

Abstract
We investigated the regulatory effect of glucosamine (GlcN) for the production of nitric oxide (NO) and expression of inducible NO synthase (iNOS) under various glucose conditions in macrophage cells. At normal glucose concentrations, GlcN dose dependently increased LPS-stimulated production of NO/iNOS. However, GlcN suppressed NO/iNOS production under high glucose culture conditions. Moreover, GlcN suppressed LPS-induced up-regulation of COX-2, IL-6, and TNF-α mRNAs under 25 mm glucose conditions yet did not inhibit up-regulation under 5 mm glucose conditions. Glucose itself dose dependently increased LPS-induced iNOS expression. LPS-induced MAPK and IκB-α phosphorylation did not significantly differ at normal and high glucose conditions. The activity of LPS-induced nuclear factor-κB (NF-κB) and DNA binding of c-Rel to the iNOS promoter were inhibited under high glucose conditions in comparison with no significant changes under normal glucose conditions. In addition, we found that the LPS-induced increase in O-GlcNAcylation as well as DNA binding of c-Rel to the iNOS promoter were further increased by GlcN under normal glucose conditions. However, both O-GlcNAcylation and DNA binding of c-Rel decreased under high glucose conditions. The NF-κB inhibitor, pyrrolidine dithiocarbamate, inhibited LPS-induced iNOS expression under high glucose conditions but it did not influence iNOS induction under normal glucose conditions. In addition, pyrrolidine dithiocarbamate inhibited NF-κB DNA binding and c-Rel O-GlcNAcylation only under high glucose conditions. By blocking transcription with actinomycin D, we found that stability of LPS-induced iNOS mRNA was increased by GlcN under normal glucose conditions. These results suggest that GlcN regulates inflammation by sensing energy states of normal and fuel excess.
Keywords: glucose, inflammation, NF-κB, O-GlcNAcylation, O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT)
Introduction
Nitric oxide (NO) has been implicated in inflammatory mediator and inducible NO synthase (iNOS) is expressed predominantly by microglia and macrophages by sensing danger or foreign signals. NO leads macrophages with cytostatic or cytotoxic activity against viruses, bacteria, fungi, and protozoa (1). However, the high levels of NO produced by activated macrophages may also harm healthy cells.
Glucosamine (2-amino-2-deoxy-d-glucose; GlcN)3 is a natural sugar that is synthesized from glucose through the hexosamine biosynthetic pathway (HBP) by virtually all cells. Entry into the HBP is catalyzed by the first and rate-limiting enzyme glutamine: fructose-6-phosphate amidotransferase (2). Exogenous GlcN, which enters the HBP bypassing fructose-6-phosphate amidotransferase is taken into cells by glucose transporters (GLUT-1, GLUT-2, and GLUT-4) after which it is phosphorylated and further metabolized (3). Increased GlcN elevates UDP-N-acetyl-GlcN (UDP-GlcNAc), a direct substrate for the transfer of single O-GlcNAc to nuclear and cytosolic proteins by O-GlcNAc transferase (4–6). GlcN or HBP has been implicated in insulin signaling through this O-GlcNAcylation mechanism (7, 8).
The immune regulatory functions of GlcN have been demonstrated: GlcN may exhibit a therapeutic effect in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis (9). At the cellular level, GlcN inhibits interleukin-1β (IL-1β)-induced nuclear factor-κB (NF-κB) activation in chondrocytes (10, 11). As well, GlcN inhibits lipopolysaccharide (LPS)-induced NO production in RAW 264.7 macrophages and microglia (10–14). Moreover, GlcN exhibits neuroprotective and anti-inflammatory effects in a model of middle cerebral artery occlusion via suppression of NF-κB activity (15). More recently, we demonstrated that GlcN inhibits the transcription of inflammatory genes via regulation of O-GlcNAcylation of transcription factors. For example, LPS-mediated iNOS induction is suppressed by GlcN by regulating c-Rel O-GlcNAcylation and subsequent NF-κB activity in both microglia and macrophage cells (16).
Activation of macrophages or microglia by LPS stimulates mitogen-activated protein (MAP) kinases and NF-κB. MAP kinases and NF-κB are both strongly implicated in the regulation of iNOS expression by LPS (17–20). NF-κB is normally sequestered in the cytoplasm by inhibitors of NF-κB, inhibitor of κB (IκB) proteins including IκBα and IκBβ. In response to stimulation, IκB becomes phosphorylated and is subjected to degradation after ubiquitination. This accompanies NF-κB translocation to the nucleus where the protein activates transcription (21). Vertebrate NF-κB transcription complexes can be any of a variety of homo- and heterodimers formed by the subunits RelA (p65), RelB, c-Rel, p50 (a product of p105), and p52 (a product of p100) (22, 23). These complexes bind to DNA regulatory sites and usually activate specific target gene expression. The target gene specificity of NF-κB is thought to arise from a number of factors. Some factors include the specific NF-κB complexes that are in different cell types, the distinct κB target site binding specificities of different NF-κB complexes, and the different protein-protein interactions and posttranslational modifications that NF-κB complexes (24, 25).
It is well established that most metabolic diseases are directly or indirectly associated with a low-grade chronic inflammation that causes complications such as nephropathy, neuropathy, retinopathy, and atherosclerosis (26). Therefore, the excessive caloric intake might be associated with the pathogenesis of both obesity-related complications and chronic inflammation. Indeed, free fatty acids have been shown to activate the LPS receptor, Toll-like receptor (TLR) 4 (27). Free fatty acid may also induce the production of proinflammatory cytokines by macrophages (27). High-glucose concentrations are also involved in inflammatory processes (28, 29). LPS modulates the generation of an inflammatory mediator in macrophages in hyperglycemic models (30). Therefore, inflammatory responses are obviously associated with glucotoxicity in diabetic conditions. Hyperglycemia-associated metabolic syndrome promotes abnormally elevated protein O-GlcNAcylation, which participates in the glucotoxicity (31–33). The association between hyperglycemia and metabolic syndrome-associated inflammation is well established yet the involvement of O-GlcNAcylation in those events is obscure. Therefore, exploring the regulation of O-GlcNAcylation and its functional implications in inflammatory regulation under different glucose settings is important. In this study, we investigated the potential of both pro- and anti-inflammatory effects of GlcN to regulate NO/iNOS induction as well as O-GlcNAcylation in macrophage cells under normal and high glucose culture conditions. GlcN demonstrated an inverse effect on LPS-mediated iNOS induction and O-GlcNAcylation and DNA binding of c-Rel between normal and high glucose conditions. Therefore, GlcN or the HBP metabolic pathway likely regulates inflammation and the related signaling pathway depends on nutrient availability.
Results
LPS-mediated NO Production Was Increased by GlcN under Normal Glucose Culture Conditions but Inhibited by GlcN under High Glucose Culture Conditions in RAW264.7 Macrophage Cells
Previous reports by our group showed that GlcN inhibits LPS-induced NO production (13, 15). In this report, we investigated the dose-dependent effect of GlcN on LPS-caused NO synthesis under increasing glucose concentrations. RAW264.7 cells were incubated with culture medium containing zero, normal (5 mm), or high glucose (25, 50, or 100 mm) in the presence or absence of LPS (100 ng/ml) and/or GlcN (5 mm). Concentrations higher than 20 mm were considered as analogous to a diabetic condition within the cell culture system. The nitrite levels (an index of NO production) in culture medium were compared after 24 h. LPS produced detectable NO generation at concentrations higher than 5 mm glucose conditions. Furthermore, LPS-induced NO generation was dose dependently increased at various glucose concentrations (Fig. 1A). The addition of GlcN 2 h before LPS administration resulted in a complicated effect on NO generation as per glucose concentration. GlcN increased LPS-mediated NO generation at normal glucose conditions but GlcN strongly inhibited LPS-mediated NO generation at concentrations higher than 25 mm glucose (Fig. 1A).
FIGURE 1.

Effects of GlcN on LPS-induced NO production and cell viability under 5 or 25 mm glucose conditions in cultured RAW264.7 cells. RAW264.7 cells were incubated with DMEM containing 0, 5, 25, 50, or 100 mm glucose for 4 h and then untreated or treated with GlcN (5 mm). 2 h after GlcN treatment, cells were stimulated with 0.1 μg/ml of LPS for 24 h (A). Cells were cultured with DMEM containing 5 or 25 mm glucose for 4 h. Cells were then treated at the indicated concentrations of GlcN for 2 h followed by stimulation with 0.1 μg/ml of LPS (B and C). NO production at 24 h was measured by measuring nitrite levels in the culture medium (B, upper panels) and cell viability at 72 h was determined by an MTT assay and expressed as a percentile compared with untreated control (B, lower panels). Cell phenotype at 48 h was observed under a phase-contrast microscope (C). Shown are representatives of three independent experiments. All values are mean ± S.E. *, denotes significantly increased from the untreated control; **, indicates significantly increased from LPS-stimulated conditions; #, indicates significantly decreased from LPS-stimulated conditions; †, indicates decreased from untreated control.
