

 This work is licensed under a
This work is licensed under a Summary
ACTH-secreting phaeochromocytoma (ASP) is a rare cause of ACTH-dependent Cushing’s syndrome (CS). We report the case of a 63-year-old female presenting with CS secondary to an ASP complicated by bowel perforation. This case report highlights ASP as an uncommon but important cause of ectopic ACTH secretion (EAS). There have been 29 cases of ASP, all of which were unilateral and benign, but associated with significant complications. Patients presenting with ASP have the potential for cure with unilateral adrenalectomy. Given this promising prognosis if recognised, ASP should be considered in the diagnostic workup of ACTH-dependent CS. As this case demonstrates, gastrointestinal complications can arise from severe hypercortisolaemia associated with CS. Early medical and surgical intervention is imperative as mortality approaches 50% once bowel perforation occurs.
Learning points
Consider phaeochromocytoma in the diagnostic workup of ACTH-dependent CS; screen with plasma metanephrines or urinary catecholamines.
Serial screening may be required if ACTH-secreting phaeochromocytoma is suspected, as absolute levels can be misleading.
Early catecholamine receptor blockade and adrenal synthesis blockade may avoid the need for rescue bilateral adrenalectomy in ACTH-secreting phaeochromocytoma.
Consider early medical or surgical management when gastrointestinal features are present in patients with CS, as bowel perforation due to severe hypercortisolaemia can occur and is associated with significant mortality.
Background
Cushing’s syndrome (CS) is a rare condition related to prolonged exposure to excess cortisol with an annual incidence of 0.2–5 cases per million people per year (1). Approximately 80% of CS cases are ACTH dependent, the majority resulting from a pituitary adenoma (Cushing’s disease) (1). Ectopic ACTH secretion (EAS) occurs in 5–10% of ACTH-dependent CS (2). The disease responsible for EAS is frequently thoracic in origin, including bronchial carcinoid, thymic carcinoid or small-cell lung cancer. However, it is often difficult to localise the EAS source, and some occult tumours are never identified. Less than 6% of EAS cases are attributed to a phaeochromocytoma (3). We present the case of a patient with CS due to an ectopic ACTH-secreting phaeochromocytoma (ASP) complicated by bowel perforation requiring hemicolectomy. Given the increased risk of complications with this dual pathology, a high index of clinical suspicion for ASP should exist in patients presenting with EAS.
Case presentation
A 63-year-old female presented to a regional hospital with peripheral oedema and worsening hypertension on a background of previously well-controlled hypertension. On examination, she was hypertensive, tachycardic, hirsute and had ecchymoses on her trunk and limbs. Laboratory investigations demonstrated new-onset hyperglycaemia and hypokalaemia (3.0mmol/L, reference range 3.5–5.1mmol/L). An elevated early morning serum cortisol (2220nmol/L, reference range <618nmol/L) failed to suppress after a 1mg dexamethasone suppression test (DST). Additionally, the 24-h urine-free cortisol excretion was elevated (24h UFC, 12 465nmol/24h, reference range 200–1250nmol/24h), and ACTH was elevated at 16pmol/L (reference range <10pmol/L), consistent with ACTH-dependent CS. The 24-h urinary adrenaline levels were elevated (326nmol/24h, reference range 5–80nmol/24h); however, 24-h urinary noradrenaline was normal (442nmol/24h, reference range 40–780nmol/24h).
Magnetic resonance imaging (MRI) of the pituitary gland was normal. Computed tomography (CT) demonstrated no intra-thoracic or pelvic pathology, but revealed abnormal adrenal glands bilaterally. The right adrenal had two adenomas measuring 10mm each. The left adrenal was enlarged measuring 20×33×34mm with attenuation values over 50 Hounsfield units (Fig. 1A). The patient was transferred to our institution for further investigation.
Figure 1.
