

Abstract
IgG isotypes can differentially bind to Fcγ receptors and complement, making the selection of which isotype to pursue for development of a particular therapeutic antibody important in determining the safety and efficacy of the drug. IgG2 and IgG4 isotypes have significantly lower binding affinity to Fcγ receptors. Recent evidence suggests that the IgG2 isotype is not completely devoid of effector function, whereas the IgG4 isotype can undergo in vivo Fab arm exchange leading to bispecific antibody and off-target effects. Here an attempt was made to engineer an IgG1-based scaffold lacking effector function but with stability equivalent to that of the parent IgG1. Care was taken to ensure that both stability and lack of effector function was achieved with a minimum number of mutations. Among the Asn297 mutants that result in lack of glycosylation and thus loss of effector function, we demonstrate that the N297G variant has better stability and developability compared with the N297Q or N297A variants. To further improve the stability of N297G, we introduced a novel engineered disulfide bond at a solvent inaccessible location in the CH2 domain. The resulting scaffold has stability greater than or equivalent to that of the parental IgG1 scaffold. Extensive biophysical analyses and pharmacokinetic (PK) studies in mouse, rat, and monkey further confirmed the developability of this unique scaffold, and suggest that it could be used for all Fc containing therapeutics (e.g. antibodies, bispecific antibodies, and Fc fusions) requiring lack of effector function or elimination of binding to Fcγ receptors.
Keywords: antibody engineering, drug development, Fc receptor, immunoglobulin G (IgG), protein stability, aglycosylated IgG, antibody developability, antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, disulfide engineering
Introduction
IgG isotypes can differentially engage Fcγ receptors and C1q binding to recruit immune effector functions and complement dependent cytotoxicity. More than 50% of all marketed and clinical candidate antibodies target cell-surface proteins (1) and do not require cytotoxicity as part of their mechanism of action. For these targets, having therapeutic antibodies with immune effector function could be detrimental and pose a safety risk. For example, the target cells could be depleted due to antibody-dependent cellular cytotoxicity (ADCC).3 The four IgG isotypes bind to the activating FcγRI, FcγRIIa, and FcγRIIIa, and inhibiting FcγRIIb and FcγRIIIb, with different affinities (2, 3). Therefore, isotype selection is critical to the safety and efficacy of therapeutic antibodies. For example, IgG2 or IgG4 isotypes are selected in cases where effector function is not desirable, whereas the IgG1 isotype is used if ADCC or complement-dependent cytotoxicity is required. Recent evidence suggests the IgG2 isotype is not completely devoid of effector function (4–6). For example, IgG2 binds to cynomolgus monkey FcγRIIa with similar affinity as IgG1 (5, 7). In addition, IgG2 hinge cysteines (Cys) can mispair, leading to disulfide isoforms with altered activity (8). IgG2 is also less stable to low pH and heat-induced denaturation compared with IgG1 (9, 10). The IgG4 scaffold has the liability of disassociating and pairing with another IgG4 antibody in vivo leading to the formation of bispecific antibodies, and potential off-target effects (11).
Antibody structure consists of two distinct active regions, the Fab domain that binds to antigen or target and the Fc domain that interacts with Fc receptors. The Fc domain imparts serum half-life through interaction with neonatal receptor FcRn. The Fc domain also interacts with Fcγ receptors leading to effector function. Although removing the Fc region would eliminate the effector function, it would also significantly reduce the serum half-life that the Fc contributes (12). Therefore, to eliminate the effector function only, mutations that significantly reduce binding to FcγRs have been attempted (13). This includes mutating the IgG hinge or Fc sequence at the FcγR interaction site or eliminating the glycosylation site (Asn297) in the CH2 domain of IgG1 to generate an aglycosylated antibody. The Asn297 glycosylation site can be eliminated by mutating the Asn at position 297 to, for example, Ala and Gln. A recent study examined the Asn to Gly (N297G) variant, along with Asn to Ala (N297A), impact on FcγR binding and pharmacokinetics for the first time and concluded that N297G and N297A variants have similar pharmacokinetics (PK) to that of the wild type antibodies (14). Aglycosylated antibodies have a very low level of binding to FcγRs (15, 16). The advantage with this approach is that it requires a single mutation in the constant domain (CH2), thus minimizing the risk of immunogenicity via the formation of new epitopes. However, aglycosylated antibodies, either produced in mammalian cells but engineered to have no sugar as described above, or produced in Escherichia coli systems, which do not have glycosylation machinery, have lower thermal stability compared with the glycosylated IgG molecules (17). The removal of glycosylation affects only the CH2 domain stability with little or no impact on the other domains in the IgG scaffold. As with any engineering of endogenous proteins, modification of the Fc for removal of glycosylation does bring in immunogenicity concerns. However, the lack of engineered Fc or hinge scaffold-related immunogenicity in the marketed products and therapeutic candidates that are in late stage clinical trials suggests that this concern is likely not a roadblock in the development of the therapeutic (16). For example, otelixizumab has a mutation of Asn297 to remove the glycosylation site in the Fc. Another example is eculizumab, in which the Fc is a combination of IgG2 and IgG4 sequences, and has low immunogenicity. It must be noted that although to date immunogenicity due to the removal of the glycosylation site or engineering of the FcγR binding site in the hinge or Fc region has not been reported, this could change as more patients are dosed with modified therapeutic molecules for longer periods.
Engineering and/or modification of the glycosylation content has also been applied to alter ADCC. For example, although removal of fucosyl moieties in the sugar structure can lead to a 10-fold increase in ADCC (18), enriching for fucose does not eliminate the FcγRs binding and could still pose concern when ADCC is not desired. Enriching sialic acid content could also lead to reduced ADCC, however, this could result in anti-inflammatory activities and may not completely eliminate the ADCC risk (19, 20). Furthermore, obtaining antibody drug substance with a consistent amount and type of glycosylation from lot to lot during GMP production is not trivial.
Here we employ structure-based Fc engineering in an attempt to develop an IgG scaffold with the following properties: (i) no or significantly reduced binding to Fcγ receptors; (ii) stability similar to that of IgG1 without the heterogeneity of the disulfide isoforms that are present in IgG2; (iii) low or no binding to cynomolgus monkey FcγRs, in particular FcγRIIa; (iv) desirable developability attributes such as storage stability and PK similar to IgG molecules (no change in binding to FcRn); and (v) can be isolated using established antibody manufacturing processes. This should be achieved with as few mutations as possible. The first property could be achieved through mutating the FcγR binding site residues in the Fc region of the antibody or by engineering aglycosylated IgG molecules. The goal of obtaining a stable, homogeneous antibody led us to choose the IgG1 aglycosylated scaffold for further engineering and optimization. Clinical and published data on aglycosylated IgG molecules (15, 21–23) show them to be well tolerated in patients (e.g. otelixizumab, MetMab, and BMS-945429 clinical trials). Further PK pharmacokinetic analysis of aglycosylated antibodies such as ALD518 in humans show a serum half-life of 25 days (24), which is similar to that observed typically for glycosylated human IgG molecules. Antibodies generated for these studies and also utilized in Ref. 25 are summarized in Table 1.
TABLE 1.