Next, we examined dose effects of GlcN on NO production and cell viability in RAW264.7 cells in response to LPS. The level of nitrite in culture medium at 24 h of LPS stimulation was increased by GlcN in a dose-dependent manner at normal glucose culture conditions. However, LPS-induced NO production was dose dependently inhibited by GlcN at 25 mm glucose conditions (Fig. 1B, upper panels) as previously demonstrated (13, 15). GlcN inhibited cell death that was induced by LPS under 25 mm glucose conditions. Conversely, GlcN yielded a minimal effect on cell death under 5 mm glucose conditions (Fig. 1B, lower panels). Morphological examinations indicated that GlcN protected cells against LPS-mediated cell death at 48 h under both 5 and 25 mm glucose conditions (Fig. 1C).
Influences of GlcN on LPS-mediated Production of iNOS and Other Inflammatory Genes under Normal and High Glucose Culture Conditions Were Examined
The effect of GlcN on LPS-mediated induction of proinflammatory genes was examined under normal or high glucose conditions. Raw264.7 cells were incubated with culture medium containing normal glucose (5 mm) or high glucose (25 mm) in the presence or absence of LPS (100 ng/ml) and/or GlcN (5 mm). LPS-initiated iNOS expression at 24 h was increased by GlcN under 5 mm glucose culture conditions. Conversely, GlcN inhibited iNOS and COX-2 protein expression in cells under 25 mm glucose culture conditions (Fig. 2A). Next, we examined the effect of GlcN on the activity of the iNOS promoter under ambient glucose conditions. GlcN suppressed the activity of the iNOS promoter at 25 mm glucose conditions, as previously described (15). In contrast, GlcN stimulated reporter activity of the iNOS promoter under 5 mm conditions (Fig. 2B).
FIGURE 2.

Effects of GlcN on LPS-induced up-regulation of proinflammatory genes in RAW264.7 cells cultured under 5 or 25 mm glucose conditions. A, RAW 264.7 cells were incubated with DMEM containing 5 or 25 mm glucose for 4 h. Cells were untreated or treated with 5 mm GlcN for 2 h and then stimulated with 0.1 μg/ml of LPS for 24 h. Protein expression of iNOS and COX-2 were measured by Western blotting (A, left), and relative densitometric intensity was quantitatively measured and normalized to α-tubulin levels (A, right). B, cells were transfected with iNOS promoter-reporter constructs and stimulated with 0.1 μg/ml of LPS with or without 5 mm GlcN in the presence of 5 or 25 mm glucose containing DMEM. Luciferase activity was measured at 24 h and shown as relative luciferase activity. C and D, RAW264.7 (C) or primary cultured mouse peritoneal macrophage (D) cells were incubated with 5 or 25 mm glucose media for 4 h, untreated or treated with 5 mm GlcN for 2 h, and then stimulated with 0.1 μg/ml of LPS for 24 h. iNOS, COX-2, IL-6, TNF-α, IL-1β, and GAPDH mRNA levels were determined by RT-PCR. Blots are representatives of three independent experiments. All values are mean ± S.E. *, denotes significantly increased from the untreated control; **, indicates significantly increased from LPS-stimulated conditions; #, indicates significantly decreased from LPS-stimulated conditions.
Levels of mRNA encoding iNOS, COX-2, IL-6, TNF-α, and IL-1β were increased by LPS and their expressions were strongly inhibited by GlcN under 25 mm conditions. At 5 mm culture conditions, GlcN stimulated LPS-induced iNOS mRNA. However, LPS-stimulated mRNA expression of COX-2, IL-6, and TNF-α were all not significantly influenced by GlcN at 5 mm glucose conditions. LPS-mediated expression of IL-1β was inhibited by GlcN at 5 mm conditions (Fig. 2C). We verified the GlcN effect on LPS-induced iNOS mRNA expression using primary cultured mouse peritoneal macrophages; GlcN was inhibited at 25 mm conditions but stimulated at 5 mm conditions (Fig. 2D). We have verified that GAPDH expression was not changed by glucose and/or GlcN (data not shown).
Time Course iNOS Induction in Response to LPS and/or GlcN under Normal or High Glucose Culture Conditions Was Examined
We examined the time course of expression of mRNA encoding iNOS, and that of protein expression per se, in RAW264.7 cells exposed to LPS (100 ng/ml) under 5 or 25 mm glucose culture conditions. Furthermore, we determined how GlcN affected induction of such expression. iNOS protein was detected as early as 6 h LPS stimulation at both 5 and 25 mm glucose. GlcN stimulated iNOS expression at 5 mm glucose but was relatively ineffective at 25 mm glucose conditions (Fig. 3A). LPS-induced expression of iNOS was more distinct at 12 h. GlcN was further stimulated at concentrations of 5 mm glucose but slightly inhibited under 25 mm glucose. Likewise, the increase in iNOS mRNA by LPS was inhibited by GlcN at 12 and 24 h of LPS stimulation under 25 mm glucose conditions. Contrastingly, LPS-induced iNOS mRNA was stimulated by GlcN at all time points examined at 5 mm glucose concentration (Fig. 3B).
FIGURE 3.

Time course regulation of LPS-induced iNOS expression by GlcN under 5 or 25 mm glucose conditions. RAW264.7 cells were incubated with DMEM containing 5 or 25 mm glucose for 4 h and then treated with 5 mm GlcN and/or 0.1 μg/ml of LPS for 0.5, 2, 6, or 12 h (for protein expression) or 6, 12, or 24 h (for mRNA expression). Whole cell lysates were prepared and iNOS and GAPDH protein levels were measured by Western blotting (A). iNOS mRNA level was determined by RT-PCR and quantitatively determined by real-time PCR (B). GAPDH protein and mRNA levels were measured as a control. Data are representatives of three independent experiments. All values are mean ± S.E. *, denotes significantly increased from the untreated control; **, indicates significantly increased from LPS-stimulated conditions; #, indicates significantly decreased from LPS-stimulated conditions.
Glucose and/or GlcN-regulated iNOS, C/EBP-δ, C/EBP-β, and GRP78 Protein Expression in an Independent Manner
To explore any dependence of possible differential regulation of GlcN for iNOS induction on glucose-regulated transcription factors or ER stress responses, we examined iNOS, C/EBP-δ, C/EBP-β, and GRP78 protein induction levels in RAW264.7 cells. Cells cultured in increasing concentrations of glucose (0, 5, 25, 50, and 100 mm) displayed increased iNOS expression in response to LPS in a dose-dependent manner. Under the same conditions, the expression of C/EBP-δ, C/EBP-β, and GRP78 were all reduced by glucose in a dose-dependent manner both in the absence and presence of LPS (Fig. 4A). C/EBP-β expression was less distinctly reduced by glucose in the presence of LPS.
FIGURE 4.
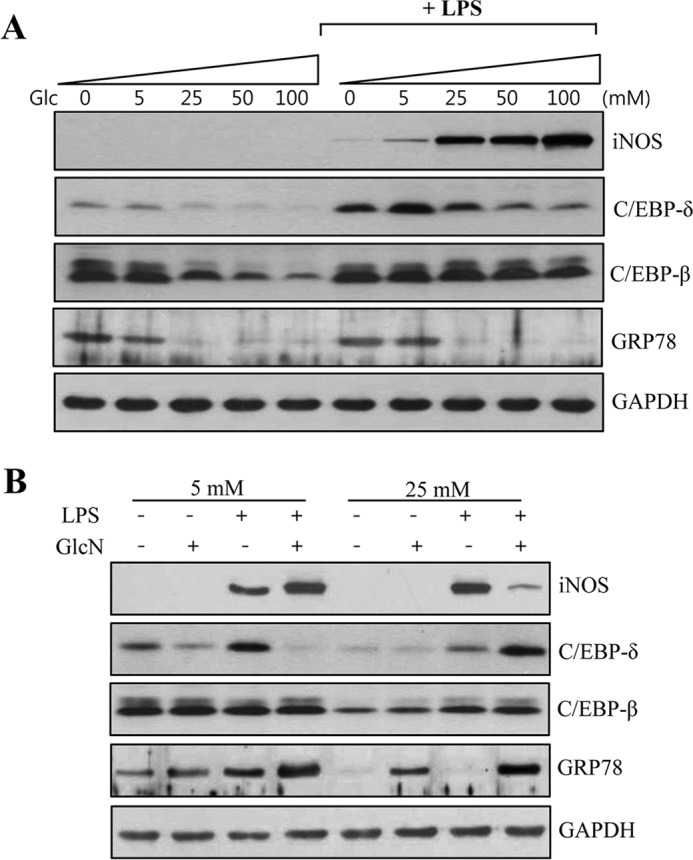
Regulations of C/EBP-δ, C/EBP-β, and GRP78 protein expression in response to LPS and/or GlcN under various glucose culture conditions. A, RAW264.7 cells were cultured with DMEM containing 0, 5, 25, 50, or 100 mm glucose for 4 h and either unstimulated or stimulated with 0.1 μg/ml of LPS for 24 h. B, cells were incubated with DMEM containing 5 or 25 mm glucose for 4 h and treated with 5 mm GlcN for 2 h and then stimulated with 0.1 μg/ml of LPS for 24 h. Whole cell lysates were prepared and iNOS, C/EBP-δ, C/EBP-β, and GRP78 protein levels were determined by Western blotting along with GAPDH as a loading control. Shown blots are representatives of three independent experiments. All values are mean ± S.E. *, denotes significantly increased from the untreated control; #, indicates significantly decreased from LPS-stimulated conditions.