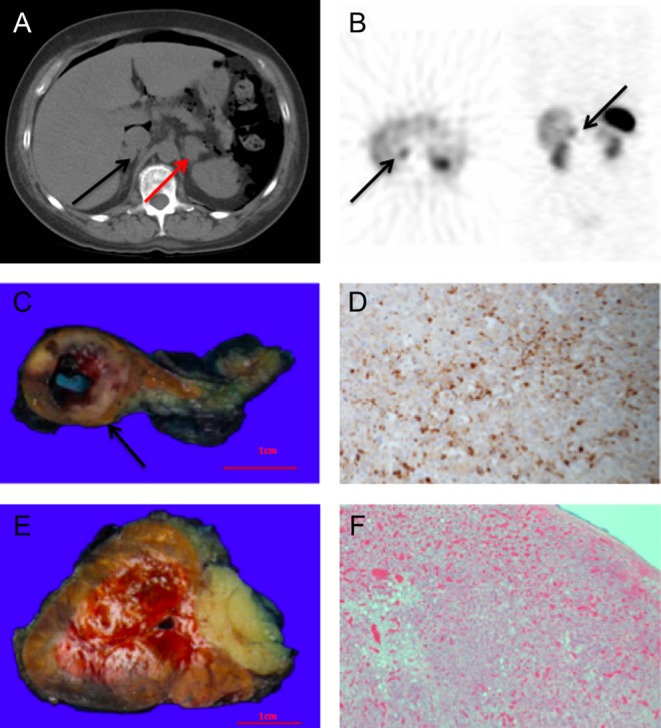
(A) CT abdomen with evidence of intra-peritoneal gas, black arrow: adenoma within the right adrenal, red arrow: enlarged left adrenal. (B) Octreotide scan demonstrating physiological uptake in the left adrenal, black arrows: increased uptake in the right adrenal. (C) Right adrenal specimen, weight 17g, black arrow: 14mm well-circumscribed phaeochromocytoma with central cystic degeneration. (D) Right adrenal, demonstrating cytoplasmic positivity for chromogranin A. (E) Left adrenal, cortical hyperplasia, weight 33g. (F) Left adrenal, infiltration of eosinophilic cell characteristic of hyperplasia.
Investigation
Initial management included commencing insulin aspart for persistent hyperglycaemia and aggressive potassium replacement for hypokalaemia. Hypertension was difficult to control, despite using five antihypertensive agents, including prazosin. The combination of pharmacologically resistant hypertension, persisting tachycardia, radiological evidence of right adrenal adenoma and the elevated urinary adrenalines raised the possibility of a phaeochromocytoma either as an ASP or a separate biochemically silent phaeochromocytoma. Repeat plasma metanephrines and urinary catecholamines were unconvincing. Plasma metanephrines were minimally elevated (665pmol/L, reference range <500pmol/L) and normetanephrines were normal (321pmol/L, reference range <900pmol/L). Repeat ACTH remained elevated (25.5pmol/L, reference range 1.6–13.9pmol/L). As up to 50% of Cushing’s disease cases have a normal MRI (1), further investigations were deemed necessary to confirm the ACTH source and validate our concerns about an adrenal source.
To differentiate between a pituitary and ectopic ACTH source, a high-dose DST was scheduled. However, the patient developed sudden, severe abdominal pain with radiological evidence of intra-abdominal gas (Fig. 1A), requiring cancellation of the high-dose DST. Emergency diagnostic laparoscopy was suspicious for micro-perforation of the bowel at the splenic flexure. In response to this complication of severe hypercortisolism, bilateral inferior petrosal sinus sampling (BIPSS) was expedited once the patient was clinically stable and performed within 2 days to definitively exclude a pituitary ACTH source. No significant ACTH gradient was demonstrated, further supporting EAS and the possibility of ectopic ACTH arising from a phaeochromocytoma itself rather than two separate disease states. To ensure that other ectopic ACTH sources were excluded and to consolidate our view about an adrenal source, an octreotide scan was performed showing focal uptake in the right adrenal (Fig. 1B), supporting suspicions of a right adrenal pathology. There was physiological uptake in the left adrenal (Fig. 1B). The sensitivity of octreotide scintigraphy when used to detect ACTH-secreting tumours is 50–57% (4). Metaiodobenzylguanidine (MIBG) may have been appropriate; however, the scan was unavailable for another week. A 68Ga-DOTA-octreotate (GaTate) positron emission tomography (PET)/CT was unavailable at our institution at that time. With the working diagnosis of ASP, alpha blockade was optimised with uptitration of prazosin to 6mg three times daily to control persistent hypertension. Prazosin was not converted to phenoxybenzamine, given that similar results can be achieved with prazosin (5). Ketoconazole (200mg twice daily) was commenced for adrenal synthesis blockade, given that metyrapone was locally unavailable.
