

Abstract
Indoleamine 2,3-dioxygenase 1 (IDO1) is a single chain oxidoreductase that catalyzes tryptophan degradation to kynurenine. In cancer, it exerts an immunosuppressive function as part of an acquired mechanism of immune escape. Recently, we demonstrated that IDO1 expression is significantly higher in all thyroid cancer histotypes compared with normal thyroid and that its expression levels correlate with T regulatory (Treg) lymphocyte densities in the tumor microenvironment. BRAFV600E- and RET/PTC3-expressing PcCL3 cells were used as cellular models for the evaluation of IDO1 expression in thyroid carcinoma cells and for the study of involved signal transduction pathways. BRAFV600E-expressing PcCL3 cells did not show IDO1 expression. Conversely, RET/PTC3-expressing cells were characterized by a high IDO1 expression. Moreover, we found that, the STAT1-IRF1 pathway was instrumental for IDO1 expression in RET/PTC3 expressing cells. In detail, RET/PTC3 induced STAT1 overexpression and phosphorylation at Ser-727 and Tyr-701. STAT1 transcriptional regulation appeared to require activation of the canonical NF-κB pathway. Conversely, activation of the MAPK and PI3K-AKT pathways primarily regulated Ser-727 phosphorylation, whereas a physical interaction between RET/PTC3 and STAT1, followed by a direct tyrosine phosphorylation event, was necessary for STAT1 Tyr-701 phosphorylation. These data provide the first evidence of a direct link between IDO1 expression and the oncogenic activation of RET in thyroid carcinoma and describe the involved signal transduction pathways. Moreover, they suggest possible novel molecular targets for the abrogation of tumor microenvironment immunosuppression. The detection of those targets is becoming increasingly important to yield the full function of novel immune checkpoint inhibitors.
Keywords: cancer; cell signaling; indoleamine-pyrrole 2,3-dioxygenase (IDO1); interferon regulatory factor (IRF); signal transducers and activators of transcription 1 (STAT1)
Introduction
There is increasing evidence that solid tumors are composed by a complex aggregation of distinct cell types. Next to cancer cells and cancer stem cells, the tumor stroma contains endothelial cells, pericytes, tumor-associated fibroblasts, myeloid progenitors, and cells belonging to both the innate and adaptive immune system (1). Overall, these cells appear to promote tumor growth and progression. The immune cells of the tumor microenvironment may include subclasses with anti-tumor activity, such as CD8+ T lymphocytes, involved in the recognition and elimination of antigens expressed by cancer cells. However, it is well established that clinically detectable cancers must have developed mechanisms to evade the immune system (2). One such mechanism includes the expression of the immune-suppressive enzyme indoleamine 2,3-dioxygenase 1 (IDO1),2 either by tumor cells or the tumor-associated dendritic cells that may move to tumor-draining lymph nodes (3, 4). IDO1 is a single chain oxidoreductase that catalyzes the degradation of the essential amino acid tryptophan to kynurenine, the first step in the biosynthesis of nicotinamide adenine dinucleotide (NAD). T cells have appeared to be preferentially sensitive to IDO1 activation, such that when starved for tryptophan they cannot divide, therefore losing the ability to be activated by specific antigens (4). Moreover, kynurenine generated by the IDO1 pathway has appeared important for the induction of Tregs and immune suppression (5). In cancer cells, kynurenine has been shown to promote tumor-cell survival and motility through the activation of the aryl hydrocarbon receptor (AHR) in an autocrine/paracrine fashion (6). Very recently, we demonstrated that IDO1 expression is significantly higher in all thyroid cancer histotypes compared with normal thyroid and that its expression levels correlate with Treg lymphocyte densities in the thyroid tumor microenvironment (7).
BRAF point mutations and RET gene rearrangements (RET/PTCs) are the two major groups of mutations involved in papillary thyroid carcinoma initiation and progression. Cells expressing BRAFV600E showed overactivation of the mitogen-activated protein kinase (MAPK) pathway with increased expression of dual-specificity phosphatase genes and a loss of extracellular signal regulated kinase (ERK) negative feedback on RAF dimerization (8). RET/PTCs have been involved in the activation of the MAPK, phosphoinositide 3-kinase-AKT (PI3K-AKT), nuclear factor-κB (NF-κB), and c-Jun N-terminal kinase (JNK) pathways and in the phosphorylation and transcriptional activation of signal transducer and activator of transcription (STATs) 1 and 3 (9). Interestingly, RET/PTCs have high MAPK pathway activation (8), consistent with preferential signaling via BRAF homodimers (10). Acute RET/PTC activation stimulates both DNA synthesis and apoptosis in thyroid cells, is associated with loss of thyroid-specific differentiation, and interferes with thyrotropin (TSH) receptor-mediated intracellular signaling at various levels (11). In human papillary thyroid carcinomas, RET rearrangements behave as weak BRAFV600E-like mutations being associated with classical papillary histology, loss of differentiation, and intermediate clinical risk (8). Analysis of RET/PTC-induced gene expression profiles showed strong modulation of genes coding for proteins involved in the immune response and in intracellular signal transduction pathways activated by cytokines and chemokines, indicating a critical role of these oncogenes in the early modulation of the immune response (9).
In this paper, we report data on the pathways that are involved in IDO1 expression in thyroid cancer cells. In detail, we show that IDO1 is induced by RET/PTC rearranged oncoproteins and that this phenomenon depends on the induction of STAT1 overexpression and full phosphorylation at the levels of Ser-727 and Tyr-701.
Results
RET/PTC3 Induces IDO1 Expression in PcCL3 Cells
To evaluate the effect of BRAFV600E and RET/PTC3 on IDO1 expression, we tested the expression of the immunomodulatory enzyme in BRAF9.6 and PTC3–5 cells characterized by the doxycycline (Dox)-inducibility of BRAF and RET/PTC3 transgenes, respectively. In BRAF9.6 cells, Dox treatment did not elicit any up-regulation of IDO1 mRNA expression (Fig. 1A). Conversely, in PTC3–5 cells acute induction of RET/PTC3 was associated with a significant increase of IDO1 mRNA expression evident at 48 h and lasting for at least 96 h (Fig. 1A).
FIGURE 1.
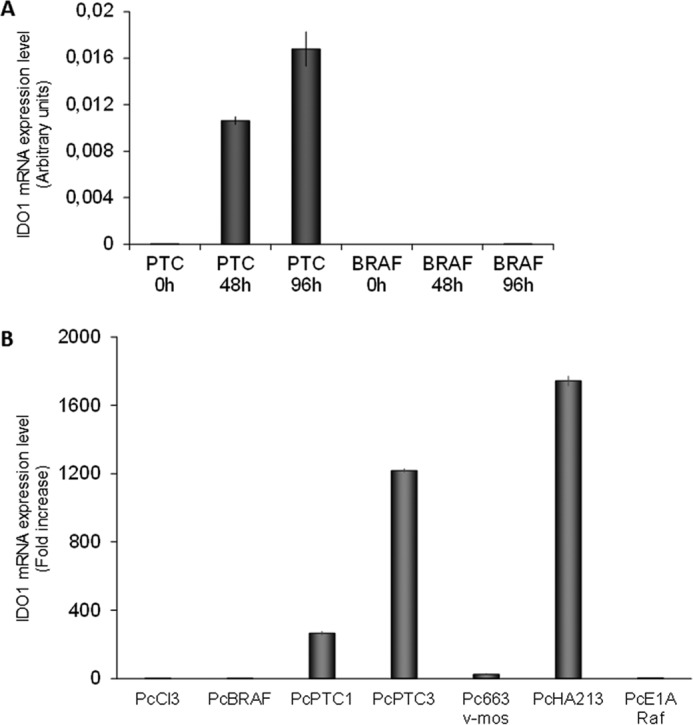
IDO1 expression in PcCL3 cells expressing BRAFV600E or RET/PTC3 oncogenes. A, PTC3–5 cells and BRAF9.6 were incubated with Dox (1 μg/ml) for the indicated times and harvested for total RNA extraction. After total RNA extraction and cDNA synthesis, IDO1 mRNA expression levels were assayed by quantitative PCR. The data are presented as means of arbitrary units. In BRAF9.6 cells Dox treatment for 48 and 96 h did not elicit any up-regulation of IDO1 mRNA expression. Conversely, in PTC3–5 cells induction of RET/PTC3 for the same time was associated with a significant increase of IDO1 mRNA expression. The experiment was repeated three times in triplicate and the more representative result is depicted. B, PcCL3-derived cell lines stably expressing, respectively, BRAFV600E (PcBRAF), RET/PTC1 (PcPTC1), RET/PTC3 (PcPTC3), v-Mos (Pc663), RET/PTC1 and H-RasG12V (PcHA213), and v-Raf (PcE1ARaf) oncogenes were harvested for total RNA extraction. After total RNA extraction and cDNA synthesis, IDO1 mRNA expression levels were assayed by quantitative PCR. Cells expressing RET/PTC1 and RET/PTC3 showed very high IDO1 expression levels compared with PcCL3 cells, whereas mRNA of the immunomodulatory molecule was barely detectable in the other cell lines. The data are presented as relative quantification compared with PcCL3 parental cells. The experiment was repeated two times in triplicate and the results are presented as mean ± S.D. (error bars) of the obtained data.
