

Abstract
Mequindox (MEQ) is a synthetic antimicrobial agent of quinoxaline-1,4-dioxide group (QdNOs). The liver is regarded as the toxicity target of QdNOs, and the role of N → O group-associated various toxicities mediated by QdNOs is well recognized. However, the mechanism underlying the in vivo effects of MEQ on the liver, and whether the metabolic pathway of MEQ is altered in response to the pathophysiological conditions still remain unclear. We now provide evidence that MEQ triggers oxidative damage in the liver. Moreover, using LC/MS-ITTOF analysis, two metabolites of MEQ were detected in the liver, which directly confirms the potential connection between N → O group reduction metabolism of MEQ and liver toxicity. The gender difference in MEQ-induced oxidative stress might be due to adrenal toxicity and the generation of M4 (2-isoethanol 1-desoxymequindox). Furthermore, up-regulation of the MAPK and Nrf2-Keap1 family and phase II detoxifying enzymes (HO-1, GCLC and NQO1) were also observed. The present study demonstrated for the first time the protein peroxidation and a proposal metabolic pathway after chronic exposure of MEQ, and illustrated that the MAPK, Nrf2-Keap1 and NF-кB signaling pathways, as well as the altered metabolism of MEQ, were involved in oxidative toxicity mediated by MEQ in vivo.
Quinoxaline-di-N-oxides (QdNOs) possessing the quinoxaline-1,4-dioxide basic structure, are known to be potent antibacterial agents with a wide range of biological properties1,2,3,4,5,6,7. They had been developed for use in livestock because of their significant antibacterial abilities and growth-promoting properties3,6,7,8,9,10. Carbadox (CBX), olaquindox (OLA) and quinocetone (QCT) are the members of QdNOs5,6. Mequindox (3-methyl-2-acetyl-N-1,4-dioxyquinoxaline, C11H10N2O3; MEQ) (Fig. 1), acting as inhibitor of DNA synthesis, is a synthetic QdNOs which acts as an antibacterial agent11,12,13. MEQ has been widely applied in pigs and chickens in China since the 1980s owing to its strong inhibitory activities against both gram-positive and -negative species14.
Figure 1. The chemical structures of mequindox (MEQ), mequindox-4-monoxide (N1-MEQ) and desoxymequindox (B-MEQ).

A lot of evidence suggested that the major metabolic pathway for QdNOs involves N → O group reduction1,2,15, and that this type of metabolism is closely related to their toxicity10,16,17. QdNOs were reported to have the ability of hypoxia–selective DNA cleavage2,18. A few studies revealed that the metabolites of CBX, OLA and QCT exhibit varying degrees of toxicity5,10,19. 2-isoethanol 4-desoxymequindox (M11), an N → O reduction metabolite of MEQ, was detected in the testis of the Wistar rats after the exposure of MEQ (25–275 mg/kg diet) for 180 days, where it was accompanied by oxidative DNA damage12. MEQ and its primary metabolites N1-desoxymequindox (N1-MEQ) and bidesoxy-mequindox (B-MEQ) (Fig. 1), were also found to be genotoxic in short-term in vitro and in vivo tests2,20. Therefore, the metabolites were thought to participate in MEQ–induced liver damage. However, to our knowledge, only two published studies have examined the effects of MEQ metabolites on the liver in vivo. One study aimed to identify the metabolism, distribution, and elimination of MEQ in pigs, chickens, and rats14, and another provided evidence that the metabolites of MEQ were related to the oxidative damage observed in Wistar rats21. It has been well established that there are generally species-specific differences in the metabolism of drugs, and the expression of drug-metabolizing enzymes may be altered by pathophysiological conditions, including oxidative stress. The metabolism of MEQ may also be altered in vivo after chronic exposure of animals. Therefore, it still needs to determine the potential toxic metabolites and the possible metabolic pathway of MEQ in the mouse liver under oxidative toxicity in the chronic toxic study of MEQ.