Progression to right adrenalectomy was delayed by recurrence of severe abdominal pain with peritonism. Emergency exploratory laparotomy resulted in a left hemicolectomy for perforation at the splenic flexure. The postoperative course was complicated by labile blood pressure, rapid atrial fibrillation, pneumonia, hypokalaemia and delirium. Plasma metanephrines taken before laparotomy were increasing (863pmol/L, reference range <500pmol/L); although normetanephrines were in normal range, they were double that measured 12 days before (644pmol/L, reference range <900pmol/L).
Treatment
Unfortunately, the patient further deteriorated, developing delirium with paranoid ideations. Repeat serum cortisol and 24-h UFC were markedly elevated (5149nmol/L, reference range <618nmol/L, and 42 435nmol/24h, reference range 25–360nmol/24h respectively), reflecting inadequate response to adrenal synthesis blockade. Rapid control of severe hypercortisolaemia with etomidate (6) was not possible due to lack of availability in Australia. The patient’s progressive deterioration thus necessitated urgent surgical intervention. Despite the octreotide scan suggesting the right adrenal as the likely site of ectopic ACTH production and our desire for a unilateral right adrenalectomy, a bilateral approach was instead deemed necessary after much deliberation with our anaesthesiologists and surgical colleagues. This decision was made in consideration of the patient’s significant clinical instability, the potential for technical difficulty due to recent abdominal surgery, and most importantly, concern was raised regarding the enlarged left adrenal gland. Despite it showing physiological uptake on octreotide scanning, there was apprehension regarding the risk of leaving this in situ as should the causative lesion not be isolated to the right adrenal, it was unanimously felt that the patient would not survive a repeat laparotomy.
Surgery was technically difficult given the recent hemicolectomy and was complicated by intra-operative splenic haemorrhage and subsequent splenectomy. Histopathology confirmed phaeochromocytoma in the right adrenal with cytoplasmic positivity for chromogranin A and ACTH (Fig. 1C and D). The left adrenal demonstrated cortical hyperplasia with no evidence of adenoma and was negative for ACTH staining (Fig. 1E and F).
Outcome and follow-up
There was improvement in the patient’s biochemistry by the 12th postoperative day. With the patient still on hydrocortisone, ACTH was <0.07pmol/L, serum cortisol 10nmol/L and 24h UFC 4067nmol. After 4 weeks, hypertension and hyperglycaemia were well controlled and hypokalaemia had resolved. The patient was discharged home on fludrocortisone and dexamethasone and had clinical resolution of her CS features.
Discussion
ACTH-secreting phaeochromocytoma
Phaeochromocytomas have a variety of presentations, typically presenting with adrenergic features including hypertension, tachycardia and diaphoresis (7). They can co-secrete hormones other than catecholamines and thus present with symptoms of that hormone excess, potentially complicating diagnosis (8). Diagnosis requires biochemical evidence of catecholamine excess followed by radiological localisation, which can be challenging (9). Our patient had mildly elevated urinary adrenaline and plasma metanephrine levels. Although stress is a cause of false-positive results (9), the patient’s clinical and radiological signs were suggestive of an ASP or a separate biochemically silent phaeochromocytoma. Biochemically silent phaeochromocytomas can cause false-negative results and may result from small (<1cm) or dopamine-producing tumours (9).
