG (reverse); IL-6 (−263 → −107), ATGGAGATGAATCAC (forward) and GTTTACTCTTAACTG (reverse).
Statistical Analysis
Data are expressed as the mean ± S.D. Analyses were performed with Student's t test using GraphPad Prism6 software. A value of p < 0.05 was considered to be significant. p < 0.05, p < 0.01, and p < 0.001 are designated by *, **, and ***, respectively.
Author Contributions
X. Z., J. R. S., L. A. B., and H. Y. K. conceived and designed the experiments. X. Z., S. K., S. C., and Y. T. performed the experiments. X. Z. and H. Y. K. analyzed and interpreted the data. X. Z., L. A. B., and H. Y. K. wrote and reviewed the manuscript. All authors reviewed the results and approved the final version of manuscript.
Acknowledgments
We thank Dr. David Samols (Case Western Reserve University) for editing the paper, Dr. Minh Lam (Case Western Reserve University) for assisting with immunofluorescence microscopy, and Dr. Ron Evans from the Salk Institute for providing the GRE-Luc reporter plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK078965. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NR
- nuclear hormone receptor
- MCD
- minimal-change disease
- FSGS
- focal segmental glomerulosclerosis
- GC
- glucocorticoid
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- HPC
- human podocyte
- ACTN
- actinin
- CREB
- cAMP response element-binding protein
- HDAC
- histone deacetylase
- NRE
- NF-κB response element
- qPCR
- quantitative PCR
- Veh
- vehicle.
References
- 1.Zahran A. M., Aly S. S., Elsayh K. I., Badawy A., and Gamal Y. (2014) Glucocorticoid receptors expression and histopathological types in children with nephrotic syndrome. Ren. Fail. 36, 1067–1072 [DOI] [PubMed] [Google Scholar]
- 2.Coppo R. (2013) Different targets for treating focal segmental glomerular sclerosis. Contrib. Nephrol. 181, 84–90 [DOI] [PubMed] [Google Scholar]
- 3.Korbet S. M. (2012) Nephrology and the percutaneous renal biopsy: a procedure in jeopardy of being lost along the way. Clin. J. Am. Soc. Nephrol. 7, 1545–1547 [DOI] [PubMed] [Google Scholar]
- 4.Bose B., Cattran D., and Toronto Glomerulonephritis Registry (2014) Glomerular diseases: FSGS. Clin. J. Am. Soc. Nephrol. 9, 626–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith L. J. (1996) Leukotrienes in asthma: the potential therapeutic role of antileukotriene agents. Arch. Intern. Med. 156, 2181–2189 [DOI] [PubMed] [Google Scholar]
- 6.Korbet S. M. (2012) Treatment of primary FSGS in adults. J. Am. Soc. Nephrol. 23, 1769–1776 [DOI] [PubMed] [Google Scholar]
- 7.Pani A. (2013) Standard immunosuppressive therapy of immune-mediated glomerular diseases. Autoimmun. Rev. 12, 848–853 [DOI] [PubMed] [Google Scholar]
- 8.Heitzer M. D., Wolf I. M., Sanchez E. R., Witchel S. F., and DeFranco D. B. (2007) Glucocorticoid receptor physiology. Rev. Endocr. Metab. Disord. 8, 321–330 [DOI] [PubMed] [Google Scholar]
- 9.Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., and Evans R. M. (1995) The nuclear receptor superfamily: the second decade. Cell 83, 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Bosscher K., Vanden Berghe W., and Haegeman G. (2000) Mechanisms of anti-inflammatory action and of immunosuppression by glucocorticoids: negative interference of activated glucocorticoid receptor with transcription factors. J. Neuroimmunol. 109, 16–22 [DOI] [PubMed] [Google Scholar]
- 11.Reichardt H. M., Tuckermann J. P., Göttlicher M., Vujic M., Weih F., Angel P., Herrlich P., and Schütz G. (2001) Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 20, 7168–7173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pratt W. B. (1993) The role of heat shock proteins in regulating the function, folding, and trafficking of the glucocorticoid receptor. J. Biol. Chem. 268, 21455–21458 [PubMed] [Google Scholar]
- 13.Wang J. C., Derynck M. K., Nonaka D. F., Khodabakhsh D. B., Haqq C., and Yamamoto K. R. (2004) Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. U.S.A. 101, 15603–15608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng X., Zhao X., Khurana S., Bruggeman L. A., and Kao H. Y. (2013) Microarray analyses of glucocorticoid and vitamin D3 target genes in differentiating cultured human podocytes. PLoS ONE 8, e60213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing C. Y., Saleem M. A., Coward R. J., Ni L., Witherden I. R., and Mathieson P. W. (2006) Direct effects of dexamethasone on human podocytes. Kidney Int. 70, 1038–1045 [DOI] [PubMed] [Google Scholar]
- 16.Eichler T. E., Ransom R. F., and Smoyer W. E. (2005) Differential induction of podocyte heat shock proteins by prolonged single and combination toxic metal exposure. Toxicol. Sci. 84, 120–128 [DOI] [PubMed] [Google Scholar]
- 17.Wada T., Pippin J. W., Marshall C. B., Griffin S. V., and Shankland S. J. (2005) Dexamethasone prevents podocyte apoptosis induced by puromycin aminonucleoside: role of p53 and Bcl-2-related family proteins. J. Am. Soc. Nephrol. 16, 2615–2625 [DOI] [PubMed] [Google Scholar]
- 18.Sjöblom B., Salmazo A., and Djinović-Carugo K. (2008) α-Actinin structure and regulation. Cell. Mol. Life Sci. 65, 2688–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan J. M., Kim S. H., North K. N., Rennke H., Correia L. A., Tong H. Q., Mathis B. J., Rodríguez-Pérez J. C., Allen P. G., Beggs A. H., and Pollak M. R. (2000) Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24, 251–256 [DOI] [PubMed] [Google Scholar]
