

Abstract
Binding of ICAP1 (integrin cytoplasmic domain-associated protein-1) to the cytoplasmic tails of β1 integrins inhibits integrin activation. ICAP1 also binds to KRIT1 (Krev interaction trapped-1), a protein whose loss of function leads to cerebral cavernous malformation, a cerebrovascular dysplasia occurring in up to 0.5% of the population. We previously showed that KRIT1 functions as a switch for β1 integrin activation by antagonizing ICAP1-mediated inhibition of integrin activation. Here we use overexpression studies, mutagenesis, and flow cytometry to show that ICAP1 contains a functional nuclear localization signal and that nuclear localization impairs the ability of ICAP1 to suppress integrin activation. Moreover, we find that ICAP1 drives the nuclear localization of KRIT1 in a manner dependent upon a direct ICAP1/KRIT1 interaction. Thus, nuclear-cytoplasmic shuttling of ICAP1 influences both integrin activation and KRIT1 localization, presumably impacting nuclear functions of KRIT1.
Keywords: cell compartmentalization, cerebral cavernous malformation, integrin, KRIT1 (Krev interaction trapped), protein targeting, ICAP1 (integrin cytoplasmic domain-associated protein)
Introduction
Integrins are heterodimeric trans-membrane adhesion receptors that transduce signals that control complex cell functions, such as survival, proliferation, tissue formation, and homeostasis (1). Integrin signaling is mediated via interactions between their short cytoplasmic tails and cytoplasmic signaling and scaffolding proteins (2, 3). In addition, binding of proteins to the integrin β tails can induce conformational changes in the integrin extracellular domains that alter integrin affinity for extracellular ligands (4). This “inside-out” signaling mechanism alters the integrin activation state and is a key regulator of cell adhesion, cell spreading, cytoskeletal rearrangement, and adhesion signaling.
Integrin activation is triggered by the direct binding of the band four-point-one, ezrin, radixin, moesin (FERM)2 domain-containing proteins talin and kindlin to two conserved NPXY motifs in the integrin β cytoplasmic tail (5, 6). Notably, negative regulators that bind the integrin cytoplasmic domain and prevent talin or kindlin binding can counteract talin and kindlin-mediated integrin activation (7, 8). Here we report our investigation of one such negative regulator, integrin cytoplasmic domain-associated protein-1 (ICAP1). ICAP1 is a phosphotyrosine binding (PTB) domain-containing protein that interacts selectively with the β1 integrin cytoplasmic domain through a canonical PTB domain/NPXY amino acid motif interaction and inhibits β1 integrin activation by competing with talin and kindlin for binding to the β1 integrin tail (7, 9).
The other well characterized ICAP1-binding protein is Krev interaction trapped-1 (KRIT1) (10, 11). KRIT1 is a multidomain, 736-amino acid protein containing an N-terminal Nudix domain, three NPX(Y/F) motifs, an ankyrin repeat domain, and a FERM domain (12). Notably, loss-of-function (typically nonsense) mutations in KRIT1 are associated with cerebral cavernous malformation (CCM), a common dysplasia of the vasculature (13, 14). CCMs consist of clusters of thin-walled, dilated blood vessels that form mulberry-shaped lesions in the brain (15). CCMs have been reported in up to 0.5% of the population (16) and are strongly associated with hemorrhagic stroke, seizure, epilepsy, and other focal neurological outcomes. CCMs are also caused by loss of function mutations in CCM2 or CCM3 genes (17), and the CCM2 protein can form the hub of a multiprotein KRIT1-CCM2-CCM3 complex: the CCM complex (12, 18, 19). Loss of KRIT1, CCM2, or CCM3 proteins is therefore directly associated with focal neurological defects, stroke, and vascular abnormalities.
Although not mutated in CCMs, ICAP1 is linked to the CCM complex through its interaction with KRIT1 (7, 20). ICAP1 binds KRIT1 in a bidentate mode, recognizing two regions: the highly conserved RR region and the first of the three KRIT1 NPX(Y/F) motifs (7). Importantly, the same binding site on ICAP1 is used to interact with either KRIT1 or integrin β1. By binding ICAP1, KRIT1 inhibits ICAP1 binding to integrins, relieving ICAP1-mediated suppression of integrin activation (7). Consistent with this, increased integrin activation is observed when increasing amounts of KRIT1 are available to bind to ICAP1 (7). In endothelial cells, KRIT1 also appears to stabilize the ICAP1 protein, so KRIT1 loss leads to decreased ICAP1 levels and consequently increased β1 integrin activation (20). In addition to its role with ICAP1, KRIT1 has also been linked to several other important signaling pathways, including Rho/ROCK (21–23), Notch/PI3K (24), reactive oxygen species/SOD2/AKT (25), and β-catenin (26).
ICAP1 and KRIT1 shuttle between the nucleus and cytoplasm, and putative nuclear localization signals (NLS) have been identified in both proteins (27–29). However, very little is understood about shuttling dynamics or its cellular significance. One hypothesis is that KRIT1 and ICAP1 regulate each other by sequestering the partner inside the nucleus, thus preventing interaction with cytoplasmic or membrane proteins, such as β1 integrin (30), but nuclear roles for KRIT1 and ICAP1 are also possible (27, 31). Here we report that ICAP1 contains a functional NLS, which is necessary and sufficient for localization to the nucleus. Deletion or mutation of the ICAP1 NLS prevents nuclear localization, and cytoplasmic ICAP1 is more effective at suppressing the activation of integrin β1. Notably, in co-expression studies, we also find that, by binding KRIT1, ICAP1 drives KRIT1 localization to the nucleus. Thus, nuclear-cytoplasmic shuttling of ICAP1 can influence both integrin activation and KRIT1 localization, presumably influencing the nuclear functions of KRIT1.
Results
The Integrin-binding PTB Domain of ICAP1 Is More Effective Than Full-length ICAP1 at Suppressing β1 Integrin Activation
Consistent with the role of ICAP1 as a direct inhibitor of β1 integrin activation (32–35), we previously demonstrated that expression of green fluorescent protein (GFP)-tagged ICAP1 PTB domain (ICAP1 residues 49–200, ICAP1PTB) suppresses β1 integrin activation and that this requires the formation of a typical PTB domain-ligand interaction between integrin β1 and ICAP1 (7). However, ICAP1 contains an additional 48 residues N-terminal to the PTB domain (Fig. 1A), and here we investigated whether this region modulates ICAP1-mediated suppression of integrins. We accomplished this by expressing GFP, GFP-tagged full-length ICAP1 (GFP-ICAPFL), or GFP-ICAP1PTB constructs in CHO-α5β1 cells and assessing integrin activation using a well validated flow cytometric assay (36). CHO-α5β1 cells are Chinese hamster ovary (CHO) cells that stably express chimeric αIIbα5β3β1 integrins. These integrins contain the cytoplasmic domains of α5β1 and thus are regulated like normal β1 integrins (37) but have the extracellular and transmembrane domains of αIIbβ3. This allowed us to assess activation by measuring binding of the ligand-mimetic anti-αIIbβ3 antibody PAC1 (36). PAC1 binding was normalized to surface integrin expression using an anti-αIIbβ3 integrin antibody that binds in an activation-independent manner, and cells were gated to have comparable GFP intensities. As shown in Fig. 1B, we validated the assay by showing that increasing expression of the GFP-tagged integrin activator talin head (5) increased β1 integrin activation, whereas GFP-ICAP1PTB produced a dose-dependent suppression of activation, as reported previously (7). Notably, although GFP-ICAP1FL also triggered dose-dependent inhibition of activation, it was less effective at repressing integrin β1 activation than GFP-ICAP1PTB (Fig. 1B). As expected, the ability of ICAP1FL to influence integrin activation depended on an intact β1-binding site because the integrin-binding defective ICAP1 mutant (ICAP1 L135A/I138A/I139A, ICAP1FL/) (7) had no impact on integrin activation (Fig. 1A).
FIGURE 1.