The phenomenon of ASPs was first described in the literature in 1960s. Many cases reported an association between hypercortisolism and an adrenomedullary tumour or paraganglioma. They failed to prove causality or exclude the possibility of dual pathology due to limitations in investigational methods. In 1979, Forman developed the first recognised diagnostic criteria for phaeochromocytoma as the cause of EAS (10). In 1995, Chen performed the first review of cases of ASP since the introduction of diagnostic criteria, reporting 14 cases since 1979 (11). Chen noted that negative ACTH staining on tumour tissue did not exclude functional ACTH secretion, as was suggested by Forman. This concept has been supported in later cases, with suggestions that immunohistochemistry should be coupled with molecular studies to identify the presence of ACTH precursors or derivatives that may support functional ACTH secretion (12). Chen revised the diagnostic criteria to reflect this, stating that a case had to report (i) clinical and laboratory evidence of hypercortisolism, (ii) elevated plasma ACTH, (iii) evidence of phaeochromocytoma by urinary catecholamines or MRI evidence of an adrenal mass with a bright T2 signal, (iv) resolution of signs and symptoms of adrenocorticoid and catecholamine excess after unilateral adrenalectomy, and (v) rapid normalisation of plasma ACTH levels after adrenalectomy (11).
By applying these criteria, there have been 29 cases of ASP described in the literature since 1977, all of which were unilateral and benign (8, 11, 13, 14). This pathology can therefore be cured with unilateral adrenalectomy (11). Except in cases of extreme medical instability, bilateral adrenalectomy should be avoided due to requirement for adrenal replacement therapy. Long-term follow-up of patients with bilateral adrenalectomy has demonstrated that 11–32% have an Addisonian crisis (15).
Since 1977, there have been two likely cases of ASP; however, death occurred before fulfilment of the diagnostic criteria – myocardial infarction in a 67-year-old female and septic cerebral emboli in a 28-year-old female (13, 16). The 29 surviving cases had multiple complications including cardiac arrest, pneumonia with respiratory failure, hypertensive encephalopathy, psychosis, cerebellar infarction, pulmonary embolus, haemorrhagic shock and sepsis (3, 15, 17, 18, 19, 20). This highlights the morbidity associated with elevated cortisol and catecholamines and the need for prompt normalisation.
A right-sided ASP was suspected in our patient, and thus, a unilateral adrenalectomy was the procedure of choice. However, the patient’s rapid deterioration due to bowel complications associated with medically refractory hypercortisolaemia and resultant significant abdominal surgeries unfortunately necessitated rescue bilateral adrenalectomy. Alternative strategies to rapidly manage severe hypercortisolaemia such as intravenous etomidate (6) were not available in Australia. Due to morbidity associated with bilateral adrenalectomy, we recommend consideration of screening for phaeochromocytoma in patients presenting with ACTH-dependent CS to facilitate earlier diagnosis, thus potentially avoiding the need for bilateral adrenalectomy.
Bowel perforation
Bowel perforation is a rare complication of CS and phaeochromocytoma. Few cases of bowel perforation complicating phaeochromocytoma have been reported in the literature (7). It is postulated that catecholamine excess causes splanchnic vasoconstriction and subsequent ileus, pseudo-obstruction, ischaemia and necrosis (7). Bowel perforation is a well-known life-threatening complication of CS and exogenous corticosteroid use, resulting from steroid-mediated thinning of the collagen comprising the colonic lamina propria (21). Severe hypercortisolism likely caused our patient’s bowel perforation given her biochemically silent phaeochromocytoma and the disproportionately elevated cortisol for the degree of ACTH elevation. ASPs have been noted to produce modest elevations in plasma ACTH with severe hypercortisolism, possibly due to the proximity of the adrenal medulla and cortex (2). Our patient had the third highest reported at 5149nmol/L, with the highest reported serum cortisol from an ASP being 8225nmol/L (20). This is the second case of bowel perforation complicating an ASP (15). Patients are at risk of perioperative hypertensive crisis and mortality if not adequately prepared for surgery.