To confirm this finding, IDO1 expression was tested in PcCL3-derived cell lines stably expressing, respectively, BRAFV600E (PcBRAF), RET/PTC1 (PcPTC1), RET/PTC3 (PcPTC3), v-Mos (Pc663), RET/PTC1 and H-RasG12V (PcHA213), and v-Raf (PcE1A Raf) oncogenes. Interestingly, cells expressing RET/PTC1 and RET/PTC3 showed very high IDO1 expression levels (RET/PTC1 about 200-fold, RET/PTC3 about 1200-fold, RET/PTC1 + H-RasG12V about 1800-fold compared with PcCL3 cells), whereas mRNA of the immunomodulatory molecule was barely detectable in the other cell lines (Fig. 1B).
RET/PTC3-induced IDO1 Expression in PTC3–5 Cells Depends on Activation of a RET/PTC3-STAT1-IRF1 Pathway
The dissection of RET/PTC3-activated signaling pathways involved in the induction of IDO1 expression focused on STAT1, a transcription factor known to play a central role in γ-interferon-induced IDO1 expression in dendritic cells (12).
In PTC3–5 cells treated with Dox for up to 96 h, characterized by robust IDO1 mRNA overexpression, RET/PTC3 induced both up-regulation of STAT1 protein expression and its full activation through the phosphorylation of tyrosine 701 and serine 727 (Fig. 2). Simultaneously, we could detect a progressive increase of IDO1 protein lasting up to 96 h (Fig. 2).
FIGURE 2.
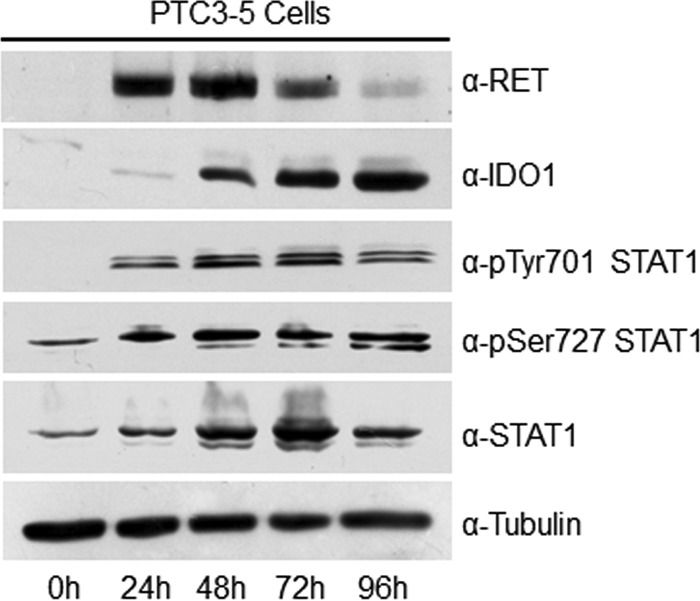
RET/PTC3 regulates STAT1 activation. PTC3–5 cell line was incubated with Dox (1 μg/ml) for the indicated times and harvested for cell lysate preparation. Expression of RET, IDO1, pTyr-701 STAT1, pSer-727 STAT1, total STAT1, and tubulin were assayed by immunoblot. RET/PTC3 induced both the up-regulation of STAT1 expression and its full activation through the simultaneous phosphorylation of Tyr-701 and Ser-727. Analysis of IDO1 confirmed RET/PTC3-induced IDO1 overexpression also as protein. All the experiments were repeated twice and the more representative results are depicted.
To demonstrate in a more consistent way the dependence of IDO1 expression on RET/PTC3-induced STAT1 activation, we performed an RNAi experiment. Silencing of STAT1 was associated with a significant reduction of RET/PTC3-induced STAT1, IRF1, and IDO1 mRNA (Fig. 3, A–C) and a loss of STAT1 and IDO1 protein up-regulation (Fig. 3D).
FIGURE 3.

STAT1 silencing impairs RET/PTC-induced IRF1 and IDO1 overexpression. PTC3–5 cells were transfected with rat STAT1 siRNA (25 nm) (siSTAT1) or with non-target siRNA (25 nm) (siNC). Twenty-four hours after transfection, cells were stimulated with Dox (1 μg/ml) for 48 h before cell harvesting for total RNA and cell lysate preparation. After total RNA extraction and cDNA synthesis, STAT1 (A), IRF1 (B), and IDO1 (C) mRNA expression levels were evaluated by quantitative PCR. STAT1, IRF1, and IDO1 mRNA expression resulted significantly in impaired STAT1-silenced cells compared with controls. The quantitative PCR data are presented as means of arbitrary units. D, immunoblottings for RET, IDO1, total STAT1, and tubulin were performed. In STAT1-silenced cells, despite a comparable expression of RET/PTC3, STAT1 and IDO1 protein expression was significantly impaired compared with controls. All experiments were repeated twice and the more representative results are depicted. p values were calculated applying the two-tailed unpaired Student's t test.
To answer the question how RET/PTC3-induced STAT1 overexpression, we evaluated the involvement of the canonical NF-κB pathway, known to be activated by RET/PTCs (13). We explored the effect of NF-κB inhibitors Bay11-7085 and JSH-23 on STAT1 and IDO1 expression, in PTC3–5 cells treated with Dox for 24 or 48 h. In detail, Bay11-7085 is an irreversible inhibitor of IκBα phosphorylation, whereas JSH-23 is an inhibitor of p65 nuclear translocation. As shown in Fig. 4, both inhibitors down-regulated RET/PTC3-induced STAT1 (Fig. 4, A–C) and IDO1 expression (Fig. 4D). To understand if RET/PTC3-induced activation of the canonical NF-κB pathway directly regulated IDO1 expression, a ChIP assay using an anti-p65 antibody was performed in PTC3–5 cells. In detail, one NF-κB binding sequence in the STAT1 promoter and three in the IDO1 promoter/enhancer regions were considered. As shown in Fig. 5, chromatin immunoprecipitation with an anti-p65 antibody of cells treated with Dox for 24 h allowed a significant enrichment in the promoter sequence of STAT1 (Fig. 5A) to be obtained, but not in the three promoter/enhancer regions of IDO1 (Fig. 5B). These data indicated that RET/PTC3-induced activation of a canonical NF-κB pathway up-regulated directly the STAT1 expression that, in turn, modulated IDO1 expression.
FIGURE 4.

RET/PTC3-induced activation of canonical NF-κB pathway is involved in STAT1 and IDO1 gene expression. PTC3–5 cells were incubated with Dox (1 μg/ml) for the indicated times in the presence or absence of the NF-κB inhibitors Bay11-7085 (2 and 5 μm) or JSH-23 (10 μm) and harvested for cell lysate preparation or total RNA extraction and cDNA synthesis. A, immunoblottings for RET, STAT1, and tubulin were performed and B, the bands of three independent experiments were analyzed by densitometry and normalized with tubulin. Inhibition of canonical NF-κB pathway or inhibition of p65 nuclear translocation down-regulated STAT1 expression in a statistically significant manner. C, STAT1 mRNA expression levels were assayed by quantitative PCR. D, IDO1 mRNA expression levels were assayed by quantitative PCR. The data are presented as means of arbitrary units. Inhibition of the canonical NF-κB pathway or inhibition of p65 nuclear translocation down-regulated STAT1 and IDO1 mRNA expression. All experiments were repeated three times and the more representative results are depicted. p values were calculated applying the two-tailed unpaired Student's t test. *, p < 0.05.
FIGURE 5.