Next, we determined the effect of GlcN on the expression of C/EBP-δ, C/EBP-β, and GRP78 under 5 or 25 mm glucose conditions. C/EBP-β was not significantly influenced by LPS and/or GlcN at 5 or 25 mm glucose. At 5 mm glucose, the expression of C/EBP-δ was stimulated by LPS but inhibited by GlcN. However, at 25 mm glucose, GlcN further increased LPS-increased C/EBP-δ protein expression (Fig. 4B). GRP78 protein was expressed in a lower level at 25 mm glucose than at 5 mm glucose conditions. However, GRP78 expression was significantly increased by 5 mm GlcN in 25 mm glucose with or without LPS. In addition, GRP78 was slightly increased by GlcN in the presence of LPS in 5 mm glucose condition.
The Activation of MAPKs and IκBα in Response to LPS and/or GlcN under 5 or 25 mm Glucose Conditions Was Investigated
Activation of mitogen-activated protein kinase (MAPK) signaling by LPS, and the effect of GlcN on such signaling, was examined 30 min, 2 h, and 12 h after LPS (100 ng/ml) addition under 5 or 25 mm glucose culture conditions. LPS increased phosphorylation of ERK1/2, p38, and JNK in RAW264.7 cells at both 5 and 25 mm glucose conditions. GlcN did not significantly affect the extent of any LPS-induced phosphorylation of ERK1/2, p38, and JNK 30 min after LPS addition under 5 or 25 mm glucose. A reduction in ERK1/2 phosphorylation by GlcN was evident 12 h after LPS addition under both 5 and 25 mm glucose conditions. LPS-induced phosphorylation of p38 and JNK was reduced in the presence of GlcN at 12 h after LPS addition at 25 mm glucose but not at 5 mm glucose (Fig. 5, A and B). IκBα was both phosphorylated and degraded by LPS at 30 min. GlcN did not influence degradation nor phosphorylation of IκBα at either 5 or 25 mm glucose conditions.
FIGURE 5.
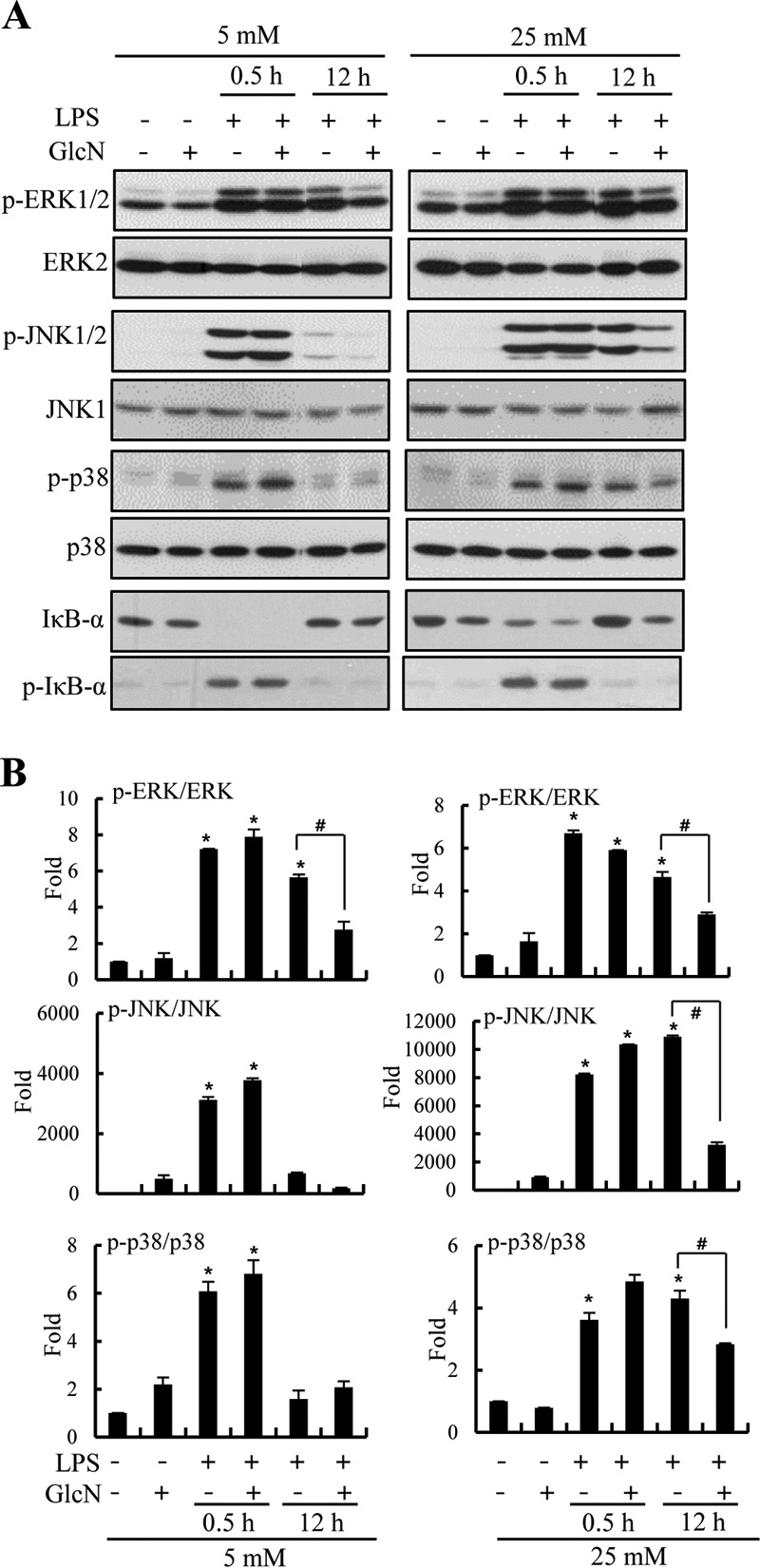
Effect of GlcN on LPS-induced signaling pathway under 5 or 25 mm glucose conditions. RAW264.7 cells were incubated with DMEM containing glucose at 5 or 25 mm for 4 h, treated with 5 mm GlcN for 2 h, and stimulated with 0.1 μg/ml of LPS for 0.5 or 12 h. A, Western blots were performed using p-ERK1/2, ERK1/2, p-JNK1/2, JNK1, p-p38, p38, IκB-α, and p-IκB-α-specific antibodies. B, relative densitometric intensity for p-ERK1/2, p-JNK1/2, or p-p38 was quantitatively measured by normalizing to ERK1/2, JNK1, or p38 levels, respectively. Blots are representatives of three independent experiments. All values are mean ± S.E. *, denotes significantly increased from the untreated control; #, indicates significantly decreased from LPS-stimulated conditions.
GlcN Suppressed NF-κB Activation in Response to LPS under 25 mm Glucose, but Not under 5 mm Glucose Environment
We compared the effect of GlcN on LPS-induced NF-κB activation between normal and high glucose conditions. LPS stimulated nuclear translocation of c-Rel and p50 in RAW264.7 cells maintained at both 5 and 25 mm glucose culture conditions. Conversely, p65 nuclear translocation was increased at only 25 mm glucose conditions (Fig. 6A). Nuclear translocations of p65, c-Rel, and p50 in response to LPS were inhibited by GlcN at 25 mm glucose, but were not significantly influenced under 5 mm glucose (Fig. 6A). Next, we examined the effects of LPS and/or GlcN on NF-κB activation using the NF-κB-reporter construct. LPS activated the NF-κB reporter at 25 mm glucose but less strongly at 5 mm glucose. GlcN inhibited NF-κB activity under 25 mm glucose conditions but did not significantly exhibit any influence at 5 mm glucose conditions (Fig. 6B).
FIGURE 6.