In previous studies, oxidative stress was found to be closely related to the damaging effects of QdNOs, such as apoptosis, DNA and lipid damage in vivo and in vitro22,23,24. MEQ exhibited mutagenicity and was a potent inducer of DNA damage via the production of reactive oxygen species(ROS)25. Oxidative-stress was also involved in the genotoxicity induced by QCT and OLA26,27. As for the mechanism of genotoxicity, the production of ROS, including superoxide anion (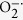 ), hydroxyl radicals (HO·) and hydrogen peroxide (H2O2) during N → O reduction of QCT by xanthine oxidoreductase (XOR), was shown to be a main factor in the DNA strand breakage and generation of 8-hydroxy deoxyguanosine (8-OHdG)5. We recently showed that MEQ could result in adrenal toxicity by oxidative stress in H295R cells that originated from a human adrenocortical carcinoma28. It has been demonstrated that oxidative stress was one of the most important toxicity mechanisms in the adrenal gland29, liver and spleen21, and endocrine and reproductive systems12 in male Wistar rats following exposure to MEQ. Although the oxidative damage invoked by QdNOs, including MEQ, caused much more attentions, the role of some related signaling pathways still remains unclear.
), hydroxyl radicals (HO·) and hydrogen peroxide (H2O2) during N → O reduction of QCT by xanthine oxidoreductase (XOR), was shown to be a main factor in the DNA strand breakage and generation of 8-hydroxy deoxyguanosine (8-OHdG)5. We recently showed that MEQ could result in adrenal toxicity by oxidative stress in H295R cells that originated from a human adrenocortical carcinoma28. It has been demonstrated that oxidative stress was one of the most important toxicity mechanisms in the adrenal gland29, liver and spleen21, and endocrine and reproductive systems12 in male Wistar rats following exposure to MEQ. Although the oxidative damage invoked by QdNOs, including MEQ, caused much more attentions, the role of some related signaling pathways still remains unclear.
The p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) are members of MAPK cascades. They are activated by oxidative stress30,31,32,33,34,35 to adjust the intracellular redox status36. Nuclear factor κB (NF-κB) is a sequence-specific transcription factor that functions in the immunological and cellular detoxifying defense systems34,37. In response to oxidative stress, Nrf2 translocates into the nucleus after dissociating from Kelch-like ECH-associated protein 1 (Keap 1), and then binds to antioxidant response elements (AREs)38,39,40. The Nrf2/Keap1/ARE pathway is a major cellular defense mechanism against oxidative stress by mediating phase II detoxifying enzymes and antioxidant enzymes41. The p38-Nrf2 signaling pathway has been shown to modulate oxidative stress in vitro42,43 and in vivo36,43. Previous studies indicated that, along with ROS generation, the phosphorylation of p38 and JNK were significantly increased in HepG2 cells after exposure to OLA44 and QCT45. Additionally, the oxidative damage caused by QCT involved the over expression of Nrf2 in rat liver46,47 and H295R cells48. However, to date, it is not clear whether the activation of the MAPK, Nrf2-Keap1 and NF-κB pathways participate in oxidative liver damage caused by MEQ though they have the same quinoxaline ring.
Based on the above information, this study was designed to investigate the hypothesis that MEQ–induced liver toxicity was associated with oxidative stress, the metabolism of MEQ and the activation of the MAPKs, NF-κB, and Nrf2-Keap1 signaling pathways. Since the liver, which is highly vulnerable to oxidative stress due to its high metabolic rate and low levels of endogenous scavengers, has been identified as one of the main target organs of QdNOs49,50,51, we evaluated the liver toxicity in Kunming mice after exposure to MEQ for 11 months. In the present study, we investigated: (1) the effect of MEQ on body and liver weight, and morphological changes in livers; (2) the effect of MEQ on the activities of albumin (ALB), alkaline phosphatase (ALP), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in the serum of mice; (3) identification the metabolites of MEQ in liver by LC/MS-ITTOF analysis; (4) the effect of MEQ on the levels of reduced glutathione (GSH) and total superoxide dismutase (T-SOD), and on oxidative damage to lipids, proteins and DNA, as measured by the levels of malondialdehyde (MDA), protein carbonyl content (PCC), and 8-OHdG; (5) whether MEQ activated the oxidative stress upstream signaling pathways involved in liver damage (e.g. p38, JNK); (6) the effect of MEQ on the mRNA expression of some cytokines related to the activation and regulation of oxidative stress (eg. NF-кB, Nrf2, Keap1, GCLC, NQO1 and HO-1).