Therapeutic mAb and engineered variants
| Molecule name | Type |
|---|---|
| mAbW.IgG1 | IgG1 Control |
| mAbW.IgG2 | IgG2 Control |
| mAbW.SEFL1.1 | IgG1 N297G |
| mAbW.SEFL1.2 | IgG1 N297A |
| mAbW.SEFL1.3 | IgG1 N297Q |
| mAbW.SEFL2.0a | IgG1 L242C, N297G, K334C |
| mAbW.SEFL2.1b | IgG1 A287C, N297G, L306C |
| mAbW.SEFL2.2b | IgG1 R292C, N297G, V302C |
| mAbW.SEFL2.3b | IgG1 N297G, V323C, I332C |
| mAbW.SEFL2.4b | IgG1 V259C, N297G, L306C |
a An intra CH2 domain disulfide bond at Leu242-Lys334 position was introduced to improve thermal stability, adapted from Ref. 29.
b These novel intra CH2 domain disulfide bonds (A287C-L306C, R292C-V302C, V323C-I332C, and V259C-L306C) were designed utilizing structural analysis.
Results
Computational Analysis
To analyze the structure and conformation of the loop containing Asn297 and determine the substitution that would result in removal of the sugars but maintain the structure and contacts as much as possible, a total of 57 crystal structures were identified from the Protein Data Bank (PDB) (26). These structures had been determined at a resolution of 2.5 Å or better. Fig. 1a shows the superposition of the CH2 domain from the known Fc crystal structures. The glycosylation site Asn297 is indicated in blue. The root mean square deviation for the Cα position is 2.0 Å indicating similar secondary and tertiary structures. Although the β-strand regions superimpose well, the loop regions show high structural flexibility. To assess the conformation variation at the Asn297 position, the Ramachandran backbone dihedral angles were calculated using the coordinates deposited in the PDB and plotted in Fig. 1b. It is evident from the Fig. 1b that Asn297 adapts multiple backbone conformations including ϕ,ψ angles that are sterically disallowed for non-Gly amino acids. This observation suggests Gly could be a better substitution for the Asn297 position, in contrast to the published analogs, which used primarily Ala or Gln.
FIGURE 1.

a, superposition of CH2 domain structures extracted from the high resolution (≤2.5 Å) crystal structures of Fc and antibody deposited in the PDB. Although the β-strand region superimposes well, the loop comprising the Asn297 position (shown in blue) shows high structural variability. b, Ramachandran φ,ψ plot showing the backbone dihedral angle distribution of Asn297. Consistent with a, the Asn297 backbone has high structural variability and adapts φ positive conformation, which is an unique feature of Gly (38), in some of the crystal structures.
Purification Characteristics
To assess the developability of the generated analogs, the Asn297 variants (mAbW.SEFL1.1, mAbW.SEFL1.2, and mAbW.SEFL1.3) were expressed in a stable production cell line. Human IgG1 and IgG2 scaffolds with the same Fab domains were also included as controls. The expression level for the three variants was similar and comparable with the parental IgG1 and IgG2 molecules (supplemental Fig. S1). The supernatants were purified using a protein A affinity column followed by cation exchange chromatography (CEX). Interestingly, mAbW.SEFL1.2 and mAbW.SEFL1.3, unlike mAbW.SEFL1.1, had a tailing peak in the CEX profile suggesting that, when Asn297 is substituted with Ala or Gln, it results in a heterogeneous population with different interactions between the protein surface and the charged resin, perhaps due to the presence of multiple conformations. To confirm this observation, the molecules were re-expressed and purified. The purification again showed similar phenomena (Fig. 2). As a result, the purification efficiency or step yield of mAbW.SEFL1.2 and mAbW.SEFL1.3 was much lower than that of mAbW.SEFL1.1 and mAbW.SEFL2.0(78–80% yield for mAbW.SEFL1.1 or mAbW.SEFL2.0 versus 41–45% for mAbW.SEFL1.2 or mAbW.SEFL1.3); this yield is well below the target typically observed for mAbs.
FIGURE 2.

CEX purification profiles for the mAbW.SEFL1.1 (c), mAbW.SEFL1.2 (f), mAbW.SEFL1.3 (e), and mAbW.SEFL2.0 (d) along with mAbW.IgG1 (a) and mAbW.IgG2 (b) controls. Interestingly, mAbW.SEFL1.2 (N297A) and mAbW.SEFL1.3 (N297Q), unlike mAbW.SEFL1.1 (N297G), had a trailing peak (indicated by red arrow).
Stability Analysis
Thermal stability can be an indication of the conformational stability of a protein. The thermal stabilities of the mAbW.SEFL1.1, mAbW.SEFL1.2, mAbW.SEFL1.3, and mAbW.SEFL2.0 along with mAbW.IgG1 and mAbW.IgG2 controls were analyzed by differential scanning calorimetry (DSC). The DSC profiles of the samples in acetate sucrose Tween buffer, pH 5.2 (A52SuT), are shown in Fig. 3, and the apparent first thermal transitions (CH2 domain) are shown in Table 2. The individual domains (CH2, CH3, and Fab) melt independently in most cases, resulting in three peaks; the resolution between these peaks depends on the difference between the transition temperatures of each domain. The overall thermal stability of a molecule is affected by both CH2 and Fab domains. The CH3 domain has much less impact on the overall thermal stability of a molecule (10, 27). In SEFL1.x cases, because the Fab Tm values change little, we can use the CH2 Tm value to estimate thermal stability of the molecules. Consistent with the more homogeneous CEX profile observed earlier, the mAbW.SEFL1.1 construct has slightly higher thermal stability as determined from the first transition (CH2 domain) (65.7 °C Tm for mAbW.SEFL1.1, compared with 62.1 °C for mAbW.SEFL1.2 and mAbW.SEFL1.3; Table 2). The mAbW SEFL2.0, which is derived from mAbW SEFL1.1 with addition of the engineered intradomain disulfide bond at the CH2, has an increased CH2 domain thermal transition temperature compared with SEFL1.1.
FIGURE 3.
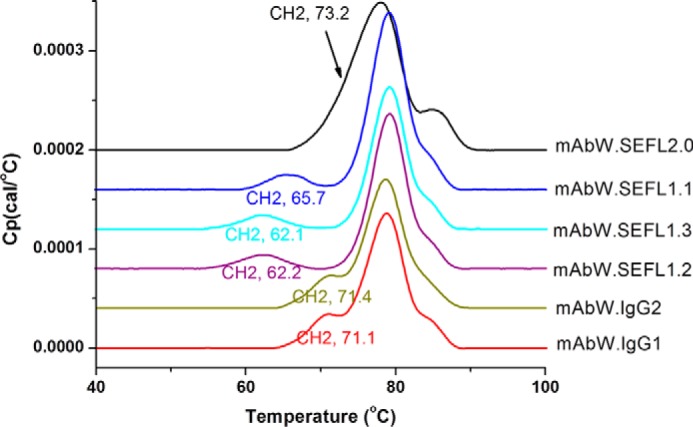
DSC profile of mAbW.SEFL1.1, mAbW.SEFL1.2, mAbW.SEFL1.3, and mAbW.SEFL2.0 along with mAbW.IgG1 and mAbW.IgG2 controls measured in A52SuT buffer.
TABLE 2.