Regulation of LPS-mediated NF-κB activation by GlcN under 5 or 25 mm glucose culture conditions. RAW264.7 cells were incubated with DMEM containing 5 or 25 mm glucose for 4 h, treated with 5 mm GlcN for 2 h, and stimulated with 0.1 μg/ml of LPS for 24 h. A, nuclear protein extracts were prepared and p65, c-Rel, or p50 expressions were measured by Western blotting and relative densitometric intensity was quantitatively measured by normalizing to HDAC1 protein. B, RAW264.7 cells were transfected with NF-κB-reporter constructs and preincubated with 5 or 25 mm glucose containing medium for 4 h and pretreated with 5 mm GlcN for 2 h. Cells were then stimulated with 0.1 μg/ml of LPS for 24 h. Luciferase activities were measured from cell lysates and relative values were demonstrated. C, cells were incubated with 5 or 25 mm glucose containing media for 4 h with or without GlcN for 2 h followed by stimulation with LPS for 24 h. Total cell lysates were prepared and binding of Rel proteins to the biotinylated NF-κB probe was examined by pulling down using streptavidin-agarose beads. Bound proteins were determined by Western blotting using specific antibodies for c-Rel, p65, and p50. D, formaldehyde cross-linked chromatin DNA was immunoprecipitated with anti-c-Rel antibody and eluted DNA was quantified by real-time PCR or standard reverse transcriptase PCR (RT-PCR) for the iNOS promoter. Input represents each PCR product obtained from 2% of the preimmunoprecipitated DNA. Blots are representatives of three independent experiments. All values are mean ± S.E. *, denotes significantly increased from the untreated control; **, indicates significantly increased from LPS-stimulated conditions; #, indicates significantly decreased from LPS-stimulated conditions.
To determine whether LPS and/or GlcN directly regulated the binding of Rel family proteins to promoter DNA, we assayed the ability of Rel proteins to bind to a biotinylated oligonucleotide corresponding to the distal NF-κB site. This site (−957 to −977 in the mouse iNOS promoter) is important regarding gene induction by LPS (34). Stimulation with LPS for 24 h induced increases in the binding levels of both c-Rel and p50 at both 5 and 25 mm glucose concentrations. Furthermore, p65 binding also increased at 25 mm glucose. LPS-stimulated binding of p65, c-Rel, and p50 were all suppressed by GlcN at 25 mm glucose yet these bindings were all not affected by GlcN at 5 mm glucose (Fig. 6C). To examine the physiological relevance of such DNA binding, we performed chromatin immunoprecipitation (ChIP) assays. LPS induced an increase in the binding of c-Rel to the endogenous iNOS promoter both in 5 and 25 mm glucose conditions. In contrast, binding was suppressed by GlcN at 25 mm glucose but not influenced under 5 mm glucose conditions (Fig. 6D). This suggests a regulatory role for GlcN in LPS-mediated iNOS induction that may be controlled by regulating the chromatin mobilization of c-Rel proteins.
LPS-dependent Increase in c-Rel O-GlcNAcylation Is Inhibited by GlcN at 25 mm Glucose but Increased by GlcN at 5 mm Glucose Conditions
We have previously reported that LPS and/or GlcN regulate iNOS induction via post-translational O-GlcNAcylation of c-Rel (16). We performed wheat germ agglutinin (WGA) pulldown assays using WGA-conjugated agarose beads that specifically precipitate proteins containing N-acetylglucosamine or N-acetylneuramic acid. RAW264.7 cells were treated with LPS (100 ng/ml) and/or GlcN (5 mm) for 24 h under 5 or 25 mm glucose conditions. O-GlcNAcylation of total cell lysates was stimulated by GlcN at 5 mm glucose compared with no significant changes by GlcN at 25 mm glucose (Fig. 7A, upper panels). The amount of c-Rel protein in the WGA precipitate was increased by LPS and this increase was inhibited by GlcN at the 25 mm glucose conditions, as previously described (16). However, WGA binding of c-Rel in response to LPS was dramatically increased by GlcN at the 5 mm glucose conditions (Fig. 7A, lower panels). These results suggest that O-GlcNAcylation of c-Rel or a protein that interacts with c-Rel is differentially regulated by GlcN depending on glucose concentrations. We further confirmed O-GlcNAcylation changes of total and c-Rel proteins in response to LPS and/or GlcN under 5 mm glucose conditions by labeling O-GlcNAc with galactosyltransferase using [3H]galactose as a substrate. O-GlcNAcylation of total nucleocytoplasmic proteins was increased by GlcN regardless of LPS presence (Fig. 7B, upper panel). O-GlcNAcylation of c-Rel determined by [3H]galactose labeling demonstrated that c-Rel O-GlcNAcylation at 5 mm glucose was slightly increased by GlcN and further increased by LPS plus GlcN (Fig. 7B, lower panels).
FIGURE 7.

Changes of O-GlcNAcylation by LPS and/or GlcN under 5 or 25 mm glucose conditions. RAW264.7 cells were incubated with DMEM containing 5 or 25 mm glucose for 4 h, treated with 5 mm GlcN for 2 h, and stimulated with 0.1 μg/ml of LPS for 24 h. A, total cell lysates were prepared and subjected to precipitation with WGA-conjugated agarose beads. The precipitates were immunoblotted by anti-O-GlcNAc (CTD 110.6) or anti-c-Rel antibody. B, cells cultured at 5 mm glucose were untreated or treated with GlcN followed by stimulation with LPS for 24 h. Whole cell lysates were immunoprecipitated with anti-c-Rel antibody. Total cell lysates or c-Rel immunoprecipitates were subjected to galactosyltransferase labeling using [3H]UDP-galactose as a substrate and exposed to X-ray film for autoradiography. C and D, cells cultured at 5 or 25 mm glucose were treated with GlcN followed by stimulation with LPS for 24 h. Nuclear and cytosolic fractions were prepared and subjected to precipitation (IP) with WGA-conjugated agarose beads (C) or anti-c-Rel antibody (D). Total or precipitated proteins were immunoblotted by c-Rel, p65, p50, and O-GlcNAc (CTD110.6) antibodies. Blots are representatives of three independent experiments.
Next, we compared O-GlcNAcylation of nuclear and cytosolic c-Rel and p65 in response to LPS and/or GlcN under 5 or 25 mm glucose. Both nuclear and cytosolic c-Rel responded similarly to LPS and/or GlcN at two different glucose conditions for O-GlcNAcylation. c-Rel O-GlcNAcylation was increased by LPS but this increase was inhibited by GlcN at 25 mm glucose conditions. However, GlcN further increased the O-GlcNAcylation of c-Rel in response to LPS at 5 mm glucose conditions (Fig. 7C). O-GlcNAcylation of p65 was increased by GlcN both in the nucleus and cytosol at 5 mm glucose, but it was not significantly influenced by LPS and/or GlcN at 25 mm glucose. These results suggest that O-GlcNAcylation is not likely associated with nuclear translocations of c-Rel and p65.
O-GlcNAcylation of c-Rel may regulate the association between other members of Rel proteins. An antibody against c-Rel co-immunoprecipitated p65 and p50 in both nuclear and cytosolic fractions. The association between c-Rel and p50 in the nucleus increased in response to LPS; GlcN inhibited this association in 25 mm glucose but GlcN did not effect this association in 5 mm glucose (Fig. 7D).
LPS-mediated NO/iNOS Increases Were Inhibited by PDTC at 25 mm Glucose but Not at 5 mm Glucose Concentrations
We investigated the effects of the NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), on activation of NF-κB as well as the induction of NO/iNOS in response to LPS and/or GlcN under ambient (5 and 25 mm) glucose conditions. PDTC suppressed LPS-induced NO generation under 25 mm glucose conditions (Fig. 8A). Similarly, LPS-induced iNOS expression was inhibited by PDTC in a dose-dependent manner at 25 mm glucose conditions. Conversely, PDTC exhibited no, or a minimal effect, on LPS- or LPS plus GlcN-mediated NO/iNOS induction under 5 mm glucose conditions (Fig. 8, B and C). LPS plus GlcN-induced expression of COX-2 proteins was inhibited by 20 μm PDTC under 5 mm glucose.
FIGURE 8.

Effect of PDTC on LPS and/or GlcN-mediated iNOS/NO production under 5 or 25 mm glucose conditions. RAW264.7 cells were incubated with DMEM containing 5 or 25 mm glucose for 4 h, treated with 10 or 20 μm PDTC and/or 5 mm GlcN for 2 h. Cells were then stimulated with 0.1 μg/ml of LPS for 24 h. A, nitrite levels were measured. B and C, whole cell lysates were prepared and analyzed by Western blotting using iNOS and COX-2 specific antibodies (B). The relative densitometric intensity was quantitatively measured by normalizing to GAPDH protein (C). D, nuclear extract was prepared and DNA binding to a 32P-labeled NF-κB probe was measured by EMSA (upper panels). The relative densitometric intensity was quantitatively measured and demonstrated (lower graphs). E, total cell lysates were prepared and subjected to precipitation with WGA-conjugated agarose beads. Precipitated proteins were immunoblotted by c-Rel, p65, and GAPDH antibodies (upper blots). Relative densitometric intensity of O-GlcNAcylation of c-Rel and p65 subunits were quantitatively measured and normalized to GAPDH levels (lower graphs). Data are representatives of three independent experiments. All values are mean ± S.E. #, indicates significantly decreased from LPS-stimulated conditions; *, denotes significantly increased from the untreated control; **, indicates significantly increased from LPS-stimulated conditions.
