

Abstract
The discovery of brown adipose tissue (BAT) as a key regulator of energy expenditure has sparked interest in identifying novel soluble factors capable of activating inducible BAT (iBAT) to combat obesity. Using a high content cell-based screen, we identified fibroblast growth factor 16 (FGF16) as a potent inducer of several physical and transcriptional characteristics analogous to those of both “classical” BAT and iBAT. Overexpression of Fgf16 in vivo recapitulated several of our in vitro findings, specifically the significant induction of the Ucp1 gene and UCP1 protein expression in inguinal white adipose tissue (iWAT), a common site for emergent active iBAT. Despite significant UCP1 up-regulation in iWAT and dramatic weight loss, the metabolic improvements observed due to Fgf16 overexpression in vivo were not the result of increased energy expenditure, as measured by indirect calorimetric assessment. Instead, a pattern of reduced food and water intake, combined with feces replete with lipid and bile acid, indicated a phenotype more akin to that of starvation and intestinal malabsorption. Gene expression analysis of the liver and ileum indicated alterations in several steps of bile acid metabolism, including hepatic synthesis and reabsorption. Histological analysis of intestinal tissue revealed profound abnormalities in support of this conclusion. The in vivo data, together with FGF receptor binding analysis, indicate that the in vivo outcome observed is the likely result of both direct and indirect mechanisms and probably involves multiple receptors. These results highlight the complexity of FGF signaling in the regulation of various metabolic processes.
Keywords: adipogenesis, bile acid, fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), intestinal metabolism, metabolism
Introduction
In recent years, the hypothesis that brown adipose tissue (BAT)4 may provide therapeutic potential to counteract obesity sparked a surge of research to better understand BAT biology and identify means to pharmacologically promote BAT-induced thermogenesis in vivo. BAT is specialized for non-shivering thermogenesis to dissipate energy as heat via the expression of a mitochondrial protein called uncoupling protein 1 (UCP1) (1). Although the distribution of BAT differs among species, years of research now support BAT as a significant regulator of energy expenditure in mammals, including humans (2). Accordingly, in mice, BAT-mediated thermogenesis suppresses high fat diet-induced obesity (3–5), and in humans the presence of active BAT inversely correlates with adiposity (6, 7). In response to stimuli such as cold or β-adrenergic signals, clusters of UCP1-expressing adipocytes can be found within the subcutaneous WAT depots of adult mice (6, 8). These adaptive cells have been described as “beige” or “brite” cells (also called inducible BAT (iBAT)) (9). iBAT can now be distinguished from classical BAT by the expression of a growing number of specific transcription factors and the enrichment of several genetic markers (9–11). Evidence for iBAT in humans was identified using 18F-fluorodeoxyglucose positron emission tomography and computed tomographic (PET-CT) imaging and biopsy analysis studies (6, 12–14). Thus, the identification of therapeutic targets to activate BAT and iBAT in obese individuals to drive weight loss through increased energy expenditure has gained attention in both the basic research and drug discovery communities.
In an effort to identify the genes encoding proteins capable of promoting adipocyte commitment and differentiation toward the iBAT profile, the mouse mesenchymal stem cell-like line C3H/10T1/2 was transiently transfected with cDNA libraries enriched for putative soluble factors. We performed a high content imaging screen in these cells to identify factors that would modulate adipogenesis, as monitored by mitochondrial abundance and neutral lipid accumulation. We subjected outliers from the screen to a highly multiplexed expression analysis including many markers of the transcriptional programs of brown and white adipocytes. We identified several genes encoding soluble proteins capable of inducing iBAT-like morphological and transcriptional profiles similar to bone morphogenetic protein (BMP4), a soluble factor with reported brite cell or iBAT-like differentiation abilities (15, 16). Among the top hits was fibroblast growth factor 16 (FGF16), one of the 22 members of the FGF family. FGF are secreted proteins that play a wide role in developmental cell processes, including differentiation and proliferation, but also drive regulatory, morphological, and endocrine effects (17). A paracrine FGF, human FGF16, is 98.5% identical at the amino acid level to rodent Fgf16 (18).
In humans, FGF16 deficiency is linked to a metacarpal 4–5 fusion defect with no other overt symptoms (19–22). In rodents, the severity of Fgf16 deficiency appears to be strain-dependent in that only minimal effects manifest on a C57BL/6 background, whereas Fgf16-deficient 129 × Swiss Black mice are embryonic lethal; the latter rescued by backcrossing to the C57BL/6 background (23, 24). In adult rodents, Fgf16 is expressed predominantly on cardiomyocytes (25) and is correlated with heart function, as evidenced by the observation that overexpression of Fgf16 via adeno-associated virus can partially rescue cryoinjury-induced cardiac hypertrophy (26).
In contrast to the adult expression profile, the examination of rat embryos at different stages of embryogenesis revealed Fgf16 expression in classical BAT depots during the latter stages of development, reaching the highest expression around stage embryonic day 20.5 and then tapering off by postnatal day 1 (27). During this time frame, maximal expression of Fgf16 was inversely correlated with Ucp1, with Ucp1 being strongly expressed at postnatal day 1. Furthermore, 8-week-old rats exposed to cold temperatures for a week exhibited increased Ucp1 expression in BAT tissue, whereas expression of Fgf16 was virtually absent. Thus, along with data demonstrating the mitogenic capacity of recombinant Fgf16 on primary rat adipocytes, the authors concluded Fgf16 was a potent embryonic BAT mitogen, with no significant post-natal role in BAT (27). However, the potential of increased expression of FGF16 to induce iBAT in adult mice has not been reported. Because our screen indicated Fgf16 was a potent driver of adipogenic differentiation in vitro, we used adeno-associated virus (AAV) to overexpress Fgf16 in adult mice (FGF16 mice) and determine whether diet-induced obese (DIO) metabolic phenotypes can be reversed via an Fgf16 iBAT-driven mechanism. Although strong metabolic improvements were observed, including increased Ucp1 gene expression and UCP1 protein expression in the inguinal WAT (iWAT) depots, the dramatic weight loss observed was not a result of increased energy expenditure in FGF16 mice. Instead, the metabolic changes observed appear to be the result of reduced food intake and changes in bile acid synthesis and lipid absorption.
Results
An in Vitro High Content Screen Identifies FGF16 as an Adipogenic Factor
To identify the factors that modulate adipocyte commitment and differentiation, the mouse mesenchymal stem cell-like line C3H/10T1/2 was transiently transfected with a collection of cDNA encoding putative secreted factors. After incubation with cDNA for 72 h, an adipogenic differentiation medium (ADM) was introduced and was then refreshed 48 h later (Fig. 1A). After an additional 48 h, the cells were stained, fixed, and imaged.
FIGURE 1.

A high content cell-based screen reveals FGF16 as an adipogenesis activator. A, a schematic describing the cell assay protocol. Cells were transfected with cDNA in 384-well format and treated with ADM after 72 h. After a second medium exchange and a total of 96 h of differentiation, cells were fixed and stained with Hoechst (nucleus), LipidTox Green (neutral lipid), and MitoTracker Deep Red (mitochondria) before imaging at ×20 on a PerkinElmer Opera QEHS. B–G, representative images of cells transfected with plasmid containing an empty vector neutral control, WNT3A, BMP4, or FGF16. B, D, and F, ADM negative media. C, E, G, and H, ADM positive media. I, outliers from the primary screen were technically validated by retesting and expression analysis by Fluidigm 96.96 dynamic arrays. J, linear correlation between screen (x axis) and validation (y axis) hits. K, the scatter plot depicts a principal component feature reduction of image and expression data for the clones and controls. Highlighted clones include fibroblastic mitogens (blue, such as WNT3A), neutral controls (clear, including empty vector controls), and pro-adipogenic factors (red, including BMP4).
In the absence of ADM, C3H/10T1/2 cells do not undergo significant adipogenic differentiation, as evidenced by the failure to develop lipid droplets (Fig. 1B). After 96 h of treatment with ADM, ∼20–30% of the cells transfected with a vector-only control develop into round, lipid droplet-containing cells resembling mature adipocytes (Fig. 1C). The treatment of cells with or without ADM but transfected with wingless-type murine mammary tumor virus (MMTV) integration site family member 3A (WNT3A), which is both a mitogen (28, 29) and a potent osteogenic factor (30–32), resulted in ∼50% more cells than neutral controls but little or no adipogenic differentiation (compare Fig. 1, D and E with B and C). In contrast, in the presence of ADM and a pro-adipogenic factor such as BMP4 (15, 33), ∼40% of the cells can be seen to have differentiated within the same time frame (compare Fig. 1, F and G with B and C). In addition, BMP4 is a mitogen, resulting not only in an increased percentage of differentiated adipocytes but a larger number of them as well (Fig. 1G). The inset image highlights, at greater magnification, the co-localization staining pattern of both abundant mitochondria and lipid in cells treated with ADM and BMP4 (Fig. 1G).
We extracted 36 features from individual images using the Columbus image analysis software suite and aggregated transfection replicates, and we performed quality control using the Genedata Screener software package. From aggregate values we identified outliers for individual features as well as multivariate metrics. As would be expected from the overexpression of potent, soluble factors in multipotent cells, there was a large number of outliers by each of these measurements. We focused our attention on 365 clones, representing 352 unique Entrez Genes, detected as hits by at least two methods for technical and orthogonal validation (Fig. 1I). Technical validation was performed in a manner similar to the primary screen, although the number of features extracted was reduced (Table 1). To assess the validation rate, each clone was characterized as in the primary screen, and an arbitrary scoring scheme was applied such that each outlying image-based value was accorded 1 point, each outlying principal component value was accorded 2 points, and 10 points were accorded for an outlying jackknife value. Each clone in the primary screen was scored similarly, and in general, higher scores in the primary screen correlated with higher validation scores (Fig. 1J, Spearman's rank-order correlation, (r) = 0.4519).
TABLE 1.