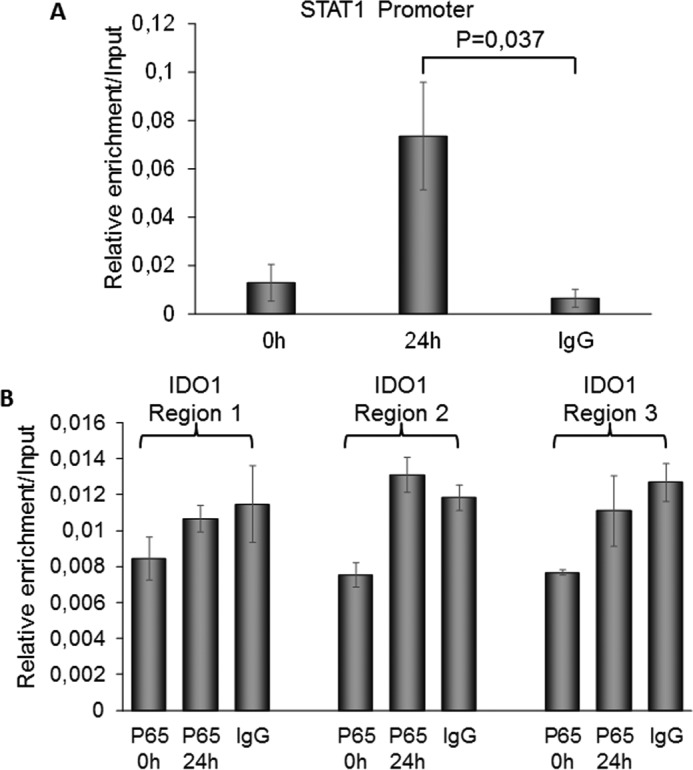
RelA/p65 regulates STAT1 but not IDO1 gene expressions. PTC3–5 cells were incubated with Dox (1 μg/ml) for 24 h to induce RET/PTC3 oncogene expression. Analysis of STAT1 and IDO1 promoters using MatInspector software allowed detection of one NF-κB binding region for STAT1 and three NF-κB binding regions for IDO1. A ChIP assay was performed using an anti-p65 antibody after dual cross-linking of the cells and the isolated DNA was amplified by Q-PCR using specific primers spanning the detected putative NF-κB binding regions of STAT1 and IDO1 genes. A, STAT1 promoter ChIP analysis showed a significant enrichment of the putative NF-κB binding region in p65 IP after 24 h of oncogene expression compared with control (IgG IP). B, IDO1 promoter ChIP analysis did not show any significant enrichment of the three putative NF-κB binding regions in p65 IP after 24 h of oncogene expression compared with control (IgG IP). The results are presented as mean ± S.D. of three independent experiments. p values were calculated applying the two-tailed unpaired Student's t test.
STAT1 Phosphorylation on Ser-727 Depends on MAPK and PI3K Activation
To explore the role of the MAPK pathway in STAT1 activation and IDO1 expression, in the first instance we analyzed BRAF9.6 cells treated with Dox for up to 96 h. As shown in Fig. 6A, BRAFV600E expression only induced phosphorylation of STAT1 Ser-727 without affecting STAT1 Tyr-701 or the expression levels of the transcription factor. Moreover, BRAFV600E did not induce IDO1 expression also at the protein level (Fig. 6A). Thus, specific MAPK activation, induced by BRAFV600E, appeared to affect only STAT1 Ser-727 phosphorylation in PcCl3 cells, with a consequent incomplete activation of STAT1 and no expression of IDO1.
FIGURE 6.

STAT1 Ser-727 phosphorylation depends on activation of the MAPK and PI3K pathways. A, BRAF9.6 cell line was incubated with Dox (1 μg/ml) for the indicated times and harvested for cell lysate preparation. Immunoblottings for pERK, IDO1, pTyr-701 STAT1, pSer-727 STAT1, total STAT1, and tubulin were performed. BRAFV600E expression only induced phosphorylation of serine 727 without affecting tyrosine 701 phosphorylation and STAT1 and IDO1 expression levels (PC: positive control). B, PTC3–5 cells were incubated with Dox (1 μg/ml) for the indicated times in the presence or absence of the MEK inhibitor U0126 (10 μm) and harvested for cell lysate preparation. Immunoblottings for RET, pTyr-701 STAT1, pSer-727 STAT1, total STAT1, pERK, and tubulin were performed. Inhibition of RET/PTC3-induced activation of the MAPK pathway appeared to strongly inhibit STAT1 serine 727 phosphorylation and, to a lower extent, STAT1 tyrosine 701 phosphorylation, without affecting the expression levels of the transcription factor. C, PTC3–5 cells were incubated with Dox (1 μg/ml) for the indicated times in the presence or absence of the PI3K inhibitor wortmannin (0.3 μm) and harvested for cell lysate preparation. Immunoblottings for RET, pTyr-701 STAT1, pSer-727 STAT1, total STAT1, pAKT, and tubulin were performed. Inhibition of RET/PTC3-induced activation of the PI3K pathway appeared to inhibit significantly STAT1 serine 727 phosphorylation and, to a lower extent, STAT1 tyrosine 701 phosphorylation, without affecting the expression levels of the transcription factor. D, the STAT1 pTyr-701 and pSer-727 bands of three independent experiments were analyzed by densitometry and normalized toward tubulin. The results indicate that either the MAPK or PI3K pathway inhibition cause a not significant light reduction of STAT1 tyrosine 701 phosphorylation, but a statistically significant strong reduction of STAT1 serine 727 phosphorylation. E, PTC3–5 cells were incubated with Dox (1 μg/ml) for the indicated times in the presence or absence of the MEK inhibitor U0126 (10 μm) and harvested for total RNA preparation. After total RNA extraction and cDNA synthesis, IDO1 mRNA expression levels were assayed by quantitative PCR. The data are presented as means of arbitrary units. Inhibition of RET/PTC3-induced activation of the MAPK pathway appeared to strongly inhibit IDO1 mRNA expression (*, p = 0.0022). F, PTC3–5 cells were incubated with Dox (1 μg/ml) for the indicated times in the presence or absence of the PI3K inhibitor wortmannin (0,3 μm) and harvested for total RNA preparation. After total RNA extraction and cDNA synthesis, IDO1 mRNA expression levels were assayed by quantitative PCR. The data are presented as means of arbitrary units. Inhibition of RET/PTC3-induced activation of the PI3K pathway appeared to strongly inhibit IDO1 mRNA expression (*, p = 0.01). All experiments were repeated twice and the more representative results are depicted. p values were calculated applying the two-tailed unpaired Student's t test.
To confirm this finding, STAT1 phosphorylation was evaluated in PTC3–5 cells treated with Dox for 48 h in the presence of the selective MEK inhibitor U0126. As shown in Fig. 6, B and D, inhibition of the MAPK pathway appeared to significantly reduce STAT1 Ser-727 phosphorylation (14.15 ± 7.31%, p = 0.04), affecting in a less pronounced way STAT1 Tyr-701 phosphorylation (65.97 ± 13.4%, p = 0.17). Conversely, RET/PTC3-induced STAT1 overexpression was completely unaffected (Fig. 6B).
Furthermore, phosphorylation of STAT1 was evaluated in PTC3–5 cells treated with Dox for 48 h in the presence of the selective PI3K inhibitor wortmannin. Inhibition of the PI3K-AKT pathway resulted in a significant inhibition of STAT1 Ser-727 phosphorylation (20.12 ± 1.9%, p = 0.011) and a less pronounced reduction in STAT1 Tyr-701 phosphorylation (54.07 ± 8.12%, p = 0.079) (Fig. 6, C and D). Conversely, no difference in STAT1 expression could be detected (Fig. 6C). Interestingly, inhibition of STAT1 full activation, caused by U0126 and wortmannin, resulted in a significant reduction of RET/PTC3-induced IDO1 mRNA overexpression (Fig. 6, E and F).