Apparent CH2 Domain thermal transition temperatures of the SEFL samples in A52SuT
| CH2 domain | mAbW.IgG1 | mAbW.IgG2 | mAbW.SEFL1.1 | mAbW.SEFL1.2 | mAbW.SEFL1.3 | mAbW.SEFL2.0a |
|---|---|---|---|---|---|---|
| Tm (°C) | 71.1 | 71.4 | 65.7 | 62.2 | 62.1 | 73.2 |
a The CH2 and Fab domain thermal transitions are not well resolved from each other due to CH2 and Fab domain co-melting.
Improving Stability of Aglycosylated IgG
Previous studies show that the carbohydrates of IgGs are located at the interface of CH2-CH2 domains in Fc, and form contacts between the two CH2 domains, resulting in a more compact structure of the molecule (28). It is also known that removing glycosylation at the CH2 domain interface results in lower thermal stability, as evident from Table 2. As expected with the removal of the carbohydrate, mAbW.SEFL1.1 has significantly lower melting temperature than the parental antibody. To improve thermal stability of the mAbW.SEFL1.1 molecule, an intradomain Cys clamp (disulfide bond) was introduced utilizing the data provided in Ref. 29. The introduction of a disulfide bond, by mutating Leu242 and Lys334 to Cys (L242C and K334C), into the CH2 domain of aglycosylated IgGs improves the thermal stability of the molecule significantly as evident from the DSC analysis (Table 2). Fig. 3 also shows that the stability of mAbW.SEFL1.1 can be improved through engineering an intradomain Cys clamp at the CH2 domain. The resulting first apparent transition temperature (CH2/Fab melting) is similar to or better than the parental mAbW.IgG1 (71.1 °C for parental mAbW.IgG1 versus 73.2 °C for mAbW.SEFL2.0).
In Vivo Sprague-Dawley Rat PK Study
The PK profiles of the SEFL antibodies were determined in adult Sprague-Dawley rats (n = 3) by subcutaneous injection at 5 mg/kg and collecting 250-μl samples of blood at 0, 2, 8, 24, 48, 96, 168, 336, 504, 672, 840, and 1008 h post-dose. Each blood sample was maintained at room temperature after collection, and following a 30–40 min clotting period, samples were centrifuged at 2 to 8 °C at 11,500 rpm for about 10 min using a calibrated Eppendorf 5417R Centrifuge System (Brinkmann Instruments, Inc., Westbury, NY). The collected serum sample was then transferred into a prelabeled (for each rat) cryogenic storage tube, and stored at −60 to −80 °C for future bioanalysis.
The PK profiles of different SEFL antibodies in Sprague-Dawley rats are shown in Fig. 4. The mAbW.SEFL1.1 and the other Asn297 variants, with the exception of mAbW.SEFL2.0, have exposures and clearance comparable with those of the parent mAbW.IgG1 antibody. The AUC for mAbW.SEFL1.1 and parent mAbW.IgG1 were 903 and 1050 μg × day/ml, respectively. The clearance for mAbW.SEFL1.1 was 0.234 and comparable with that for the parent mAbW.IgG1, which is 0.204 ml/h/kg. The mAbW.SEFL2.0 had the lowest exposure with an AUC of 332 μg × day/ml and a clearance rate of 0.641 ml/h/kg, which is the highest among all the variants despite having better thermal stability (Table 3).
FIGURE 4.

PK profiles of SEFL antibodies were determined following subcutaneous administration to Sprague-Dawley rats at 5 mg/kg (n = 3). Antibody concentrations were measured over the 6-week study by sandwich ELISA with an LLOQ of 10 ng/ml.
TABLE 3.
Rat PK parameters
| Rat PK study (5 mg/kg SC) | T1/2 | AUC | CL/F |
|---|---|---|---|
| Day | μg × day/ml | ml/h/kg | |
| mAbW.IgG1 | 12 ± 3 | 1050 ± 216 | 0.204 ± 0.044 |
| mAbW.SEFL1.1 | 12 ± 1 | 903 ± 126 | 0.234 ± 0.035 |
| mAbW.SEFL1.2 | 12 ± 1 | 790 ± 55 | 0.265 ± 0.018 |
| mAbW.SEFL1.3 | 10 ± 3 | 866 ± 141 | 0.245 ± 0.041 |
| mAbW.SEFL2.0 | 5 ± 3 | 332 ± 56 | 0.641 ± 0.120 |
| mAbW.IgG2 | 19 ± 3 | 1477 ± 266 | 0.144 ± 0.027 |
Thus, although mAbW.SEFL2.0 had all the desired molecular physical properties, in particular the thermal stability equivalent to that of the parent mAbW.IgG1, it showed faster clearance in the rat PK study. However, it must be noted here that cynomolgus, human, and mouse FcRn-antibody binding analysis revealed no difference between the IgG control and SEFL molecules (supplemental Fig. S2).
Improving Thermal Stability without Negatively Impacting PK or Other Biophysical Properties
Although mAbW.SEFL2.0 successfully improved the yield and stability of the aglycosylated protein, the PK in rat was compromised; this would impact the ability to use rat as a model for biology, and could also potentially affect PK in humans. Examination of the structure shows the engineered disulfide bond is not near the FcRn binding site. Therefore, the impact on the PK could not be readily explained. We decided to explore other potential sites for the introduction of an intra-CH2 domain disulfide bond. Multiple high-resolution structures deposited in the Protein Data Bank were examined. We utilized structural parameters, described in Ref. 30 to arrive at four potential sites: Ala287-Leu306 (mAbW.SEFL2.1), Arg292-Val302 (mAbW.SEFL2.2), Val323-Ile332 (mAbW.SEFL2.3), and Val259-Leu306 (mAbW.SEFL2.4) in the CH2 domain for mutating to Cys. The predicted four sites met the following criteria: (i) away from loops, (ii) not at or near the FcRn binding site, and (iii) partially or fully buried, and (iv) linking two β-strands (Fig. 5). The constructs were cloned, expressed from a CHO transient system, and purified. All of the disulfide-engineered molecules expressed well and fit to the protein A purification platform except mAbW.SEFL2.4. The mAbW.SEFL2.4 antibody could not be purified using protein A and therefore protein G was utilized during the purification step. The disulfide linkage of the five constructs, mAbW.SEFL2.0, mAbW.SEFL2.1, mAbW.SEFL2.2, mAbW.SEFL2.3, and mAbW.SEFL2.4, were evaluated by peptide mapping with the endoprotease Lys-C under non-reducing conditions. To cleave the mAb under non-reducing conditions, a high guanidine hydrochloride concentration (∼5.6 m) was used to disrupt the strong non-covalent interactions. Lys-C was chosen due to its relatively high activity in high concentration guanidine hydrochloride solution. The denaturing step was performed under mildly acidic conditions in the presence of N-ethylmaleimide to block free Cys residues, and therefore prevent disulfide scramble. The non-reduced Lys-C digests of the mAb were analyzed by on-line LC-ESI-MS/MS. As an example, Fig. 6 shows the non-reduced Lys-C peptide maps of mAbW.SEFL2.0 (top trace in red) and parental mAbW.IgG1 (bottom trace in blue). mAbW.SEFL2.0 contains two mutated cysteines, L242C and K334C. The two-engineered cysteines formed a new intra-CH2 domain disulfide bond (shown as the highlighted disulfide-linked peptides in Fig. 6). All the other native cysteines in mAbW.SEFL2.0 formed the exact same disulfide bonds as the parental mAbW.IgG1. Because the highlighted peptides contained multiple cysteines, the disulfide connectivity of the two-engineered cysteines (L242C and K334C) could not be unambiguously assigned. Therefore, those two peptides were collected and further cleaved with the protease Glu-C (supplemental Fig. S3). By reducing the complex peptides to ones containing a single L242C and K334C, respectively, the connectivity between the two engineered cysteines was correctly assigned. Of the five designed molecules, four had the correct disulfide linkage as designed without disruption of the native disulfide bonds (e.g. CH2 intradomain and hinge disulfide connectivity). The mAbW.SEFL2.4 disulfide engineered variant affected the native disulfide linkages (supplemental Fig. S4). Further examination of the structure suggested that the mAbW.SEFL2.4 site is too close to the existing native intradomain disulfide bond in the CH2 domain, which could be a potential reason for the observed heterogeneity in the non-reduced Lys-C peptide mapping analysis. Note that Protein A could not be used to purify this analog, further indicating a significant structural change at the CH3 and possibly in the CH2 domain.