Results
Body weight and liver coefficients
The final body weight and liver coefficients of mice after administration of MEQ for 11 months are shown in Fig. 2. The liver coefficients was expressed as (wet weight of liver, mg)/(total body weight, g). Significant reductions in body weight were observed in the 25 mg/kg MEQ group (p < 0.05), and the 55, 110 mg/kg groups (p < 0.01) as compared with the control. Furthermore, there was a significant increase in the liver coefficients in the 25, 55 mg/kg MEQ (p < 0.05), and 110 mg/kg MEQ (p < 0.01) groups as compared with controls.
Figure 2. Body weight and liver coefficients of mice after administration of MEQ for 11 months.

*p < 0.05, and **p < 0.01. Values represent means ± SD (n = 10).
Histopathological evaluation
As shown in Fig. 3, obvious histopathological changes were observed in the liver after administration of 55 and 110 mg/kg MEQ. The hepatic cells exhibited degeneration and necrosis (a necrotic lesion) in the 55 mg/kg MEQ group. In the 110 mg/kg MEQ group, there was marked neutrophilic infiltration within and around the bile duct and most bile duct epithelium examined showed proliferation, in contrast to control group.
Figure 3. Selected microphotographs of liver (200×, 400×).

(A) Control liver (200×). (B) Liver of the 110 mg/kg MEQ group showing degeneration and necrosis (200×). (C) Liver of the control group, the bile duct was normal (400×). (D) Liver of the 110 mg/kg MEQ group, the bile duct epithelium was marked with proliferation and neutrophil infiltration (400×).
Hepatocellular enzymes and liver function indices
In comparison with the control group, three liver function indices – ALB, ALT and ALP – showed increased serum activity in all the MEQ–treated groups (Fig. 4).The 25 mg/kg MEQ group also showed significantly increased levels of AST as compared to controls (p < 0.05).This result was consistent with histopathological examination of liver, indicating the liver damage induced by MEQ.
Figure 4. Effects of MEQ on the activity of ALB, ALP, ALT and AST in the mice serum after administration of MEQ for 11 months.

*p < 0.05, and **p < 0.01. Values represent means ± SD (n = 10).
MEQ and its metabolites in liver
The prototype and metabolites of MEQ were determined according to the retention times and fragment ions. The results showed that noprototype of MEQ was found, while only two metabolites (M4 and M8)appeared in all treated groups (Fig. 5). Accurate MS2 spectra of M4 and M8 identified them as 2-isoethanol 1-desoxymequindox (the hydrogenation of the 1-desoxymequindox) and 2-isoethanol 4-desoxymequindox (the hydrogenation of the 4-desoxymequindox), respectively.
Figure 5. Accurate EIC of prototype and metabolites of MEQ in mice liver.

(A) Liver of control; (B) Liver of 25 mg/kg MEQ diet; (C) Liver of 55 mg/kg MEQ diet; (D) Liver of 110 mg/kg MEQ diet; (E) The chemical structure of M4 is 2-isoethanol 1-desoxymequindox, and M8 is 2-isoethanol 4-desoxymequindox.