Phosphorylation of STAT1 Tyr-701 Depends on Direct Phosphorylation by RET/PTC3
The data obtained thus far indicated that full activation of STAT1 depended not only on MAPK and PI3K-AKT pathways, able to mainly induce STAT1 Ser-727 phosphorylation, and on the canonical NF-κB pathway, involved in STAT1 overexpression, but on at least another signal transduction pathway simultaneously activated by RET/PTC3 and involved in STAT1 Tyr-701 phosphorylation. Thus, we hypothesized that STAT1 tyrosine 701 phosphorylation could be delivered by independent mechanisms/pathways including: JAK2 activation or Src tyrosine-protein kinase involvement or RET/PTC3 physical interaction with STAT1 followed by a direct tyrosine phosphorylation event. To confirm these hypotheses, in the first instance, we compared the degree of STAT1 phosphorylation (at Tyr-701 and at Ser-727) in PTC3–5 cells treated with Dox with or without different doses of the specific JAK2 inhibitor AG490 for 24 h. As shown in Fig. 7A, any dose of the inhibitor did not affect RET/PTC3 activation or STAT1 phosphorylation. Consistently, treatment with the JAK2 inhibitor did not change RET/PTC3-induced IDO1 expression (Fig. 7B). Similarly, silencing of JAK2 expression did not affect RET/PTC3-induced STAT1 Tyr-701 or Ser-727 phosphorylation (Fig. 7C) or IDO1 expression (Fig. 7D). These findings excluded the involvement of JAK2 in RET/PTC3-induced activation of STAT1.
FIGURE 7.

JAK2 does not mediate RET/PTC3-induced activation of STAT1. PTC3–5 cell line was incubated with Dox (1 μg/ml) or with Dox (1 μg/ml) and JAK2 inhibitor AG490 (5, 10, and 50 μm) for 24 h and harvested for cell lysate preparation or for total RNA extraction and cDNA synthesis. A, immunoblottings for RET, pTyr-905 RET, pSer-727 STAT1, pTyr-701 STAT1, total STAT1, and tubulin were performed. Treatment with AG490 did not appear to influence RET activation and STAT1 phosphorylation and expression. B, IDO1 mRNA expression levels were assayed by quantitative PCR. The data are presented as means of arbitrary units. Inhibition of JAK2 did not significantly influence IDO1 expression. C, the PTC3–5 cell line was transfected with rat JAK2 siRNA (50 nm) (siJAK2) or with non-target siRNA (50 nm) (siNC). Twenty-four hours after transfection, cells were stimulated with Dox (1 μg/ml) for 48 h before cell harvesting for RNA and cell lysate preparation. Immunoblottings for RET, pTyr-701 STAT1, pSer-727 STAT1, total STAT1, JAK2, and tubulin were performed. JAK2 silencing did not appear to influence RET activation and STAT1 phosphorylation and expression. D, IDO1 mRNA expression levels were assayed by quantitative PCR. The data are presented as means of arbitrary units. Silencing of JAK2 did not significantly influence IDO1 expression. All the experiments were repeated twice and the more representative results are depicted.
In the second instance, we tested the effect of three Src inhibitors in PTC3–5 cells. Unfortunately, all of them, namely SU6656, PP1, and SRCI-1, are characterized by a weak selectivity (14, 15) and inhibited simultaneously STAT1 tyrosine 701 and serine 727 phosphorylation, ERK phosphorylation, and STAT1 expression in relationship to a concurrent inhibitory effect not only on Src but also on RET/PTC3, demonstrated by the down-regulation of RET tyrosine 905 phosphorylation (data not shown). Thus, we changed the strategy to demonstrate the role of Src in RET/PTC3-induced full activation of STAT1. In detail, we transiently transfected HEK293T cells with two RET/PTC3 mutant constructs, namely RET/PTC3-V804M, almost completely resistant to the PP1 inhibitory effect (16), and RET/PTC3-Y981F, characterized by the inability to bind Src (17). Interestingly, HEK293T cells transfected with the RET/PTC3-V804M construct showed STAT1 Tyr-701 phosphorylation levels that were superimposable to those observed in HEK293T cells transfected with RET/PTC3WT (Fig. 8). Conversely, STAT1 Tyr-701 phosphorylation was almost completely abrogated in the RET/PTC3WT-transfected cells after treatment with the RET and Src inhibitor PP1, whereas it was almost completely maintained in cells transfected with the RET/PTC3 PP1-resistant mutant (Fig. 8). Similarly, no difference in STAT1 Tyr-701 phosphorylation levels could be detected in HEK293T cells transfected with the RET/PTC3WT construct and those transfected with the RET/PTC3-Y981F mutant deprived of the capacity to bind Src (Fig. 8). These experiments clearly indicated that Src does not play a key role in RET/PTC3-induced STAT1 Tyr-701 phosphorylation.
FIGURE 8.

Src does not mediate RET/PTC3-induced activation of STAT1. HEK293T cells, transiently transfected with pcDNA3, pcDNA3-RET/PTC3 WT, and the mutant pcDNA3-RET/PTC3-V804M Myc-tagged, were treated with the Src inhibitor PP1 (5 μm) for 24 h and harvested for cell lysate preparation. Immunoblotting for RET, pTyr-905 RET, pTyr-701 STAT1, pSer-727 STAT1, total STAT1, and tubulin were performed. Treatment with PP1 caused a marked reduction of RET Tyr-905 and STAT1 Tyr-701 phosphorylations in cells transfected with RET/PTC WT (pR/P3 WT), but not in cells transfected with the mutant RET/PTC3-V804M (pR/P3 V804M), which is resistant to PP1. Furthermore, HEK293T cells were transiently transfected with pcDNA3, pcDNA3-RET/PTC3 WT, and the mutant pcDNA3-RET/PTC3-Y918F, which is unable to bind Src, and harvested for cell lysate preparation, 24 h after transfection. Immunoblotting for RET, pTyr-905 RET, pTyr-701 STAT1, pSer-727 STAT1, total STAT1, and tubulin were performed. In cells transfected whit RET/PTC3-Y918F (pR/P3 Y918F) phosphorylation of STAT1 Tyr-701 was superimposable to that of RET/PTC3 WT (pR/P3 WT). These results indicate that Src is not involved in RET/PTC3-induced STAT1 Tyr-701 phosphorylation. All the experiments were repeated twice and the more representative results are depicted.
Finally, we performed co-immunoprecipitation experiments on cell lysates of HEK293T cells transiently cotransfected with RET/PTC3 and STAT1 or RET/PTC3 and Src or RET/PTC3, STAT1, and Src. As shown in Fig. 9A, immunoprecipitation of RET/PTC3 with an anti-RET antibody allowed a specific co-immunoprecipitation of STAT1. Similarly, immunoprecipitation of STAT1 with an anti-Myc antibody allowed a specific co-immunoprecipitation of RET/PTC3, but not of Src. Furthermore, immunoprecipitation of Src with an anti-Src antibody did not allow a specific co-immunoprecipitation of STAT1 (Fig. 9A). These data indicate that RET/PTC3 and STAT1 aggregate one with the other, whereas Src and STAT1 do not.
FIGURE 9.

RET/PTC3 interacts with STAT1 and phosphorylates STAT1 on Tyr-701. A, co-immunoprecipitation experiment in HEK293T cells. HEK293T cells were transiently co-transfected with expression vectors for RET/PTC3 and Myc-tagged STAT1 or RET/PTC3 and Src or RET/PTC3, Myc-tagged STAT1 and Src. Twenty-four hours after transfection, cells were harvested for cell lysate preparation. Five hundred μg of total lysate were immunoprecipitated (IP) using an anti-RET antibody or an anti-Myc antibody (used for STAT1 IP) or an anti-Src antibody. Immunoprecipitates were then subjected to Western blotting using the anti-Myc antibody in RET IP, the anti-RET and anti-Src antibodies in STAT1 IP, and the anti-Myc antibody in Src IP. To verify binding specificity, the presence of STAT1, RET, and Src was searched on the protein complexes immunoprecipitated with normal goat (control of RET IP), normal mouse (control of Myc IP), or normal rabbit (control of Src IP) IgG mixtures. Immunoprecipitation of RET allowed a specific co-immunoprecipitation of STAT1 in the cells expressing RET/PTC3 and STAT1 or RET/PTC3, STAT1, and Src. Similarly, immunoprecipitation of STAT1 (with an anti-Myc antibody) allowed a specific co-immunoprecipitation of RET in the cells expressing RET/PTC3 and STAT1 or RET/PTC3, STAT1, and Src. Conversely, Src was not detected in the STAT1 IP and STAT1 could not be found in Src IP. These data indicate that STAT1 and RET/PTC3 aggregate one with the other, whereas Src and STAT1 do not. B, RET/PTC3 in vitro kinase assay. HEK293T cells were transiently transfected with expression vector for RET/PTC3 or Myc-tagged STAT1. Twenty-four hours after transfection, cells were harvested for cell lysate preparation. Five hundred μg of total lysate were immunoprecipitated using an anti-RET antibody or an anti-Myc antibody (used for STAT1 IP). The two IPs were pulled together and resuspended in kinase buffer. The reaction was carried out for 30 min at 30 °C and stopped by adding Laemmli buffer. The kinase reactions were submitted to Western blotting and the presence of RET, pTyr-701 STAT1, pSer-727 STAT1, and total STAT1 were assayed using the specific antibodies. RET/PTC3 phosphorylated STAT1 Tyr-701, but not STAT1 Ser-727. Furthermore, RET/PTC3 and STAT1 were incubated alone in the kinase buffer. No evidence of RET/PTC3-driven pulldown of phosphorylated STAT1 from HEK293T cell lysates or STAT1 autophosphorylation could be detected. All experiments were repeated twice and the more representative results are depicted.