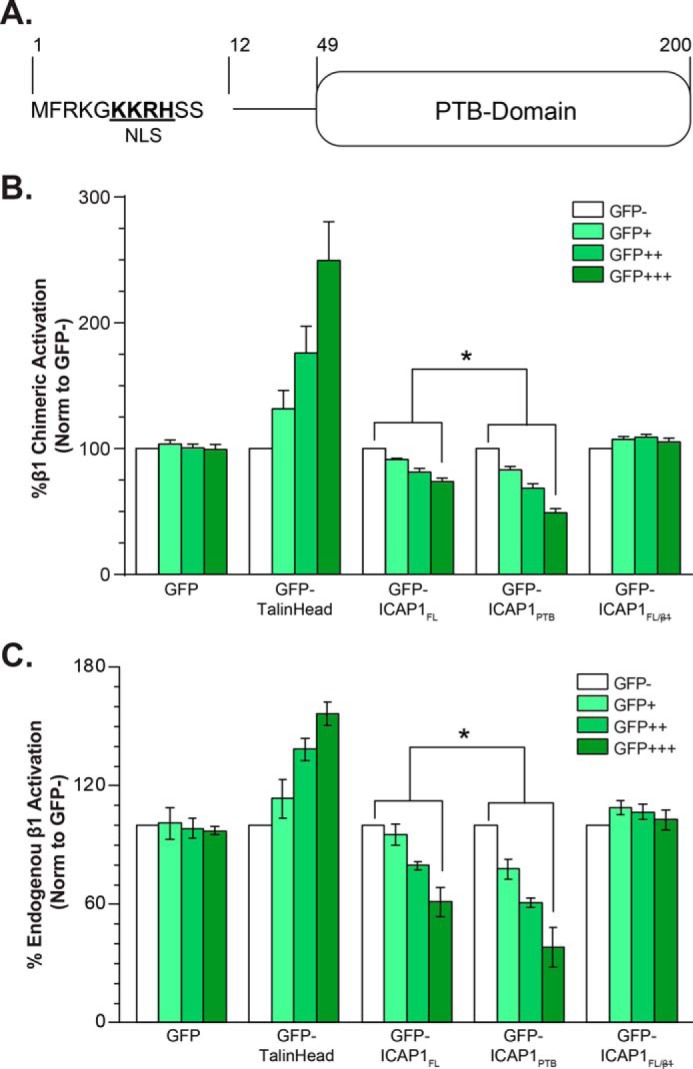
ICAP1FL is less efficient than ICAP1PTB at repressing β1 integrin activation in CHO cells. A, schematic of ICAP1 noting the NLS sequence and PTB-domain boundaries. B and C, CHO-α5β1 cells (B) or CHO cells (C) were transfected with GFP, GFP-ICAP1FL, GFP-ICAP1PTB, the integrin binding-defective mutant GFP-ICAP1FL/, or GFP-talin head, and the activation of stably expressed chimeric αIIbα5β3β1 (B) or endogenous α5β1 (C) integrins was assessed by flow cytometry. Gating on cell populations with different GFP intensities permits analysis of dose-dependent effects. The activation index of each gated population was expressed as the percentage of that in the GFP-negative population. Results are presented as mean percentage ± S.E. (error bars) of the activation index in the untransfected (GFP−) population from 4–8 independent experiments. *, p ≤ 0.01 as determined by a two-way ANOVA with Tukey's correction for multiple tests.
We have previously shown that the ICAP1 PTB domain regulates chimeric αIIbα5β3β1 integrins in the same way as endogenous α5β1 integrins (7). However, to validate the results in Fig. 1B, we also assessed the effect of ICAP1 and ICAP1 mutants on the activation of endogenous CHO cell α5β1 using a recombinant fragment of fibronectin as a reporter (36). With endogenous integrin (Fig. 1C), we obtained results very similar to those with chimeric integrins (Fig. 1B), and notably again full-length ICAP1 produced significantly less inhibition of integrin activation than did the ICAP1 PTB domain.
The preceding data indicate that the N-terminal portion of ICAP1 reduces the ability of the ICAP1 PTB domain to suppress integrin activation. It has been suggested that ICAP1 can adopt a closed conformation where the N-terminal and C-terminal portions of the protein associate, masking the integrin-binding site in the ICAP1 PTB domain (38). Deletion of the ICAP1 N-terminal region might disrupt the inhibitory closed conformation, resulting in increased integrin binding, and this could explain why the ICAP1PTB is a more potent inhibitor of β1 integrin activation than ICAP1FL. To test this hypothesis, we compared ICAP1FL and ICAP1PTB binding to integrin β1. However, pull-down binding assays using cell lysates expressing GFP-ICAP1 constructs and recombinant His-tagged integrin cytoplasmic tails immobilized on beads (39) indicated that there are no major differences between the relative binding of ICAP1FL and ICAP1PTB to integrin β1 tails (Fig. 2, A and B). The αIIb beads served to control for background binding. As expected, ICAP1FL/ did not bind to β1 tails. Our data therefore suggest that deletion of the N terminus of ICAP1 does not alter integrin binding, but to further explore this question, binding curves were determined by incubating increasing amounts of purified recombinant ICAP1 (either FL or PTB) with constant levels of immobilized integrin tails. Beads were washed, and the amount of ICAP1 bound was determined by immunoblot analysis (Fig. 2C). The bands were quantified, and curve-fitting analysis demonstrated no significant increase in ICAP1PTB affinity for integrin β1 tail compared with ICAP1FL (Fig. 2D). The 95% confidence interval of KD for ICAP1PTB-β1 was 0.17–0.44 μm compared with 0.12–0.31 μm for ICAP1FL-β1. These data establish that ICAP1FL retains integrin-binding activity that can be disrupted by mutations in the PTB domain. They further suggest that the N-terminal portion of ICAP1 influences ICAP1-mediated suppression of integrin activation in ways other than directly altering integrin binding affinity.
FIGURE 2.

ICAP1FL and ICAP1PTB bind integrin β1 tails equally well. A, pull-down of GFP-tagged ICAP1 constructs from CHO cell lysates with purified recombinant αIIb or β1 integrin tails. Tail loading was assessed by Coomassie Blue staining. The input lane indicates 5% of input lysate. B, ICAP1 binding to purified recombinant αIIb and β1 tails was quantified and expressed as a percentage of input (mean ± S.E. (error bars); n = 4). C, increasing amounts of purified ICAP1FL or ICAP1PTB were pulled down with His-tagged integrin tails (either αIIb or β1a) immobilized on beads. Protein was detected by immunoblot analysis. D, a binding curve was generated by quantifying the bands, normalizing to the input control, and plotting the relative signal versus the input of purified protein (mean ± S.D. (error bars), n = 3).
GFP-ICAP1FL and GFP-ICAP1PTB Show Differential Localization to the Nucleus
To investigate potential mechanisms by which the N-terminal region of ICAP1 might regulate ICAP1 function, we compared the subcellular localization of GFP-ICAP1FL and GFP-ICAP1PTB. CHO cells were transfected with either GFP or GFP-tagged ICAP1, plated on fibronectin-coated coverslips, fixed, stained with DAPI (to identify the nucleus), and examined by fluorescence microscopy. It was immediately apparent that GFP-ICAP1FL and GFP-ICAP1PTB differ in their localization to the nucleus (Fig. 3A). Whereas GFP exhibited a fairly even distribution between the nucleus and the cytoplasm, and GFP-ICAP1PTB had a similar or perhaps more cytoplasmic distribution, GFP-ICAPFL was strongly enriched in the nucleus, leaving very little GFP-ICAP1FL in the cytosol. Analysis of multiple images from replicate experiments using CellProfiler version 2.0 (40) allowed us to calculate the relative intensity of the GFP signal co-localizing with the nucleus (identified using the DAPI stain) versus the whole cell. When quantified, ∼56% of the GFP-ICAP1FL signal was nuclear, whereas GFP-ICAP1PTB was statistically significantly lower with ∼32% of the signal nuclear (Fig. 3B). To validate our image-based findings using a biochemical assay, CHO cells infected by lentivirus to stably express GFP or GFP-tagged ICAP1 constructs were fractionated into nuclear and cytoplasmic fractions and analyzed by immunoblotting (28% of the cytoplasmic fractions and 80.0% of the nuclear fractions were loaded on the gel). Markers of nucleus (HDAC-1) and cytosol (carbonyl reductase) were used to verify the purity of nuclear and cytoplasmic fractions. Consistent with our microscopy data, a greater percentage of ICAP1FL than ICAP1PTB was found in the nuclear fraction (Fig. 3C). When quantified across multiple independent replicates, these differences were statistically significant (Fig. 3D). We do note that, as observed by other investigators (27, 41, 42), percentages of nuclear ICAP1 measured by fractionation were lower than those obtained by quantitative fluorescence microscopy. Nonetheless, both fluorescence microscopy and cell fractionation indicated that ICAP1FL is more nuclear than the ICAP1PTB domain.
FIGURE 3.

ICAP1FL is more nuclear than ICAP1PTB. A, CHO cells overexpressing GFP-tagged ICAP1 constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. Representative images are shown; bar, 10 μm. B, percentage of GFP intensity in the nucleus compared with the integrated GFP intensity of the entire cell was calculated using CellProfiler version 2.0. Boxes, 25th through 50th and 50th through 75th percentile; whiskers, 5th through 95th percentile (n = 97–130 cells from 5 independent experiments). *, p ≤ 0.0001 as determined by a one-way ANOVA with Tukey's correction for multiple tests. C, representative fractionation of CHO cells overexpressing GFP-tagged ICAP1 constructs. C, 28% of the cytoplasmic fraction; N, 80% of the nuclear fraction. Carbonyl reductase (CBR1) and histone deacetylase (HDAC1) represent quality controls for cytoplasmic and nuclear fractions, respectively. D, quantification of cell fractionation data, where the percentage nuclear = total nuclear/(total nuclear N + total cytoplasmic) × 100 (bar, mean percentage nuclear value; n = 10). *, p ≤ 0.001 as determined by a one-way ANOVA with Tukey's correction for multiple tests.
Preferential localization of GFP-ICAP1FL in the nucleus, away from β1 integrins, provides a potential mechanism for the reduced ability of ICAP1FL to suppress β1 activation. Loss of β1 binding had little effect on ICAP1 localization (Fig. 3, B and D). Collectively, our data indicate that enhanced suppression of β1 activation correlates with decreased nuclear targeting of ICAP1.