FIGURE 5.
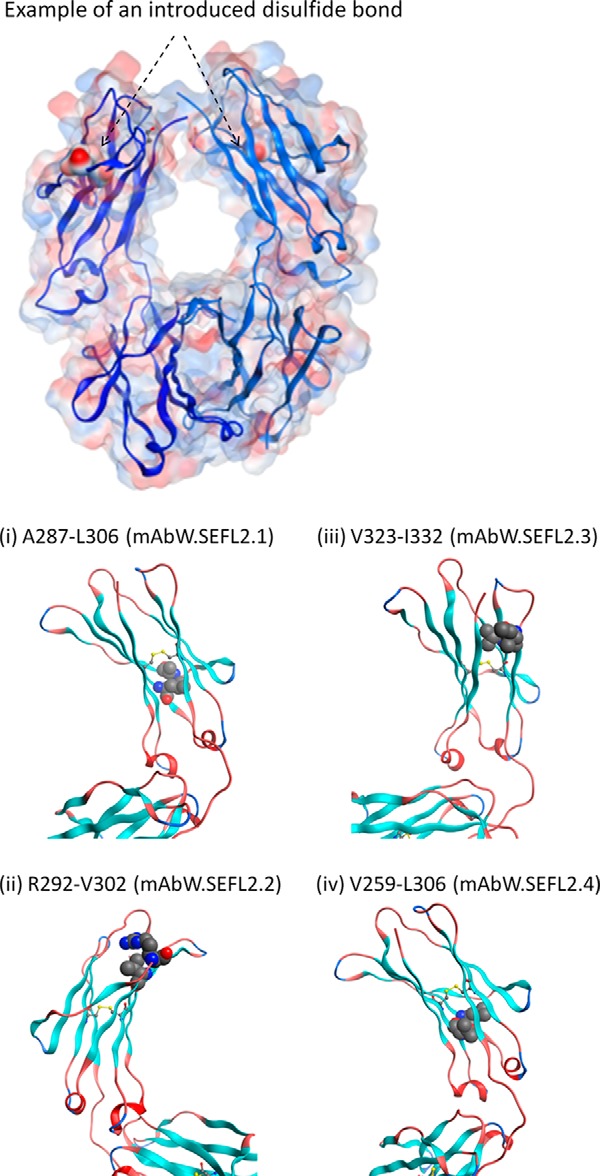
Top panel, transparent surface representation of Fc with an example of an introduced disulfide bond. The residue pair (Ala287-Leu306) that is mutated to Cys are shown in solid surface representation with an arrow indicating its location. Note that Fc is a homodimer. Bottom panel, ribbon representation of the CH2 domain showing the predicted 4 sites for engineering the disulfide bond to improve stability. The sites were identified based on structural parameters such as distance between Cα-Cα and Cβ-Cβ atoms (described in Ref. 30), solvent exposure (prefer buried sites), secondary structure (avoid loops), and away from FcRn binding sites. The Fc and CH2 domain coordinates are derived from Protein Data Bank entry 1L6X, a high resolution (1.65 Å) crystal structure of Fc fragment of Rituximab (39). The disulfide modeling as well as the generation of figures shown here were carried out using the Molecular Operating Environment software (Chemical Computing Group Inc.).
FIGURE 6.

LC-ESI-MS/MS analysis of Lys-C digests of mAbW.SEFL2.0 (top trace in red) and parental mAbW.IgG1 (bottom trace in blue) under non-reducing conditions. The two engineered cysteines, L242C and K334C, formed an intra-CH2 domain disulfide bond involving the highlighted peptides. To confirm the new disulfide bond by reducing the complex peptides to that containing a single L242C and K334C, the two highlighted peptides were collected and further cleaved with Glu-C protease. The LC-ESI-MS/MS analysis of Glu-C digest of the two disulfide linked peptides are shown in supplemental Fig. S3.
To assess whether the engineered disulfide bond is buried, two of the four disulfide engineered molecules (mAbW.SEFL2.1 and mAbW.SEFL2.2) were subjected to thioether stress (pH 9.1, 57 °C for 2 days). The study revealed thioether modifications for the disulfide bonds connecting the heavy and light chains, but not at the introduced engineered disulfide bond (thioether linkage level was about 14 to 16% for both disulfide engineered molecules and the IgG control). The data suggest that the engineered disulfide bond is buried as designed and that its addition did not dramatically alter the conformation of the protein, as there was little difference in the exposure of residues that are buried in the parental molecule. The thioether level is about 14–16% at the heavy-light chain or hinge disulfide bonds for both parental and the disulfide engineered molecules.
Differential Scanning Calorimetry of Disulfide Engineered Antibodies
The CH2 domain melting temperature (Tm) for the disulfide engineered N297G molecules was determined by differential scanning calorimetry (Fig. 7). The CH2 domain Tm of the disulfide-engineered antibodies, mAbW.SEFL2.1, mAbW.SEFL2.2, and mAbW.SEFL2.3, is much higher (79 °C) than that of parental mAbW.IgG1 (71 °C) or N297G mutation (mAbW.SEFL1.1) alone (66 °C) (Table 4). In other words, introduction of the buried disulfide bond in the N297G scaffold resulted in an increase of ∼13 °C in melting temperature. It must be noted that for the disulfide-engineered molecules, Fab co-melting along with CH2 domain makes it difficult to quantitatively estimate the improvement in Tm. However, it is clear that the introduced disulfide significantly improved the thermal stability of aglycosylated IgG, with the CH2 transition occurring near the melting temperature for the Fab domain.
FIGURE 7.
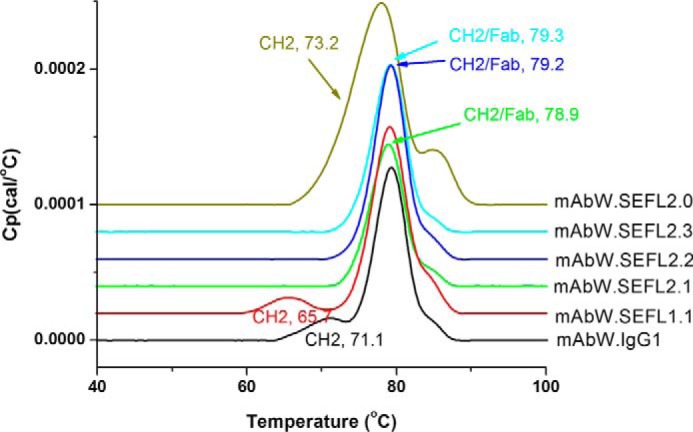
DSC profiles of mAbW.SEFL2.0, mAbW.SEFL2.1, mAbW.SEFL2.2, and mAbW.SEFL2.3 along with mAbW.SEFL1.1 and mAbW.IgG1 controls in A52SuT buffer.