Summary
Although ASP is associated with significant morbidity, it has the potential for cure with unilateral adrenalectomy. Thus, we recommend consideration of screening patients presenting with ACTH-dependent CS with plasma metanephrines. As this case study has demonstrated, the compilation of clinical symptoms, imaging and trend in serial biochemistry is more significant than absolute levels of these results. Once the diagnosis of ASP is suspected, early catecholamine receptor blockade and adrenal synthesis blockade may avoid the development of complications such as intestinal perforation and the subsequent need for rescue bilateral adrenalectomy.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This case report did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author contribution statement
Dr Elise Flynn, Dr Sara Baqar, Dorothy Liu, Prof Zajac and Prof Seeman assisted in the concept, design and editing of the article. Prof Seeman and Dr De Luise were the physicians overseeing the case. Mr Stephen Farrell was the surgeon involved in the case. Dr Ekinci and Prof De Luise assisted with the final draft and approved the content.
References
- 1.Lacroix A, Feelders RA, Stratakis CA, Nieman LK. 2015. Cushing’s syndrome. Lancet 386 913–927. ( 10.1016/S0140-6736(14)61375-1) [DOI] [PubMed] [Google Scholar]
- 2.Salgado LR, Fragoso MCBV, Knoepfelmacher M, Machado MC, Domenice S, Pereira MAA, de Mendonça BB. 2006. Ectopic ACTH syndrome: our experience with 25 cases. European Journal of Endocrinology 155 725–733. ( 10.1530/eje.1.02278) [DOI] [PubMed] [Google Scholar]
- 3.Ballav C, Naziat A, Mihai R, Karavitaki N, Ansorge O, Grossman AB. 2012. Mini-review: pheochromocytomas causing the ectopic ACTH syndrome. Endocrine 42 69–73. ( 10.1007/s12020-012-9646-7) [DOI] [PubMed] [Google Scholar]
- 4.Zemskova MS, Gundabolu B, Sinaii N, Chen CC, Carrasquillo JA, Whatley M, Chowdhury I, Gharib AM, Nieman LK. 2010. Utility of various functional and anatomic imaging modalities for detection of ectopic adrenocorticotropin-secreting tumors. Journal of Clinical Endocrinology and Metabolism 95 1207–1219. ( 10.1210/jc.2009-2282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weingarten TN, Cata JP, O’Hara JF, Prybilla DJ, Pike TL, Thompson GB, Grant CS, Warner DO, Bravo E, Sprung J. 2010. Comparison of two preoperative medical management strategies for laparoscopic resection of pheochromocytoma. Urology 76 508.e6–508.e11. ( 10.1016/j.urology.2010.03.032) [DOI] [PubMed] [Google Scholar]
- 6.Allolio B, Schulte HM, Kaulen D, Reincke M, Jaursch-Hancke C, Winkelmann W. 1988. Nonhypnotic low-dose etomidate for rapid correction of hypercortisolaemia in Cushing’s syndrome. Klinische Wochenschrift 66 361–364. ( 10.1007/BF01735795) [DOI] [PubMed] [Google Scholar]
- 7.Karri V, Khan S, Wilson Y. 2005. Bowel perforation as a presenting feature of pheochromocytoma: case report and literature review. Endocrine Practice 11 385–388. ( 10.4158/ep.11.6.385) [DOI] [PubMed] [Google Scholar]
- 8.Kirkby-Bott J, Brunaud L, Mathonet M, Hamoir E, Kraimps J-L, Trésallet C, Amar L, Rault A, Henry JF, Carnaille B. 2012. Ectopic hormone-secreting pheochromocytoma: a francophone observational study. World Journal of Surgery 36 1382–1388. ( 10.1007/s00268-012-1488-1) [DOI] [PubMed] [Google Scholar]