Oxidative stress
To establish whether oxidative stress was elevated in the injured mouse liver, we measured the generation of T-SOD, GSH, MDA, PCC, and 8-OHdG in liver tissue as oxidative indices. As shown in Fig. 6, significant increases in the levels of 8-OHdG (p < 0.05) and PCC (p < 0.01) were noted in all treated groups as compared to controls. The levels of GSH, MDA and T-SOD were significantly higher in the 110 mg/kg than in controls (p < 0.01 or p < 0.05). Interestingly, there were significant gender differences in the levels of T-SOD, GSH and MDA induced by MEQ. At dose of 25 and 55 mg/kg MEQ, significantly increased levels of MDA and GSH were also observed in both genders (p < 0.01). However, after treatment with25 and 55 mg/kg MEQ, a significantly increased level of T-SOD was observed in females (p < 0.05, p < 0.01), but not in males. After treatment with MEQ, females showed higher levels of MDA and T-SOD as compared to males, while males showed higher levels of GSH as compared to females.
Figure 6.

Effects of MEQ on (A) Peroxidation of DNA, (B) Protein carbonyl content, (C) Lipid peroxidation, (D) GSH, (E) T-SOD in mouse liver after administration of MEQ for 11 months. *p < 0.05, and **p < 0.01. Values represent means ± SD (n = 10).
Activation of the NF-кB, MAPK and Nrf2-Keap1 signaling pathways
To confirm whether MEQ–induced oxidative damage in the mouse liver induced various signaling pathways, the expression of some oxidative stress–related genes (e.g. p38, JNK, Nrf2, Keap1, NF-кB, HO-1, NQO1, GCLC) were evaluated using real-time quantitative RT-PCR (Fig. 7). Figure 7 shows that exposure to 110 mg/kg MEQ induced significant increases in p38 and JNK expression in the liver (p < 0.01). With increasing MEQ doses, there was a significant increase in Nrf2 expression (p < 0.01), and a marked reduction in Keap1 expression (p < 0.05 or p < 0.01). Exposure to MEQ significantly induced the expression of NF-кB and NQO1 in all the treated groups (p < 0.01). Increased mRNA expression of HO-1 was observed after treatment with 110 mg/kg MEQ (p < 0.01), and GCLC was significantly increased in both 55 and 110 mg/kg MEQgroups (p < 0.01). These results show a cellular protection response to oxidative stress mediated by chronic toxic study of MEQ.
Figure 7. Alterations of P38, JNK, Nrf2, Keap1, NF-KB, HO-1, GCLC and NQO1 expression in mouse liver after administration of MEQ for 11 months.

*p < 0.05, and **p < 0.01. Values represent means ± SD (n = 10).
Discussion
Previous studies have demonstrated that oxidative stress is associated with the in vitro toxicity of QdNOs, including cytotoxicity26,46,52, adrenal toxicity24,26, genotoxicity5,6,25,27,46,53 and apoptosis8,44,45,52,54,55,56,57. In vivo studies in rats11,12,21,29,47,58,59 and mice60,61 have confirmed these findings. However, relatively little is known about the metabolism and the molecular mechanisms that mediate their toxicity. In the present study, we have demonstrated that the MAPK pathway, Nrf2-Keap1 family and NF-кB are closely related to the oxidative damage induced by MEQ in mouse liver. A higher sensitivity to oxidative damage was observed in females, as compared to males, after chronic administration of MEQ for up to 11 months. Furthermore, we identified that M4 and M8 were critical metabolites responsible for the liver toxicity induced by MEQ. Interestingly, the pathways of MEQ metabolism may be altered under a toxic environment via the altered expression of drug–metabolizing enzymes. Additionally, besides the traditional 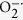 and HO·, the intermediate radicals of MEQ may be the key steps in triggering oxidative damage and activating the MAPK and Nrf2-Keap1 pathways.
and HO·, the intermediate radicals of MEQ may be the key steps in triggering oxidative damage and activating the MAPK and Nrf2-Keap1 pathways.