Phenotypic features extracted from imaging analysis
Mito, mitochondia; SER, Saddle-Edge-Ridge; px, pixel.
| Phenotypic features | Screen | Validation |
|---|---|---|
| Nuclei | ||
| Number of objects |  |
|
| Cell area (μm2), mean/well | |
|
| Cell roundness, mean/well | |
|
| Mito intensity mean, mean/well | |
|
| Mito intensity sum, mean/well | |
|
| Lipid intensity mean, mean/well | |
|
| Lipid intensity sum, mean/well | |
|
| Total spot area, mean/well | |
|
| Relative spot to background intensity, mean/well | |
|
| Number of spots, mean/well | |
|
| Mito SER spot 1 px, mean/well | |
|
| Mito SER hole 1 px, mean/well | |
|
| Mito SER edge 1 px, mean/well | |
|
| Mito SER ridge 1 px, mean/well | |
|
| Mito SER valley 1 px, mean/well | |
|
| Mito SER saddle 1 px, mean/well | |
|
| Mito SER bright 1 px, mean/well | |
|
| Mito SER dark 1 px, mean/well | |
|
| Lipid SER spot 2 px, mean/well | |
|
| Lipid SER hole 2 px, mean/well | |
|
| Lipid SER edge 2 px, mean/well | |
|
| Lipid SER valley 2 px, mean/well | |
|
| Lipid SER saddle 2 px, mean/well | |
|
| Lipid SER dark 2 px, mean/well | |
|
| Lipid spots | ||
| Number of objects | |
|
| Relative spot to background intensity, mean/well | |
|
| Corrected spot intensity, mean/well | |
|
| Uncorrected spot peak intensity, mean/well | |
|
| Spot contrast, mean/well | |
|
| Spot background intensity, mean/well | |
|
| Spot area (px2), mean/well | |
|
| Region intensity, mean/well | |
|
| Intensity spot Channel 3 mean, mean/well | |
|
| Intensity spot Channel 3 sum, mean/well | |
|
| Spot to region intensity, mean/well | |
|
| Spot roundness, mean/well | |
|
The highest scoring 336 clones, plus 12 controls, were further characterized by qPCR using the Fluidigm 96.96 dynamic array platform. Gene expression levels for 88 markers of cell fate, in addition to eight housekeeping controls for normalization, were assessed. The 72 of 88 assays that had detectable expression (Ct < 28) in at least 10 samples combined with the imaging features were subjected to feature reduction by principal component analysis. A scatterplot depicting the first two principal components, representing 27 and 13% of the total variations in the data for the 336 clones, plus highlighted controls, demonstrates the breadth of adipogenic differentiation phenotypes observed in the screen (Fig. 1K) By several measures, the strongest adipogenic factor identified in the screen was FGF16 (Fig. 1H).
Although previous studies identified Fgf16 as a potent BAT mitogen during embryogenesis (27), we were unable to find published data describing a role for Fgf16 in adult BAT or WAT. Thus, because of the profile of Fgf16 from the high content cell-based screen, and the previously known role for Fgf16 in embryogenesis, we sought to more closely examine Fgf16 in the adult DIO mouse model and evaluate whether overexpression of Fgf16 in this context could induce metabolic effects via an iBAT-mediated mechanism.
Overexpression of Fgf16 in Obese Mice Significantly Improves Metabolic Phenotypes and Induces UCP1-expressing Adipocytes in WAT
Using AAV as a gene delivery method, we treated B6-DIO male mice with a single intravenous injection of AAV-Fgf16 or with an empty vector (EV) as a negative control. By 4 weeks post-injection, the body weight of FGF16 mice was substantially lower than that of EV mice (Fig. 2A). Accordingly, fasting blood glucose levels were lowered significantly compared with the control group (Fig. 2B). At 3.5 weeks post-AAV injection, mice were fasted and challenged with a single bolus of glucose to evaluate changes in glucose tolerance. FGF16 mice exhibited a dramatic improvement in glucose tolerance compared with EV mice (Fig. 2C). Serum analysis at this time revealed a significant decline in the fasting insulin levels of FGF16 mice (Fig. 2D), suggesting Fgf16 overexpression in DIO mice improves insulin resistance, as predicted from the dramatic weight loss observed. Upon harvest we confirmed increased Fgf16 expression in the liver, in both brown and white adipose tissue, and the ileum, compared with the vehicle group (Fig. 2E). Although the overall levels of Ucp1 message were not changed in the BAT of FGF16 mice, Ucp1 expression was significantly increased in the iWAT of FGF16 mice (Fig. 2F), an indication of iBAT. Immunohistochemical comparison for UCP1 protein expression in the adipose tissue from FGF16 mice versus EV mice not only revealed a significant reduction in the size of adipocytes in both BAT and iWAT but also showed increased instances of clusters of UCP1+ areas in the iWAT of FGF16 mice (Fig. 2, G–J). Altogether, these results demonstrate that overexpression of Fgf16 in B6-DIO mice results in up-regulated gene and protein expression of the principle brown adipocyte regulator, UCP1, in iWAT, thus promoting an iBAT phenotype in vivo.
FIGURE 2.

Overexpression of Fgf16 in vivo reduces body weight, improves glucose metabolism, and induces Ucp1 expression. C57BL/6 DIO mice were injected with AAV containing either Fgf16 (blue circles) or EV (red circles). Body weight (A) and fasting blood glucose levels (B) were measured at time 0 and 2 and 4 weeks post-AAV injection. n = 10 animals/group; data are presented for each animal with the average and S.D. for each group indicated. Statistical significance was determined by two-way ANOVA. ****, p < 0.0001. C, a glucose tolerance test was performed 4 weeks post-AAV injection. Average values and S.D. for blood glucose levels at 0, 15, 30, and 60 min post-intraperitoneal injection of glucose. n = 10 animals/group; statistical significance was determined by two-way ANOVA. **, p = 0.002; ****, p < 0.0001. D, fasting serum insulin levels were determined at 4 weeks post-AAV injection. n = 10 animals/group; data are presented for each animal with the average and S.D. for each group indicated. Statistical significance was determined by an unpaired 2-tailed t test with Welch's correction; ****, p < 0.0001. E, qPCR analysis for Fgf16 mRNA expression in liver, BAT, iWAT, eWAT, and ileum from FGF16 mice versus EV mice. n = 6 animals/group; data are presented for each animal with the average and S.D. for each cohort indicated. Statistical significance was determined by two-way ANOVA. ****, p < 0.0001. F, qPCR analysis for Ucp1 mRNA expression in BAT and iWAT from FGF16 mice versus EV mice. n = 6 animals/group; data are presented for each animal with the average and S.D. for each cohort indicated. Statistical significance was determined by two-way ANOVA. ****, p < 0.0001. G and H, immunohistochemical analysis of UCP1 expression in BAT from EV mice versus FGF16 mice, respectively. I and J, immunohistochemical analysis of UCP1 expression in iWAT from EV mice versus FGF16 mice, respectively. Images are representative of 6 animals/cohort.
The Fat Mass Reduction Associated with Fgf16 Overexpression Is Independent of Energy Expenditure
To determine whether the significant up-regulation of Ucp1 gene and UCP1 protein expression and body weight loss correlated with altered energy expenditure, we employed several measures, including indirect calorimetry, to more thoroughly characterize FGF16 mice versus EV mice. Using an Oxymax Comprehensive Lab Animal Monitoring System (CLAMS), the mice were acclimated for 2 weeks in metabolic cages and then randomized into groups based on body weight, body composition analysis, and fasting serum lipid analysis. On the first day of the study, mice received a single injection of AAV containing either FGF16 or EV. Thereafter, the body weight of each mouse was recorded every day (Fig. 3A). By 6 days post-AAV injection, FGF16 mice exhibited a significant reduction in body weight compared with EV mice. At this point, the system was recalibrated, and metabolic parameter recordings were initiated. For each animal, both light and dark cycle measurements, at 12-minute increments, were recorded for 6 complete days. The results represent the average values observed for the animals within a group for both the light and dark cycles per day. Although no significant change was observed between the FGF16 mice and EV mice in the rate of oxygen consumption (VO2) or the rate of carbon dioxide production (VCO2) during the course of the day (Fig. 3, B and C, respectively), the overall respiratory exchange ratio differed significantly between the FGF16 mice and EV mice during both the light and dark cycles (Fig. 3D). Consistent with the observed VO2 and VCO2 levels, no change in heat production (energy expenditure) was observed between cohorts (Fig. 3E). Analysis of both vertical motion and horizontal motion (Fig. 3F) revealed a significant lack of movement in FGF16 mice during the active dark cycle. Furthermore, FGF16 mice consumed less water and less food (Fig. 3G) during this time frame.
FIGURE 3.

Fgf16 overexpression in vivo reduces body weight independently of increased energy expenditure. A, daily body weight changes for EV mice (red circles) versus FGF16 mice (blue circles). n = 6 animals/group. The average and S.D. for each group is indicated for each day post-AAV injection. Statistical significance was determined by two-way ANOVA. *, p = 0.03; ***, p = 0.003; ****, p < 0.0001. B–F, indirect calorimetry analysis of EV mice (red bars) versus FGF16 mice (blue bars) for VO2, VCO2, respiratory exchange ratio, rate of heat production, vertical (rearing) motion, and x axis IR beam breaks, respectively, collected between days 6 and 13 post-AAV injection. Data from each animal were collected at 12-min intervals and averaged, and then values from each cohort were averaged for light versus dark cycles. Statistical significance was determined by one-way ANOVA and a Sidak's multiple comparison test.*, p = 0.03; ****, p < 0.0001. NS, not significant. G, cumulative water consumption measured from days 6–13 post-AAV injection. n = 6 animals/group; statistical significance was determined by an unpaired 2-tailed t test with Welch's correction. *, p < 0.03. Daily average food consumption was measured from EV mice (red bars) and FGF16 mice (blue bars) under normal housing conditions between days 6 and 10 post-AAV injection. n = 16 animals/cohort; statistical significance was determined by an unpaired two-tailed Student's t test with Welch's correction. ****, p < 0.0001. H–J, 15 days post-AAV injection, EV mice (red bars) and FGF16 mice (blue bars) were weighed, and body mass composition was determined using NMR analysis. Percent body composition for fat mass, fluid mass, and lean mass (upper panels) and total fat, fluid, and lean mass (grams) (lower panels) are shown. For each cohort, n = 6 mice were housed under the CLAMS cage system, and n = 4 mice were housed in standard cages. Values collected from both AAV cohorts were averaged. Statistical significance was determined by two-way ANOVA. ***, p = 0.0004 (I) and 0.0008 (J); ****, p < 0.0001. K, 14 days post-AAV injection, fasting serum leptin and adiponectin levels were determined. L, upon harvest, the amount of triglyceride per gram of liver and glycogen content per gram of liver were determined for EV mice (red bars) and FGF16 mice (blue bars). M, representative images of feces collected from FGF16 mice and EV mice, with similar magnification used for both images. N, fasting serum cholesterol, triglyceride, and NEFA levels. O, fecal cholesterol, triglyceride, NEFA, and bile acids from EV mice (red bars) and FGF16 mice (blue bars). n = 10 animals/group (K–O). Statistical significance was determined by an unpaired 2-tailed t test with Welch's correction. *, p = 0.02 and ***, p = 0.003 (O); ****, p ≤ 0.0001 (K, L, and O).