PDTC suppressed LPS-induced NF-κB/DNA-binding activity at 25 mm glucose conditions. However, PDTC did not inhibit LPS-induced NF-κB/DNA-binding activity under 5 mm glucose conditions. Furthermore, PDTC potentiated the LPS plus GlcN-induced NF-κB/DNA-binding activity at 5 mm glucose (Fig. 8D).
Next, we examined O-GlcNAcylation changes of c-Rel and p65 by PDTC in response to LPS and/or GlcN under ambient glucose culture conditions. The O-GlcNAcylation of c-Rel in response to LPS plus GlcN was slightly (less than 2-fold) inhibited by PDTC at 5 mm glucose conditions. However, the O-GlcNAcylation of c-Rel by LPS was dramatically (more than 15-fold) decreased by PDTC at 25 mm glucose conditions (Fig. 8E). O-GlcNAcylation of p65 was not significantly influenced by PDTC under both 5 and 25 mm glucose conditions.
GlcN Increased the Stability of iNOS mRNA under 5 mm Glucose Culture Conditions
Because the expression of iNOS is regulated post-transcriptionally as well as transcriptionally, we examined the iNOS mRNA degradation. As an inhibitor of transcription, actinomycin D (ActD) was added to RAW264.7 cells to inhibit new synthesis of iNOS at 12 h of LPS or LPS plus GlcN stimulation. Cells were harvested at 2, 4, 6, and 8 h after the addition of ActD. At 5 mm glucose, iNOS mRNA was reduced in LPS-stimulated conditions in a time-dependent manner by an addition of ActD, whereas the iNOS mRNA level was not significantly changed by ActD in LPS plus GlcN-stimulated conditions (Fig. 9A, left graph). Contrarily, LPS-induced iNOS mRNA was not significantly changed by ActD, whereas iNOS mRNA in response to LPS plus GlcN displayed a time course decrease at 25 mm glucose conditions (Fig. 9A, right graph).
FIGURE 9.

Changes of iNOS mRNA stability by LPS and/or GlcN. A, RAW264.7 cells were incubated with DMEM containing 5 or 25 mm glucose for 4 h, and stimulated with 0.1 μg/ml of LPS in the presence or absence of 5 mm GlcN. ActD (10 μg/ml) was then treated at 12 h of LPS addition. Cells were harvested at the indicated time points and the iNOS mRNA levels were determined by quantitative real-time PCR. B, RAW264.7 cells were incubated with DMEM containing 5 mm glucose for 4 h and treated with GlcN and/or LPS for 6 h. The expression of TTP mRNA was measured by RT-PCR (left blot) and quantitative real-time PCR (right graph). Data are representatives of three independent experiments. All values are mean ± S.E. *, denotes significantly increased from LPS-treated conditions; **, indicates significantly increased from the untreated control; #, indicates significantly decreased from LPS-stimulated conditions.
Tristetraprolin (TTP) is involved in mRNA degradation of iNOS (35). TPP mRNA was increased 6 h after LPS stimulation at 5 mm glucose conditions. GlcN significantly reduced the LPS-mediated increase of TTP (Fig. 9B).
Discussion
Among the most critical factors of survival are the ability to withstand starvation and an effective immune response to pathogens. Inflammation is basically triggered by nutrient surpluses, and inflammatory responses and metabolic disorders share common signaling molecules and pathways. A large body of emerging evidence suggests that nutrient signaling through the HBP serves an important role in cellular adaptation to fluctuations in nutrient availability even at physiological glucose concentrations (36). Our previous study (37) discussed the distinct role of the GlcN, in which metabolic processes are allowed to adapt to nutritional states of normal and fuel excess: GlcN impaired the insulin response when fed to normal diet-fed mice yet it alleviated insulin resistance in high fat diet-fed mice. The results in this report indicate that increased GlcN addition served an inverse role in regulating LPS-mediated inflammation in macrophage depending on glucose availability. GlcN increased LPS-mediated iNOS induction under normal (5 mm) glucose culture conditions, whereas it strongly inhibited LPS-mediated iNOS induction under high (25 mm) glucose conditions. The direct effect of glucose availability on inflammatory response at the cellular level has been described. LPS-induced NO/iNOS induction was increased at high glucose-cultured cells than normal glucose-cultured cells via enhancing activation of NF-κB (30, 38, 39). We newly propose in this report that glucose availability sensed by HBP flux influences the regulation of inflammation.
We postulate that the endoplasmic reticulum (ER) might be a point for the sensing of metabolic stress and the translation of that stress into inflammatory signaling and consequent responses. Indeed, the ER could be considered an essential point of integration between nutrient and pathogen responses as ER is very sensitive to glucose and energy availability, lipids, pathogens, and pathogen-associated components. It has been demonstrated that elevated concentrations of glucose induce ER stress by increasing HBP flux in skeletal muscle and aortic endothelial cells (40). According to our results, whereas GlcN increased GRP78 expression, exposure to high glucose reduced the expression of GlcN-induced GRP78 in macrophage cells. Therefore, metabolic stress and HBP may impose a complicated effect on ER stress in macrophage cells. Currently, any consequent link between ER stress and inflammatory regulation under normal or high glucose conditions is elusive.
Several transcriptional factors, such as NF-κB, Fos/Jun, CREB, C/EBP, GAS, and IRF, have been described to be involved in the regulation of iNOS gene expression (41–44). Among these transcriptional factors, C/EBP-δ may be a critical mediator in glucose-mediated regulation of iNOS gene expression in response to LPS (39). We found that C/EBP-δ protein expression was reduced as the glucose concentration increased with or without LPS. This demonstrated an inverse correlation with an LPS-induced increase of iNOS expression. In addition, the increased expression of C/EBP-δ in response to LPS is reduced by GlcN at 5 mm conditions but increased by GlcN under 25 mm conditions. This suggests that C/EBP-δ imparts a negative role in regulating inflammation in response to LPS and/or GlcN. Anti-inflammatory function of C/EBP-δ has been studied: a C/EBP-δ deficiency increases up-regulation of inflammatory genes in pancreatic beta cells (45). However, the role of C/EBP-δ in glucose- and/or GlcN-mediated inflammatory regulation may depend on type of cell or type and duration of the insult and has to be clarified in further research.
In our previous report (13, 16), GlcN inhibited LPS-induced NF-κB and MAPK activation in response to LPS under high glucose conditions. In this study, we found that LPS-induced phosphorylation of MAPKs and IκBα were not influenced by GlcN under 5 mm glucose conditions. Instead, GlcN differentially regulated the LPS-induced NF-κB activation between 5 and 25 mm glucose conditions. GlcN suppressed the LPS-induced NF-κB activation under 25 mm glucose conditions, whereas GlcN did not influence the activity of NF-κB caused by LPS under 5 mm glucose culture conditions. These results imply a distinct role of GlcN in regulating NF-κB activity according to glucose availability.
Increased GlcN results in O-GlcNAcylation, which mediates important features of transcriptional regulation. Previously, we have shown that GlcN inhibits LPS-induced NF-κB activation by regulating O-GlcNAcylation of p65 or c-Rel in response to LPS (15, 16). O-GlcNAcylation of p65 or c-Rel and their subsequent transcriptional activities may be differentially regulated by GlcN under ambient glucose conditions. O-GlcNAcylation and DNA binding of c-Rel in response to LPS was dramatically increased by GlcN under 5 mm glucose conditions. However, increased O-GlcNAcylation did not impose any increased association with other Rel proteins. A major question that arises then is whether the increased c-Rel binding to an iNOS promoter is actually sufficient to induce increased iNOS expression. The O-GlcNAcylation changes or recruitment of other transcription factors to the iNOS promoter might also be required for increased iNOS induction. Although this remains a possibility, our attempts to obtain evidence for changed recruitment or O-GlcNAcylation of RNAPII, CBP, p300, or mSin3A (in response to LPS plus GlcN under normal glucose conditions) proved unsuccessful (data not shown).