Liver was identified as one of the main target organs for toxicity mediated by CYA49, QCT50 and MEQ11. MEQ induced disorganized hepatic cord patterning, cellular swelling and centrilobular liver cell necrosis in Wistar rats11. The altered levels of ALB, ALT, ALP and AST indicate chronic liver disease11,49. In the present study, a significant reduction in body weight and a significant increase in the liver coefficients were observed (Fig. 2). The histopathological evaluation showed marked liver damage, including extensive proliferation of the bile duct epithelium, and neutrophilic infiltrate within and around the bile duct. In biochemical analysis, MEQ–mediated increase in serum ALP exhibited disrupted plasma membrane bilayer and then resulted in efflux of serum ALT (cytosolic) and AST (mitochondrial). These results were consistent with previous reports suggesting that liver damage occurs after sub-chronic oral administration of MEQ in Wistar rats11,21,29.
A previous in vitro study reported that MEQ could be metabolized into ten metabolites after incubation with liver microsomes62. Recently, quantitative analysis of the in vivo metabolism of MEQ revealed that M4 was a common metabolite of MEQ in liver models of pigs, chickens and rats14. In another study of the relationship between the metabolites and toxicity, M11 was identified as the main toxic metabolite in the liver and spleen of Wistar rats after exposure to MEQ for 180 days21. The different metabolites detected in liver demonstrated the changed metabolic pathway of MEQ under the toxic environment, and therefore, M11 could be regarded as a biomarker of liver damage induced by MEQ. It is generally accepted that the metabolic activity of the enzymes such as aldo-keto reductases (AKR) and glutathione transferases (GST) has important consequences for the cellular redox status, and in turn, these enzymes could be regulated by oxidative stress7. Expression of drug–metabolizing enzymes may be altered in response to the pathophysiological conditions of the imbalance oxidative stress (Fig. 8). Thus, it was presumed that the alteration of MEQ–metabolizing enzymes such as carbonyl reductase 1 (CBR1), xanthine oxidoreductase (XOR), porcine aldehyde oxidase (SsAOX1) and cytochrome P450 (CYP)34 was possible a reason for the altered metabolic pathway of MEQ under liver damage status. Further study should focus on the expression and activity of these metabolizing enzymes after chronic exposure to MEQ. Herein, the detection of M4 and M8 not only directly confirmed the potential connection between N → O group reduction metabolism of MEQ and its organ toxicity, but also, indicated that these metabolites may act as biomarkers of liver toxicity in mice.
Figure 8. The proposed mechanisms of oxidative stress in mouse liver caused by MEQ.

The oxidative damage occurs via the reduction of the N → O group of MEQ, which activate the MAPKs (e.g. P38 and JNK) signaling pathway, as well as Nrf2-Keap1 family, NF-κB and II phase detoxifying enzymes (e.g. HO-1, GCLC and NQO1). The sex difference in oxidative stress and genetoxicity caused by MEQ and N1-MEQ, respectively, may be derived from the adrenal toxicity along with the sex hormone secretion disorder.
Apart from the deoxidation rate of QdNOs6, the appearance of ROS and unstable oxygen–sensitive radical intermediates during N → O reduction of QdNOs is considered to play a critical role in the DNA damage1,18,63,64. Previously, the production of oxygen–sensitive radical intermediates during QdNOs metabolism was thought to explain the mutagenicity18, antibacterial activity1 and hypoxic cytotoxin induced by these compounds63,65. The varying degrees of genotoxicity caused by QdNOs depends on the persistence of the ROS species6 and the stability of the radical intermediates65. Therefore, MEQ undergo intracellular one-electron enzymatic reduction to yield an oxygen–sensitive drug-radical intermediate and then generate highly reactive secondary radicals including 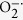 , HO· and MEQ radical intermediates that ultimately cause oxidative stress and DNA damage in the mouse liver (Figs 8 and 9). The metabolism of MEQ to M8 requires two steps, during which two radical intermediates were formed (Fig. 9). The proposed metabolic pathway in mice after chronic exposure of MEQ (Fig. 9) confirmed the critical role of radical intermediates in oxidative toxicity.
, HO· and MEQ radical intermediates that ultimately cause oxidative stress and DNA damage in the mouse liver (Figs 8 and 9). The metabolism of MEQ to M8 requires two steps, during which two radical intermediates were formed (Fig. 9). The proposed metabolic pathway in mice after chronic exposure of MEQ (Fig. 9) confirmed the critical role of radical intermediates in oxidative toxicity.