ICAP1 Contains a Functional N-terminal Nuclear Localization Signal
ICAP1 is a relatively small protein (200 amino acids) with a known C-terminal PTB domain and an N terminus that is predicted to be unstructured (Fig. 1A). ICAP1 has previously been shown to shuttle in and out of the nucleus, and a putative N-terminal NLS was identified (27). Because this sequence is outside of the PTB domain, this would explain why the PTB domain alone has a localization different from that of the full-length protein. To explore the role of this sequence in ICAP1 localization, we generated three GFP-tagged ICAP1 constructs: a truncation of the first 17 amino acids removing the putative NLS (ICAP1 residues 18–200, ICAP1ΔNLS), a mutation of NLS lysine residues to alanine (ICAP1 K6A/K7A, ICAP1NLSmut), and the introduction of a premature stop codon after the NLS (ICAP1 residues 1–10, ICAP1NLS). After transfection into CHO cells and plating on FN-coated coverslips, the cellular localization of GFP-ICAP1 was analyzed using fluorescence microscopy. Whereas GFP-ICAP1FL preferentially targeted to the nucleus, both GFP-ICAP1ΔNLS and GFP-ICAP1NLSmut demonstrated a clear shift to the cytoplasm, exhibiting less nuclear localization than control GFP (Fig. 4A). These data demonstrate that the ICAP1 NLS is necessary for nuclear localization, and they are consistent with prior analyses (27). Additionally, we showed that the first 10 amino acids of ICAP1, which contain the ICAP1NLS, are sufficient to strongly localize GFP to the nucleus (Fig. 4A). Quantification of multiple images revealed that disrupting the NLS sequence by either truncation or mutation causes a statistically significant reduction of nuclear ICAP1, whereas the addition of the ICAP1 NLS to GFP significantly increases localization of GFP to the nucleus (Fig. 4B). Again, we validated our findings biochemically by fractionating CHO lines that stably express GFP or GFP-tagged ICAP1 constructs (Fig. 4, C and D). Collectively, these data suggest that the ICAP1 NLS is both necessary and sufficient for nuclear localization.
FIGURE 4.

ICAP1 contains a functional NLS. A, CHO cells overexpressing GFP-tagged ICAP1 constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. Representative images are shown; bar, 10 μm. B, relative amount of GFP intensity in the nucleus compared with the integrated GFP intensity of the entire cell. Boxes, 25th through 50th and 50th through 75th percentile; whiskers, 5th through 95th percentile (n = 88–139 cells from 5 independent experiments). *, p ≤ 0.005 as determined by a one-way ANOVA with Tukey's correction for multiple tests. C, representative fractionation of CHO cells overexpressing GFP-tagged ICAP1 constructs. C, 28% of the cytoplasmic fraction; N, 80.0% of the nuclear fraction. Carbonyl reductase (CBR1) and histone deacetylase (HDAC1) represent quality controls for cytoplasmic and nuclear fractions, respectively. D, quantification of cell fractionation data where the percentage nuclear = total nuclear/(total nuclear N + total cytoplasmic) × 100 (bar, mean percentage nuclear value; n = 9). *, p ≤ 0.02 as determined by a one-way ANOVA with Tukey's correction for multiple tests.
Disrupting ICAP1 Nuclear Localization Enhances Suppression of β1 Integrin
To test whether altering ICAP1 localization to the nucleus influences its ability to suppress integrin activation, we transiently expressed GFP-ICAP1 mutants in CHO-α5β1 cells and assessed β1 integrin activation using our flow cytometric assay. Notably, whereas ICAP1FL, ICAP1ΔNLS, and ICAP1NLSmut each produced dose-dependent suppression of integrin activation, ICAP1 with a disrupted NLS (ICAP1ΔNLS and ICAP1NLSmut) exhibited a significantly greater suppression of integrin β1 activation (Fig. 5A). The increased suppression of β1 integrin cannot be explained by changes in the level of integrin expression because no significant alteration in integrin levels was observed, and PAC1 binding was normalized to surface integrin expression. Differences in ICAP1-mediated β1 integrin suppression cannot be due to variations in expression of the ICAP1 constructs because populations were gated for equivalent GFP expression levels, ensuring comparison of cells with comparable levels of GFP-ICAP1. Furthermore, both GFP-ICAP1ΔNLS and GFP-ICAP1NLSmut appeared to bind β1 cytoplasmic tails to the same extent as GFP-ICAP1FL when assessed in pull-down assays (Fig. 5B). As expected, GFP-ICAP1NLS, which lacks the integrin-binding PTB domain, did not bind β1 tails in pull-down assays and did not impact integrin activation (Fig. 5, A and B). These data suggest the ICAP1 NLS diminishes β1 suppression by translocating ICAP1 to the nucleus, away from β1 integrins.
FIGURE 5.
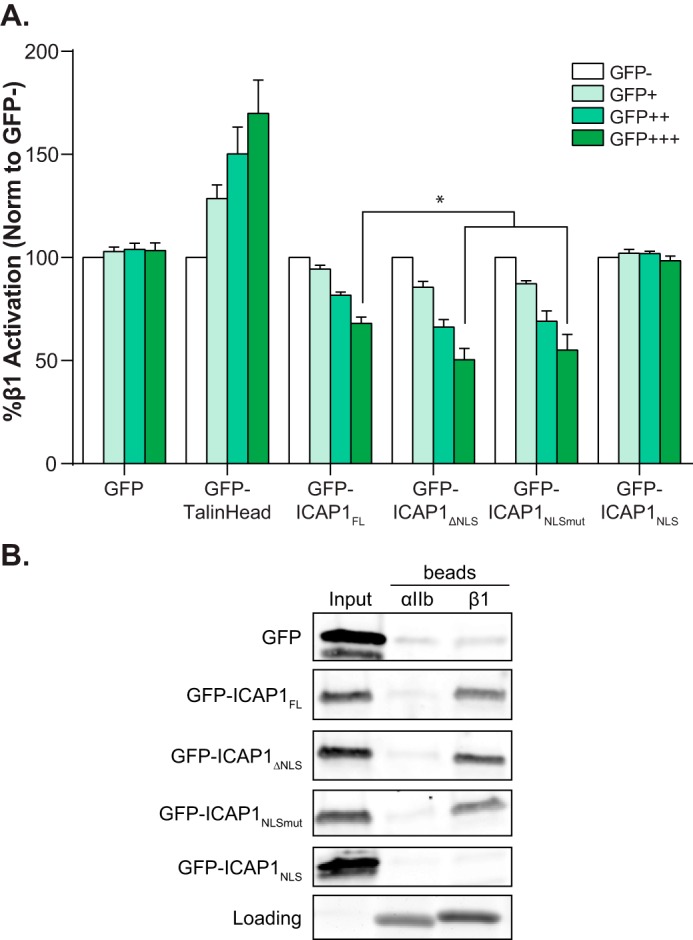
Localization of ICAP1 to the nucleus diminishes its suppression of β1-integrin activation. A, CHO cells were transfected with GFP, GFP-talin head, or GFP-tagged ICAP1 constructs, and the activation of stably expressed chimeric αIIbα5β3β1 integrins was assessed by flow cytometry. Gating on cell populations with different GFP intensities permits analysis of the dose dependence of effects. Results are the mean percentage ± S.E. (error bars) of the untransfected (GFP−) population from 6–8 independent experiments. *, p ≤ 0.001 as determined by a two-way ANOVA with Tukey's correction for multiple tests. B, pull-down of GFP-tagged ICAP1 constructs from CHO cell lysates with purified recombinant αIIb or β1 integrin tails. Tail loading was assessed by Coomassie Blue staining. The input lane indicates 5% of input lysate. Results shown are representative of four independent experiments.
Binding to ICAP1 Drives Nuclear Localization of the N-terminal Fragment of KRIT1
We have shown previously that the N-terminal fragment of KRIT1 (KRIT1 residues 1–198, KRIT1Nterm) inhibits ICAP1-mediated suppression of integrin β1 activation by competing with β1 integrin for binding to ICAP1 (7). Additionally, data from other laboratories suggest that KRIT1 localization drives ICAP1 localization (28). To test whether cellular localization might play a role in the reversal of ICAP1-mediated integrin suppression by KRIT1, we co-expressed GFP or GFP-tagged KRIT1Nterm with DsRed or DsRed-tagged ICAP1 in CHO cells and evaluated nuclear localization in double positive cells. As expected, GFP co-expression had little impact on the localization of DsRed-ICAP1 or DsRed-ICAP1 mutants (Fig. 6A). DsRed-ICAP1FL exhibited greater nuclear localization than DsRed, whereas DsRed-ICAP1ΔNLS and DsRed-ICAP1NLSmut exhibited less nuclear localization, a pattern very similar to that seen for GFP-tagged proteins in Fig. 4B. However, when KRIT1Nterm (which contains the ICAP1-binding site) was expressed, the mean nuclear fraction of DsRed-ICAP1FL statistically significantly increased from 49 to 61% (Fig. 6A). Notably, a direct ICAP1FL/KRIT1Nterm interaction was required for this increase in nuclear ICAP1, because co-expression of KRIT1Nterm containing mutations that inhibit ICAP1 binding (7) (KRIT1 R179A/R185A/N192A/Y195A, KRIT1Nterm/) did not have this effect (Fig. 6A). These data are consistent with reports that KRIT1 drives ICAP1 nuclear localization (28). However, neither DsRed-ICAP1ΔNLS nor DsRed-ICAP1NLSmut showed any significant changes in localization when co-expressed with either GFP-KRIT1Nterm or GFP-KRIT1Nterm/ (Fig. 6A). Thus, KRIT1Nterm was unable to enhance nuclear targeting of ICAP1 lacking a functional NLS, showing that binding to KRIT1Nterm is insufficient to target ICAP1 to the nucleus.