PDTC, an antioxidant and NF-κB inhibitor, strongly inhibited LPS-mediated NO/iNOS induction and NF-κB activation when cells were cultured under 25 mm glucose conditions, as we and others previously reported (13, 46). However, LPS-mediated NO/iNOS induction was not significantly altered by PDTC under 5 mm glucose conditions. Furthermore, PDTC did not inhibit the DNA-binding activity of NF-κB to the binding site of the iNOS promoter at 5 mm glucose conditions. This may be attributable to the differential regulation of O-GlcNAcylation of Rel proteins by PDTC under ambient glucose conditions. PDTC did not influence O-GlcNAcylation of p65 and c-Rel in response to LPS plus GlcN under 5 mm glucose conditions yet it inhibited O-GlcNAcylation of c-Rel at 25 mm glucose conditions. Therefore, PDTC presumably suppressed the LPS-induced activation of NF-κB by a mechanism that inhibits O-GlcNAcylation of NF-κB. GlcN under 5 mm glucose conditions may protect NF-κB from PDTC-regulated O-GlcNAcylation and/or transcriptional activity. The potential effect of PDTC to regulate expression or activities of O-GlcNAc transferase (OGT) or O-GlcNAcase under ambient glucose conditions needs to be further examined.
Although an increased responsiveness of LPS-induced NF-κB activity by GlcN under 5 mm glucose conditions seems to contribute to the increased iNOS induction, other additional regulatory mechanisms may exist as well. We proposed the changes of iNOS mRNA stability as an additional scenario responsible for a GlcN-dependent increase in LPS-mediated iNOS induction under 5 mm glucose conditions. The expression of iNOS must be tightly regulated by both transcriptional and post-transcriptional mechanisms. Post-transcriptional regulation of gene expression is often dependent on sequences located in the 3′-untranslated region (3′-UTR) of mRNAs (47, 48). The zinc-finger protein TTP, KH-type splicing regulatory protein, and HuR all bind the iNOS mRNA 3′-UTR and recruits the exosome to the mRNA (49). LPS transiently up-regulated and GlcN inhibited such increases of the expression of TTP without affecting the other proteins, KH-type splicing regulatory protein or HuR under both 5 and 25 mm glucose conditions (data not shown). Therefore, up-regulation of TTP by LPS in our experimental model may contribute to an increased mRNA degradation. Although the present data suggest an involvement of this protein in iNOS expression, additional experiments are required to address the role of TTP in iNOS mRNA regulation under various glucose conditions.
The concentration of GlcN in the plasma of normal subjects is ∼0.04 mmol/liter, rising up to 0.06 mmol/liter in those taking GlcN supplements (50). Intravenous administration of GlcN in rats induces much higher plasmatic concentrations than oral administration: infusion of 30.45 g of GlcN produced GlcN concentrations of ∼1.42 mmol/liter (51). Because of the limited information regarding HBP flux, which is accelerated by GlcN infusion in humans, in vivo physiological dosing might be an important issue to be addressed in the future.
In summary, we have demonstrated that the combination of increased GlcN flux and nutrient availability from extracellular culture medium plays a distinct role in the induction of iNOS and potentially other proinflammatory mediators in response to LPS. This adds to the growing body of evidence stressing the importance of HBP as a nutrient-sensing pathway at both excess nutrient and normal physiology conditions. HBP may operate as a master regulator of inflammation signaling by governing the rapid switch from inflammatory activation to repression or vice versa. This master regulator depends on nutrient availability for inflammatory regulation by integrating transcriptional regulation and mRNA stability.
Experimental Procedures
Reagents
Reagents were purchased from Sigma or Amresco (Cochran Road, OH) unless otherwise noted.
Cell Cultures
RAW264.7 murine macrophage cells (purchased from the Korean Cell Line Bank) were maintained at 37 °C at 5% CO2 in DMEM supplemented with 5% FBS (Hyclone, Logan, UT), 1% streptomycin and penicillin. For peritoneal macrophage isolation, 6-week-old male BALB/c mice were intraperitoneally injected with 200 μl of 3.5% thioglycollate, and cells were isolated from the peritoneal cavity 3 days after injection. The cells were then incubated for 6 h at 37 °C and cells were washed twice with PBS. The remaining adherent cells were used as the peritoneal macrophages described in the experiments. Overnight cultured cells were washed twice with PBS and replaced DMEM containing 5 or 25 mm glucose. After 4 h, cells were treated with 5 mm GlcN for 2 h and then stimulated with 0.1 μg/ml of LPS for 24 h.
Nitrite Assay
NO production was determined by measuring the amount of nitrite, as described with minor modifications (15). The 50 μl of cultured medium was reacted with 50 μl of Griess reagent (1% sulfanilamide, 0.1% naphthylenediamine, and 5% phosphoric acid). The optical density at 540 nm (OD540) was measured with a microplate reader. Nitrite concentrations were calculated by comparison with observance of standard of NaN2 solution.
Cell Viability
Cell viability was measured by the mitochondria-dependent reduction of MTT solution (3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to formazan. Cells were incubated with 5 mg/ml of MTT solution for 1 h at 37 °C. Medium was removed and cells were dissolved in DMSO:EtOH (1:1, v/v). The optical density at 540 nm (OD540) was measured using microplate reader.
Reverse Transcription PCR
Total RNA from cells was extracted with TRIzolTM (Invitrogen) reagent. Complementary DNA was synthesized from 1 μg of RNA with GoScriptTM Reverse Transcriptase (Promega, Madison, WI) according to the manufacturer's protocol. PCR was performed using specific primers of mouse GAPDH, F, TCATTGACCTCAACTACAGGT, R, CTAAGCAGTTGGTGGTGCAG; iNOS, F, GCATCCCAAGTACGAGTGGT, R, CCATGATGGTCACATTCTGC; COX-2, F, GCTGTACAAGCAGTGGCAAA, R, GTCTGGAGTGGGAGGCACT; IL-6, F, CCGGAGAGGAGACTTCACAG, R, TGGTCTTGGTCCTTAGCCAC; IL-1β, F, GGAGAAGCTGTGGCAGCTA, R, GCTGATGTACCAGTTGGGGA; TNF-α, F, GACCCTCACACTCAGATCAT, R, TTGAAGAGAACCTGGGAGTA. The final PCR products were electrophoresed in 1% agarose gel. For real-time PCR experiments, cDNAs were amplified with SYBR Green Real-time PCR Master Mix (TOYOBO, Osaka, Japan). All results were normalized to the expression of GAPDH.
Western Blotting Analysis and Immunoprecipitation
Cells were lysed with in Nonidet P-40 lysis buffer (150 mm NaCl, 50 mm Tris-HCl (pH 8.0), 0.5% Nonidet P-40 and containing protease and phosphatase inhibitors). For immunoprecipitation, 1 mg of total proteins were incubated with anti-c-Rel antibodies for 1 h at 4 °C. Antibody-protein complex was precipitated with protein G-Sepharose beads and precipitated proteins were analyzed by Western blotting. For WGA pulldown assays, 1 mg of cell lysate was incubated with WGA-agarose beads for 1 h at 4 °C and precipitated proteins were analyzed by Western blotting. The proteins (20–40 μg for each) were separated by SDS-PAGE and transferred onto nitrocellulose membranes (Amersham Biosciences), which were blocked with 5% nonfat milk or 5% BSA within TBST for 1 h at room temperature. Membranes were incubated with antibodies against anti-phospho-Akt (Ser-473), anti-Akt (Cell Signaling Technology, Danvers, MA, numbers 9271 and 9272), anti-phospho-JNK1/2 (Invitrogen, 44682G), anti-phospho p38, anti-JNK, and anti-ERK1/2 (Cell Signaling Technology, numbers 4511, 9252, and 4695), anti-O-GlcNAc CTD110.6 (Covance, Berkeley, CA, MMS-248R), anti-iNOS (BD Transduction, Lexington, KY, 610107), anti-cyclooxygenase-2 (COX-2) (Cayman, Ann Arbor, MI, 160107), anti-p65, anti-c-Rel, anti-p50, anti-IκBα, anti-phospho-IκBα, HDAC1, anti-phospho-ERK1/2, and anti-p38 (Santa Cruz Biotechnology, Santa Cruz, CA, sc-372, sc-71, sc-114, sc-371, sc-101713, sc-6298, sc-7383, and sc-535), α-tubulin (Calbiochem Merck KGaA, Darmstadt, Germany, ABT-170), and GAPDH (Cell Signaling, number 2118) overnight at 4 °C. After washing with TBST, horseradish peroxidase (HRP)-conjugated secondary antibodies (Amersham Biosciences) (1:10,000 dilution in TBST) were applied and developed by the Enhanced Chemiluminescence (ECL) detection system (Amersham Biosciences). Densitometric quantification of protein bands were detected using ImageJ (NIH).
Transient Transfection and Luciferase Assay
The NF-κB reporter contained three copies of the NF-κB binding site fused to the firefly luciferase gene (Clontech). Cells were transfected with LipofectamineTM and PLUSTM reagents (Invitrogen) according to the manufacturer's protocol. After 3 h, medium was replaced with DMEM containing 5 or 25 mm glucose and stimulated with 0.1 μg/ml of LPS in the presence or absence of 5 mm GlcN. Luciferase activity was measured using a Turner Designs luminometer (TD-20/20) at 24 h and normalized to transfection efficiency using a cotransfected β-galactosidase.