Figure 9. The proposed metabolic pathways of MEQ in mouse liver.

The production of six MEQ intermediate radicals, as well as  and OH· occurs via the reduction of the N → O group of MEQ.
and OH· occurs via the reduction of the N → O group of MEQ.
In order to demonstrate that exposure to MEQ caused oxidative stress in the mouse liver, we assayed five oxidative stress-related indicators. The results showed that MEQ caused significant increases in the levels of 8-OHdG, PCC, MDA, T-SOD and GSH. Previous studies also reported that the organ toxicity of MEQ in vivo was subjected to oxidative stress. In a study to investigate the metabolic response of mice after exposure to MEQ at 15, 75 and 350 mg/kg for 7 days, the oxidative damage was elevated by high doses of MEQ61. When rats were exposed to MEQ (25, 55, 110 and 275 mg/kg) for 180 days, high doses of MEQ caused testicular, liver, kidney,and adrenal toxicity, along with oxidative damage12,21,29. Oxidative stress could impair the antioxidant defense system and result in serious damage to cellular macromolecules such as DNA, lipids and protein24. However, to our knowledge, apart from MDA and 8-OHdG, no study about PCC induced by QdNOs was investigated. Here, we tested the ability of MEQ to induce protein peroxidation in the mouse liver, and we firstly found that protein was also one of the major targets of MEQ–induced oxidative damage.
As the most abundant intracellular antioxidant, SOD can serve as a redox biomarker of the antioxidant state66, and the role of GSH is to protect cells against oxidative damage67. It was reported that administration of MEQ (275 mg/kg diet) for 180 days in Wistar rats caused testicular, liver, kidney, and adrenal toxicity along with a significant increase in SOD activity and significant decrease in GSH levels12,21,29. The increased levels of T-SOD and GSH may be related to the activation of the upstream signaling pathway responsible for regulating oxidative stress. This hypothesis was confirmed by further study on the Nrf2-Keap1 family in mouse liver. Interestingly, there was an obvious sex difference in the levels of T-SOD, GSH and MDA. The levels of MDA and T-SOD were higher in females, while the activity of GSH was higher in males, suggesting more serious oxidative stress in females. These results were similar to a previous study that high doses of MEQ could significantly increase SOD activities in females when rats were treated with MEQ (55, 110 and 275 mg/kg) for 90 days11. Additionally, a few studies have reported adrenal toxicity caused by MEQ in vivo26,29 and in vitro28,48. Previous findings have shown that long-term MEQ treatment induced reproductive toxicity via alterations in the expression of some genes responsible for cholesterol transport and testosterone synthesis12, demonstrating the altered sex hormone secretion induced by MEQ. The changes in sex hormone secretion may be an indirect effect of adrenal toxicity induced by MEQ. These difference, in turn may explain the different sensitivities of males and females to MEQ – induced oxidative damage.
It was reported that the primary metabolites of MEQ – N1-MEQ and B-MEQ –were partially and completely reduced derivatives of MEQ, respectively2,14,68. Our recent research revealed that N1-MEQ showed higher adrenal toxicity than B-MEQ28. In amicronucleus test, the mice were treated with N1-MEQ at dose levels of 0.08, 0.16, 0.31 mg/kg, N1-MEQ (0.08 mg/kg) induced a significant increase in the ratio of micronucleated polychromatic erythrocytes (MN-PCEs) in females while not in males2, indicating an higher sensitivity of female mice to N1-MEQ-induced genotoxicity. The sex differences in N1-MEQ-induced genotoxicity may correspond to the levels of MEQ-induced oxidative stress (Fig. 8). Thus, we suspected that the occurrence of M4 (hydroxylation of N1-MEQ) may be another reason for the sex differences in MEQ–induced oxidative damage (Fig. 8).