To demonstrate that RET/PTC3 phosphorylates STAT1, we performed an in vitro kinase assay using immunoprecipitated RET/PTC3 and STAT1 proteins. As shown in Fig. 9B, RET/PTC3 phosphorylated STAT1 on Tyr-701, whereas the Ser-727 was not affected.
Discussion
Very recently, we demonstrated that IDO1 expression is significantly higher in all thyroid cancer histotypes compared with normal thyroid and that its expression levels correlate with Treg lymphocyte density in the tumor microenvironment (7). Interestingly, IDO1 expression was higher in BRAFV600E-negative papillary thyroid carcinomas and in RET mutation-positive medullary thyroid carcinomas (7). This observation prompted us to explore the preferential signal transduction pathways involved in the regulation of IDO1 expression in thyroid cancer cells. For this purpose, PcCL3 cells characterized by the doxycycline-inducible expression of BRAFV600E and RET/PTC3 were used. Interestingly, whereas BRAFV600E-expressing PcCL3 cells did not show IDO1 overexpression, RET/PTC3-expressing ones demonstrated a dramatic increase of the enzyme expression levels. This finding was confirmed also in BRAFV600E- and RET/PTC-stably expressing PcCL3 cell lines.
For the dissection of RET/PTC3-activated signaling pathways involved in the induction of IDO1 expression, we concentrated on STAT1 activation, known to play a central role in the regulation of γ-interferon-induced IDO1 expression in dendritic cells (18). We could demonstrate that RET/PTC3 induces both expression up-regulation of STAT1 and its full phosphorylation at Tyr-701 and Ser-727. Simultaneously, we could demonstrate a prolonged activation of IDO1 expression lasting longer and happening at higher levels than that of RET/PTC3. This finding could be explained by the fact that RET/PTC3 induced the expression of IDO1 via STAT1. Once STAT1 was in the active form, it could maintain a prolonged expression of IDO1, independently from RET/PTC3.
The central role of STAT1 on IDO1 expression was demonstrated by its silencing in PTC3–5 cells, which resulted in down-regulation of RET/PTC3-induced IRF1 mRNA and IDO1 mRNA and protein expression. Thus, we could confirm that, downstream of STAT1, RET/PTC3 induces up-regulation of IRF1 expression through STAT1 itself that binds the GAS promoter region of the IRF1 gene (18). STAT1 and IRF1 are then expected to cooperate for IDO1 expression, binding to the ISRE/GAS promoter region of the IDO1 gene (18) (Fig. 10).
FIGURE 10.

Proposed mechanism whereby RET/PTC3 regulates IDO1 expression in thyroid cancer cells. RET/PTC3 induces STAT1 expression through activation of the canonical NF-κB pathway. The activation of STAT1 is achieved by phosphorylation of Ser-727 and Tyr-701. RET/PTC3 induces phosphorylation of Ser-727 through activation of the MAPK and PI3K pathways, via PKCδ (23, 24); whereas RET/PTC3 phosphorylates STAT1 Tyr-701 directly. STAT1 translocates into the nucleus and induces the expression of IRF1. STAT1 and IRF1 bind to ISRE/GAS elements on the IDO1 promoter and promote the expression of the immunomodulatory enzyme. Activation of the canonical NF-κB pathway does not appear to modulate directly IDO1 gene expression.
The notion that RET/PTC3 can activate the canonical NF-κB pathway (13) prompted us to explore also the effect of this transcription factor on the STAT1 pathway using two commercially available inhibitors. Interestingly, inhibition of IκBα phosphorylation, with the inhibitor Bay11-7085, and inhibition of p65 nuclear translocation, with the inhibitor JSH-23, were both associated with a significant reduction of STAT1 and IDO1 expression. These data indicate that canonical NF-κB contributes to RET/PTC3-induced expression of IDO1 driving STAT1 overexpression (Fig. 10). Unfortunately, this experimental approach could not exclude a direct contribution of the RET/PTC3-induced NF-κB pathway activation to IDO1 expression, eventually through the binding of specific κB response elements postulated to be present on the IDO1 gene promoter (19). To clarify this aspect, we performed a ChIP assay in PTC3–5 cells exploring the binding of RelA/p65 to the promoter region of IDO1. Although we could demonstrate p65 binding to the STAT1 promoter after 24 h of RET/PTC3 activation, in the same conditions, we had no enrichment of the putative NF-κB binding sites detected on IDO1 promoter/enhancer regions of DNA. In summary, this experiment confirmed the direct regulatory role of the canonical NF-κB pathway on STAT1 expression and excluded its direct action on IDO1 promoter. In line with our findings, thus far IDO1 expression has been demonstrated to depend only on the activation of the non-canonical NF-κB pathway (20, 21), which has not appeared to be regulated by RET/PTCs (13).
Furthermore, we explored the mechanisms of RET/PTC3-induced STAT1 phosphorylation. Interestingly, STAT1 Ser-727 phosphorylation has been attributed to MAPK and PI3K-AKT pathway activation (22). Because RET/PTC3 is a strong activator of the MAPK pathway, in the first instance, we analyzed the effects of this pathway on STAT1 activation. Interestingly, Dox-inducible expression of BRAFV600E in PcCL3 cells induced only STAT1 Ser-727 phosphorylation, without affecting STAT1 Tyr-701 or STAT1 expression. The incapacity of BRAFV600E to induce IDO1 expression could be ascribed to the lack of full activating potential on STAT1. However, the evidence that BRAFV600E induces Ser-727 phosphorylation indicated that this phosphorylation event is particularly dependent on activation of the MAPK pathway. Indeed, treatment of PTC3–5 cells with the MEK inhibitor U0126 resulted in a significant reduction in STAT1 phosphorylation, especially at the level of Ser-727. In the second instance, we also evaluated the effect on STAT1 phosphorylation of the other key signal transduction pathway activated by RET/PTC, namely the PI3K-AKT pathway. Interestingly, also inhibition of PI3K, using the selective inhibitor wortmannin, resulted in a significant reduction of STAT1 phosphorylation and again the strongest effect was on Ser-727. These data confirm that the RET/PTC3-induced MAPK and PI3K-AKT pathways contribute to activate PKCδ, which has been shown to associate with STAT1 and mediate its Ser-727 phosphorylation in the contest of γ-interferon signaling (23, 24) (Fig. 10).