FIGURE 6.

Localization of KRIT1Nterm is driven by ICAP1. A and B, CHO cells co-overexpressing GFP-tagged KRIT1 constructs and DsRed-tagged ICAP1 constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. Images were analyzed using CellProfiler. Percentage nuclear fluorescence compared with the total integrated fluorescence intensity of the entire cell for DsRed (A) and GFP (B) was calculated. Boxes, 25th through 50th and 50th through 75th percentile; whiskers, 5th through 95th percentile (n = 62–138 cells from 3 independent experiments). *, p ≤ 0.0001 as determined by a one-way ANOVA with Tukey's correction for multiple tests.
In the process of assessing the impact of KRIT1Nterm on ICAP1 localization, we noticed that ICAP1 profoundly altered KRIT1Nterm localization. When GFP-KRIT1Nterm was co-expressed with DsRed, GFP-KRIT1Nterm had an even distribution throughout the cell, comparable with the distribution of GFP alone (Fig. 6B). However, when co-expressed with DsRed-ICAP1FL, which localizes strongly to the nucleus, GFP-KRIT1Nterm localization shifted to the nucleus (Fig. 6B). Additionally, when co-expressed with either DsRed-ICAP1ΔNLS or DsRed-ICAP1NLSmut, which exhibit more cytoplasmic localization, GFP-KRIT1Nterm also localized more to the cytoplasm (Fig. 6B). Thus, GFP-KRIT1Nterm appears to follow the localization of ICAP1, and mutations that alter ICAP1 localization also influence that of KRIT1. The effect of ICAP1 on KRIT1Nterm localization appears to depend on a direct KRIT1-ICAP1 interaction because GFP-KRIT1Nterm/ localization was not affected by co-expression of any of the DsRed-ICAP1 constructs (Fig. 6B). Overall, our data suggest that, whereas binding to the N-terminal portion of KRIT1 enhances nuclear localization of ICAP1, KRIT1Nterm is insufficient to drive ICAP1 localization; instead, ICAP1 drives KRIT1 localization in a binding-dependent manner.
Mutations in KRIT1 Lead to Changes in Cellular Localization
The preceding experiments investigated the role of the N-terminal portion of KRIT1 encompassing the Nudix domain and the first NPXY motif. However, KRIT1 is a multidomain protein that, in addition to having several other binding partners, contains two putative NLS and one predicted nuclear export signal (NES) (Fig. 7A) (17, 28, 30, 43, 44). To determine the localization of full-length KRIT1 (KRIT1FL) and the role of the predicted NLS and NES in the absence of co-expressed ICAP1, GFP-tagged KRIT1 was overexpressed in CHO cells (which do not express either endogenous ICAP1 or KRIT1 (45)). We generated mutants that disrupted either the NLS1 (KRIT1 K46A/K47A/K48A/R49A/K50A/K51A, KRIT1FL-NLS1mut), NLS2 (KRIT1 K569A/K570A/H571A/K572A, KRIT1FL-NLS2mut), or NES (KRIT1 L557A/L558A/L559A, KRIT1FL-NESmut). KRIT1FL constructs were then expressed in CHO cells by lentiviral transduction. GFP-KRIT1FL, like GFP-KRIT1Nterm, distributed evenly between the nucleus and the cytoplasm (Fig. 7B). Interestingly, only GFP-KRIT1NLS1mut showed a change in localization, whereas the cellular localization of GFP-KRIT1FL-NESmut and GFP-KRIT1FL-NLS2mut was indistinguishable from GFP-KRIT1FL (Fig. 7B). When quantified, GFP-KRIT1FL-NLS1mut was statistically less nuclear than KRIT1FL (24% compared with 35%, respectively), whereas the other mutants showed no change (Fig. 7B).
FIGURE 7.

Evaluation of putative KRIT1 localization sequences. A, schematic of KRIT1 noting the sequence of NLS1, NES, and NLS2 as well as domain boundaries. B, CHO cells overexpressing GFP-tagged KRIT1 constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. Representative images are shown; bar, 10 μm. C, relative amount of GFP intensity in the nucleus compared with the integrated GFP intensity of the entire cell. Boxes, 25th through 50th and 50th through 75th percentile; whiskers, 5th through 95th percentile (n = 85–157 cells from 4 independent experiments). *, p ≤ 0.0001 as determined by a one-way ANOVA with Tukey's correction for multiple tests.
ICAP1 Drives KRIT1FL Localization in a Binding-dependent Fashion
To test whether ICAP1 could drive the localization of KRIT1FL, as it did for KRIT1Nterm, KRIT1FL constructs were overexpressed in CHO cells by lentiviral transduction, and the ICAP1 constructs were transfected the next day. Double-positive cells were then imaged and analyzed by CellProfiler version 2.0. GFP localization was not affected by ICAP1 expression (Fig. 8A), but DsRed-ICAP1FL was able to drive significant nuclear localization of GFP-KRIT1FL (Fig. 8A). This result is very similar to that obtained with GFP-KRIT1Nterm (Fig. 6B). Furthermore, co-expression of either DsRed-ICAP1ΔNLS or DsRed-ICAP1NLSmut with KRIT1FL decreased the nuclear fraction of KRIT1FL (Fig. 8A). As seen for KRIT1Nterm, the ability of ICAP1 to influence KRIT1 localization is dependent on their interaction because the binding-defective GFP-KRIT1FL/ was evenly distributed throughout the cell and was not affected by ICAP1 expression (Fig. 8A). To evaluate the contribution of KRIT1 NLS1, the ICAP1 constructs were co-expressed with GFP-KRIT1FL-NLS1mut, a mostly cytoplasmic KRIT1 construct. Most interestingly, the nuclear fraction of GFP-KRIT1FL-NLS1mut increased when co-expressed with DsRed-ICAP1FL, reaching levels comparable with that of KRIT1FL when co-expressed with ICAP1FL (Fig. 8A). Together, our data suggest that the ICAP1 NLS is sufficient to take both ICAP1 and KRIT1FL to the nucleus, indicating that ICAP1 drives KRIT1FL localization.
FIGURE 8.

Localization of KRIT1FL is driven by ICAP1. CHO cells co-overexpressing GFP or GFP-tagged KRIT1 constructs and DsRed-tagged ICAP1 constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. A and B, percentage nuclear fluorescence compared with the integrated fluorescence intensity of the entire cell for GFP (A) and DsRed (B) was calculated. Boxes, 25th through 50th and 50th through 75th percentile; whiskers, 5th through 95th percentile (n = 47–103 cells from three independent experiments). *, p ≤ 0.0001 as determined by a one-way ANOVA with Tukey's correction for multiple tests.
KRIT1 Needs Its NLS1 to Enhance Nuclear Accumulation of ICAP1
We previously noted that although GFP-KRIT1Nterm was insufficient to support ICAP1NLSmut or ICAP1ΔNLS nuclear localization (Fig. 6A), it did nevertheless increase nuclear targeting of DsRed-ICAP1FL. We therefore examined the effect of KRIT1FL on ICAP1 localization. As was seen before, GFP did not affect the localization of any of the ICAP1 constructs (Fig. 8B). However, co-expression with GFP-KRIT1FL caused a statistically significant increase of nuclear DsRed-ICAP1FL (46.7% versus 56.5%), and this did not occur when DsRed-ICAP1FL was co-expressed with GFP-KRIT1FL/. This validates the earlier finding that the ICAP1/KRIT1 interaction increases nuclear ICAP1. Intriguingly, co-expression of GFP-KRIT1FL-NLS1mut did not cause an increase in nuclear DsRed-ICAP1FL. This suggests that, whereas the ICAP1 NLS is necessary and sufficient for the nuclear localization of the ICAP1-KRIT1 complex and the KRIT1 NLS1 is dispensable for nuclear accumulation of the complex, the NLS1 of KRIT1 is nonetheless required to maintain the highest levels of nuclear ICAP1.