TABLE 4.
Apparent CH2 or CH2/Fab domain thermal transition temperatures of the SEFL samples in A52SuT
| CH2 domain parameter | mAbW.IgG1 | mAbW.SEFL1.1 | mAbW.SEFL2.1 | mAbW.SEFL2.2 | mAbW.SEFL2.3 |
|---|---|---|---|---|---|
| Tm (°C) | 71.1 | 65.7 | 78.9 | 79.2 | 79.3 |
In Vivo Rat and Cynomolgus Monkey PK Study
To assess the impact of the mutations made to improve thermal stability on the PK, rat (n = 3; Fig. 8a) and cynomolgus monkey (n = 2; Fig. 8b) PK studies were carried out. The rat PK profiles of the SEFL antibodies were determined in adult Sprague-Dawley rats (n = 3) by subcutaneous injection at 5 mg/kg and as described above. Of the three disulfide engineered scaffolds, SEFL2.3 had higher clearance and shorter half-life (Fig. 8a). Based on this observation, the SEFL2.3 was not considered further. The PK profiles of the SEFL2.1 and SEFL2.2 antibodies were determined in the cynomolgus monkey (n = 2) by subcutaneous injection at a 5 mg/kg dose. Serum samples were collected pre-dose and 0.5, 2, 8, 24, 48, 96, 168, 336, 504, 672, 840, 1008, 1176, and 1344 h post-dose. Fig. 8b depicts the PK profiles of different SEFL antibodies in cynomolgus monkeys. The newly engineered disulfide bond molecules, mAbW.SEFL2.1 and mAbW.SEFL2.2, have better PK profiles compared with the one identified through the literature (mAbW.SEFL2.0) (29). The mAbW.SEFL2.2 also has slightly better PK attributes as compared with the parent mAbW.IgG1 antibody. The AUC of mAbW.SEFL2.2 and parent mAbW.IgG1 antibodies were 1337 and 1037 μg × day/ml, respectively. The clearance for the mAbW.SEFL2.2 was 0.159 ml/h/kg, slightly lower than that of the parent mAbW.IgG1, which is 0.201 ml/h/kg. The mAbW.SEFL2.0 had the lowest exposure with AUC of 595 μg × day/ml and a clearance rate around 0.351 ml/h/kg, which is the highest among all the SEFL antibodies. The mAbW.SEFL2.1 and mAbW.SEFL1.1 have better PK attributes than mAbW.SEFL2.0, but have slightly lower exposures than that of the mAbW.SEFL2.2 and parent mAbW.IgG1 (see Table 5). This strongly suggests that the second generation SEFL molecules have not only improved conformational and thermal stability, but that they have improved PK in the rat and cynomolgus monkey (and similar PK in mouse; supplemental Fig. S5) animal models tested when compared with the previously available aglycosylated molecules. It must be noted that the observation made for the N297G variant here is consistent with a recent study that reported aglycosylated antibodies have PK profiles similar to that of the wild type in cynomolgus monkey (14).
FIGURE 8.

a, PK profiles of second generation SEFL antibodies were determined following subcutaneous administration to Sprague-Dawley rats (n = 3) at 5 mg/kg. Antibodies concentrations were measured over the 6-week study by sandwich ELISA with an LLOQ of 10 ng/ml. b, PK profiles of SEFL antibodies were determined following subcutaneous administration to cynomolgus monkeys (n = 2) at 5 mg/kg. Antibodies concentrations were measured over the 8-week study by sandwich ELISA with an LLOQ of 10 ng/ml.
TABLE 5.
Cynomolgus monkey PK parameters
| 5 mg/kg SC dosing | T1/2 | AUC | CL/F |
|---|---|---|---|
| Day | μg × day/ml | ml/h/kg | |
| mAbW.IgG1 | 17 | 1,037 | 0.201 |
| mAbW.SEFL1.1 | 14 | 784 | 0.286 |
| mAbW.SEFL2.0 | 7 | 595 | 0.351 |
| mAbW.SEFL2.1 | 12 | 1,006 | 0.207 |
| mAbW.SEFL2.2 | 16 | 1,337 | 0.159 |
Molecule Assessment of Disulfide-engineered Antibodies
To further test if the second generation SEFL molecules (SEFL 2.1 and 2.2) provide viable scaffold on which to build therapeutic candidates, they were assessed in production yield and biophysical properties. The molecule assessments results (Table 6 and supplemental Fig. S1) show that both molecules had good expression levels and good yield during the production, as well as the higher thermal stability discussed above. Both molecules have CH2 melting temperatures around 79 °C and acceptable viscosities in formulation buffer (A52Su: 10 mm acetic acid, 9% sucrose, pH 5.2). The CEX profiles of SEFL2.1 and -2.2, consistent with aforementioned observation on N297G, did not reveal a tailing peak unlike the N297Q or -A (supplemental Fig. S6).
TABLE 6.
Summary of molecule assessment results
| Parameter | mAbW.SEFL2.1 | mAbW.SEFL2.2 |
|---|---|---|
| Percent main peak in A52Sua | 98.1 | 99.5 |
| CH2/Fab Tm (°C) in A52SuTb | 78.9 | 79.2 |
| Viscosity @150 mg/ml (cP) in A52SuTb | 8.4 | 7.1 |
| Percent main peak in PBS buffera | 97.1 | 98.5 |
a Percent main peak or purity as determined by the size exclusion chromatography. A52Su, 10 mm acetic acid, 9% sucrose, pH 5.2.
b A52SuT, 10 mm acetate, 9% sucrose, 0.004% polysorbate 20, pH 5.2.
Discussion
The purpose of this effort was to generate a stable effector functionless (SEFL) molecule that could be used to generate therapeutic proteins. In other words, to create an IgG scaffold with no residual effector function, whereas minimizing the effect of removal of the sugar moieties on PK and stability, and minimizing heterogeneity of the protein, including in the hinge disulfide bonds formed. Although IgG2 has significantly less effector function than IgG1, low levels of ADCC have recently been reported (4–6). Due to differences in the hinge region disulfide architecture, the IgG2 molecules can also exist in three different isoforms, resulting in innate heterogeneity of the protein that can occur in vivo, but that must be controlled throughout production (8, 31). The IgG2s also have decreased thermal stability relative to the IgG1s (10). Because of this heterogeneity and decreased stability, the IgG1 subtype was chosen as the basis for our protein engineering efforts.