Streptavidin-Agarose Pulldown Assay
Streptavidin-agarose pulldown assays were performed as described previously (13). The procedure allows for quantitative binding of transcription factors or molecules of interest to a specific probe: a 20-nucleotide sequence containing the NF-κB binding site (5′-GCTAGGGGGATTTTCCCTCT-3′) at position −957/−977 of the iNOS promoter. Total protein lysate was prepared with lysis buffer (10 mm HEPES, pH 8.0, 1.5 mm MgCl2, 200 mm sucrose, 0.5% Nonidet P-40, 10 mm KCl and protease inhibitors) and then nuclear proteins were extracted in lysis buffer (1 mm EDTA, pH 8.0, in PBS) with sonication. The nuclear protein samples were incubated with 2 μg of biotinylated iNOS-κB DNA probe and 25 μl of steptavidin-conjugated agarose beads for 1 h at 4 °C. DNA-protein complexes were determined by Western blotting using the indicated antibodies.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear protein extracts were prepared as described previously (13). The double-stranded DNA oligonucleotide probe containing the consensus NF-κB binding site (Promega) was labeled by T4 polynucleotide kinase (New England Biolabs, Beverly, MA). Nuclear protein (10 μg) were incubated with labeled oligonucleotide probe in binding buffer (50 mm KCl, 12.5 mm HEPES, pH 7.6, 6.25 mm MgCl2, 0.05 mm EDTA, 0.5% Nonidet P-40, 0.5 mm DTT, 5% glycerol, and 2 μg of poly(dI-dC)) for 15 min on ice. Probe-protein complexes were separated on a 4.5% polyacrylamide gel.
Galactosyltransferase Labeling
O-GlcNAcylated proteins were labeled by galactosyltransferase using its radiolabeled substrate (UDP-[3H]galactose) as described previously (16). Protein lysates were immunoprecipitated with anti-c-Rel antibody and mixed with labeling buffer (5 mm MnCl2, 10 mm galactose, 50 mm HEPES, pH 7.4). Reactions were initiated by addition of 2 μCi of UDP-[3H]galactose (American Radiolabeled Chemicals, St. Louis, MO) in 5′-AMP solution (2.5 mm 5′-AMP) and 50 milliunits of galactosyltransferase (Sigma) followed by incubation at 37 °C for 1 h. Proteins were separated by SDS-PAGE, intensified with EN3HANCE fluor (PerkinElmer Life Sciences, Waltham, MA), dried, and exposed to X-ray film.
Chromatin Immunoprecipitation Assay
ChIP assays were performed as described previously (15). Briefly, cells were treated with formaldehyde (1%) to the culture medium for cross-linking, and cell lysates were prepared. Samples were sonicated on ice in a Bioruptor (COSMO Bio Co., Ltd., Tokyo, Japan). Supernatant was precleared with 20 μg of sheared salmon sperm DNA, 5 μg of normal IgG, and 50 μl of protein G-beads for 2 h, and immunoprecipitated by addition of c-Rel antibody (Santa Cruz Biotechnology). After being washed, the immunoprecipitates were eluted with elution buffer. The eluted immunoprecipitates were treated with RNase A overnight at 65 °C, and proteins were removed by treatment with EDTA, 1 m Tris-HCl (pH 6.5), and proteinase K at 42 °C for 1 h. The DNA was extracted using a DNA purification kit (Qiagen, Hilden, Germany). The gene promoter sequences in the immunoprecipitates were amplified by PCR using the primers for iNOS promoter, F (−904), GTGTACCTCAGACAAGGGC, and R (−1058), CACACATGGCATGGAATTTT. The resulting products were separated by 1% agarose gel electrophoresis or quantitatively measured by real-time PCR.
Statistical Analysis
The data are expressed as the mean ± S.E. and analyzed for statistical significance using analysis of variance, followed by Scheffe's test for multiple comparisons and paired Student's t test for comparing two. A p value <0.05 was considered significant.
Author Contributions
I. O. H. and E. S. O. designed the study and wrote the paper. J. S. H., M. Y. K., K. H. K., and Y. K. L. performed experiments. I. K. L. and J. K. contributed to designing experimental procedures. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Research Federation of Korea Grants NRF-2013R1A1A2058244 and NRF-2012R1A5A1048236 and The Fire Fighting Safety & 119 Rescue Technology Research and Development Program supported by the Ministry of Public Safety and Security (MPSS-Fire Fighting Safety-2016–86). The authors declare that they have no conflicts of interest with the contents of this article.
- GlcN
- 2-amino-2-deoxy-d-glucose
- HBP
- hexosamine biosynthetic pathway
- ActD
- actinomycin D
- TTP
- tristetraprolin
- ER
- endoplasmic reticulum
- MTT
- 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- WGA
- wheat germ agglutinin.
References
- 1.MacMicking J., Xie Q. W., and Nathan C. (1997) Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350 [DOI] [PubMed] [Google Scholar]
- 2.Marshall S., Bacote V., and Traxinger R. R. (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 266, 4706–4712 [PubMed] [Google Scholar]
- 3.Shikhman A. R., Brinson D. C., Valbracht J., and Lotz M. K. (2009) Differential metabolic effects of glucosamine and N-acetylglucosamine in human articular chondrocytes. Osteoarthritis Cartilage 17, 1022–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawkins M., Barzilai N., Liu R., Hu M., Chen W., and Rossetti L. (1997) Role of the glucosamine pathway in fat-induced insulin resistance. J. Clin. Invest. 99, 2173–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Love D. C., and Hanover J. A. (2005) The hexosamine signaling pathway: deciphering the “O-GlcNAc code,” Sci STKE 2005, re13. [DOI] [PubMed] [Google Scholar]
- 6.Traxinger R. R., and Marshall S. (1991) Coordinated regulation of glutamine:fructose-6-phosphate amidotransferase activity by insulin, glucose, and glutamine: role of hexosamine biosynthesis in enzyme regulation. J. Biol. Chem. 266, 10148–10154 [PubMed] [Google Scholar]
- 7.Parker G. J., Lund K. C., Taylor R. P., and McClain D. A. (2003) Insulin resistance of glycogen synthase mediated by O-linked N-acetylglucosamine. J. Biol. Chem. 278, 10022–10027 [DOI] [PubMed] [Google Scholar]
- 8.Yang X., Ongusaha P. P., Miles P. D., Havstad J. C., Zhang F., So W. V., Kudlow J. E., Michell R. H., Olefsky J. M., Field S. J., and Evans R. M. (2008) Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 451, 964–969 [DOI] [PubMed] [Google Scholar]
- 9.Zhang G. X., Yu S., Gran B., and Rostami A. (2005) Glucosamine abrogates the acute phase of experimental autoimmune encephalomyelitis by induction of Th2 response. J. Immunol. 175, 7202–7208 [DOI] [PubMed] [Google Scholar]
- 10.Gouze J. N., Bianchi A., Bécuwe P., Dauça M., Netter P., Magdalou J., Terlain B., and Bordji K. (2002) Glucosamine modulates IL-1-induced activation of rat chondrocytes at a receptor level, and by inhibiting the NF-κB pathway. FEBS Lett. 510, 166–170 [DOI] [PubMed] [Google Scholar]
- 11.Largo R., Alvarez-Soria M. A., Díez-Ortego I., Calvo E., Sánchez-Pernaute O., Egido J., and Herrero-Beaumont G. (2003) Glucosamine inhibits IL-1β-induced NFκB activation in human osteoarthritic chondrocytes. Osteoarthritis Cartilage 11, 290–298 [DOI] [PubMed] [Google Scholar]
- 12.Meininger C. J., Kelly K. A., Li H., Haynes T. E., and Wu G. (2000) Glucosamine inhibits inducible nitric oxide synthesis. Biochem. Biophys. Res. Commun. 279, 234–239 [DOI] [PubMed] [Google Scholar]
- 13.Hwang S. Y., Hwang J. S., Kim S. Y., and Han I. O. (2013) Glucosamine inhibits lipopolysaccharide-stimulated inducible nitric oxide synthase induction by inhibiting expression of NF-κB/Rel proteins at the mRNA and protein levels. Nitric Oxide 31, 1–8 [DOI] [PubMed] [Google Scholar]
- 14.Yi H. A., Yi S. D., Jang B. C., Song D. K., Shin D. H., Mun K. C., Kim S. P., Suh S. I., and Bae J. H. (2005) Inhibitory effects of glucosamine on lipopolysaccharide-induced activation in microglial cells. Clin. Exp. Pharmacol. Physiol. 32, 1097–1103 [DOI] [PubMed] [Google Scholar]
- 15.Hwang S. Y., Shin J. H., Hwang J. S., Kim S. Y., Shin J. A., Oh E. S., Oh S., Kim J. B., Lee J. K., and Han I. O. (2010) Glucosamine exerts a neuroprotective effect via suppression of inflammation in rat brain ischemia/reperfusion injury. Glia 58, 1881–1892 [DOI] [PubMed] [Google Scholar]