- 20.Yao J., Le T. C., Kos C. H., Henderson J. M., Allen P. G., Denker B. M., and Pollak M. R. (2004) α-Actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol. 2, e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng D., DuMontier C., and Pollak M. R. (2015) The role of α-actinin-4 in human kidney disease. Cell Biosci. 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai S., Wang Z., Pan X., Chen X., Wang W., Ren H., Feng Q., He J. C., Han B., and Chen N. (2009) ACTN4 gene mutations and single nucleotide polymorphisms in idiopathic focal segmental glomerulosclerosis. Nephron Clin. Pract. 111, c87-c94 [DOI] [PubMed] [Google Scholar]
- 23.Dai S., Wang Z., Pan X., Wang W., Chen X., Ren H., Hao C., Han B., and Chen N. (2010) Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 25, 824–835 [DOI] [PubMed] [Google Scholar]
- 24.Liu Z., Blattner S. M., Tu Y., Tisherman R., Wang J. H., Rastaldi M. P., Kretzler M., and Wu C. (2011) α-Actinin-4 and CLP36 protein deficiencies contribute to podocyte defects in multiple human glomerulopathies. J. Biol. Chem. 286, 30795–30805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakraborty S., Reineke E. L., Lam M., Li X., Liu Y., Gao C., Khurana S., and Kao H. Y. (2006) α-Actinin 4 potentiates myocyte enhancer factor-2 transcription activity by antagonizing histone deacetylase 7. J. Biol. Chem. 281, 35070–35080 [DOI] [PubMed] [Google Scholar]
- 26.An H. T., Kim J., Yoo S., and Ko J. (2014) Small leucine zipper protein (sLZIP) negatively regulates skeletal muscle differentiation via interaction with α-actinin-4. J. Biol. Chem. 289, 4969–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao X., Hsu K. S., Lim J. H., Bruggeman L. A., and Kao H. Y. (2015) α-Actinin 4 potentiates nuclear factor κ-light-chain-enhancer of activated B-cell (NF-κB) activity in podocytes independent of its cytoplasmic actin binding function. J. Biol. Chem. 290, 338–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grgic I., Hofmeister A. F., Genovese G., Bernhardy A. J., Sun H., Maarouf O. H., Bijol V., Pollak M. R., and Humphreys B. D. (2014) Discovery of new glomerular disease-relevant genes by translational profiling of podocytes in vivo. Kidney Int. 86, 1116–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khurana S., Chakraborty S., Cheng X., Su Y. T., and Kao H. Y. (2011) The actin-binding protein, actinin α 4 (ACTN4), is a nuclear receptor coactivator that promotes proliferation of MCF-7 breast cancer cells. J. Biol. Chem. 286, 1850–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khurana S., Chakraborty S., Lam M., Liu Y., Su Y. T., Zhao X., Saleem M. A., Mathieson P. W., Bruggeman L. A., and Kao H. Y. (2012) Familial focal segmental glomerulosclerosis (FSGS)-linked α-actinin 4 (ACTN4) protein mutants lose ability to activate transcription by nuclear hormone receptors. J. Biol. Chem. 287, 12027–12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khurana S., Chakraborty S., Zhao X., Liu Y., Guan D., Lam M., Huang W., Yang S., and Kao H. Y. (2012) Identification of a novel LXXLL motif in α-actinin 4-spliced isoform that is critical for its interaction with estrogen receptor α and co-activators. J. Biol. Chem. 287, 35418–35429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villa L., Boor P., Konieczny A., Kunter U., van Roeyen C. R., Denecke B., Gan L., Neusser M. A., Cohen C. D., ERCB Consortium, Eitner F., Scholl T., Ostendorf T., and Floege J. (2013) Late angiotensin II receptor blockade in progressive rat mesangioproliferative glomerulonephritis: new insights into mechanisms. J. Pathol. 229, 672–684 [DOI] [PubMed] [Google Scholar]
- 33.Kirschke E., Goswami D., Southworth D., Griffin P. R., and Agard D. A. (2014) Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barish G. D., Yu R. T., Karunasiri M., Ocampo C. B., Dixon J., Benner C., Dent A. L., Tangirala R. K., and Evans R. M. (2010) Bcl-6 and NF-κB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 24, 2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schäcke H., Döcke W. D., and Asadullah K. (2002) Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 96, 23–43 [DOI] [PubMed] [Google Scholar]
- 36.Huang Y., Border W. A., Yu L., Zhang J., Lawrence D. A., and Noble N. A. (2008) A PAI-1 mutant, PAI-1R, slows progression of diabetic nephropathy. J. Am. Soc. Nephrol. 19, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clement L. C., Avila-Casado C., Macé C., Soria E., Bakker W. W., Kersten S., and Chugh S. S. (2011) Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat. Med. 17, 117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thorn C. F., Lu Z. Y., and Whitehead A. S. (2003) Tissue-specific regulation of the human acute-phase serum amyloid A genes, SAA1 and SAA2, by glucocorticoids in hepatic and epithelial cells. Eur. J. Immunol. 33, 2630–2639 [DOI] [PubMed] [Google Scholar]
- 39.Gershoni-Baruch R., Brik R., Zacks N., Shinawi M., Lidar M., and Livneh A. (2003) The contribution of genotypes at the MEFV and SAA1 loci to amyloidosis and disease severity in patients with familial Mediterranean fever. Arthritis Rheum. 48, 1149–1155 [DOI] [PubMed] [Google Scholar]