Conversely, MAPK and PI3K pathway inhibition affected less significantly phosphorylation of STAT1 Tyr-701, which we postulated to depend from an independent mechanism/pathway. We explored the possibility that STAT1 Tyr-701 phosphorylation could happen through JAK2 or tyrosine kinase Src or the occurrence of a physical interaction of RET/PTC3 and STAT1 followed by a tyrosine phosphorylation step directly performed by the oncogene. The obtained data point to the direct kinase action of RET/PTC3 on STAT1 Tyr-701 (Fig. 10). Indeed, JAK2 pharmacological inhibition and JAK2 silencing in PTC3–5 cells did not affect STAT1 phosphorylation and IDO1 expression. Unfortunately, in PTC3–5 cells the use of Src pharmacological inhibitors to demonstrate a role of the kinase in STAT1 phosphorylation was weakened by the low selectivity of SU6656, PP1, and SRCI-1 (14, 15) and the concurrent inhibition of Src and RET/PTC3. Thus, we had to resort to HEK293T cells transiently transfected with RET/PTC3 mutants. Although we expected differences in the signaling network between these cells and rat thyroid cells, we thought that they could represent an appropriate container to explore molecular interactions and to make predictions on molecular functions. Indeed, the use of the RET/PTC3-V804M mutant, resistant to the RET and Src inhibitor PP1, and of the RET/PTC3-Y981F mutant, characterized by the inability to bind Src, allowed a marginal role of Src kinase in STAT1 Tyr-701 phosphorylation to be predicted. Conversely, immunoprecipitation experiments on cell lysates of HEK293T cells, transiently transfected with RET/PTC3, STAT1, and Src constructs, clearly showed the occurrence of a RET/PTC3 and STAT1 aggregation. Moreover, kinase assay co-incubating RET/PTC3 and STAT1 proteins showed that RET/PTC3 directly phosphorylates Tyr-701 of STAT1. A previous study, which analyzed the dynamics of RET/PTCs and STAT1 interactions in thyroid carcinoma cell lines, demonstrated that RET/PTC1 and RET/PTC3 activate STAT1 inducing Tyr-701, but apparently not Ser-727, phosphorylation. Moreover, after having excluded a role of JAK2 and Src, as we did, the authors postulated the existence of a physical interaction between RET/PTCs and STAT1, as previously observed for STAT3, although they were unable to demonstrate it by immunoprecipitation experiments (25). On the contrary, we were able to demonstrate the importance of both STAT1 phosphorylations (Tyr-701 and Ser-727) and STAT1 expression up-regulation in RET/PTC3-induced STAT1 activation in thyroid cells. Moreover, we were able to demonstrate the occurrence of a physical interaction of RET/PTC3 and STAT1 followed by a tyrosine phosphorylation step directly performed by the oncogene (Fig. 10). It is possible that the differences between our study and the previous one might be related to differences of the adopted cellular models and experimental approaches. In the present study, we primarily used a fairly physiologic model characterized by a well differentiated rat thyroid cell line modified to deliver doxycycline-inducible expression of RET/PTC3. Furthermore, for the pulldown experiments, we used the human embryonic kidney cell line HEK293T transfected with RET/PTC3, STAT1, and Src expression vectors. In the latter study, all the experiments were carried out in thyroid cancer (TPC1) and colon cancer (ARO, containing a BRAFV600E mutation) cell lines and mouse fibroblasts transfected with RET/PTC and STAT1 expression vectors. Thus, our study might refine and complete more gross, although not less important, acquisitions coming from the previous one. In accordance with the previous study, we could exclude that RET/PTC3 stimulates γ-interferon synthesis and secretion as Dox treatment did not modify the concentration of the cytokine in the cell culture media (data not shown).
Very recently, tryptophan catabolite kynurenine was identified as an endogenous ligand of the human AHR (6). In detail, kynurenine produced by intracellular dioxygenases appeared to suppress antitumor immune responses and to promote tumor-cell survival and motility through AHR in an autocrine/paracrine fashion. Interestingly, these types of pathways were demonstrated to be active in human brain tumors and to be associated with malignant progression and poor survival.
Our data extend the carcinogenetic role of dioxygenases-derived kynurenine to thyroid carcinoma. Indeed, we present a novel genetic model supporting in vitro the dependence of IDO1 expression from an oncogene, namely RET/PTC, in thyroid cells.
In conclusion, this work shows for the first time that the RET/PTC oncogenes induce expression of IDO1 through the full activation (overexpression and full phosphorylation) of STAT1. This event might represent an initiation step of thyroid tumorigenesis. Conversely, in the absence of RET/PTC rearrangements, STAT1 activation might be a later event during cancer progression regulated by other genetic or epigenetic mechanisms. Independently of its timing, STAT1-driven IDO1 overexpression ultimately results in a gain in immune evasion and tumorigenic potential of cancer cells. Further studies will be needed to completely understand the role of IDO1 in thyroid tumorigenesis and to propose the use of IDO1 inhibitors in the treatment of advanced thyroid carcinomas.
Experimental Procedures
Plasmids
pcDNA3-RET/PTC3R51, pcDNA3-RET/PTC3-V804MR51 Myc-tagged and pCEFL-Src plasmids were available in our laboratory and obtained as described previously (26); pCMV6-STAT1 Myc- and FLAG-tagged plasmids were purchased from OriGene Technologies (Rockville, MD). pcDNA3-RET/PTC3-Y981F was obtained by site-directed mutagenesis of pcDNA3-RET/PTC3, using the QuikChange XL mutagenesis kit (Stratagene, Agilent Technologies, Santa Clara, CA), according to the manufacturer's instructions.
Cell Cultures
BRAF9.6 cells, characterized by Dox-inducible expression of the BRAFV600E mutation, and PTC3–5 cells, characterized by Dox-inducible expression of the RET/PTC3 rearrangement, were derived from the PcCL3 line (a clonal rat thyroid line requiring TSH for growth), as described elsewhere (11, 27), and were kindly provided by Dr. James A. Fagin (Memorial Sloan Kettering Cancer Center, New York). BRAF9.6 and PTC3–5 cells were grown to near confluence in 10-cm dishes and then transferred to TSH-free medium for 3 days before the addition of 1 μg/ml of Dox. At the designated time points, the cells were harvested for total RNA and cell lysates preparation. Signal transduction dissection experiments using specific inhibitors were performed by treating the cells for 24 or 48 h with the inhibitor. At the end, cells were harvested and used to prepare cell lysates or total mRNA according to standard procedures. U0126 was purchased from Cell Signaling (Danvers, MA), wortmannin from Life Technologies (Thermo Scientific, Waltham, MA), and AG490, Src inhibitor-1, SU6656, PP1, Bay11-7085, and JSH-23 from Sigma (Sigma-Aldrich, Milan, Italy). PcCL3-derived cell lines stably expressing BRAFV600E, RET/PTC1, RET/PTC3, v-Mos, RET/PTC1, and H-RasG12V, and v-Raf oncogenes were generated and grown as described previously (29). HEK293T cells were from American Type Culture Collection (ATCC, Manassas, VA) and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Thermo Scientific).
Transient transfections were carried out with the Lipofectamine 2000 reagent according to manufacturer's instructions (Thermo Scientific). In detail, cells were seeded at a density of 1.5 × 106/100-mm dish the day before transfection, transfected with 5 μg of DNA, and harvested 24 or 48 h later.
Quantitative PCR
Total RNA was extracted with TRIzol (Thermo Scientific) and first-strand cDNA synthesis was performed using 2 μg of each RNA sample primed with random hexamers with 200 units of SuperScript III reverse transcriptase (Thermo Scientific), according to the manufacturer's instructions.
To evaluate expression of IDO1, STAT1, IRF1, and JAK2 mRNA, quantitative PCR amplifications were performed using Platinum SYBR GreenER qPCR SuperMix UDG Universal (Life Technologies) according to the manufacturer's instructions (IDO F, GATGTGGGCTTTGCTCTACC; IDO R, GCTTCCCATTCTCAATCAGC; STAT1 F, TGAAGCTGAGACTGTTGGTG; STAT1 R, GAACTCAGCTGCCAAACTTC; IRF1 F, GACGGACTGAGCAGCTCTAC; IRF1 R, CTGCATATGCCACTCAGAGA; JAK2 F, GCCGAAGAAATTTGTGTGGC; JAK2 R, TGGAAGACATGATTGGGTGGA; β-actin F, TCATCACTATCGGCAATGAG; β-actin R, CTTTACGGATGTCAACGTCA) and the results were analyzed as described previously (30). In detail, the cycle threshold (CT) value coupled with individualized amplification efficiencies for each primer set was used to calculate the normalized expression of the indicated gene mRNA using the Q-Gene software (31). p values were calculated applying the two-tailed unpaired Student's t test and all differences were considered significant when p < 0.05.
Immunoblotting Experiments
Immunoblotting experiments were performed according to standard procedures. Polyclonal anti-RET antibody (1:1000) and anti-total ERK (1:1000) were from Santa Cruz Biotechnology. Anti-rat IDO1 (1:200), anti-pTyr-701 STAT1 (1:1000), and anti-pSer-727 STAT1 (1:200) were from Abcam (Abcam Cambridge, UK). Anti-α-tubulin (1:5000) and anti-Myc (clone 9E10; 1:5000) were from Sigma. Anti-total STAT1 (1:1000), anti-pERK (1:1000), anti-pAKT (1:1000), anti-JAK2 (1:1000), anti-pTyr-905 RET (1:1000), and anti-Src (1:1000) were from Cell Signaling Technology. Secondary anti-mouse, anti-rabbit, and anti-goat antibodies coupled to horseradish peroxidase were purchased from Sigma. The densitometric analysis of the results was performed using the ImageJ processing program.