KRIT1FL Localization Changes When Endogenous ICAP1 Is Lost
The preceding experiments all relied on expression of fluorophore-tagged ICAP1 in CHO cells, a line that apparently lacks endogenous ICAP1 and KRIT1 (45). We therefore assessed ICAP1 nuclear localization in EA.hy926 cells, a widely used cell line generated by fusing human umbilical vein endothelial cells with the lung carcinoma A549. EA.hy926 cells retain many endothelial features and can be stably cultured for long periods (46). We have been unable to identify anti-ICAP1 antibodies suitable for immunofluorescence microscopy and therefore used cell fractionation to investigate the nuclear localization of endogenous ICAP1. This revealed that, as observed for exogenously expressed GFP-ICAP1, endogenous ICAP1 is present in both the cytosol and the nucleus (Fig. 9A).
FIGURE 9.

Localization of exogenous KRIT1 in EAhy926 cells changes upon loss of ICAP1. A, representative fractionation of endogenous ICAP1 in EAhy926 cells. C, 28% of the cytoplasmic fraction; N, 80% of the nuclear fraction. Carbonyl reductase (CBR1) and histone deacetylase (HDAC1) represent quality controls for cytoplasmic and nuclear fractions, respectively. Results are representative of three independent experiments. B, immunoblot of EAhy926 lysates that overexpress either GFP or GFP-tagged KRIT1 constructs and have been infected with either an shSCR or shRNAs targeting ICAP1 (shICAP1-21, shICAP1-23). Vinculin (VCL) was used as a loading control. C–E, EAhy926 cells infected with either shSCR or shICAP1 and overexpressing GFP or GFP-tagged KRIT1 constructs were plated on fibronectin, fixed 24 h later, and stained with DAPI to identify nuclei. Representative images are shown; bar, 10 μm. F, percentage of GFP intensity in the nucleus compared with the integrated GFP intensity of the entire cell. Boxes, 25th through 50th and 50th through 75th percentile; whiskers, 5th through 95th percentile (n = 88–93 cells from 3 independent experiments). *, p ≤ 0.0001 as determined by a one-way ANOVA with Tukey's correction for multiple tests.
Having established that endogenous ICAP1 is found in the nucleus of EA.hy926 cells, we next wished to evaluate how the loss of endogenous ICAP1 affects the localization of KRIT1. Unfortunately, stable knockdown of ICAP1 results in loss of KRIT1 protein (20, 43), preventing us from evaluating the effect of ICAP1 knockdown on endogenous KRIT1 localization. Therefore, we introduced GFP, GFP-KRIT1FL, or GFP-KRIT1FL/ into EA.hy926 cells by lentiviral transduction. These cells were then infected with one of two different lentiviral shRNA constructs targeting ICAP1 (shICAP1-21 and shICAP1-23) or a scrambled shRNA (shSCR) control. Cells were treated with puromycin (the shRNA selection marker) and either plated on FN-coated coverslips for fluorescence microscopy or lysed for immunoblot analysis to assess knockdown. As shown in Fig. 9B, shICAP1-21 and shICAP1-23 both reduced ICAP1 protein levels, although shICAP1-23 consistently resulted in greater loss of endogenous ICAP1. Unsurprisingly, GFP nuclear localization was unaffected by any of the shRNAs either visually or quantitatively (Fig. 9, C and F). Interestingly, in ICAP1 knockdown cells, KRIT1FL became more cytoplasmic (Fig. 9, E and F). Notably, this effect was most pronounced with shICAP1-23, which produced the greatest ICAP1 knockdown. Importantly, the ICAP1 binding-defective mutant GFP-KRIT1FL/ was less nuclear than GFP-KRIT1FL (Fig. 9, E and F) and was comparable with GFP-KRIT1FL in the absence of ICAP1 (Fig. 9F). Furthermore, the localization of GFP-KRIT1FL/ was not impacted by ICAP1 knockdown, and nuclear levels were comparable with GFP-KRITFL in cells lacking ICAP1 (Fig. 9, E and F). Collectively, these data indicate that exogenous KRIT1 localization is affected by direct binding to endogenous ICAP1.
Discussion
ICAP1 has only two well validated binding partners: KRIT1 and integrin β1. Through the direct binding of its PTB domain to the cytoplasmic tail of integrin β1, ICAP1 represses β1 integrin activation and thus modulates downstream signaling (32–35). KRIT1 competes with β1 integrin for binding to ICAP1 and hence can reverse ICAP1-mediated integrin β1 repression (7). However, as KRIT1 protects ICAP1 from proteasomal degradation, it can also potentiate ICAP1-dependent processes (20), but impacts beyond the regulation of integrin β1 activation have not yet been determined. Here we investigated the control of ICAP1-mediated inhibition of integrin activation and found that nuclear localization of ICAP1 diminishes repression of integrin β1 activation. We further revealed that ICAP1 drives KRIT1 localization to the nucleus, suggesting that ICAP1 has important roles in determining KRIT1 subcellular localization.
ICAP1 Localization Affects Cellular Functions
Differential localization of signaling molecules is an important mechanism for regulation of signaling pathways. We demonstrate that ICAP1 contains a functional NLS, which is both necessary and sufficient for localization of ICAP1 to the nucleus. Nuclear accumulation reduces cytosolic ICAP1 levels, thereby diminishing the efficiency of ICAP1 repression of β1 integrin activation. Because integrin β1 is linked to a wide range of downstream signaling pathways, tight regulation is essential for proper cellular function. For example, increased β1 signaling leads to RhoA activation, and the RhoA activation and cell contractility seen upon the loss of KRIT1 or ICAP1 protein levels is dependent on integrin β1 (20, 47). Conversely, increased cytoplasmic ICAP1 leads to greater repression of integrin β1 activation, and thus general defects in cell adhesion and cell spreading would be expected. This could lead to a loss of endothelial cell polarity and lumen formation (48), phenotypes also seen in CCM patient samples (49). In addition to influencing levels of ICAP1 in the cytoplasm and thus impacting cytoplasmic functions of ICAP1, as discussed further below, nuclear localization of ICAP1 may enhance nuclear roles of ICAP1. Thus, controlling subcellular localization permits switching between nuclear and cytoplasmic roles of ICAP1.
Signals that regulate ICAP1 nuclear localization are currently unknown but will be important subjects for future study. It will be interesting to inhibit the shuttling of endogenous ICAP1 and to determine the impact on integrin activation and integrin-mediated cellular processes, such as cell adhesion and migration. Unfortunately, we do not currently have sufficient information on the mechanisms or regulation of ICAP1 shuttling between the nucleus and the cytoplasm to experimentally disrupt it in a specific manner. For this reason, our current investigation of the effects of ICAP1 on KRIT1 localization and integrin activation has relied on overexpression of ICAP1 mutants with perturbed localization or on ICAP1 knockdown.
ICAP1 Drives the Localization of KRIT1
Our data demonstrate that the localization of ICAP1 strongly influences KRIT1 localization. Nuclear ICAP1 is capable of recruiting KRIT1 to the nucleus, even if the KRIT1 NLS is ablated. Furthermore, knockdown of endogenous ICAP1 reduces nuclear localization of exogenously expressed KRIT1. An important role of ICAP1 nuclear localization may therefore be the recruitment of KRIT1 to the nucleus. Consequently, it is likely that aberrant ICAP1 localization would lead to some KRIT1-related phenotypes. For example, the loss of KRIT1 leads to decreased β-catenin and VE-cadherin in cell-cell junctions, and this leads to increased nuclear β-catenin and the up-regulation of its transcriptional targets (26). Additionally, the loss of KRIT1 leads to the mislocalization of the polarity complex Tiam, Par3, and PKCζ and thus defects in directed migration and vascular lumen formation (49). It would be interesting to determine whether ICAP1-directed KRIT1 mislocalization would lead to similar phenotypes. We note that other KRIT1 binding partners also influence KRIT1 localization. In addition to shuttling between the nucleus and cytoplasm, KRIT1 also localizes to cell-cell junctions (22, 50, 51) and microtubules (52, 53). The KRIT1 FERM domain forms a ternary complex with membrane anchor protein HEG1 and Rap1, and KRIT1 mutants defective in binding to either HEG1 or Rap1 do not localize to cell-cell junctions (51, 54). The loss of CCM2 also leads to the loss of KRIT1 from cell-cell junctions, but it is unclear whether this is related to a direct interaction between the two proteins.
KRIT1 Influences the Localization of ICAP1
In addition to ICAP1 driving localization of KRIT1, our data demonstrate that, conversely, KRIT1 binding increases the levels of nuclear ICAP1 and that this effect is dependent on an intact KRIT1 NLS1. It is likely that although the ICAP1 NLS is sufficient to take both ICAP1 and KRIT1 into the nucleus, the KRIT1 NLS1 cooperates to allow increased nuclear import. Alternatively, KRIT1 NLS1 could decrease the nuclear export of the KRIT1-ICAP1 complex via an unknown mechanism.
Potential Roles for ICAP1-KRIT1 within the Nucleus
Neither the cellular stimulus that leads to a change in ICAP1-KRIT1 localization nor its functional significance has been determined. Our data support the hypothesis that nuclear retention of ICAP1 is a tool to modulate the cytoplasmic role of ICAP1 (suppression of β1 integrin activation), but ICAP1 may also have independent roles in the nucleus. The ICAP1 NLS has been implicated in the regulation of c-Myc mRNA expression (27), but it is currently unclear whether this is due to a specific nuclear role of ICAP1 or is secondary to changes in integrin β1 signaling. Our data suggest that one key role of nuclear ICAP1 could be to drive KRIT1 into the nucleus. KRIT1 expression has been linked to increased SOD2 and FOXO1 mRNA levels (25), although whether the nuclear localization of KRIT1 is required for the up-regulation is unknown. Intriguingly, one recent report demonstrated that KRIT1 localizes to actively transcribing regions of the nucleus (31), raising the possibility that KRIT1 has specific roles in the nucleus that would then be regulated by ICAP1 localization.