On day 14 post-AAV injection, mice were analyzed by NMR for body mass composition. FGF16 mice exhibited a significant decrease in fat as a measure of both percent body composition (Fig. 3H, top panel) and as total fat mass (bottom panel). A similar trend was detected for fluid as a measure of percent body composition (Fig. 3I, top panel) and as total fluid mass (bottom panel). Although the percent lean mass composition increased in FGF16 mice (Fig. 3J, top panel), the actual lean mass decreased (bottom panel) compared with EV mice. Altogether, these results suggest that Fgf16 overexpression, despite iBAT induction, leads to a loss in fat mass in the absence of obvious changes in energy expenditure.
Fgf16 Overexpression Results in Significant Lipid Excretion
Despite significant weight loss, FGF16 mice generally appeared normal during each study performed, particularly during the light cycle when their activity levels paralleled that of EV mice. Fasting serum leptin and adiponectin levels were significantly reduced in FGF16 mice (Fig. 3K), consistent with decreased fat mass. The level of triglycerides and glycogen (Fig. 3L) per gram of liver was markedly reduced.
One striking difference was the appearance of feces from FGF16 mice; their stools appeared darker and oilier than feces from EV mice, which were typically pale and dry (Fig. 3M). Although analysis of fasting serum from FGF16 mice indicated reduced cholesterol levels, there was no change in triglycerides or non-esterified fatty acids (NEFA) (Fig. 3N) compared with EV mice. In contrast, feces from FGF16 mice contained significantly higher concentrations of cholesterol, triglycerides, and NEFA compared with EV mice (Fig. 3O). Furthermore, the amount of bile acids in the feces was dramatically increased (Fig. 3O). Thus, these findings suggest that Fgf16 overexpression may affect lipid and bile acid synthesis, excretion, and or absorption. To understand the underlying mechanism for these changes, gene expression analysis was carried out in the major metabolic tissues.
Gene Expression Analysis Reveals Alteration in Lipid and Bile Acid Metabolism Post-FGF16 Treatment
As shown by qPCR (Fig. 2F), Ucp1 gene expression increased significantly in the iWAT of FGF16 mice. Additionally, BAT cell markers, Cidea and Ppargc1a, were also increased in iWAT (Fig. 4A). The regulation of these genes appeared to be specific to iWAT, in that expression was unchanged in the epididymal WAT (eWAT) of FGF16 mice (Fig. 4B). Consistent with reduced fat mass in FGF16 mice, gene expression for the enzymes regulating triacylglycerol synthesis and lipid droplet formation, diglyceride acyltransferases 1 and 2 (Dgat1 and Dgat2), and acetyl-CoA carboxylase (Acaca) were significantly reduced in eWAT, as well as the genes encoding lipoprotein lipase (Lpl) (Fig. 4B). Dgat2 and Lpl were also significantly reduced in iWAT (Fig. 4A).
FIGURE 4.

Expression profiles of relevant genes involved in Fgf16 signaling, iBAT activation, bile acid metabolism, and lipid absorption. Two weeks post-AAV injection, iWAT (A), eWAT (B), liver (C), ileum (D), and pancreas (E) tissue from EV mice (red bars) and FGF16 mice (blue bars) were collected and analyzed for gene expression changes. Data are presented as the average relative mRNA ± S.E. Statistical significance was determined using an unpaired Student's t test, correcting for multiple comparisons using the Holm-Sidak method with α = 5.000%. *, p < 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
Consistent with reduced liver triglyceride content, gene expression analysis of livers collected from FGF16 mice showed suppressed sterol regulatory element-binding transcription factor 1c (Srebp1c) and reductions in genes involved in gluconeogenesis and glycogenolysis, pyruvate carboxylase (Pcx, alias Pc), and glucose 6-phosphatase (G6p), consistent with reduced liver glycogen levels and improvement in overall glucose handling (Fig. 4C). Although there was a reduction in serum cholesterol levels, the expression of the enzyme responsible for the rate-limiting step in cholesterol synthesis, 3-hydroxy-3-methylglutaryl-CoA reductase (Hmgcr), was increased 2.7-fold with Fgf16 overexpression (Fig. 4C). Hepatic expression of the ATP-binding cassette subfamily G transporters 5 and 8 (Abcg5 and Abcg8), which heterodimerize to form a cholesterol transporter, also showed a significant increase in expression or a trend toward increased expression, respectively (Fig. 4C). Because liver is the site of bile acid synthesis, genes involved in maintaining bile acid homeostasis were also examined. A dramatic reduction in the expression of the principal genes encoding enzymes involved in both the classical and alternative bile acid synthesis pathways was observed, including cytochrome P450, family 7, subfamily A, polypeptide 1 (Cyp7a1), cytochrome P450, family 27, subfamily a, polypeptide 1 (Cyp27a1), and the associated downstream enzymes, cytochrome P450, family 8, subfamily b, polypeptide 1 (Cyp8b1), and cytochrome P450, family 7, subfamily b, polypeptide 1 (Cyp7b1). Nuclear receptor subfamily 1, group H, member 4 (Fxr, alias Nr1h4) was also reduced. Consistent with the reduced bile acid synthesis, expression of the bile salt export pump, Bsep (alias Abcb11), expressed at the canalicular membrane of hepatocytes, was down-regulated (Fig. 4C). Expression of transporters involved in portal bile acid uptake, Ntcp and Oatp4 (alias Slca10), showed a reduction in FGF16 mice, whereas Mrp4 (alias Abcc4), located on the basolateral membrane and important for systemic efflux of bile acids, was significantly increased (Fig. 4C).
In the ileum, the expression of basolateral bile acid transporters, the organic solute transporters Ost-α and Ost-β, were unchanged, whereas the apical solute carrier family 10, member 2, Slc10a2 (alias Asbt), responsible for bile acid absorption, showed a trend toward increased expression that did not reach statistical significance (Fig. 4D). Abcg5 and Abcg8 expression in the ileum was reduced in FGF16 mice (Fig. 4D). Thus, taken together, these data suggest alterations in several steps of bile acid metabolism, including hepatic synthesis and reabsorption.
Key pancreatic amylolytic, proteolytic, and lipolytic enzyme (34) expression was also examined. No significant changes were observed in FGF16 mice, except in chymotrypsin-like elastase family, member 2A (Cela2a) and pancreatic lipase-related protein 1 (Plrpl), which were both significantly reduced (Fig. 4E). This may suggest incomplete digestion of lipid-bile acid droplets, which could contribute to the increased lipid and bile acid excretion observed in the FGF16 mice.
Because liver FGF signaling plays a significant role in bile acid metabolism, FGFR expressions in the liver were also analyzed. FGF16 mice revealed a significant increase in Fgfr1 and Fgfr2 but not Fgfr3 or Fgfr4. There was no change in expression of Fgf21 or the co-receptor β-Klotho in FGF16 mice, suggesting that FGF16 overexpression did not induce endocrine FGF activity (Fig. 4C).
Pair Feeding Reveals an Effect of Fgf16 Overexpression-mediated Weight Loss Beyond Starvation and Independent of Leptin
Because a significant reduction in food intake was observed in the CLAMS study, pair-feeding studies were carried out to understand whether food intake reduction was primarily responsible for the observed metabolic changes. Thus, B6-DIO mice were subjected to pair-feeding analysis for 2 weeks. Ad libitum fed FGF16 mice were paired with EV mice (EV-PF), the latter of which received per day only the amount of food consumed by its FGF16 partner on the previous day. Ad libitum fed EV mice were included as a control for normal eating behavior. Food intake levels were recorded daily (Fig. 5A), and body weights were measured 4 days prior to AAV injection, the day of AAV injection (D1), and on days 4, 7, 11, and 15 post-AAV injection (Fig. 5B). NMR analysis was recorded 1 day prior to AAV injection and on day 15 (Fig. 5, C and D). Despite their similar food intake, the data revealed a noticeable further reduction in body weight in the FGF16 mice compared with their EV-PF counterparts, Comparing the total weights of fat mass, lean mass, and fluid mass, again FGF16 overexpression had a stronger effect on these parameters over the pair-fed counterparts.
FIGURE 5.

Pair-feeding studies reveal Fgf16 overexpression-mediated weight loss is distinct from reduced food intake and is leptin-independent. A–D, single-housed B6-DIO mice were injected with either AAV-Fgf16 or AAV-EV and subjected to pair feeding (PF), where the daily food amount given to the EV group (EV-PF, red circles) was adjusted to the food intake amount of ad libitum fed FGF16 mice (blue circles) from the previous day. A control cohort of EV mice fed ad libitum was included (black circles). A and B, mice were monitored every day for food intake and body weight, respectively. C and D, 15 days post-AAV injection, FGF16 mice (blue bars), EV-PF mice (red bars), and EV mice (black bars) were weighed, and body mass composition was determined using NMR analysis. C, total fat, fluid, and lean mass (grams) are shown, respectively. D, percent body composition for fat mass, fluid mass, and lean mass are shown, respectively. E–H, in parallel to the B6-DIO mice, a PF study was conducted on B6.V-Lepob/J mice. The mice were harvested after only 11 days post-AAV injection because of significant fecal impaction observed in 11 of the 12 FGF16 mice. For each cohort, n = 12 mice. The values collected from cohorts were averaged. A–H, statistical significance was determined by two-way ANOVA.*, p < 0.01; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. NS, not significant.