STAT1 and JAK2 Silencing
For STAT1 silencing, PTC3–5 cells were transfected with 25 nm rat STAT1 siRNA (ON-TARGETplus SMART pool, L-080050-01, Thermo Scientific) or with 25 nm nontarget siRNA (ON-TARGETplus Non-Targeting Pool, Thermo Scientific) using Lipofectamine 2000 (Thermo Scientific). Twenty-four hours after transfection, cells were stimulated with Dox for 48 h before preparation of total RNA or cell lysates. STAT1, IRF1, and IDO1 expressions were evaluated by Q-PCR and Western blotting, as described in the previous paragraphs. For JAK2 silencing, PTC3–5 cells were transfected with 50 nm rat JAK2 siRNA (ON-TARGETplus SMART pool, L-088340-02, Dharmacon) or 50 nm non-target siRNA using Lipofectamine 2000. Twenty-four hours after transfection, cells were stimulated with Dox for 48 h before preparation of total RNA or cell lysates. JAK2, RET, STAT1, pSer-727-STAT1 and pTyr-701-STAT were evaluated by Western blotting. IDO mRNA expression was evaluated by Q-PCR, as previously described.
Chromatin Immunoprecipitation (ChIP) Assay
Rat IDO1 and STAT1 promoters were analyzed using MatInspector software (Genomatix) (32) for the identification of putative binding sites for RelA/p65. In detail, for STAT1, 5000 bp of the promoter, localized upstream of the starting codon, were analyzed. At variance, for IDO1, 2000 bp of the promoter, localized upstream of the starting codon, and the sequence of the first intron, were analyzed.
A dual cross-linking ChIP assay (28) was used. Briefly, PTC3.5 cells were treated for 24 h with Dox to induce expression of the RET/PTC3 oncogene. The cells were washed twice with PBS at room temperature and resuspended in PBS containing 1 mm MgCl2. In the first instance, disuccinimidyl glutarate (Sigma) was added to a final concentration of 2 mm and cells were incubated 45 min at room temperature for cross-linking. In the second instance, formaldehyde was added to a final concentration of 1% (v/v) and cells were incubated for 15 min at room temperature for dual cross-linking. Finally, glycine was added to a final concentration of 125 mm and cells incubated for 5 min at room temperature to quench formaldehyde cross-linking. After harvesting, cells were lysed in 700 μl of ice-cold cell lysis buffer (5 mm HEPES, pH 8.0, 85 mm KCl, 0.5% Nonidet P-40, 1 mm PMSF, 1× protease inhibitor mixture (Roche)) for 10 min on ice. Nuclei were precipitated by centrifugation (3,000 × g for 5 min) resuspended in 600 μl of ice-cold RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mm PMSF, 1× protease inhibitor mixture (Roche)) and incubated on ice for 20 min. Sonication with Soniprep 150 (Sanyo) was performed to obtain DNA fragments of 100–600 bp in length (15 cycles each of 10 min with 30 min pause, power 70%). An anti-p65 antibody (1:100) (Cell Signaling Technology) was used to recover protein-DNA complexes. Normal rabbit IgG mixtures (Santa Cruz Biotechnology) were used as negative control. The chromatin pulldown was performed using 20 μl of protein G-Sepharose (GE Healthcare, Milan, Italy).
Quantitative PCR amplifications were performed using 90 ng of DNA and Platinum SYBR GreenER Q-PCR SuperMix UDG Universal (Life Technologies) according to the manufacturer's instructions. Primers used were: STAT1 promoter F, GGAGCCCCACGTTTCTCAC; STAT1 promoter R, TTTCCTCCTCGGCCAATCAG (for the amplification of a DNA fragment spanning from −3639 to −3504 bp upstream of the ATG); IDO1 promoter region 1 F, CTCCACCGCAAGCATAGACC; IDO1 promoter region 1 R, CTCATGCCACACCCACTCC (for the amplification of a DNA fragment spanning from −1695 to −1545 bp upstream of the ATG); IDO1 intron 1 region 2 F, ACTACGGTCTAGCGTGCAG; IDO1 intron 1 region 2 R, AGGTTGAGAACTGCTGCACA (for the amplification of a DNA fragment spanning from +1480 to +1638 bp downstream of the ATG); IDO1 intron 1 region 3 F, ATAAGGTGGTGCGCACTTGA; IDO1 intron 1 region 3 R, CTGTGTGTGTAGGCCAGAGG (for the amplification of a DNA fragment spanning from +2438 to +2592 bp downstream of the ATG). The results were analyzed using the ΔCt method with the input (10% of sonicated DNA) as endogenous control.
Coimmunoprecipitations
Five hundred μg of total lysate were used for each immunoprecipitation assay. In HEK293T cells transiently transfected with RET/PTC3 and Src or RET/PTC3 and STAT1 or RET/PTC3 and Src and STAT1, molecular complexes assembled with RET, Src, and STAT1 were immunoprecipitated using an anti-RET antibody (1:100), an anti-Src antibody (1:100), or an anti-Myc (1:100) (anti-Myc-tagged STAT1), respectively. The protein pulldown was performed using 40 μl of protein G-Sepharose (GE Healthcare, Milan Italy) in the case of goat or mouse antibodies, or protein A-Sepharose (GE Healthcare) in the case of rabbit antibody. Obtained protein complexes were resuspended in 30 μl of 2× Laemmli buffer. Immunoprecipitates were then subjected to 10% SDS-gel electrophoresis. The presence of STAT1, Src, and RET was searched by Western blotting using the specific antibodies. To verify binding specificity, the presence of STAT1, Src, and RET was searched on the protein complexes immunoprecipitated with normal mouse, normal rabbit, or normal goat IgG mixtures (Santa Cruz Biotechnology), respectively.
In Vitro Kinase Assay
HEK293T cells transiently transfected with RET/PTC3 or STAT1 plasmid were cultured for 24 h and lysed in a buffer containing 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, 150 mm NaCl, 1% Triton X-100, 0.5% Nonidet P-40, 50 mm NaF, 1 mm sodium vanadate, 2 mm phenylmethylsulfonyl fluoride (PMSF), and 1× complete protease inhibitor mixture (Roche Diagnostics). RET/PTC3 and STAT1 were immunoprecipitated with anti-RET and anti-Myc antibodies, respectively, as described in the previous paragraph. To demonstrate direct STAT1 phosphorylation by RET/PTC3, the RET/PTC3-washed immunoprecipitate and the STAT1-washed immunoprecipitate were pulled together and resuspended in 40 μl of a kinase buffer containing 25 mm Tris-HCl, pH 7.5, 1 m NaF, 2 mm 1,4-dithiothreitol (DTT), 0.1 mm sodium vanadate, 10 mm MgCl2, and 0.2 mm ATP. As positive control of the reaction, the RET/PTC3-washed immunoprecipitate was incubated with 2 μm poly(GT) (poly-l-glutamic acid-l-tyrosine) (Sigma). Conversely, to rule out the occurrence of STAT1 autophosphorylation or RET/PTC3-driven pulldown of phosphorylated STAT1 from HEK293T cell lysates, the STAT1-washed immunoprecipitate or the RET/PTC3-washed immunoprecipitate were incubated alone in the kinase buffer. The kinase reactions were incubated 30 min at 30 °C in agitation, stopped by adding 10 μl of 2× Laemmli buffer and 5 μl were subjected to 8% SDS-gel electrophoresis. The degree of STAT1 phosphorylation was analyzed using anti-pTyr-701 STAT1 (1:1000) and anti-pSer-727 STAT1 (1:200). At variance, the degree of positive control phosphorylation was analyzed using anti-Tyr(P) (PY20) (1:1000) (Santa Cruz Biotechnology).
Author Contributions
F. F., A. M., N. A., and E. P. conceived and coordinated the study and wrote the paper. So. M., E. M., N. N., P. V., R. C., and Si. M. designed, performed, and analyzed the experiments shown in Figs. 1A and 2–9. R. M. M., F. L., and M. S. tested IDO expression in PcCL3-derived cell lines stably expressing the thyroid oncogenes and designed and constructed expression vectors of RET mutants. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by grants from the Fondazione Cassa di Risparmio di Perugia, Associazione Italiana per la Ricerca sul Cancro (IG 9338), and a liberal contribution from the Beadle Family Foundation (San Antonio, TX) and the Brunello Cucinelli Foundation (Corciano, Italy) (all to E. P.). The authors declare that they have no conflicts of interest with the contents of this article.
- IDO1
- indoleamine 2,3-dioxygenase 1
- AHR
- aryl hydrocarbon receptor
- Treg
- T regulatory
- TSH
- thyrotropin
- Dox
- doxycycline
- Q-PCR
- quantitative PCR.