Potential Regulation of ICAP1-KRIT1 Localization
The balance between nuclear export and import determines the extent of nuclear accumulation. Changes in that equilibrium could dictate the different biological effects of either ICAP1 or KRIT1. The ICAP1 NLS lies within a predicted unstructured region N-terminal to the PTB domain. It is possible that a conformational change in ICAP1 or the direct binding of another protein could either expose or mask the NLS. Data indicating that the binding to integrin β1 leads to increased cytoplasmic ICAP1 potentially support this hypothesis (27). Additionally, phosphorylation has been shown to both enhance and suppress nuclear export (55). Because the ICAP1 NLS is just N-terminal to a serine/threonine-rich motif, this is another possible mechanism for ICAP1 localization regulation. Finally, whereas the ICAP1 NLS is sufficient to take KRIT1 into the nucleus even in the absence of the only functional KRIT1 NLS, KRIT1 increases ICAP1 nuclear localization, suggesting that the KRIT1 NLS may have a secondary role on both KRIT1 and KRIT1-ICAP1 complex localization, and this too may be regulated by additional binding partners, post-translational modifications, or conformational changes. In addition, although we saw no changes in nuclear localization when we mutated either the NES or the second NLS of KRIT1, it is possible that conformational changes and/or the binding of other partners would better expose or cooperate with these signals. There is evidence suggesting that KRIT1 may have both an “open” and a “closed” conformation due to head-tail interactions and that this affects subcellular localization (28, 52). Additionally, some reports suggest that the KRIT1/CCM2 interaction also influences KRIT1 localization (30), but it is still unclear how ICAP1 would affect this dynamic.
Implications for Understanding the CCM Disease
Mutations in one of the three CCM genes occurs in ∼95% of familial CCM cases and ∼67% of sporadic CCM cases with multiple lesions (56, 57). However, CCM gene mutations have been identified in only 5–20% of sporadic cases (58), despite familial and sporadic lesions being pathologically indistinguishable (15). One interesting possibility is that, in these lesions, CCM proteins are being functionally repressed, perhaps through mislocalization, yet the role and mechanism of CCM protein localization have not been extensively studied. Our work highlights the importance of KRIT1/ICAP1 interactions in determining subcellular localization of the complex, influencing cell adhesion and potentially additional functions.
Experimental Procedures
Antibodies/cDNA
Ligand-mimetic anti-αIIbβ3 PAC1 (BD Biosciences), anti-GFP (Rockland, catalogue no. 600-101-215), anti-ICAP-1 (R&D Systems, catalogue no. AF6805), anti-HDAC-1 (Santa Cruz Biotechnology, catalogue no. sc-7872), anti-carbonyl reductase (CBR1, Santa Cruz Biotechnology, Inc., catalogue no. sc-70212), and anti-vinculin (Sigma, V-9131) were purchased. The anti-ICAP-1 antibody was validated by knockdown. All other antibodies gave only bands of the expected size (or sizes) consistent with the manufacturers' validations. Wild-type and mutant ICAP1 and KRIT1 cDNAs previously used for protein expression/purification (7) were subcloned into pDsRed-C1 and pEGFP-C1 (Clontech). Additional mutations were introduced by QuikChange site-directed mutagenesis (Stratagene). Lentiviral ICAP1 and KRIT1 expression constructs were generated by the protocol of Fu et al. (61). In brief, attL1/attL2 sites were added to GFP-tagged cDNA by two rounds of PCR, and the final product, purified by agarose gel extraction, was used in a Gateway cloning reaction (Life Technologies) to insert the GFP-tagged construct into CMV-pLENTI-Hygro (Addgene). Lentiviral constructs in the pLKO vector (U6 promoter) containing shRNAs (target sequences in brackets) against ICAP1 (TRCN0000122921 [sequence 21, CCTGTGCAGAATTTCGAATAA] and TRCN0000122923 [sequence 23, TGAAGGGCCATTAGACCTGAT]), along with a scrambled control (TRC library-based lentiviral scramble; catalogue no. SHC002) were obtained from a TRC shRNA library (Sigma-Aldrich). All constructs used were authenticated by DNA sequencing.
Cell Culture
HEK 293T cells, CHO cells, and a previously described CHO cell line stably expressing chimeric αIIbα5β3β1 integrins (37) were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 1% sodium pyruvate, and 1% nonessential amino acids (Gibco). EaHy926 cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 1% sodium pyruvate, and 2% hypoxanthine-aminopterin-thymidine supplement (Gibco). Cells were regularly tested for the presence of mycoplasma using the MycoAlertTM mycoplasma detection kit (Lonza).
Lentiviral Knockdown and Overexpression
Lentiviruses were produced by co-transfecting packaging vectors psPAX2 (viral proteins Gag and Rev under the SV40 promoter; Addgene plasmid 12260, a gift from D. Trono, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) and pMD2.G (viral protein VSV-G expressed under the CMV promoter; Addgene plasmid 12259, a gift from D. Trono) into HEK 293T cells with the shRNA construct. Viral supernatant was collected 48 and 72 h after transfection and filtered with a 0.45-μm filter.
To establish polyclonal knockdown or overexpression lines, cells were incubated with viral supernatant and 8 μg/ml Polybrene for 18 h. Cells were either analyzed 48–72 h postinfection or selected with 2 μg/ml puromycin (pLKO) or 50 μg/ml hygromycin (CMV-pLENTI-Hygro) as appropriate.
Knockdown was assessed by immunoblotting of stable lines lysed in radioimmune precipitation assay buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) containing cOmplete protease inhibitor mixture tablets (Roche Applied Science). Immunoblotting was performed with primary antibodies diluted in 5% milk in TBS-T (1:100 of α-ICAP1, 1:1000 of α-GFP, 1:10,000 of α-vinculin) fluorescent secondary antibodies (IR Dye800 or IR Dye680; LI-COR Biosciences; diluted 1:20,000 in 5% milk in TBS-T) and scanned on the Odyssey CLx infrared imaging system. Quantification was performed on the raw images in Image Studio (LI-COR Biosciences); lanes were defined, and bands were automatically identified. The profile feature was used to identify band boundaries, and lane background was subtracted. The signal for the protein of interest was normalized to that of the loading control. Optimization of images for publication was performed by adjusting the brightness and contrast in Image Studio (LI-COR Biosciences).
Integrin Activation Assays
The activation state of stably overexpressed αIIbα5β3β1 chimeric integrin in CHO cells transiently expressing GFP-tagged ICAP1 constructs was assessed in multicolor flow cytometry assays using a modification of methods described previously (36). CHO cells expressing chimeric αIIbα5β3β1 integrins were transfected with the indicated GFP expression constructs using linear polyethyleneimine, Mr 25,000 (Polysciences, Inc.) following the manufacturer's instructions. 24 h later, the cells were suspended and incubated with ligand-mimetic anti-αIIbβ3 monoclonal antibody IgM-PAC1 (BD Biosciences) in the presence or absence of 10 mm EDTA. Chimeric integrin expression was assessed in parallel by staining with anti-αIIbβ3 antibody D57 (59). Bound PAC1 and D57 were detected using Alexa 647 fluorophore-conjugated goat anti-mouse IgM and anti-mouse IgG (Invitrogen), respectively. The activation of endogenous α5β1 was assessed in CHO cells using methods described previously (36). Briefly, 5 × 105 cells were suspended and incubated with biotinylated GST-fibronectin type III repeats 9–11 (FN9–11) in the presence or absence of 10 mm EDTA. Integrin expression was assessed by parallel staining with PB1, an anti-α5 antibody (Developmental Studies Hybridoma Bank). Bound FN9–11 and PB1 were detected using allophycocyanin-conjugated streptavidin and Alexa-647 fluorophore-conjugated goat anti-mouse IgG (Invitrogen), respectively. Cells were analyzed on an LSRII flow cytometer (BD Biosciences), and data analysis was completed using FlowJo software. GFP-positive cells were gated such that the mean fluorescence intensities (MFIs) for GFP-positive cells were the same across all samples, ensuring that analysis was performed on populations expressing equivalent levels of exogenous protein. The activation index (AI) of cells gated to have equivalent mean GFP intensity was calculated as AI = (F − F0)/Fintegrin, where F is the geometric MFI of PAC1 or FN9–11 binding, F0 is the MFI of PAC1 or FN9–11 binding in the presence of EDTA, and Fintegrin is the MFI of D57 or PB1 binding to transfected cells. To compare across experiments, data were expressed as the percentage of AI in the GFP-negative population within each sample. Data were charted, and statistical analyses were performed using GraphPad Prism.