To determine whether the reduction in food intake was via a leptin-dependent mechanism, pair-feeding analysis was also performed on B6.V-Lepob/J mice. Because of an unforeseen presentation of profound fecal impaction in 11 of the 12 mice in the FGF16 cohort, the study was terminated after only 11 days. In this time, however, we observed significant food reduction in the ad libitum fed FGF16 mice compared with the ad libitum fed EV mice (Fig. 5E). Similar to B6-DIO mice, by 1 week post-AAV injection, a stronger reduction in body weight was observed in the FGF16 mice compared with the EV-PF mice, which persisted until day 11 (Fig. 5F), thus suggesting a Fgf16-mediated weight loss mechanism independent of leptin and beyond food intake alone. NMR analysis at day 11 indicated a small but significant difference in both fat mass and lean mass between FGF16 mice and EV-PF mice (Fig. 5, G and H).
Plasma and fecal lipid levels were also measured. Compared to the EV group, in B6-DIO mice the FGF16 group showed no significant changes in plasma TG or NEFA levels but a significant reduction in cholesterol levels, while in B6.V-Lepob/J mice, the FGF16 group showed no significant changes in cholesterol or NEFA levels, and a significant increase in plasma triglyceride levels (Fig. 6, A–C, H–J). However, in both models, effects on both TG and cholesterol were different between FGF16 and pair-fed groups. Similar to the CLAMS study shown in Fig. 3, in B6-DIO, feces from FGF16 mice contained higher amounts of triglyceride and cholesterol, whereas the pair-fed group showed no difference from the ad libitum EV group (Fig. 6, F and G). The fecal impaction observed in the B6.V-Lepob/J mice hindered attempts to collect enough feces for analysis; however, visual inspection of the bedding from the B6.V-Lepob/J cages indicated a similar phenomenon of darker, oilier feces among the FGF16 mice compared with EV-PF and EV mice.
FIGURE 6.

Fgf16 overexpression results in significant lipid and bile acid excretion. Upon harvesting, serum from the PF B6-DIO and PF B6.V-Lepob/J cohorts was evaluated for lipid and liver enzyme levels. A–E, triglyceride, cholesterol, NEFA, ALT, and AST levels from the B6-DIO cohorts. F and G, feces collected from the B6-DIO cohorts were evaluated for triglyceride and cholesterol content. H–L, triglyceride, cholesterol, NEFA, ALT, and AST levels from the B6.V-Lepob/J cohorts. A–L, n = 10–12 animals/group. Statistical significance was determined by unpaired 2-tailed t test with Welch's correction. *, p < 0.01; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. M–Q, upon harvesting, tissues and feces from the PF B6-DIO and PF B6.V-Lepob/J cohorts were processed and evaluated for total bile acid content. M, the total bile acid content from liver (L), gall bladder (GB), small intestine (SI), and colon (C) from the PF B6-DIO cohorts was determined individually and presented as the average amount of each tissue per body weight (top panel) or per animal (bottom panel). N, for each cohort the average amount of bile acid pool (liver, gall bladder, and small intestine combined) was calculated per body weight (top panel) and per animal (bottom panel). O, evaluation of total bile acid content from feces collected from the PF B6-DIO cohorts shows significant bile acid excretion in FGF16 mice compared with EV-PF and EV mice; data indicate total bile acid content per gram of feces (top row) and per animal (bottom row). n = 6 animals/group for tissues and n = 10/group for feces. P, the total bile acid content for each tissue type per gram of body weight (top panel) and per animal (bottom panel) from the PF B6.V-Lepob/J cohorts. Q, bile acid pool (liver, gall bladder, and small intestine combined) per body weight (top panel) and per animal (bottom panel) from the PF B6.V-Lepob/J cohorts. M–Q, n = 6 animals/group for tissues. Statistical significance was determined by unpaired 2-tailed t test with Welch's correction; *, p < 0.01; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. NS, not significant.
Because perturbation of bile acid metabolism was observed in the CLAMS study, the effects of FGF16 on bile acid levels from various tissues were also examined in the pair-feeding studies. The bile acid content from liver, gallbladder, small intestine, colon, and feces was determined and calculated based on amount per body weight (Fig. 6, M–Q, top panels) and amount per animal (Fig. 6, M–Q, bottom panels). In both B6-DIO and B6.V-Lepob/J mice, Fgf16 overexpression led to a drastic reduction in bile acids across tissues, which led to a strong reduction in bile acid pool (including liver, gallbladder, and small intestine; Fig. 6, N and Q). Total bile acid content was also determined for the feces samples from the B6-DIO cohorts. The data showed that, in addition to massive lipid excretion, Fgf16 overexpression also resulted in massive bile acid excretion (Fig. 6O). In contrast, bile acid levels in pair-fed mice showed either no change or an increase compared with the ad libitum EV group. These results again suggest that the effects of Fgf16 overexpression on lipid and bile acid homeostasis are not due simply to changes in food intake.
Gene expression analysis was also performed on tissue from the pair-feeding experiments. Although the effects of FGF16 overexpression were largely consistent with those observed from the CLAMS study (compare Fig. 7 with Fig. 4), for example, increased Ucp1 expression in iWAT and suppression of the bile acid synthesis genes Cyp7a1, Cyp27a1, Cyp7b1, and Cyp8b1 in the liver, they were distinct from those of the pair-fed group, which showed either no change to the ad libitum EV group or an effect opposite of FGF16 (Figs. 7 and 8). These results further suggest that the effects observed with Fgf16 overexpression were not solely the result of reduced food intake.
FIGURE 7.

Expression profiles of relevant genes involved in Fgf16 signaling, iBAT activation, and lipid and bile acid metabolism from PF B6-DIO cohorts. Two weeks post-AAV injection, iWAT (A), eWAT (B), liver, (C), ileum (D), and pancreas (E) tissues from FGF16 mice (blue bars), EV-PF mice (red bars), and EV mice (black bars) were collected and analyzed for gene expression changes. All tissues from each cohort were also analyzed for Fgf16 expression (F). Data are presented as the average relative mRNA ± S.E. Statistical significance was determined using an unpaired Student's t test, correcting for multiple comparisons using the Holm-Sidak method, with α = 5.000%. *, p < 0.01; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
FIGURE 8.

Expression profiles of relevant genes involved in Fgf16 signaling, iBAT activation, and lipid and bile acid metabolism from PF B6.V-Lepob/J cohorts. Eleven days post-AAV injection, iWAT (A), eWAT (B), liver, (C), ileum (D), and pancreas (E) tissues from FGF16 mice (blue bars), EV-PF mice (red bars), and EV mice (black bars) were collected and analyzed for gene expression changes. All tissues from each cohort were also analyzed for Fgf16 expression (F). Data are presented as the average relative mRNA ± S.E. Statistical significance was determined using an unpaired Student's t test, correcting for multiple comparisons using the Holm-Sidak method, with α = 5.000%. *, p < 0.01; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
In addition to serum lipid levels, serum from each cohort was analyzed for alanine and aspartate aminotransferase activity (ALT and AST, respectively) to assess possible induction of liver damages. Fgf16 overexpression in B6-DIO mice revealed a modest 2-fold increase in both ALT and AST levels compared with EV-PF mice and EV mice (Fig. 6, D and E), whereas no differences among cohorts in the B6.V-Lepob/J mice were observed (Fig. 6, K and L).
Receptor Specificity for FGF16 Suggests an Ability to Activate Multiple FGFRs
To understand which receptors may contribute to the effects observed with FGF16, we next evaluated the receptor specificity of FGF16. The rat myoblast cell line L6, which expresses very low levels of endogenous FGF receptors, was transfected with one of the seven major FGFR. Receptor activation was determined by analysis of phospho-ERK levels using a Meso Scale Discovery (MSD) system. In this artificial system, except for FGFR1b, recombinant FGF16 activated all of the receptors to varying degrees, with the highest activity observed on FGFR2c (Fig. 9D). Although weaker, significant activity was also observed on FGFR4 by FGF16 treatment (Fig. 9G), consistent with a previous report (35).
FIGURE 9.

Receptor specificity for FGF16. A–G, L6 cells were transfected with expression vectors for FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c, and FGFR4, respectively. Transfected cells were then treated with increasing doses of recombinant FGF16 protein (0.30, 0.11, 0.33, 1.0, 3.0, and 9.0 nm, open squares) or FGF1 protein as a positive control (1 nm, open diamonds). Cell lysates were prepared for an MSD assay to measure pERK/total ERK, and data are presented as % phosphoprotein. Cells transfected with vector only were used as a negative control (open circles).
Overexpression of Fgf16 in Obese Mice Results in Profound Intestinal Abnormalities
To visually identify intestinal changes that may be associated with Fgf16 overexpression, tissues from B6-DIO FGF16 mice and EV mice were collected and analyzed for histologic abnormalities. Representative images for EV mice (Fig. 10, A, C, and E) and FGF16 mice (Fig. 10, B, D, and F) are shown. Together, Masson's trichrome stain, which identifies collagen deposits, and Alcian blue, which identifies acidic mucins, revealed a pronounced disparity between the cohorts in that ileum from FGF16 mice display abnormal amounts of extracellular matrix components in the lamina propria (Fig. 10, A–D). The yellow arrowheads (Fig. 10B) indicate areas of excessive collagen deposition, whereas the black arrowheads (D) highlight excessive deposition of sulfated and carboxylated acid mucopolysaccharides and sialumucins within the lamina propria. Throughout the ileum of FGF16 mice, instances of crypt dropout, or crypt degeneration/abscesses, were also detected (Fig. 10D, red arrow). The periodic acid-Schiff stain, used to identify glycogen, did not reveal any notable difference between cohorts (Fig. 10, E and F). Thus, histopathological analysis of small intestine tissue from FGF16 mice versus EV mice revealed profound morphological abnormalities as the result of Fgf16 overexpression, indicating that these differences may significantly affect intestinal function, including digestion, transport, and absorption.
FIGURE 10.