Flexibility in the loop containing the Asn297 glycosylation site was identified as a key attribute of the CH2 domain, through comparison of multiple IgG1 structures. Glycine was identified as the amino acid that best retains the flexibility seen in the glycosylated molecule. This is in contrast to the Ala or Gln that is usually substituted for Asn, based on the similarities of the individual amino acids, in other published aglycosylated versions of the IgGs. Based on this flexibility and conformational analyses, variants were made with N297G, N297A, or N297Q substitutions; the latter two have been extensively described in the literature (32–34). When these molecules were expressed, purified, and tested for stability, the N297G analog had the highest melting temperature of these three; it also fit the platform purification process the best. Thus, the use of molecular modeling resulted in a superior scaffolding. However, as expected, the thermal melting transition, and thus the stability, of the CH2 domain in all three constructs was significantly lower than that of the glycosylated molecule, although the N297G was the most stable of the three. Conformational stability of protein therapeutics is an important consideration, because they need to be generated at a high expression level in the cell, purified at high yield, and maintain stability during storage with expected shelf lives of up to 2 years (35). One way to increase the stability of a protein, in lieu of the carbohydrate, is to engineer in a disulfide bond (or Cys clamp). However, adding more Cys to protein that already has disulfide bonds can result in disulfide scrambling (or mispairing), with the incorrect disulfide bonds formed during the protein production. This is especially a concern with the CH2 domain of the IgGs, where the hinge disulfides are proximal and solvent exposed, and indeed this is what results in the disulfide isoforms seen with the IgG2, as mentioned above. Adding Cys to the interior of the protein carries its own liabilities, as there is the possibility of perturbing the hydrophobic core structure of the protein and changing not only the stability but also the interactions between the surface of the IgG and other molecules. The use of a Cys clamp to improve stability of the aglyco CH2 domain has been reported previously, and so this was the first disulfide bond added to the N297G (mAbW.SEFL1.1) molecule. The resulting protein (mAbW.SEFL2.0) does indeed have increased stability, but when tested for PK in several preclinical animal models, it was found to have lower exposure and higher clearance in rat. Because the goal of this work was to create an improved “universal” effector functionless scaffolding, molecular modeling was again used to identify four sites where adding a Cys clamp should be less disruptive. Several second generation SEFL molecules were then made and their stability, manufacturability, and PK properties were assessed. Three of the four formed the appropriate disulfide bonds as designed, did not impact the native disulfide bonds in the hinge region as well as at the CH2 domain, and could be purified with the platform process.
Once again, the analogs based on molecular modeling proved to be the superior protein, showing PK properties similar to the parent glycosylated molecule, with the highest stability among the proteins lacking carbohydrate. The A287C,N297G,L306C (mAbW.SEFL2.1) and R292C,N297G,V302C (mAbW.SEFL2.2) proteins had improved stability, decreased clearance, and longer half-lives in both the rat and cynomolgus monkey when compared with the N297G (mAbW.SEFL1.1) alone. Indeed, the properties of these second-generation SEFL molecules approach that of the glycosylated IgG1 protein, but have no effector function and do not bind the Fcγ receptors. Thus by using a combination of molecular modeling and empirical assessment, we have been able to create an analog of the human IgG1 that is stable, and lacks effector function, a true SEFL molecule. Having this scaffolding in our toolbox should enable the generation of improved therapeutic proteins for use in situations where the effector function of the IgG1 is not desired.
Experimental Procedures
Computational Analysis and Engineering of Disulfide Bonds
Antibody and Fc (fragment crystallizable) crystal structures determined at 2.5-Å resolution or better were obtained from the PDB (26). Molecular operating environment modeling software from the Chemical Computing Group, Canada, was used to extract coordinates for the backbone atoms, superimpose CH2 domain structures, and analyze flexibility and conformation. To improve stability of aglycosylated antibodies, in particular the N297G variant, intradomain disulfides were engineered at the CH2 domain. The positions for the introduction of a Cys clamp (disulfide bond) were identified utilizing literature (29) and further structural analysis based on disulfide bond parameters described in Ref. 30. The engineered intradomain Cys clamps are listed in Table 1. Note that the positions identified for mutation to Cys are either fully or partially buried (solvent inaccessible) and are away from loops and the FcRn binding site.
Generation of SEFL Monoclonal Antibodies (mAbs)
The SEFL antibody heavy chain and light chain gene DNAs were generated by site-directed polymerase chain reaction mutagenesis from hybridoma-derived antibody DNAs. The SEFL DNAs were cloned into mammalian expression vectors that were then co-transfected into CHO cells using a standard electroporation method. Post-transfection, the cell lines were recovered in selective media to >90% viability and screened for levels of antibody expression. To provide representative material for further evaluation, the recovered cell lines were run in a bioreactor production process.
The SEFL mAbs were recovered from the clarified CHO cell condition media using a three-step process. First, the mAbs were affinity captured using MabSelect SuRe (36) (GE Healthcare Life Sciences) by directly loading the conditioned media on the column, followed by a wash using Dulbecco's phosphate-buffered saline and elution using 100 mm sodium acetate, pH 3.6. The elution pool was then brought to pH 5.0 using Tris base and then loaded onto an SP-HP column (GE Healthcare Life Sciences) and washed with buffer A (20 mm acetic acid, pH 5.0) followed by elution using a linear gradient to 20 mm acetic acid, 600 mm NaCl, pH 5.0. Finally, the mAbs were dialyzed into sodium acetate buffer (A52Su: 10 mm acetic acid, 9% sucrose, pH 5.2) for long-term stability.
PK Assay to Measure Total Human IgG in Rat or Cynomolgus Monkey Serum by ELISA
To measure the human antibody in Sprague-Dawley rat or cynomolgus monkey serum samples, a half-area black plate (Corning 3694, Corning, NY) was coated with 2 μg/ml of anti-human Fc antibody (clone 1.35.1, Amgen Inc., Thousand Oaks, CA) in PBS and then incubated 12–24 h at 4 °C. The plate was then washed and blocked with I-BlockTM (Life Technologies) overnight at 4 °C. The standards and quality control samples (QC) were prepared in rat or cynomolgus monkey serum, and serum samples were diluted in naive rat or cynomolgus monkey serum if dilutions were required. The standards, QCs, and samples were then diluted 1:20 in a buffer containing PBS, 1 m NaCl, 0.5% Tween 20, and 1% bovine serum albumin buffer (5% final rat or cynomolgus monkey serum was used in the assay). The plate was washed three times with ∼200 μl of 1× KPL buffer (KPL, Gaithersburg, MD), and subsequently 50-μl samples of diluted standards, QCs, and samples were transferred into the anti-human Fc antibody-coated plate and incubated for 1.5 h at room temperature (∼25 °C). After washing the plate, 50 μl of 100 ng/ml of an orthogonal anti-human Fc antibody horseradish peroxidase (HRP) conjugate (clone 21.1 Amgen Inc.) in I-BlockTM containing 5% BSA was added and incubated for 1.5 h at room temperature (∼25 °C). The plate was washed six times with ∼200 μl of 1× KPL wash buffer, followed by addition of 50 μl of Pico substrate (ThermoFisher, Rockford, IL), and the chemiluminescent signal was measured using a SpectraMax (Molecular Devices, Sunnyvale, CA) plate reader. Serum concentration data were analyzed using non-compartmental methods with WinNonLin® (Enterprise version 5.1.1, 2006, Pharsight® Corp., Mountain View, CA).
Differential Scanning Calorimetry
The DSC experiments were done on a MicroCal Capillary VP-DSC (Piscataway, NJ) in which temperature differences between the reference and sample cell were continuously measured, and calibrated to power units. This data channel is referred to as the DP signal, or the differential power between the reference (buffer) and sample cell. Baseline subtraction was performed by measuring buffer (sample cell) against buffer (reference cell), then further analyzed using ORIGIN 7 software. The samples were heated from 4 to 110 °C at a heating rate of 60 °C/h. The pre-scan was 15 min and the filtering period was 10 s. The concentration used in the DSC experiments was ∼1 mg/ml. In cases where two domains were co-melting and deconvolution was possible (e.g. SEFL2.0), we employed the MicorCal Origin 7 (Malvern Inc.) built-in non-2-state model.