His Tag-Integrin β Tail Pull-down Assays
GFP-tagged ICAP1 was expressed in CHO cells, and cells were lysed in 1 mm Na3VO4, 50 mm NaF, 40 mm NaPPi, 50 mm NaCl, 150 mm sucrose, 10 mm Pipes, 0.5% Triton X-100, 0.1% deoxycholate, pH 6.8, and clarified by centrifugation. pGEX-6P-1 ICAP1PTB and ICAP1FL constructs were obtained as described previously (7). Both constructs were transformed in BL21 (DE3) RosettaTM (Novagen) cells and were cultivated at 37 °C to an A600 of 0.6. ICAP expression was induced by 0.1 mm isopropyl β-d-1-thiogalactopyranoside at 16 °C for 20 h. Cell pellets were then lysed in 50 mm Tris-HCl, pH 8.2, 100 mm NaCl, 5% glycerol, 0.2 mm tris(2-carboxyethyl) phosphine, and 1× cOmpleteTM EDTA free protease inhibitor mixture (Roche Applied Science). GST-ICAP was purified from the lysate on Sepharose-4B glutathione beads, and ICAP was eluted by incubation with recombinant 3C protease. Monomeric ICAP was further purified by size exclusion chromatography on a SuperdexTM S200 16/600pg column (GE Healthcare) in 50 mm Tris-HCl, pH 8.2, 100 mm NaCl, 5% glycerol, 0.1 mm tris(2-carboxyethyl) phosphine and concentrated using Amicon® Ultra-4 (Millipore) centrifugal filters. ICAP1 binding to purified recombinant His-tagged integrin tails was assessed in pull-down assays as described previously (36, 39).
Microscopy
Transfected cells were stained with CellTrackerTM Deep Red Dye (Thermo Fisher Scientific) per the manufacturer's instructions. Cells were plated on coverslips coated with 10 μg/ml fibronectin (Sigma-Aldrich). 24 h after replating, cells were fixed in 4% paraformaldehyde in PBS, pH 7.4, for 30 min. Coverslips were washed with PBS and mounted using ProLong Diamond with 25 ng/ml DAPI added (Invitrogen). Images were acquired using a Nikon Eclipse Ti-S microscope with a ×100 objective using the μManager acquisition software (60). A minimum of 50 cells (from ≥3 replicates) were analyzed for each single-expression experiment, and a minimum of 75 cells (from ≥3 replicates) were analyzed for each double-expression experiment. We verified by immunoblot that exogenous proteins were of the expected size (data not shown).
Quantification of ICAP1-KRIT1 nuclear-cytoplasmic localization was performed using CellProfiler version 2.0 (40). Nuclear GFP and/or DsRed was quantified as the integrated intensity of the fluorophore within the nucleus, as defined by DAPI staining. The outer boundary of the cell was determined by the propagation method within CellProfiler version 2.0 using the CellTracker fluorescence signal (40). Data were charted, and statistical analyses were performed using GraphPad Prism.
Nuclear/Cytoplasmic Fractionation
Fractionation of cytoplasmic and nuclear proteins was performed using the NE-PERTM kit from Thermo Fisher Scientific following the manufacturer's instructions. 3 × 106 CHO cells or 2 × 106 EAhy.926 cells were used in each fractionation. After fractionation, cytoplasmic and nuclear fractions were each mixed with 4× Laemmli sample buffer and boiled for 5 min, and a fixed volume of each fraction was loaded onto 12 or 16% SDS-polyacrylamide gels. Separated proteins were transferred onto nitrocellulose or PVDF membranes, probed by immunoblotting with antibodies diluted in 5% milk in TBS-T (1:1000 of α-GFP, 1:1000 of α-HDAC1, 1:1000 of α-CBR1), and scanned with an Odyssey CLx infrared imaging system. For a given protein, the percentage of protein extracted from the nucleus was calculated by extrapolating the total amounts detected in cytoplasmic and nuclear fractions.
Author Contributions
K. M. D. and D. A. C. conceived and designed the research and prepared the manuscript. K. M. D. performed most experiments and data analyses. C. H.-C. performed the cellular fractionation assays seen in Figs. 3 (C and D) and 4 (C and D). B. S. performed the binding curve experiments seen in Fig. 2 (C and D). All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank past and present members of the Calderwood laboratory and Titus Boggon for helpful insights and discussion.
This work was supported by National Institutes of Health Grants R01NS085078 (to D. A. C.) and F32HL127948 (to K. M. D.) and by American Cancer Society Grant 122171-PF-12-051-01-CSM (to K. M. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- FERM
- four-point-one, ezrin, radixin, moesin
- PTB
- phosphotyrosine binding
- CCM
- cerebral cavernous malformation
- NLS
- nuclear localization signal(s)
- NES
- nuclear export signal
- shSCR
- scrambled shRNA
- MFI
- mean fluorescence intensity
- AI
- activation index
- ANOVA
- analysis of variance.
References
- 1.Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 2.Harburger D. S., and Calderwood D. A. (2009) Integrin signalling at a glance. J. Cell Sci. 122, 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morse E. M., Brahme N. N., and Calderwood D. A. (2014) Integrin cytoplasmic tail interactions. Biochemistry 53, 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwamoto D. V., and Calderwood D. A. (2015) Regulation of integrin-mediated adhesions. Curr. Opin. Cell Biol. 36, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calderwood D. A., Campbell I. D., and Critchley D. R. (2013) Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye F., Snider A. K., and Ginsberg M. H. (2014) Talin and kindlin: the one-two punch in integrin activation. Front. Med. 8, 6–16 [DOI] [PubMed] [Google Scholar]
- 7.Liu W., Draheim K. M., Zhang R., Calderwood D. A., and Boggon T. J. (2013) Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 49, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiema T., Lad Y., Jiang P., Oxley C. L., Baldassarre M., Wegener K. L., Campbell I. D., Ylänne J., and Calderwood D. A. (2006) The molecular basis of filamin binding to integrins and competition with talin. Mol. Cell 21, 337–347 [DOI] [PubMed] [Google Scholar]
- 9.Millon-Frémillon A., Bouvard D., Grichine A., Manet-Dupé S., Block M. R., and Albiges-Rizo C. (2008) Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent β1-integrin affinity. J. Cell Biol. 180, 427–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J., Clatterbuck R. E., Rigamonti D., Chang D. D., and Dietz H. C. (2001) Interaction between krit1 and icap1α infers perturbation of integrin β1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum. Mol. Genet. 10, 2953–2960 [DOI] [PubMed] [Google Scholar]
- 11.Zawistowski J. S., Serebriiskii I. G., Lee M. F., Golemis E. A., and Marchuk D. A. (2002) KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum. Mol. Genet. 11, 389–396 [DOI] [PubMed] [Google Scholar]
- 12.Fisher O. S., and Boggon T. J. (2014) Signaling pathways and the cerebral cavernous malformations proteins: lessons from structural biology. Cell Mol. Life Sci. 71, 1881–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pozzati E., Acciarri N., Tognetti F., Marliani F., and Giangaspero F. (1996) Growth, subsequent bleeding, and de novo appearance of cerebral cavernous angiomas. Neurosurgery 38, 662–669; discussion 669–670 [PubMed] [Google Scholar]
- 14.Rigamonti D., Hadley M. N., Drayer B. P., Johnson P. C., Hoenig-Rigamonti K., Knight J. T., and Spetzler R. F. (1988) Cerebral cavernous malformations: incidence and familial occurrence. N. Engl. J. Med. 319, 343–347 [DOI] [PubMed] [Google Scholar]
- 15.Tanriover G., Sozen B., Seker A., Kilic T., Gunel M., and Demir N. (2013) Ultrastructural analysis of vascular features in cerebral cavernous malformations. Clin. Neurol. Neurosurg. 115, 438–444 [DOI] [PubMed] [Google Scholar]
- 16.Revencu N., and Vikkula M. (2006) Cerebral cavernous malformation: new molecular and clinical insights. J. Med. Genet. 43, 716–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Draheim K. M., Fisher O. S., Boggon T. J., and Calderwood D. A. (2014) Cerebral cavernous malformation proteins at a glance. J. Cell Sci. 127, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voss K., Stahl S., Schleider E., Ullrich S., Nickel J., Mueller T. D., and Felbor U. (2007) CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8, 249–256 [DOI] [PubMed] [Google Scholar]
- 19.Stahl S., Gaetzner S., Voss K., Brackertz B., Schleider E., Surucu O., Kunze E., Netzer C., Korenke C., Finckh U., Habek M., Poljakovic Z., Elbracht M., Rudnik-Schöneborn S., Bertalanffy H., et al. (2008) Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum. Mutat. 29, 709–717 [DOI] [PubMed] [Google Scholar]