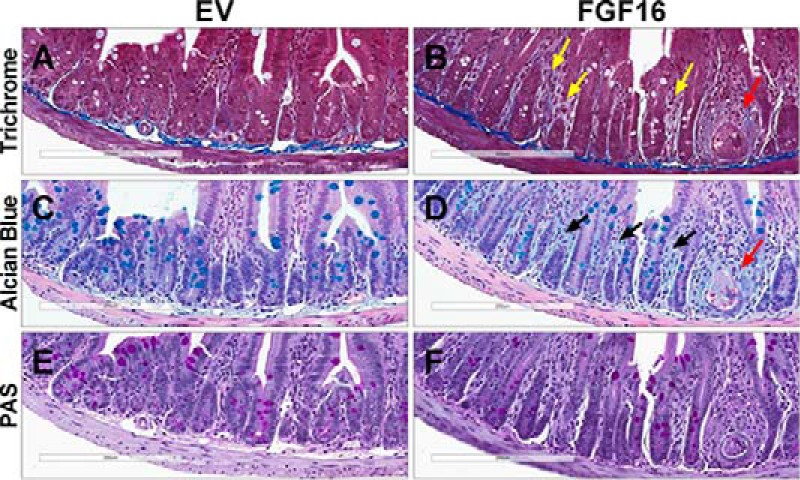
Fgf16 overexpression results in intestinal abnormalities. A–F, histopathological analysis of ileum collected from EV mice (A, C, and E) and FGF16 mice (B, D, and F). A and B, Masson's trichrome staining identifies irregular collagen deposits within the lamina propria of FGF16 mice (yellow arrowheads). C and D, Alcian blue identifies increased deposition of acidic mucins within the lamina propria of FGF16 mice (black arrowheads). B and D, ileum from FGF16 mice also exhibit instances of crypt degeneration/abscesses (red arrowheads). E and F, no overt difference in periodic acid-Schiff staining was observed in ileum tissue from EV mice versus FGF16 mice. Images are representative of n = 8 animals/cohort.
Discussion
Over the last several years, the identification of pro-adipogenic targets that promote iBAT activation, increase energy expenditure, and subsequently promote weight loss has remained center stage as a possible therapeutic strategy for treating obesity. Using a high content cell-based screen to identify pro-adipogenic soluble factors, we identified Fgf16 as a factor that significantly increased C3H/10T1/2 adipogenic differentiation in vitro and induced several features consistent with an iBAT-like phenotype. Consistent with the cell screening data, overexpression of Fgf16 in adult B6-DIO mice promoted increased Ucp1 message and UCP1 protein expression in iWAT, supporting the hypothesis of Fgf16 as an iBAT inducer.
In addition to UCP1 induction, overexpression of Fgf16 in B6-DIO mice resulted in significant decreases in body weight and blood glucose levels, a robust improvement in glucose tolerance, and decreased fasting serum insulin levels. Although these phenotypes are consistent with the hypothesis that FGF16 could be beneficial for obesity and diabetes, indirect calorimetry analysis revealed that, despite a dramatic increase in UCP1 in iWAT and a decrease in fat mass, energy expenditure was not increased in FGF16 mice. In fact, a reduction in the respiratory exchange ratio was observed with no detectable change in heat production. Measurement of both water and food consumption during the time of rapid body weight loss suggested a phenotype more consistent with starvation. The observed decrease in vertical rearing and x axis motions during the dark cycle was also consistent with starvation.
To understand whether reduced food intake was primarily responsible for the observed metabolic changes, pair-feeding studies were performed in both B6-DIO and B6.V-Lepob/J mice, the latter included also to assess whether a leptin-dependent mechanism was involved. Pair feeding revealed a role for Fgf16 beyond reduced food intake; both B6-DIO and B6.V-Lepob/J FGF16 cohorts lost more weight than the pair-fed controls. The observed intestinal phenotype and malabsorption might have additionally contributed to the weight loss in the FGF16 groups. NMR analysis indicated that, when comparing EV mice with FGF16 mice, all measures were reduced, including fat mass, lean mass, and fluid mass. Furthermore, FGF16 mice exhibited more overall loss than the EV-PF mice, indicating an effect beyond reduced food intake alone (Fig. 5). This was further supported by gene expression analysis in several metabolic tissues including fat, liver, intestine, and pancreas, where the overall changes induced by FGF16 were quite distinct compared with the pair-fed groups (Figs. 7 and 8). The reduced food intake observed in B6.V-Lepob/J mice also suggests that the Fgf16 overexpression-mediated weight loss mechanism was perhaps independent of leptin. Whether the food-intake reduction is through a direct action of Fgf16 or through an indirect effect such as malabsorption, as suggested by the intestinal phenotype, is unclear and worth further investigation.
Some differences between B6-DIO and B6.V-Lepob/J mice in response to Fgf16 overexpression were observed. For example, the difference between FGF16 and pair-fed group on NMR tissue analysis were less obvious in B6.V-Lepob/J mice than in B6-DIO mice (Fig. 5). This may suggest a level of leptin dependence. Differences in serum lipid levels between these two models further support the possibility that some aspects of FGF16 effects may be leptin-dependent. However, it cannot be ruled out that the shorter duration of the study due to the unanticipated early termination of the B6.V-Lepob/J mice might have contributed to differences in the endpoints between the two models. Overall, the effects of Fgf16 overexpression in B6-DIO and B6.V-Lepob/J mice were largely similar.
The most striking observation with FGF16 mice was the dramatic increase in lipid and bile acid content in the feces. Despite the minimal change in mRNA levels for Asbt, the major bile acid reuptake pump in the ileum, and a reduced bile acid synthesis and bile acid pool, there was an elevated excretion of bile acids and lipids. Histological examination of intestinal tissues supported malabsorption defects, including excessive deposition of collagen-like substances, sulfated and carboxylated acid mucopolysaccharides and sialumucins, and morphological changes indicative of enterocyte apoptosis. The intestinal morphology, coupled with the observed reduction in some pancreatic lipases and the reduction in the intestinal sterol uptake transporter Npc1l1, suggests that malabsorption may be the main contributor to the excessive fecal lipid content observed in the FGF16 mice and that other changes may be secondary. For example, the induction of the cholesterol synthesis pathway in the liver may be a compensatory mechanism to the intestinal phenotype. The overall effects on lipid and bile acid changes appeared specific to the FGF16 group and were not seen in the pair-fed group, suggesting again that the metabolic effects of FGF16 were due not only to reduced food intake.
The suppression of bile acid synthesis genes in the liver may have been due to the direct activity of Fgf16 on the liver (Figs. 4C and 7C). It is well established that the activation of liver FGFR4 by FGF15/FGF19 results in suppression of genes involved in the bile acid synthesis, supporting a critical role for the FGF15/19-FGFR4 axis in the enterohepatic regulation of bile acid metabolism (36, 37). FGF16 does activate FGFR4 in vitro, albeit not as strongly as on other FGFR (Fig. 9G). Given that liver is the highest expressing tissue for Fgf16 post-AAV gene delivery (Fig. 2E, 7F, and 8F), the effects on liver bile acid synthesis genes are most likely the result of hepatic FGFR4 activation by Fgf16. Therefore, in this context, Fgf16 seems to have dual effects on bile acid metabolism: a direct effect to suppress synthesis in the liver and perhaps an indirect effect to increase excretion through intestinal malabsorption.
There are 18 structurally related members of the mammalian FGF family that can be classified as either paracrine or endocrine depending on their mode of action (38). Endocrine FGF include FGF15/19, FGF21, and FGF23. Both FGF19 and FGF21 induce metabolic phenotypes, such as lowering body weight, improving glucose metabolism, and promoting advantageous lipid profile changes (39, 40). These metabolic effects are believed to be mediated mainly through FGFR1c and adipose tissue (40). Because FGF16 also strongly activates FGFR1c (Fig. 9B) and is highly expressed in fat by AAV gene delivery (Fig. 2E), the observed effects on body weight and glucose by Fgf16 overexpression may also come from a mechanism similar to the endocrine FGF molecules. Therefore, the overall phenotype from Fgf16 overexpression could come from both direct and indirect mechanisms and involve different receptors. Although this complicates the interpretation of whether iBAT directly modulates energy expenditure and subsequently weight loss, it does suggest that if a molecule can activate several important metabolic mechanisms, the ultimate effects could be quite profound. Both the desired and undesired effects seen with Fgf16 overexpression due to its ability to engage several different receptors also highlight the need to carefully consider a therapeutic development program, especially when paracrine FGF are considered as therapeutics (41).
Experimental Procedures
Adipogenic Differentiation Screen
A cDNA library consisting of 1406 human cDNA encoding putative secreted factors was cloned into the expression-ready family of CMV6 constructs (Origene). In addition, 2512 cDNA clones, both mouse and human, were assembled from a variety of sources and subcloned into a proprietary expression vector driven by a Ubiquitin C promoter. DNA for this collection was prepared by a variety of techniques ranging from endotoxin-free to molecular biology-grade methods.
The mouse cell line C3H/10T1/2, obtained from the American Type Culture Collection (ATCC), was maintained in DMEM supplemented with 10% mesenchymal stem cell-qualified fetal bovine serum (Life Technologies) and 1% antibiotic/antimycotic (Cellgro). Cells were seeded at 2000 cells/cm2 and split before reaching confluence using the TrypLE dissociation reagent (Life Technologies) in accordance with ATCC guidelines.
Transfections were performed in a 384-well format using the FuGene HD transfection reagent (Promega). In accordance with the supplier's guidelines, FuGene HD and OptiMem (Life Technologies) were equilibrated to room temperature before 0.09 μl of transfection reagent was added to 8.91 μl of serum-free medium per reaction. This mixture was incubated for 5 min at room temperature before being transferred to a 384-well polypropylene intermediate plate (ThermoFisher Scientific) with sufficient volume per well for four replicates using a Bravo liquid-handling robot (Agilent Technologies). 4 μl of plasmid DNA at a target concentration of 20 ng/μl was then immediately added to each well from library source plates and mixed thoroughly before incubation at room temperature for at least 30 min. During the DNA-lipid incubation, C3H/10T1/2 cells at passage 5 were harvested, counted, and resuspended at a concentration of 50 cells/μl in complete medium. After incubation, the DNA-lipid complexes were once again mixed thoroughly before 10 μl was aliquoted into one of three replicate collagen I-coated CellCarrier 384-well plates (PerkinElmer Life Sciences). Next, 20 μl of cell mixture was added to each well using a WellMate dispenser (ThermoFisher Scientific). Plates were stored at room temperature for 30–60 min before being placed in a humidity-controlled CO2 incubator at 37 °C.