References
- 1.Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2.Gajewski T. F., Schreiber H., and Fu Y. X. (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–102224048123 [Google Scholar]
- 3.Uyttenhove C., Pilotte L., Théate I., Stroobant V., Colau D., Parmentier N., Boon T., and Van den Eynde B. J. (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 9, 1269–1274 [DOI] [PubMed] [Google Scholar]
- 4.Mellor A. L., and Munn D. H. (2004) IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 4, 762–774 [DOI] [PubMed] [Google Scholar]
- 5.Munn D. H., and Mellor A. L. (2007) Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 117, 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Opitz C. A., Litzenburger U. M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M., Jugold M., Guillemin G. J., Miller C. L., Lutz C., Radlwimmer B., et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 [DOI] [PubMed] [Google Scholar]
- 7.Moretti S., Menicali E., Voce P., Morelli S., Cantarelli S., Sponziello M., Colella R., Fallarino F., Orabona C., Alunno A., de Biase D., Bini V., Mameli M. G., Filetti S., Gerli R., et al. (2014) Indoleamine 2,3-dioxygenase 1 (IDO1) is up-regulated in thyroid carcinoma and drives the development of an immunosuppressant tumor microenvironment. J. Clin. Endocrinol. Metab. 99, 832–840 [DOI] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network. (2014) Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menicali E., Moretti S., Voce P., Romagnoli S., Avenia N., and Puxeddu E. (2012) Intracellular signal transduction and modification of the tumor microenvironment induced by RET/PTCs in papillary thyroid carcinoma. Front. Endocrinol. (Lausanne) 3, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitsutake N., Miyagishi M., Mitsutake S., Akeno N., Mesa C. Jr., Knauf J. A., Zhang L., Taira K., and Fagin J. A. (2006) BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology 147, 1014–1019 [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Knauf J. A., Basu S., Puxeddu E., Kuroda H., Santoro M., Fusco A., and Fagin J. A. (2003) Conditional expression of RET/PTC induces a weak oncogenic drive in thyroid PCCL3 cells and inhibits thyrotropin action at multiple levels. Mol. Endocrinol. 17, 1425–1436 [DOI] [PubMed] [Google Scholar]
- 12.Huang L., Baban B., Johnson B. A. 3rd, and Mellor A. L. (2010) Dendritic cells, indoleamine 2,3-dioxygenase and acquired immune privilege. Int. Rev. Immunol. 29, 133–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neely R. J., Brose M. S., Gray C. M., McCorkell K. A., Leibowitz J. M., Ma C., Rothstein J. L., and May M. J. (2011) The RET/PTC3 oncogene activates classical NF-κB by stabilizing NIK. Oncogene 30, 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlomagno F., Vitagliano D., Guida T., Napolitano M., Vecchio G., Fusco A., Gazit A., Levitzki A., and Santoro M. (2002) The kinase inhibitor PP1 blocks tumorigenesis induced by RET oncogenes. Cancer Res. 62, 1077–1082 [PubMed] [Google Scholar]
- 16.Carlomagno F., Guida T., Anaganti S., Vecchio G., Fusco A., Ryan A. J., Billaud M., and Santoro M. (2004) Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 23, 6056–6063 [DOI] [PubMed] [Google Scholar]
- 17.Encinas M., Crowder R. J., Milbrandt J., and Johnson E. M. Jr. (2004) Tyrosine 981, a novel ret autophosphorylation site, binds c-Src to mediate neuronal survival. J. Biol. Chem. 279, 18262–18269 [DOI] [PubMed] [Google Scholar]
- 18.Jeong Y. I., Kim S. W., Jung I. D., Lee J. S., Chang J. H., Lee C. M., Chun S. H., Yoon M. S., Kim G. T., Ryu S. W., Kim J. S., Shin Y. K., Lee W. S., Shin H. K., Lee J. D., and Park Y. M. (2009) Curcumin suppresses the induction of indoleamine 2,3-dioxygenase by blocking the Janus-activated kinase-protein kinase Cδ-STAT1 signaling pathway in interferon-γ-stimulated murine dendritic cells. J. Biol. Chem. 284, 3700–3708 [DOI] [PubMed] [Google Scholar]
- 19.Ogasawara N., Oguro T., Sakabe T., Matsushima M., Takikawa O., Isobe K., and Nagase F. (2009) Hemoglobin induces the expression of indoleamine 2,3-dioxygenase in dendritic cells through the activation of PI3K, PKC, and NF-κB and the generation of reactive oxygen species. J. Cell. Biochem. 108, 716–725 [DOI] [PubMed] [Google Scholar]
- 20.Tas S. W., Vervoordeldonk M. J., Hajji N., Schuitemaker J. H., van der Sluijs K. F., May M. J., Ghosh S., Kapsenberg M. L., Tak P. P., and de Jong E. C. (2007) Noncanonical NF-κB signaling in dendritic cells is required for indoleamine 2,3-dioxygenase (IDO) induction and immune regulation. Blood 110, 1540–1549 [DOI] [PubMed] [Google Scholar]
- 21.Puccetti P., and Grohmann U. (2007) IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-κB activation. Nat. Rev. Immunol. 7, 817–823 [DOI] [PubMed] [Google Scholar]
- 22.Nguyen H., Ramana C. V., Bayes J., and Stark G. R. (2001) Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J. Biol. Chem. 276, 33361–33368 [DOI] [PubMed] [Google Scholar]
- 23.Deb D. K., Sassano A., Lekmine F., Majchrzak B., Verma A., Kambhampati S., Uddin S., Rahman A., Fish E. N., and Platanias L. C. (2003) Activation of protein kinase C delta by IFN-γ. J. Immunol. 171, 267–273 [DOI] [PubMed] [Google Scholar]
- 24.Uddin S., Sassano A., Deb D. K., Verma A., Majchrzak B., Rahman A., Malik A. B., Fish E. N., and Platanias L. C. (2002) Protein kinase C-δ (PKC-δ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 277, 14408–14416 [DOI] [PubMed] [Google Scholar]
- 25.Hwang E. S., Kim D. W., Hwang J. H., Jung H. S., Suh J. M., Park Y. J., Chung H. K., Song J. H., Park K. C., Park S. H., Yun H. J., Kim J. M., and Shong M. (2004) Regulation of signal transducer and activator of transcription 1 (STAT1) and STAT1-dependent genes by RET/PTC (rearranged in transformation/papillary thyroid carcinoma) oncogenic tyrosine kinases. Mol. Endocrinol. 18, 2672–2684 [DOI] [PubMed] [Google Scholar]
- 26.Iavarone C., Acunzo M., Carlomagno F., Catania A., Melillo R. M., Carlomagno S. M., Santoro M., and Chiariello M. (2006) Activation of the Erk8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutively active form of the RET proto-oncogene. J. Biol. Chem. 281, 10567–10576 [DOI] [PubMed] [Google Scholar]
- 27.Mitsutake N., Knauf J. A., Mitsutake S., Mesa C. Jr, Zhang L., and Fagin J. A. (2005) Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid PCCL3 cells. Cancer Res. 65, 2465–2473 [DOI] [PubMed] [Google Scholar]
- 28.Nowak D. E., Tian B., and Brasier A. R. (2005) Two-step cross-linking method for identification of NF-κB gene network by chromatin immunoprecipitation. BioTechniques. 39, 715–725 [DOI] [PubMed] [Google Scholar]
- 29.Fusco A., Berlingieri M. T., Di Fiore P. P., Portella G., Grieco M., and Vecchio G. (1987) One- and two-step transformations of rat thyroid epithelial cells by retroviral oncogenes. Mol. Cell. Biol. 7, 3365–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puxeddu E., Knauf J. A., Sartor M. A., Mitsutake N., Smith E. P., Medvedovic M., Tomlinson C. R., Moretti S., and Fagin J. A. (2005) RET/PTC-induced gene expression in thyroid PCCL3 cells reveals early activation of genes involved in regulation of the immune response. Endocr. Relat. Cancer 12, 319–334 [DOI] [PubMed] [Google Scholar]
- 31.Muller P. Y., Janovjak H., Miserez A. R., and Dobbie Z. (2002) Processing of gene expression data generated by quantitative real-time RT-PCR. BioTechniques 32, 1372–1374, 1376,, 1378–1379 [PubMed] [Google Scholar]
- 32.Cartharius K., Frech K., Grote K., Klocke B., Haltmeier M., Klingenhoff A., Frisch M., Bayerlein M., and Werner T. (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21, 2933–2942 [DOI] [PubMed] [Google Scholar]