- 20.Faurobert E., Rome C., Lisowska J., Manet-Dupé S., Boulday G., Malbouyres M., Balland M., Bouin A. P., Kéramidas M., Bouvard D., Coll J. L., Ruggiero F., Tournier-Lasserve E., and Albiges-Rizo C. (2013) CCM1-ICAP-1 complex controls β1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 202, 545–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borikova A. L., Dibble C. F., Sciaky N., Welch C. M., Abell A. N., Bencharit S., and Johnson G. L. (2010) Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J. Biol. Chem. 285, 11760–11764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stockton R. A., Shenkar R., Awad I. A., and Ginsberg M. H. (2010) Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 207, 881–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitehead K. J., Chan A. C., Navankasattusas S., Koh W., London N. R., Ling J., Mayo A. H., Drakos S. G., Jones C. A., Zhu W., Marchuk D. A., Davis G. E., and Li D. Y. (2009) The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 15, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wüstehube J., Bartol A., Liebler S. S., Brütsch R., Zhu Y., Felbor U., Sure U., Augustin H. G., and Fischer A. (2010) Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 12640–12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goitre L., Balzac F., Degani S., Degan P., Marchi S., Pinton P., and Retta S. F. (2010) KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS One 5, e11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glading A. J., and Ginsberg M. H. (2010) Rap1 and its effector KRIT1/CCM1 regulate β-catenin signaling. Dis. Model. Mech. 3, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fournier H. N., Dupé-Manet S., Bouvard D., Luton F., Degani S., Block M. R., Retta S. F., and Albiges-Rizo C. (2005) Nuclear translocation of integrin cytoplasmic domain-associated protein 1 stimulates cellular proliferation. Mol. Biol. Cell 16, 1859–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Francalanci F., Avolio M., De Luca E., Longo D., Menchise V., Guazzi P., Sgrò F., Marino M., Goitre L., Balzac F., Trabalzini L., and Retta S. F. (2009) Structural and functional differences between KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. Exp. Cell Res. 315, 285–303 [DOI] [PubMed] [Google Scholar]
- 29.Bouvard D., Millon-Fremillon A., Dupe-Manet S., Block M. R., and Albiges-Rizo C. (2006) Unraveling ICAP-1 function: toward a new direction? Eur. J. Cell Biol. 85, 275–282 [DOI] [PubMed] [Google Scholar]
- 30.Zawistowski J. S., Stalheim L., Uhlik M. T., Abell A. N., Ancrile B. B., Johnson G. L., and Marchuk D. A. (2005) CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 14, 2521–2531 [DOI] [PubMed] [Google Scholar]
- 31.Marzo S., Galimberti V., and Biggiogera M. (2014) Unexpected distribution of KRIT1 inside the nucleus: new insight in a complex molecular pathway. Eur. J. Histochem. 58, 2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouvard D., Aszodi A., Kostka G., Block M. R., Albigès-Rizo C., and Fässler R. (2007) Defective osteoblast function in ICAP-1-deficient mice. Development 134, 2615–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouvard D., Vignoud L., Dupé-Manet S., Abed N., Fournier H. N., Vincent-Monegat C., Retta S. F., Fassler R., and Block M. R. (2003) Disruption of focal adhesions by integrin cytoplasmic domain-associated protein-1 α. J. Biol. Chem. 278, 6567–6574 [DOI] [PubMed] [Google Scholar]
- 34.Brunner M., Millon-Frémillon A., Chevalier G., Nakchbandi I. A., Mosher D., Block M. R., Albigès-Rizo C., and Bouvard D. (2011) Osteoblast mineralization requires beta1 integrin/ICAP-1-dependent fibronectin deposition. J. Cell Biol. 194, 307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W., and Boggon T. J. (2013) Cocrystal structure of the ICAP1 PTB domain in complex with a KRIT1 peptide. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 494–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouaouina M., Harburger D. S., and Calderwood D. A. (2012) Talin and signaling through integrins. Methods Mol. Biol. 757, 325–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Toole T. E., Katagiri Y., Faull R. J., Peter K., Tamura R., Quaranta V., Loftus J. C., Shattil S. J., and Ginsberg M. H. (1994) Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell Biol. 124, 1047–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Millon-Frémillon A., Brunner M., Abed N., Collomb E., Ribba A. S., Block M. R., Albigès-Rizo C., and Bouvard D. (2013) Calcium and calmodulin-dependent serine/threonine protein kinase type II (CaMKII)-mediated intramolecular opening of integrin cytoplasmic domain-associated protein-1 (ICAP-1α) negatively regulates β1 integrins. J. Biol. Chem. 288, 20248–20260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lad Y., Harburger D. S., and Calderwood D. A. (2007) Integrin cytoskeletal interactions. Methods Enzymol. 426, 69–84 [DOI] [PubMed] [Google Scholar]
- 40.Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., Golland P., and Sabatini D. M. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu X., Batta P., Innocent N., Prisco M., Casaburi I., Belletti B., and Baserga R. (2002) Nuclear translocation of insulin receptor substrate-1 by oncogenes and Igf-I: effect on ribosomal RNA synthesis. J. Biol. Chem. 277, 44357–44365 [DOI] [PubMed] [Google Scholar]
- 42.Shaiken T. E., and Opekun A. R. (2014) Dissecting the cell to nucleus, perinucleus and cytosol. Sci. Rep. 4, 4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J., Basu S., Rigamonti D., Dietz H. C., and Clatterbuck R. E. (2008) Krit1 modulates β1-integrin-mediated endothelial cell proliferation. Neurosurgery 63, 571–578; discussion 578 [DOI] [PubMed] [Google Scholar]
- 44.Zhang J., Rigamonti D., Dietz H. C., and Clatterbuck R. E. (2007) Interaction between krit1 and malcavernin: implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery 60, 353–359; discussion 359 [DOI] [PubMed] [Google Scholar]
- 45.Baycin-Hizal D., Tabb D. L., Chaerkady R., Chen L., Lewis N. E., Nagarajan H., Sarkaria V., Kumar A., Wolozny D., Colao J., Jacobson E., Tian Y., O'Meally R. N., Krag S. S., Cole R. N., et al. (2012) Proteomic analysis of Chinese hamster ovary cells. J. Proteome Res. 11, 5265–5276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouïs D., Hospers G. A., Meijer C., Molema G., and Mulder N. H. (2001) Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis 4, 91–102 [DOI] [PubMed] [Google Scholar]
- 47.Huveneers S., Truong H., Fässler R., Sonnenberg A., and Danen E. H. (2008) Binding of soluble fibronectin to integrin α5 β1: link to focal adhesion redistribution and contractile shape. J. Cell Sci. 121, 2452–2462 [DOI] [PubMed] [Google Scholar]
- 48.Zovein A. C., Luque A., Turlo K. A., Hofmann J. J., Yee K. M., Becker M. S., Fassler R., Mellman I., Lane T. F., and Iruela-Arispe M. L. (2010) β1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev. Cell 18, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lampugnani M. G., Orsenigo F., Rudini N., Maddaluno L., Boulday G., Chapon F., and Dejana E. (2010) CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J. Cell Sci. 123, 1073–1080 [DOI] [PubMed] [Google Scholar]
- 50.Glading A., Han J., Stockton R. A., and Ginsberg M. H. (2007) KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 179, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gingras A. R., Liu J. J., and Ginsberg M. H. (2012) Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J. Cell Biol. 199, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Béraud-Dufour S., Gautier R., Albiges-Rizo C., Chardin P., and Faurobert E. (2007) Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J. 274, 5518–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gunel M., Laurans M. S., Shin D., DiLuna M. L., Voorhees J., Choate K., Nelson-Williams C., and Lifton R. P. (2002) KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc. Natl. Acad. Sci. U.S.A. 99, 10677–10682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gingras A. R., Puzon-McLaughlin W., and Ginsberg M. H. (2013) The structure of the ternary complex of Krev interaction trapped 1 (KRIT1) bound to both the Rap1 GTPase and the heart of glass (HEG1) cytoplasmic tail. J. Biol. Chem. 288, 23639–23649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nardozzi J. D., Lott K., and Cingolani G. (2010) Phosphorylation meets nuclear import: a review. Cell Commun. Signal. 8, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cavalcanti D. D., Kalani M. Y., Martirosyan N. L., Eales J., Spetzler R. F., and Preul M. C. (2012) Cerebral cavernous malformations: from genes to proteins to disease. J. Neurosurg. 116, 122–132 [DOI] [PubMed] [Google Scholar]
- 57.Riant F., Cecillon M., Saugier-Veber P., and Tournier-Lasserve E. (2013) CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics 14, 133–141 [DOI] [PubMed] [Google Scholar]
- 58.McDonald D. A., Shi C., Shenkar R., Gallione C. J., Akers A. L., Li S., De Castro N., Berg M. J., Corcoran D. L., Awad I. A., and Marchuk D. A. (2014) Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum. Mol. Genet. 23, 4357–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tozer E. C., Liddington R. C., Sutcliffe M. J., Smeeton A. H., and Loftus J. C. (1996) Ligand binding to integrin αIIbβ3 is dependent on a MIDAS-like domain in the β3 subunit. J. Biol. Chem. 271, 21978–21984 [DOI] [PubMed] [Google Scholar]
- 60.Edelstein A., Amodaj N., Hoover K., Vale R., and Stuurman N. (2010) Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol. 10.1002/0471142727.mb1420s92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu C., Wehr D. R., Edwards J., and Hauge B. (2008) Rapid one-step recombinational cloning. Nucleic Acids Res. 36, e54. [DOI] [PMC free article] [PubMed] [Google Scholar]