Seventy-two hours post transfection, plates were moved from the incubator, and 15 μl of supernatant was removed followed by the addition of 25 μl of adipogenic differentiation medium. The adipogenic differentiation stock consisted of 500 μm isobutylmethylxanthine (Cayman Chemical), 125 μm indomethacin, 1 nm 3,3′,5-triiodo-l-thyronine sodium salt, 20 nm insulin, and 5 μm dexamethasone (Sigma) in complete medium (42). The transfected plates were incubated an additional 2 days before a 50% medium change was performed, again using the same adipogenic differentiation stock as before. After the medium change, plates were returned to the incubator.
After a total of 96 h of incubation in the presence of adipogenic differentiation medium, the plates were removed from the incubator and the medium replaced with warmed OptiMem containing 250 nm MitoTracker Deep Red (Life Technologies). The plates were returned to the incubator for 1 h before being removed and fixed by the addition of 3.7% formaldehyde (Sigma) to a final concentration of 1.9%. The plates were fixed at room temperature for at least 30 min before being washed three times with Dulbecco's phosphate-buffered saline (Corning) on a plate washer (BioTek). Finally, LipidTox Green HCS and Hoechst 33342 (Life Technologies) were both added to the plates in Dulbecco's phosphate-buffered saline at a 1:1000 and 1:10000 dilution, respectively, before the plates were sealed and stored overnight at 4 °C. The plates were removed from the refrigerator and allowed to equilibrate at room temperature before 8 fields for each well were imaged on an Opera QEHS (PerkinElmer Life Sciences) confocal imaging system at ×20 magnification.
High Content Screen Image Analysis
Image analysis was performed using Columbus software (PerkinElmer Life Sciences) with 36 features extracted for each field and then aggregated across an individual well as either the mean or the sum. After feature extraction, the well level data were exported to Screener (Genedata) for quality control, plate normalization by robust z-score, and replicate aggregation by median. Median-aggregated data for individual cDNA replicates were then exported to Spotfire (Tibco) for visualization and JMP (SAS) for statistical analyses.
To identify cDNA that either positively or negatively affected adipogenic differentiation, we extracted data for 36 features from each image (Table 1) and selected clones with aggregated values greater than Q3 + 1.5* IQR (interquartile range) or less than Q1 − 1.5*IQR from their respective distributions. In addition, the aggregated clone data were subjected to feature reduction by principal component analysis. Next, outliers from the first 10 principal components, representing >90% of the total variation in the data, were selected in a similar fashion to the underlying individual features. Finally, we employed a multivariate outlier metric, the Jackknife distance, and identified clones with values that were Q3 + 1.5* IQR for that distribution. As would be expected, there was a large degree of overlap between the outliers identified by each method, as they are all based on the same underlying data. To minimize false positives, we selected clones detected as outliers by at least two methods for technical and orthogonal validation.
Validation Analysis
Clones from each of our collections were prepared for validation in one of the following two ways. First, plasmid DNA for clones of interest from the Origene collection was “cherry-picked” from source plates using a Freedom EVO (Tecan) and rearrayed into a single plate for validation. At the same time, 1 μl of sample was taken from each well for rolling circle amplification and Sanger sequence confirmation. Concentrations from the individual wells were checked using the Quant-iT broad-range assay kit (Life Technologies) read out on a Safire II (Tecan), and strong positive outliers were reduced in concentration to a target of 20 ng/μl. Second, clones from the in-house cDNA collection were first retransformed and single colonies picked and confirmed by rolling circle amplification followed by Sanger sequencing. DNA was prepared for each validated clone with the use of the Plasmid Plus 96 (Qiagen) transfection-grade miniprep kit. Following DNA purification and Sanger sequence confirmation, we assessed plasmid concentration as described above and normalized to 20 ng/μl before rearraying validated plasmids into a separate 384-well source plate.
Together, 163 clones from the Origene collection and 202 clones from the in-house collection were retested in the differentiation assay as described above. In addition to the 365 clones identified for validation, representing 352 unique Entrez gene and 335 HomoloGene ID numbers, 115 additional clones were included to provide a set of putative neutrals to aid in interpretation of the data.
In parallel with the image-based technical validation, an additional set of three replicates were transfected, subjected to the same treatment protocol, and then lysed and stabilized with the Cells-to-CT reagent (Life Technologies). After completing the technical validation, we selected 336 clones for expression analysis and rearrayed all three replicates of these samples along with controls into 96-well plates. We transferred 12 μl of the 55-μl lysate for selected clones into a 50-μl first-strand synthesis reaction in a Tetrad 2 thermal cycler (Bio-Rad).
For expression analysis, we used the 96.96 dynamic array on the BioMark HD (Fluidigm). Predesigned PrimeTime qPCR assays for 96 mouse genes were purchased (Integrated DNA Technologies) along with a pool of unlabeled primers for the preamplification necessary for the Fluidigm platform. 16 cycles of preamplification were performed in a Tetrad 2 using TaqMan preamp master mix (Life Technologies) from 3.4 μl of first-strand cDNA in a 9-μl reaction. Following preamplification, samples were prepared in accordance with Fluidigm's recommendations utilizing the TaqMan universal PCR master mix (Life Technologies). Data were analyzed in real-time PCR analysis software (Fluidigm) using a quality threshold of 0.3 and automatic Ct threshold detection. ΔCt normalization was performed in Excel (Microsoft) using the geometric mean of Ct values for Actb, Ipo8, Rpl13a, Tbp, and Ywhaz (43). ΔCt values were then averaged across replicates. Assays detected above background in less than 1% of samples and those used for ΔCt normalization were excluded from subsequent analyses.
Animal Studies
All animal experiments were approved by the Institutional Animal Care and Use Committee of Amgen and cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition (44). Mice were single-housed in an air-conditioned room at 22 ± 2 °C with a 12-h light/12-h dark cycle (6:00 a.m.–6:00 p.m.). At termination, mice were euthanized following AAALAC Inc. guidelines using CO2 inhalation followed by exsanguination as a secondary method.
C57BL/6J mice, approximately 20 weeks old, and B6.V-Lepob/J mice, approximately 7 weeks old, were purchased from The Jackson Laboratory. The animals had ad libitum access to pelleted feed D12492 60% high fat diet (Research Diets) or regular chow feed, respectively, and water (reverse osmosis-purified) via automatic watering system. All C57BL/6J mice received the D12492 60% high fat diet for at least 12 weeks prior to experiments and then throughout the course of the studies.
Before in vivo studies commenced, mice were randomized into treatment groups based on body weight, body composition NMR analysis (Bruker), and/or fasting blood glucose levels, fasting serum insulin, and cholesterol, triglyceride, and NEFA levels. Blood glucose levels were measured using an Accu-Chek Aviva blood glucose meter (Roche).
AAV containing vector alone (empty vector or EV) or mouse Fgf16 driven by the EF1α promoter was administered via the tail vein using a U-100 insulin syringe. Each mouse received 4 × 1012 virus particles/100 μl PBS.
For the glucose tolerance test, mice were fasted for 4 h beginning at 7:00 a.m. on the day of the experiment. Blood samples to determine blood glucose levels were obtained from the tail vein immediately before (0 min) and 15, 30, and 60 min post-intraperitoneal injection of glucose (10 mg/kg of glucose).
Indirect Calorimetry
Oxymax CLAMS was used for automated noninvasive monitoring of animals (indirect calorimetry). The results were calculated using the CLAMS examination tool (CLAX, Columbus Instruments).
Twenty 2-week-old DIO mice were acclimated to the metabolic cages for 2 weeks and then randomized into cohorts based on body weight, body composition analysis, fasting serum cholesterol, triglyceride, and NEFA levels and fasting blood glucose levels. Mice were injected with AAV-EV or AAV-Fgf16 on day −1. Body weights were monitored every day for 15 days. The volume of oxygen consumption (VO2), volume of carbon dioxide production (VCO2), respiratory exchange ratio, heat production, water consumption, and physical activity (vertical rearing motions and x axis IR beam breaks) were monitored between days 6 and 13.
Food consumption was monitored manually between days 6 and 10. Feces were collected on days 10–14 and pooled for analysis. Body composition NMR analysis was performed on day 15, and terminal fasting serum samples were collected on day 16 post-AAV injection.
Pair-feeding Studies
Four days before study onset, single-housed mice were randomized into three groups (n = 12) based on body weight and NMR analysis: ad libitum fed FGF16 mice, pair-fed EV mice, and ad libitum fed EV mice. The pair-fed cohort began pair feeding 1 day prior to AAV injection. During the course of the studies, food intake was measured daily from all three groups, and body weights were taken on day −4, and days 1, 4, 7, 11, and 15 post-AAV injection. The pair-fed EV mice received food amounts equivalent to that consumed the previous day by his matched FGF16 mouse partner.
Sample Analysis
All serum samples analyzed were collected from animals fasted for 4 h. Kits for the following were used: cholesterol (ThermoFisher Scientific, TR13421), cholesterol standard (Pointe Scientific, C7509-STD), triglycerides (ThermoFisher Scientific, TR22421), triglycerides standard (Pointe Scientific, T7531-STD), NEFA (Wako HR series NEFA-HR2), NEFA standard solution (Wako, 276-76491), ALT and AST activity assays (Sigma-Aldrich, MAK052 and MAK055, respectively), leptin (EMD Millipore EZML-82K), adiponectin (R&D Systems, MRP300), and insulin (ALPCO, STELLUX chemi rodent insulin ELISA).
Liver triglyceride and cholesterol content was determined as described previously (37) using the same triglyceride and cholesterol kits as used for serum analysis. Liver glycogen content was determined by homogenizing the tissue in 0.3 normality (N) HCl, collecting the supernatant, and then measuring the glucose concentration using an Amplex Red kit (Invitrogen).
To determine feces lipid content and feces and total bile acid pools, the feces were prepared as described (37). Lipids were measured using the same triglyceride and cholesterol kits as used for serum analysis, and bile acids were measured enzymatically (Crystal Chem 80470).