LC-ESI-MS/MS Analysis of Lys-C Enzymatic Digest of Non-reduced mAb
To denature the antibodies under non-reducing conditions, 3 μl of antibody sample (∼200 μg) was mixed with 7 μl of denaturing buffer consisting of 8 m guanidine hydrochloride, 10 mm N-ethylmaleimide, 0.1 m sodium acetate, pH 5.0, and the mixture was incubated at 37 °C for 3 h. The denatured antibody mixture was then diluted into 400 μl of digestion buffer consisting of 4 m urea, 20 mm NH2OH, 0.1 m Tris, pH 7.0. Ten μg of Lys-C was added into the solution to achieve a protein to enzyme ratio of ∼20:1 (w/w), and the mixture was incubated at 37 °C overnight. To quench the reaction, trifluoroacetic acid (TFA) was added to the sample digests to a final concentration of 0.1%. The non-reduced Lys-C digest was separated using a reversed-phase column (ACQUITY UPLC BEH300 C4 Column, 1.7 μm, 2.1 × 150 mm). The temperature of the column was maintained at 75 °C. Mobile phase A was 0.1% (v/v) TFA in water and mobile phase B was 0.1% (v/v) TFA and 90% acetonitrile in water. Digested samples (∼30 μg) were injected and separated using a gradient (hold at 2% B for 5 min, 2–22% B for 25 min, then 22–42% B within 95 min). A flow rate of 200 μl/min was employed.
The on-line LC-ESI-MS/MS analyses were performed using a Waters Acuity ultra performance liquid chromatography (UPLC) system (Milford, MA) directly coupled with a Thermo Scientific LTQ Orbitrap Velos high resolution mass spectrometer equipped with an ESI source (San Jose, CA). The ESI source voltage was set at 4.5 kV, and the capillary temperature was set at 250 °C. The mass spectrometer was set up to acquire one high-resolution full scan at 60,000 resolution (at m/z 400), followed by five concurrent data-dependent MS/MS scans of the top five most abundant ions. Peptides were identified using Mass Analyzer, an in-house developed software application, which correlates the experimental tandem mass spectra with the theoretical tandem mass spectra generated from known peptide sequences.
Thioether Modification Analysis
Stressed thioether samples were prepared by incubating antibodies in glycine/NaOH buffer at pH 9.1 for 2 days at 57 °C. Thioether quantification at the protein level was determined using RP-HPLC and LC-MS as described previously (37). To enable separation of LC, HC, and thioether-linked LC-HC, RP-HPLC was performed on an Agilent 1100 HPLC using C8 column maintained at 75 °C. An online intact mass analysis was conducted on a Waters LCT Premier TOF instrument and the raw data were processed using Masslynx MaxEnt 1 software.
FcRn Binding Analysis
Neonatal receptor (FcRn) binding analysis was carried out on the immobilized CHO huFc surface by BIAcore. 10 nm cyno, human, or mouse FcRn was incubated with 0.1- 2000 nm effector functionless antibodies along with controls in the formulation buffer (20 mm sodium acetate, pH 5.5, 150 mm NaCl, 0.005% P20, 0.1 mg/ml of BSA) for 1 h before injection over the immobilized CHO huFc surface for 3 min at 10 μl/min. The running buffer in the BIAcore instrument was 20 mm sodium phosphate, pH 6.0, 150 mm NaCl, 0.005% polysorbate 20. Following the sample injection, bound cyno, human, or mouse FcRn was regenerated from the huFc surface in PBS, pH 7.4, containing 0.005% polysorbate 20 for 3 min. The signal obtained in the absence of any sample (or antibody) is considered 100% for cyno, human, or mouse FcRn binding. A decreased binding response with increasing concentrations of the antibodies indicated that the antibodies bound to FcRn in solution by blocking the binding of FcRn to the immobilized huFc surface.
Author Contributions
F. W. J., R. S., L. N., and K. G. designed the experiments and coordinated data generation and collection. C. L. and J. Wen carried out the biophysical analyses. H. S-M. did the pharmacokinetic analysis. Q. L. and J. Wypych designed and analyzed mass spectrometry experiments. L. L., K. D., and L. B. carried out the DNA construct generation and stable cell line development. S. M. and K. W. provided protein biochemistry and purification support. S.-Y. H., W. W., and Q. C. did the binding analysis reported in the supplemental section. K. G. carried out the computational analyses and engineering. L. N. and K. G. wrote most of the paper.
Supplementary Material
Acknowledgments
We thank Qingchun Zhang and Gregory Flynn for carrying out thioether linkage analysis on stressed samples to determine stability of the engineered disulfide bonds. We also thank Jeanine Bussiere and Margaret Karow for guidance and support throughout this project.
This work was supported by Amgen. All authors work or have worked at Amgen.

This article contains supplemental Figs. S1–S6.
- ADCC
- antibody-dependent cellular cytotoxicity
- PK
- pharmacokinetics
- cyno
- cynomolgus
- LLOQ
- lower limit of quantification
- DSC
- differential scanning calorimetry
- SEFL
- stable effector functionless
- QC
- quality control
- PDB
- Protein Data Bank
- AUC
- area under curve.