- 9.van Berkel A, Lenders JWM, Timmers HJLM. 2014. Diagnosis of endocrine disease: biochemical diagnosis of phaeochromocytoma and paraganglioma. European Journal of Endocrinology 170 R109–R119. ( 10.1530/EJE-13-0882) [DOI] [PubMed] [Google Scholar]
- 10.Forman BH, Marban E, Kayne RD, Passarelli NM, Bobrow SN, Livolsi V, Merino M, Minor M, Farber LR. 1979. Ectopic ACTH syndrome due to pheochromocytoma: case report and review of the literature. Yale Journal of Biology and Medicine 52 181. [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, Doppman JL, Chrousos GP, Norton JA, Nieman LK, Udelsman R. 1995. Adrenocorticotropic hormone-secreting pheochromocytomas: the exception to the rule. Surgery 118 988–995. ( 10.1016/S0039-6060(05)80104-7) [DOI] [PubMed] [Google Scholar]
- 12.Cassarino MF, Ambrogio AG, Pagliardini L, De Martin M, Barresi V, Cavagnini F, Pecori Giraldi F. 2012. ACTH-secreting pheochromocytoma with false-negative ACTH immunohistochemistry. Endocrine Pathology 23 191–195. ( 10.1007/s12022-012-9212-0) [DOI] [PubMed] [Google Scholar]
- 13.Berenyi M, Singh G, Gloster E, Davidson M, Woldenberg D. 1977. ACTH-producing pheochromocytoma. Archives of Pathology & Laboratory Medicine 101 31–35. [PubMed] [Google Scholar]
- 14.Schroeder JO, Asa SL, Kovacs K, Killinger D, Hadley GL, Volpé R. 2014. Report of a case of pheochromocytoma producing immunoreactive ACTH and beta-endorphin. Journal of Endocrinological Investigation 7 117–122. ( 10.1007/BF03348400) [DOI] [PubMed] [Google Scholar]
- 15.Lutgers HL, Vergragt J, Dong PV, de Vries J, Dullaart RP, van den Berg G, Ligtenberg JJ. 2010. Severe hypercortisolism: a medical emergency requiring urgent intervention. Critical Care Medicine 38 1598–1601. ( 10.1097/CCM.0b013e3181e47b7a) [DOI] [PubMed] [Google Scholar]
- 16.Oh HC, Koh J-M, Kim MS, Park JY, Shong YK, Lee K-U, Kim GS, Hong SJ, Koo HL, Kim WB. 2003. A case of ACTH-producing pheochromocytoma associated with pregnancy. Endocrine Journal 50 739–744. ( 10.1507/endocrj.50.739) [DOI] [PubMed] [Google Scholar]
- 17.Loh K-C, Gupta R, Shlossberg AH. 1996. Spontaneous remission of ectopic Cushing’s syndrome due to pheochromocytoma: a case report. European Journal of Endocrinology 135 440–443. ( 10.1530/eje.0.1350440) [DOI] [PubMed] [Google Scholar]
- 18.Sakurai H, Yoshiike Y, Isahaya S, Matsushita H, Yamanaka K, Okubo T, Kanamori H, Takahashi H, Fu T. 1987. A case of ACTH-producing pheochromocytoma. American Journal of the Medical Sciences 294 258–261. ( 10.1097/00000441-198710000-00008) [DOI] [PubMed] [Google Scholar]
- 19.Negro A, Manicardi E, Grasselli C, Babini M, Santi R, Pugni V, Spaggiari L, Tagliavini E. 2013. Severe ectopic Cushing’s syndrome due to ACTH-secreting pheochromocytoma. International Journal of Clinical Medicine 4 228–231. ( 10.4236/ijcm.2013.44040) [DOI] [Google Scholar]
- 20.Bernardi S, Grimaldi F, Finato N, De Marchi S, Proclemer A, Sabato N, Bertolotto M, Fabris B. 2011. A pheochromocytoma with high adrenocorticotropic hormone and a silent lung nodule. American Journal of the Medical Sciences 342 429–432. ( 10.1097/maj.0b013e3182260551) [DOI] [PubMed] [Google Scholar]
- 21.Kaya T, Karacaer C, Acikgoz SB, Aydemir Y, Tamer A. 2016. Severe hypokalaemia, hypertension, and intestinal perforation in ectopic adrenocorticotropic hormone syndrome. Journal of Clinical and Diagnostic Research 10 OD09–OD11. ( 10.7860/jcdr/2016/17198.7127) [DOI] [PMC free article] [PubMed] [Google Scholar]