For mRNA expression analysis by Fluidigm, total RNA from tissue samples was isolated using QIAcube and an RNeasy kit (Qiagen). cDNA was made from total RNA using a high capacity cDNA reverse transcription kit (Applied Biosystems Inc. (ABI)) according to the manufacturer's directions. cDNA samples were preamplified using the TaqMan PreAmp Master Mix (ABI) according to the manufacturer's directions. Preamplified cDNA was added to the sample loading reagent and Master Mix from TaqMan Universal PCR Master Mix (ABI) with a final concentration of RNA at 90 ng/ml. 20× gene assays (Integrated DNA Technologies) were diluted to 10× using the DA assay loading reagent (Fluidigm).
96.96 and 48.48 dynamic array chips were from Fluidigm Corp. Prior to loading of the samples and assay reagents into the inlets, the chips were primed in the Fluidigm IFC controller. 5 μl of sample prepared as described above was then loaded into each sample inlet of the dynamic array chip, and 5 μl of 10× gene expression assay mix was loaded into each detector inlet. The chip was placed on the Fludign IFC controller for loading and mixing and then placed on the BioMark real-time PCR system for thermal cycling and detection of the reaction products. All reactions were performed in duplicate. Data were analyzed using the BioMark gene expression data analysis software to obtain Ct values. The relative mRNA level was calculated by the comparative threshold cycle method using the geometric mean of GAPDH, β-actin, Tbp, and Ipo8 as internal controls. The genes screened by Fluidigm analysis are detailed in Table 2.
TABLE 2.
Genes evaluated by qPCR and/or Fluidigm analysis
| Gene | Sourcea | Catalog No. | Analysis |
|---|---|---|---|
| Abcg5 | ABI | Mm.PT.56a.8809476 | Fluidigm |
| Abcg8 | ABI | Mm.PT.56a.7910478 | Fluidigm |
| Acaca | ABI | Mm.PT.58.12492865 | Fluidigm |
| Actb | ABI | Mm.PT.56a.33540333 | Fluidigm |
| Amylase1 | ABI | Mm00651524_m1 | Fluidigm |
| Asbt (Slc10a2) | ABI | Mm.PT.56a.41743474 | Fluidigm |
| Atgl | IDT | Mm.PT.56a.13182140 | Fluidigm |
| Beta klotho | ABI | Mm.PT.56a.8497961 | Fluidigm |
| Bsep (abcb11) | ABI | Mm.PT.56a.43710058 | Fluidigm |
| Cela1 | ABI | Mm00712898_m1 | Fluidigm |
| Cela2a | ABI | Mm00468213_m1 | Fluidigm |
| Cela3b | ABI | Mm00840378_m1 | Fluidigm |
| Cidea | ABI | Mm.PT.56a.13017856 | Fluidigm |
| Cyp27a1 | ABI | Mm.PT.56a.42377566 | Fluidigm |
| Cyp7a1 | ABI | Mm.PT.56a.17448793 | Fluidigm |
| Cyp7b1 | ABI | Mm.PT.56a.31179802 | Fluidigm |
| Cyp8b1 | ABI | Mm00501637_s1 | Fluidigm |
| Dgat1 | ABI | Mm.PT.58.32759507 | Fluidigm |
| Dgat2 | ABI | Mm.PT.58.29947465 | Fluidigm |
| Fasn | IDT | Mm.PT.58.14276063 | Fluidigm |
| Fgf16 | ABI | Mm00651404_m1 | Fluidigm, qPCR |
| Fgf21 | ABI | Mm00840165_g1 | Fluidigm |
| Fgfr1 | IDT | Mm.PT.56a.13356831 | Fluidigm |
| Fgfr2 | IDT | Mm.PT.56a.12775563 | Fluidigm |
| Fgfr3 | IDT | Mm.PT.56a.12920207.g | Fluidigm |
| Fgfr4 | ABI | Mm.PT.56a.9855233 | Fluidigm |
| G6pc | ABI | Mm.PT.58.11964858 | Fluidigm |
| Gapdh | ABI | Mm.PT.39a.1 | Fluidigm, qPCR |
| Hmg-CoAR (Hmgcr) | ABI | Mm.PT.56a.13325212 | Fluidigm |
| Ipo8 | ABI | Mm.PT.39a.22214844 | Fluidigm |
| Lipe | IDT | Mm.PT.58.6342082 | Fluidigm |
| Lpl | ABI | Mm.PT.58.46006099 | Fluidigm |
| Mrp2 (abcc2) | ABI | Mm.PT.56a.5554359 | Fluidigm |
| Mrp3 (abcc3) | ABI | Mm.PT.56a.28882380 | Fluidigm |
| Mrp4 (abcc4) | ABI | Mm.PT.56a.23407634.g | Fluidigm |
| Ntcp (Slc10a1) | ABI | Mm.PT.56a.43000030 | Fluidigm |
| Oatp1 (Slco1a1) | ABI | Mm.PT.56a.32911698 | Fluidigm |
| Oatp4 (Slco1b2) | ABI | Mm.PT.56a.30581202 | Fluidigm |
| Ost-a | ABI | Mm.PT.56a.17102339 | Fluidigm |
| Ost-b | ABI | Mm.PT.56a.23668920 | Fluidigm |
| Pck1 | ABI | Mm.PT.58.42805932 | Fluidigm |
| Pcx | ABI | Mm.PT.58.31161924 | Fluidigm |
| Plrp1 | ABI | Mm00479741_m1 | Fluidigm |
| Plrp2 | ABI | Mm00448214_m1 | Fluidigm |
| Pnlip | ABI | Mm00813468_m1 | Fluidigm |
| Pparg | IDT | Mm.PT.58.9374886 | Fluidigm |
| Ppargc1a | ABI | Mm.PT.58.21954529 | Fluidigm |
| Srebp1c | ABI | Mm01138344_m1 | Fluidigm |
| Tbp | ABI | Mm.PT.39a.22214839 | Fluidigm |
| Ucp1 | ABI | Mm01244861_m1 | Fluidigm, qPCR |
a ABI, Applied Biosystems Inc.; IDT, Integrated DNA Technologies.
mRNA Expression Analysis by qPCR
Total tissue RNA was isolated using QIAcube and the RNeasy kit (Qigen). All reactions were performed in duplicate on an ABI QuantStudio 7 Flex real-time PCR system, and relative mRNA levels were calculated by the comparative threshold cycle method using Gapdh and Tbp as internal controls. The genes screened by qPCR analysis are detailed in Table 2.
pERK Activation of L6 Cells
L6 myoblast cells (ATCC) were maintained in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin. Using Lipofectamine 2000 (Invitrogen), cells (8 × 103 cells/well) were transfected with expression vectors harboring different FGF receptor isoforms (FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c, and FGFR4) as described previously (37). The day after transfection, cells were serum-starved overnight. The following day, cells were treated with recombinant mouse FGF16 (R&D Systems) or recombinant mouse FGF1 (Preprotech) proteins for 15 min and then snap-frozen in liquid nitrogen. Cell lysates were prepared by thawing the plates at 4 °C and adding 70 μl of complete MSD lysis buffer with 0.1% SDS to each well. Plates were shaken at 4 °C for 30 min on an orbital shaker. 25 μl of cell lysate was transferred to wells of phospho/total ERK1/2 whole cell lysate plates (Meso Scale Discovery) and pERK and total ERK were measured according to the manufacturer's protocol.
Immunohistochemistry and Histology
BAT, iWAT, and intestine were collected from mice, fixed with 10% neutral buffered formalin, and paraffin-embedded. Blocks were sectioned at 5-μm thickness, processed for immunohistochemistry, and stained with H&E for sample quality evaluation. Paraffin was removed from the tissue sections with xylene, and the sections were rehydrated with ethanol and immersed in distilled water. The deparaffinized sections were immersed in Diva Decloaker (Biocare Medical) and placed in a decloaking chamber (Biocare Medical) for 30 s at 125 °C and 10 s at 90 °C for antigen retrieval. Immunohistochemistry was performed with an IntelliPATH Autostainer (Biocare Medical) using a rabbit anti-human UCP1 (Abcam). The UCP1 antibody was detected with Envision Plus polymer (Dako Norden) followed by DAB Plus chromogen (Dako Norden). Alcian blue (American MasterTech), Masson's trichrome (American Master Tech), and periodic acid-Schiff (ThermoFisher Scientific) stains were performed according to the manufacturers' protocols. Slides were scanned using ScanScope XT (Aperio Digital Pathology) and ImageScope software (Leica Biosystems).
Author Contributions
I. C. R., P. C., M. O., W. G. R., and Y. L. conceived and designed the experiments, and I. C. R., P. C., L. M., D. N., M. H., J. G., C. C., and K. S. performed the experiments. K. J. L., K. H., and J. B. R. contributed reagents and materials and/or contributed to the analysis of the data. I. C. R., P. C., and Y. L. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Paul Kassner, Chris Paszty, Kevin Corbit, and Jae Lee for thoughtful advice and discussion, Jen Weiszmann, Yan Gong, Alysha Hoffman, and Joshua Dobroff for technical support, and David Lloyd for critical review of the manuscript.
All authors are or were Amgen employees and thus have an actual or perceived conflict of interest with the contents of this article.
- BAT
- brown adipose tissue
- iBAT
- inducible BAT
- UCP1
- uncoupling protein 1
- WAT
- white adipose tissue
- iWAT
- inguinal WAT
- eWAT
- epididymal WAT
- BMP
- bone morphogenetic protein
- AAV
- adeno-associated virus
- DIO
- diet-induced obese
- ADM
- adipogenic differentiation medium
- qPCR
- quantitative PCR
- EV
- empty vector
- PF
- pair-fed
- EV-PF
- pair-fed EV mice
- NEFA
- non-esterified fatty acid(s)
- CLAMS
- Comprehensive Lab Animal Monitoring System
- FGFR
- fibroblast growth factor receptor
- MSD
- Meso Scale Discovery
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase.