References
- 1.Nelson A. L., Dhimolea E., and Reichert J. M. (2010) Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9, 767–774 [DOI] [PubMed] [Google Scholar]
- 2.Jefferis R. (2009) Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 30, 356–362 [DOI] [PubMed] [Google Scholar]
- 3.Nimmerjahn F., and Ravetch J. V. (2011) FcgammaRs in health and disease. Curr. Top. Microbiol. Immunol. 350, 105–125 [DOI] [PubMed] [Google Scholar]
- 4.Schneider-Merck T., Lammerts van Bueren J. J., Berger S., Rossen K., van Berkel P. H., Derer S., Beyer T., Lohse S., Bleeker W. K., Peipp M., Parren P. W., van de Winkel J. G., Valerius T., and Dechant M. (2010) Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J. Immunol. 184, 512–520 [DOI] [PubMed] [Google Scholar]
- 5.Warncke M., Calzascia T., Coulot M., Balke N., Touil R., Kolbinger F., and Heusser C. (2012) Different adaptations of IgG effector function in human and nonhuman primates and implications for therapeutic antibody treatment. J. Immunol. 188, 4405–4411 [DOI] [PubMed] [Google Scholar]
- 6.Everds N. E., and Tarrant J. M. (2013) Unexpected hematologic effects of biotherapeutics in nonclinical species and in humans. Toxicol. Pathol. 41, 280–302 [DOI] [PubMed] [Google Scholar]
- 7.Jacobsen F. W., Padaki R., Morris A. E., Aldrich T. L., Armitage R. J., Allen M. J., Lavallee J. C., and Arora T. (2011) Molecular and functional characterization of cynomolgus monkey IgG subclasses. J. Immunol. 186, 341–349 [DOI] [PubMed] [Google Scholar]
- 8.Dillon T. M., Ricci M. S., Vezina C., Flynn G. C., Liu Y. D., Rehder D. S., Plant M., Henkle B., Li Y., Deechongkit S., Varnum B., Wypych J., Balland A., and Bondarenko P. V. (2008) Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J. Biol. Chem. 283, 16206–16215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hari S. B., Lau H., Razinkov V. I., Chen S., and Latypov R. F. (2010) Acid-induced aggregation of human monoclonal IgG1 and IgG2: molecular mechanism and the effect of solution composition. Biochemistry 49, 9328–9338 [DOI] [PubMed] [Google Scholar]
- 10.Ito T., and Tsumoto K. (2013) Effects of subclass change on the structural stability of chimeric, humanized, and human antibodies under thermal stress. Protein Sci. 22, 1542–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aalberse R. C., and Schuurman J. (2002) IgG4 breaking the rules. Immunology 105, 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman A. P., Antoniw P., Spitali M., West S., Stephens S., and King D. J. (1999) Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 17, 780–783 [DOI] [PubMed] [Google Scholar]
- 13.Strohl W. R. (2009) Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 20, 685–691 [DOI] [PubMed] [Google Scholar]
- 14.Leabman M. K., Meng Y. G., Kelley R. F., DeForge L. E., Cowan K. J., and Iyer S. (2013) Effects of altered FcgammaR binding on antibody pharmacokinetics in cynomolgus monkeys. MAbs. 5, 896–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hristodorov D., Fischer R., and Linden L. (2013) With or without sugar? (A)glycosylation of therapeutic antibodies. Mol. Biotechnol. 54, 1056–1068 [DOI] [PubMed] [Google Scholar]
- 16.Jung S. T., Kang T. H., Kelton W., and Georgiou G. (2011) Bypassing glycosylation: engineering aglycosylated full-length IgG antibodies for human therapy. Curr. Opin. Biotechnol. 22, 858–867 [DOI] [PubMed] [Google Scholar]
- 17.Mimura Y., Church S., Ghirlando R., Ashton P. R., Dong S., Goodall M., Lund J., and Jefferis R. (2000) The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: properties of a series of truncated glycoforms. Mol. Immunol. 37, 697–706 [DOI] [PubMed] [Google Scholar]
- 18.Shields R. L., Lai J., Keck R., O'Connell L. Y., Hong K., Meng Y. G., Weikert S. H., and Presta L. G. (2002) Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J. Biol. Chem. 277, 26733–26740 [DOI] [PubMed] [Google Scholar]
- 19.Nimmerjahn F., and Ravetch J. V. (2007) The antiinflammatory activity of IgG: the intravenous IgG paradox. J. Exp. Med. 204, 11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naso M. F., Tam S. H., Scallon B. J., and Raju T. S. (2010) Engineering host cell lines to reduce terminal sialylation of secreted antibodies. MAbs. 2, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reusch D., and Tejada M. L. (2015) Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 25, 1325–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aronson R., Gottlieb P. A., Christiansen J. S., Donner T. W., Bosi E., Bode B. W., Pozzilli P., and DEFEND Investigator Group. (2014) Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 37, 2746–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clarke S. J., Smith J. T., Gebbie C., Sweeney C., and Olszewski N. (2009) A phase I, pharmacokinetic (PK), and preliminary efficacy assessment of ALD518, a humanized anti-IL-6 antibody, in patients with advanced cancer. J. Clin. Oncol. 27, 3025 [Google Scholar]
- 24.Keymeulen B., Walter M., Mathieu C., Kaufman L., Gorus F., Hilbrands R., Vandemeulebroucke E., Van de Velde U., Crenier L., De Block C., Candon S., Waldmann H., Ziegler A. G., Chatenoud L., and Pipeleers D. (2010) Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia 53, 614–623 [DOI] [PubMed] [Google Scholar]
- 25.Liu L., Jacobsen R., Everds N., Zhuang Y., Yu Y., Bin, Li N., Clark D., Nguyen M. P., Fort M., Narayanan P., Stevenson R., Narhi L., Gunasekaran K., and Bussiere J. L. (2017) Biological characterization of a stable effector functionless (SEFL) monoclonal antibody scaffold in vitro. J. Biol. Chem. 292, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernstein F. C., Koetzle T. F., Williams G. J., Meyer E. F. Jr., Brice M. D., Rodgers J. R., Kennard O., Shimanouchi T., and Tasumi M. (1978) The protein data bank: a computer-based archival file for macromolecular structures. Arch. Biochem. Biophys. 185, 584–591 [DOI] [PubMed] [Google Scholar]
- 27.Demarest S. J., and Glaser S. M. (2008) Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr. Opin. Drug Discov. Devel. 11, 675–687 [PubMed] [Google Scholar]
- 28.Guddat L. W., Herron J. N., and Edmundson A. B. (1993) Three-dimensional structure of a human immunoglobulin with a hinge deletion. Proc. Natl. Acad. Sci. U.S.A. 90, 4271–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong R., Vu B. K., Feng Y., Prieto D. A., Dyba M. A., Walsh J. D., Prabakaran P., Veenstra T. D., Tarasov S. G., Ishima R., and Dimitrov D. S. (2009) Engineered human antibody constant domains with increased stability. J. Biol. Chem. 284, 14203–14210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sowdhamini R., Srinivasan N., Shoichet B., Santi D. V., Ramakrishnan C., and Balaram P. (1989) Stereochemical modeling of disulfide bridges: criteria for introduction into proteins by site-directed mutagenesis. Protein Eng. 3, 95–103 [DOI] [PubMed] [Google Scholar]
- 31.Wypych J., Li M., Guo A., Zhang Z., Martinez T., Allen M. J., Fodor S., Kelner D. N., Flynn G. C., Liu Y. D., Bondarenko P. V., Ricci M. S., Dillon T. M., and Balland A. (2008) Human IgG2 antibodies display disulfide-mediated structural isoforms. J. Biol. Chem. 283, 16194–16205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnold J. N., Wormald M. R., Sim R. B., Rudd P. M., and Dwek R. A. (2007) The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 25, 21–50 [DOI] [PubMed] [Google Scholar]
- 33.Shields R. L., Namenuk A. K., Hong K., Meng Y. G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., Fox J. A., and Presta L. G. (2001) High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604 [DOI] [PubMed] [Google Scholar]
- 34.Tao M. H., and Morrison S. L. (1989) Studies of aglycosylated chimeric mouse-human IgG: role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 143, 2595–2601 [PubMed] [Google Scholar]
- 35.Liu H. F., Ma J., Winter C., and Bayer R. (2010) Recovery and purification process development for monoclonal antibody production. MAbs. 2, 480–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishihara T., Nakajima N., and Kadoya T. (2010) Evaluation of new affinity chromatography resins for polyclonal, oligoclonal and monoclonal antibody pharmaceuticals. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 2141–2144 [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q., Schenauer M. R., McCarter J. D., and Flynn G. C. (2013) IgG1 thioether bond formation in vivo. J. Biol. Chem. 288, 16371–16382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gunasekaran K., Ramakrishnan C., and Balaram P. (1996) Disallowed Ramachandran conformations of amino acid residues in protein structures. J. Mol. Biol. 264, 191–198 [DOI] [PubMed] [Google Scholar]
- 39.Idusogie E. E., Presta L. G., Gazzano-Santoro H., Totpal K., Wong P. Y., Ultsch M., Meng Y. G., and Mulkerrin M. G. (2000) Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J. Immunol. 164, 4178–4184 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.