- 16.Hwang S. Y., Hwang J. S., Kim S. Y., and Han I. O. (2013) O-GlcNAcylation and p50/p105 binding of c-Rel are dynamically regulated by LPS and glucosamine in BV2 microglia cells. Br. J. Pharmacol. 169, 1551–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aderem A., and Ulevitch R. J. (2000) Toll-like receptors in the induction of the innate immune response. Nature 406, 782–787 [DOI] [PubMed] [Google Scholar]
- 18.Kawai T., and Akira S. (2007) Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469 [DOI] [PubMed] [Google Scholar]
- 19.Han I. O., Kim K. W., Ryu J. H., and Kim W. K. (2002) p38 mitogen-activated protein kinase mediates lipopolysaccharide, not interferon-γ, -induced inducible nitric oxide synthase expression in mouse BV2 microglial cells. Neurosci. Lett. 325, 9–12 [DOI] [PubMed] [Google Scholar]
- 20.Dasgupta S., Jana M., Zhou Y., Fung Y. K., Ghosh S., and Pahan K. (2004) Antineuroinflammatory effect of NF-κB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J. Immunol. 173, 1344–1354 [DOI] [PubMed] [Google Scholar]
- 21.Karin M., and Ben-Neriah Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18, 621–663 [DOI] [PubMed] [Google Scholar]
- 22.Ghosh S., May M. J., and Kopp E. B. (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225–260 [DOI] [PubMed] [Google Scholar]
- 23.Vallabhapurapu S., and Karin M. (2009) Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733 [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann A., Natoli G., and Ghosh G. (2006) Transcriptional regulation via the NF-κB signaling module. Oncogene 25, 6706–6716 [DOI] [PubMed] [Google Scholar]
- 25.Perkins N. D. (2006) Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene 25, 6717–6730 [DOI] [PubMed] [Google Scholar]
- 26.Hotamisligil G. S., and Erbay E. (2008) Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 8, 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi H., Kokoeva M. V., Inouye K., Tzameli I., Yin H., and Flier J. S. (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116, 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esposito K., Nappo F., Marfella R., Giugliano G., Giugliano F., Ciotola M., Quagliaro L., Ceriello A., and Giugliano D. (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106, 2067–2072 [DOI] [PubMed] [Google Scholar]
- 29.Mohanty P., Hamouda W., Garg R., Aljada A., Ghanim H., and Dandona P. (2000) Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J. Clin. Endocrinol. Metab. 85, 2970–2973 [DOI] [PubMed] [Google Scholar]
- 30.Hua K. F., Wang S. H., Dong W. C., Lin C. Y., Ho C. L., and Wu T. H. (2012) High glucose increases nitric oxide generation in lipopolysaccharide-activated macrophages by enhancing activity of protein kinase C-α/δ and NF-κB. Inflamm. Res. 61, 1107–1116 [DOI] [PubMed] [Google Scholar]
- 31.Issad T., Masson E., and Pagesy P. (2010) O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 36, 423–435 [DOI] [PubMed] [Google Scholar]
- 32.Baudoin L., and Issad T. (2015) O-GlcNAcylation and Inflammation: a vast territory to explore. Front. Endocrinol. (Lausanne) 5, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma J., and Hart G. W. (2013) Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteomics 10, 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie Q. W., Kashiwabara Y., and Nathan C. (1994) Role of transcription factor NF-κB/Rel in induction of nitric-oxide synthase. J. Biol. Chem. 269, 4705–4708 [PubMed] [Google Scholar]
- 35.Korhonen R., Linker K., Pautz A., Förstermann U., Moilanen E., and Kleinert H. (2007) Post-transcriptional regulation of human inducible nitric-oxide synthase expression by the Jun N-terminal kinase. Mol. Pharmacol. 71, 1427–1434 [DOI] [PubMed] [Google Scholar]
- 36.Soesanto Y. A., Luo B., Jones D., Taylor R., Gabrielsen J. S., Parker G., and McClain D. A. (2008) Regulation of Akt signaling by O-GlcNAc in euglycemia. Am. J. Physiol. Endocrinol. Metab. 295, E974–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang J. S., Park J. W., Nam M. S., Cho H., and Han I. O. (2015) Glucosamine enhances body weight gain and reduces insulin response in mice fed chow diet but mitigates obesity, insulin resistance and impaired glucose tolerance in mice high-fat diet. Metabolism 64, 368–379 [DOI] [PubMed] [Google Scholar]
- 38.Chen Y. J., Hsu K. W., and Chen Y. L. (2006) Acute glucose overload potentiates nitric oxide production in lipopolysaccharide-stimulated macrophages: the role of purinergic receptor activation. Cell Biol. Int. 30, 817–822 [DOI] [PubMed] [Google Scholar]
- 39.Won J. S., Im Y. B., Key L., Singh I., and Singh A. K. (2003) The involvement of glucose metabolism in the regulation of inducible nitric oxide synthase gene expression in glial cells: possible role of glucose-6-phosphate dehydrogenase and CCAAT/enhancing binding protein. J. Neurosci. 23, 7470–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan M. I., Pichna B. A., Shi Y., Bowes A. J., and Werstuck G. H. (2009) Evidence supporting a role for endoplasmic reticulum stress in the development of atherosclerosis in a hyperglycaemic mouse model. Antioxid. Redox Signal. 11, 2289–2298 [DOI] [PubMed] [Google Scholar]
- 41.Gao J., Morrison D. C., Parmely T. J., Russell S. W., and Murphy W. J. (1997) An interferon-γ-activated site (GAS) is necessary for full expression of the mouse iNOS gene in response to interferon-γ and lipopolysaccharide. J. Biol. Chem. 272, 1226–1230 [DOI] [PubMed] [Google Scholar]
- 42.Eberhardt W., Plüss C., Hummel R., and Pfeilschifter J. (1998) Molecular mechanisms of inducible nitric oxide synthase gene expression by IL-1β and cAMP in rat mesangial cells. J. Immunol. 160, 4961–4969 [PubMed] [Google Scholar]
- 43.Marks-Konczalik J., Chu S. C., and Moss J. (1998) Cytokine-mediated transcriptional induction of the human inducible nitric-oxide synthase gene requires both activator protein 1 and nuclear factor κB-binding sites. J. Biol. Chem. 273, 22201–22208 [DOI] [PubMed] [Google Scholar]
- 44.Zhang H., Chen X., Teng X., Snead C., and Catravas J. D. (1998) Molecular cloning and analysis of the rat inducible nitric oxide synthase gene promoter in aortic smooth muscle cells. Biochem. Pharmacol. 55, 1873–1880 [DOI] [PubMed] [Google Scholar]
- 45.Moore F., Santin I., Nogueira T. C., Gurzov E. N., Marselli L., Marchetti P., and Eizirik D. L. (2012) The transcription factor C/EBPδ has anti-apoptotic and anti-inflammatory roles in pancreatic beta cells. PLoS ONE 7, e31062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kengatharan M., De Kimpe S. J., and Thiemermann C. (1996) Analysis of the signal transduction in the induction of nitric oxide synthase by lipoteichoic acid in macrophages. Br. J. Pharmacol. 117, 1163–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pautz A., Linker K., Hubrich T., Korhonen R., Altenhöfer S., and Kleinert H. (2006) The polypyrimidine tract-binding protein (PTB) is involved in the post-transcriptional regulation of human inducible nitric-oxide synthase expression. J. Biol. Chem. 281, 32294–32302 [DOI] [PubMed] [Google Scholar]
- 48.Laroia G., Sarkar B., and Schneider R. J. (2002) Ubiquitin-dependent mechanism regulates rapid turnover of AU-rich cytokine mRNAs. Proc. Natl. Acad. Sci. U.S.A. 99, 1842–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linker K., Pautz A., Fechir M., Hubrich T., Greeve J., and Kleinert H. (2005) Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucleic Acids Res. 33, 4813–4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anderson J. W., Nicolosi R. J., and Borzelleca J. F. (2005) Glucosamine effects in humans: a review of effects on glucose metabolism, side effects, safety considerations and efficacy. Food Chem. Toxicol. 43, 187–201 [DOI] [PubMed] [Google Scholar]
- 51.Monauni T., Zenti M. G., Cretti A., Daniels M. C., Targher G., Caruso B., Caputo M., McClain D., Del Prato S., Giaccari A., Muggeo M., Bonora E., and Bonadonna R. C. (2000) Effects of glucosamine infusion on insulin secretion and insulin action in humans. Diabetes 49, 926–935 [DOI] [PubMed] [Google Scholar]