References
- 1.Lowell B. B., and Spiegelman B. M. (2000) Towards a molecular understanding of adaptive thermogenesis. Nature 404, 652–660 [DOI] [PubMed] [Google Scholar]
- 2.Cypess A. M., and Kahn C. R. (2010) The role and importance of brown adipose tissue in energy homeostasis. Curr. Opin. Pediatr. 22, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowell B. B., S-Susulic V., Hamann A., Lawitts J. A., Himms-Hagen J., Boyer B. B., Kozak L. P., and Flier J. S. (1993) Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 366, 740–742 [DOI] [PubMed] [Google Scholar]
- 4.Kopecky J., Clarke G., Enerbäck S., Spiegelman B., and Kozak L. P. (1995) Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J. Clin. Invest. 96, 2914–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldmann H. M., Golozoubova V., Cannon B., and Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 [DOI] [PubMed] [Google Scholar]
- 6.Saito M., Okamatsu-Ogura Y., Matsushita M., Watanabe K., Yoneshiro T., Nio-Kobayashi J., Iwanaga T., Miyagawa M., Kameya T., Nakada K., Kawai Y., and Tsujisaki M. (2009) High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58, 1526–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cypess A. M., and Kahn C. R. (2010) Brown fat as a therapy for obesity and diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 17, 143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vitali A., Murano I., Zingaretti M. C., Frontini A., Ricquier D., and Cinti S. (2012) The adipose organ of obesity-prone C57BL/6J mice is composed of mixed white and brown adipocytes. J. Lipid Res. 53, 619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harms M., and Seale P. (2013) Brown and beige fat: development, function and therapeutic potential. Nat. Med. 19, 1252–1263 [DOI] [PubMed] [Google Scholar]
- 10.Sharp L. Z., Shinoda K., Ohno H., Scheel D. W., Tomoda E., Ruiz L., Hu H., Wang L., Pavlova Z., Gilsanz V., and Kajimura S. (2012) Human BAT possesses molecular signatures that resemble beige/brite cells. PloS One 7, e49452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J., Boström P., Sparks L. M., Ye L., Choi J. H., Giang A. H., Khandekar M., Virtanen K. A., Nuutila P., Schaart G., Huang K., Tu H., van Marken Lichtenbelt W. D., Hoeks J., Enerbäck S., Schrauwen P., and Spiegelman B. M. (2012) Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cypess A. M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A. B., Kuo F. C., Palmer E. L., Tseng Y. H., Doria A., Kolodny G. M., and Kahn C. R. (2009) Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nedergaard J., Bengtsson T., and Cannon B. (2007) Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 293, E444–E452 [DOI] [PubMed] [Google Scholar]
- 14.van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D., Schrauwen P., and Teule G. J. (2009) Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360, 1500–1508 [DOI] [PubMed] [Google Scholar]
- 15.Qian S. W., Tang Y., Li X., Liu Y., Zhang Y. Y., Huang H. Y., Xue R. D., Yu H. Y., Guo L., Gao H. D., Liu Y., Sun X., Li Y. M., Jia W. P., and Tang Q. Q. (2013) BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 110, E798–E807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elsen M., Raschke S., Tennagels N., Schwahn U., Jelenik T., Roden M., Romacho T., and Eckel J. (2014) BMP4 and BMP7 induce the white-to-brown transition of primary human adipose stem cells. Am. J. Physiol. Cell Physiol. 306, C431–C440 [DOI] [PubMed] [Google Scholar]
- 17.Ornitz D. M., and Itoh N. (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyake A., Konishi M., Martin F. H., Hernday N. A., Ozaki K., Yamamoto S., Mikami T., Arakawa T., and Itoh N. (1998) Structure and expression of a novel member, FGF-16, on the fibroblast growth factor family. Biochem. Biophys. Res. Commun. 243, 148–152 [DOI] [PubMed] [Google Scholar]
- 19.Jamsheer A., Zemojtel T., Kolanczyk M., Stricker S., Hecht J., Krawitz P., Doelken S. C., Glazar R., Socha M., and Mundlos S. (2013) Whole exome sequencing identifies FGF16 nonsense mutations as the cause of X-linked recessive metacarpal 4/5 fusion. J. Med. Genet. 50, 579–584 [DOI] [PubMed] [Google Scholar]
- 20.Jones B., Byers H., Watson J. S., and Newman W. G. (2014) Identification of a novel familial FGF16 mutation in metacarpal 4–5 fusion. Clin. dysmorphol. 23, 95–97 [DOI] [PubMed] [Google Scholar]
- 21.Laurell T., Nilsson D., Hofmeister W., Lindstrand A., Ahituv N., Vandermeer J., Amilon A., Annerén G., Arner M., Pettersson M., Jäntti N., Rosberg H. E., Cattini P. A., Nordenskjöld A., Mäkitie O., et al. (2014) Identification of three novel FGF16 mutations in X-linked recessive fusion of the fourth and fifth metacarpals and possible correlation with heart disease. Mol. Genet. Genomic Med. 2, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamsheer A., Smigiel R., Jakubiak A., Zemojtel T., Socha M., Robinson P. N., and Mundlos S. (2014) Further evidence for FGF16 truncating mutations as the cause of X-linked recessive fusion of metacarpals 4/5. Birth Defects Res. A Clin. Mol. Teratol. 100, 314–318 [DOI] [PubMed] [Google Scholar]
- 23.Matsumoto E., Sasaki S., Kinoshita H., Kito T., Ohta H., Konishi M., Kuwahara K., Nakao K., and Itoh N. (2013) Angiotensin II-induced cardiac hypertrophy and fibrosis are promoted in mice lacking Fgf16. Genes Cells 18, 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu S. Y., Sheikh F., Sheppard P. C., Fresnoza A., Duckworth M. L., Detillieux K. A., and Cattini P. A. (2008) FGF-16 is required for embryonic heart development. Biochem. Biophys. Res. Commun. 373, 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotta Y., Sasaki S., Konishi M., Kinoshita H., Kuwahara K., Nakao K., and Itoh N. (2008) Fgf16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev. Dyn. 237, 2947–2954 [DOI] [PubMed] [Google Scholar]
- 26.Yu W., Huang X., Tian X., Zhang H., He L., Wang Y., Nie Y., Hu S., Lin Z., Zhou B., Pu W., Lui K. O., and Zhou B. (2016) GATA4 regulates Fgf16 to promote heart repair after injury. Development 143, 936–949 [DOI] [PubMed] [Google Scholar]
- 27.Konishi M., Mikami T., Yamasaki M., Miyake A., and Itoh N. (2000) Fibroblast growth factor-16 is a growth factor for embryonic brown adipocytes. The J. Biol. Chem. 275, 12119–12122 [DOI] [PubMed] [Google Scholar]
- 28.Boland G. M., Perkins G., Hall D. J., and Tuan R. S. (2004) Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell. Biochem. 93, 1210–1230 [DOI] [PubMed] [Google Scholar]
- 29.Cho H. H., Kim Y. J., Kim S. J., Kim J. H., Bae Y. C., Ba B., and Jung J. S. (2006) Endogenous Wnt signaling promotes proliferation and suppresses osteogenic differentiation in human adipose derived stromal cells. Tissue Eng. 12, 111–121 [DOI] [PubMed] [Google Scholar]
- 30.Gong Y., Slee R. B., Fukai N., Rawadi G., Roman-Roman S., Reginato A. M., Wang H., Cundy T., Glorieux F. H., Lev D., Zacharin M., Oexle K., Marcelino J., Suwairi W., Heeger S., et al. (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107, 513–523 [DOI] [PubMed] [Google Scholar]
- 31.De Boer J., Wang H. J., and Van Blitterswijk C. (2004) Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng. 10, 393–401 [DOI] [PubMed] [Google Scholar]
- 32.Qiu W., Andersen T. E., Bollerslev J., Mandrup S., Abdallah B. M., and Kassem M. (2007) Patients with high bone mass phenotype exhibit enhanced osteoblast differentiation and inhibition of adipogenesis of human mesenchymal stem cells. J. Bone Miner. Res. 22, 1720–1731 [DOI] [PubMed] [Google Scholar]
- 33.Tang Q. Q., Otto T. C., and Lane M. D. (2004) Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. U.S.A. 101, 9607–9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pandol S. J. (2010) The Exocrine Pancreas, Morgan and Claypool Life Sciences, San Rafael, CA: [PubMed] [Google Scholar]
- 35.Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M., and Ornitz D. M. (2006) Receptor specificity of the fibroblast growth factor family: the complete mammalian FGF family. J. Biol. Chem. 281, 15694–15700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., Gerard R. D., Repa J. J., Mangelsdorf D. J., and Kliewer S. A. (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2, 217–225 [DOI] [PubMed] [Google Scholar]
- 37.Ge H., Zhang J., Gong Y., Gupte J., Ye J., Weiszmann J., Samayoa K., Coberly S., Gardner J., Wang H., Corbin T., Chui D., Baribault H., and Li Y. (2014) Fibroblast growth factor receptor 4 (FGFR4) deficiency improves insulin resistance and glucose metabolism under diet-induced obesity conditions. J. Biol. Chem. 289, 30470–30480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itoh N., and Ornitz D. M. (2011) Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J. Biochem. 149, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itoh N., Ohta H., and Konishi M. (2015) Endocrine FGFs: evolution, physiology, pathophysiology, and pharmacotherapy. Front. Endocrinol. 6, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J., and Li Y. (2014) Fibroblast growth factor 21, the endocrine FGF pathway and novel treatments for metabolic syndrome. Drug Discov. Today 19, 579–589 [DOI] [PubMed] [Google Scholar]
- 41.Suh J. M., Jonker J. W., Ahmadian M., Goetz R., Lackey D., Osborn O., Huang Z., Liu W., Yoshihara E., van Dijk T. H., Havinga R., Fan W., Yin Y. Q., Yu R. T., Liddle C., et al. (2014) Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 513, 436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tseng Y. H., Kokkotou E., Schulz T. J., Huang T. L., Winnay J. N., Taniguchi C. M., Tran T. T., Suzuki R., Espinoza D. O., Yamamoto Y., Ahrens M. J., Dudley A. T., Norris A. W., Kulkarni R. N., and Kahn C. R. (2008) New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., and Speleman F. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.National Research Council (U. S.) Committee for the Update of the Guide for the Care and Use of Laboratory Animals (2011) Guide for the Care and Use of Laboratory Animals, 8th Ed., National Academies Press, Washington, D. C. [Google Scholar]