To confirm whether the oxidative stress caused by MEQ is associated with the activation of some signaling pathway, we investigated MAPK signal transduction, which showed higher levels of p38 and JNK expression following exposure to MEQ (Fig. 7). In two studies investigating MAPK pathways in OLA–induced apoptosis45, and the molecular mechanisms of apoptosis induced by QCT58, a significant increase in p38 and JNK were observed. In the nucleus, Nrf2 first forms dimers with c-Jun before combining with ARE. The increased expression of JNK could activate c-Jun and indirectly stimulate the Nrf2-ARE pathway (Fig. 8). Moreover, after treatment with MEQ, the overexpressed of p38 and JNK in the plasma promote phosphorylation of Nrf2 (Fig. 8). NF-кB is a redox–sensitive transcription factor involved in the immunological and cellular detoxifying defense systems36,59,69. Previously, Yu et al. discovered that QCT-induced nephrotoxicity was accompanied by an increase in Nrf2 expression47, and the NF-кB and Nrf2/HO-1 pathways could protect cells against the oxidative stress induced by QCT59. The Nrf2-Keap1 signaling pathway plays a significant role in regulating the expression of phase II detoxifying enzymes and antioxidant enzymes that protect cells from endogenous and exogenous stresses34,59. Here, treatment with MEQ resulted in a significant increase in NF-кB and Nrf2 gene expression, and a significant decrease in Keap1 gene expression (Fig. 7). These findings indicated that the high level of p38, as well as JNK, NF-кB and Nrf2 expressions may be a cellular protective response to MEQ oxidative stress (Fig. 8).
To further reveal the possible downstream genes of Nrf2-Keap1 family following exposure to MEQ, we measured its target gene products including NQO1, HO-1 and GCLC. These enzymes protect against oxidative stress-induced toxicity either via scavenging ROS directly or via catalyzing the synthesis of antioxidant and detoxifying substrates indirectly70. Our data showed that MEQ exposure induced marked increases in HO-1 and GCLC levels (Fig. 7). These results indicated that HO-1 and GCLC may mediate a protective effect against MEQ–induced oxidative damage. NQO1 has been reported to play an important role in xenobiotic metabolism and in protecting against oxidative stress induced by the intermediates produced during xenobiotic detoxification71. Here, a significant dose-dependent increase in the induction of NQO1 was observed in all MEQ–treated groups. NQO1 may involved in the metabolic reaction of hydroxy and hydrogenated production of M4 and M8. In the present study, the increased expression of NQO1 was suggested to be associated with the generation of M4, M8 and MEQ radical intermediates. This result illustrates the important role of NQO1 in the metabolism of MEQ.
In conclusion, the current study demonstrated that M4 and M8 may represent the major toxic metabolites of MEQ causing oxidative liver damage in vivo, and that oxidative stress plays a key role in the liver toxicity. The pathways governing the metabolism of MEQ may be altered by changes in the activity of the MEQ–metabolizing enzymes under the imbalance of cellular redox status. Further study should be conducted to investigate the activity of the MEQ-metabolizing enzymes and the roles of MEQ radical intermediates under this case. Sex–specific differences in oxidative stress were observed in mice after administration of MEQ. It seems that the adrenal toxicity was possible an indirect reason for sex difference in oxidative stress caused by MEQ. The protein was firstly identified as the major target of MEQ–induced oxidative damage. The present findings also reveal that NF-кB, as well as MAPK (p38 and JNK) signaling pathways and the Nrf2-Keap1 family are involved in MEQ induced–redox imbalance damage in the mouse liver.
Materials and Methods
Chemical reagents
Mequindox (C11H10N2O3, molecular weight 218.21 g/mol, CAS No: 60875-16-3, purity 98%) was obtained from Beijing Zhongnongfa Pharmaceutical Co. Ltd. (Huanggang, PR China). MDA, PCC, SOD, GSH and 8-OHdG kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, P.R. China). All other chemicals were purchased from Sigma (St. Louis, USA) unless otherwise stated.
Animals and diets
Forty in total specific pathogen-free (SPF) Kunming mice (6–7 weeks old, weighting 25–36 g) were purchased from the Center of Laboratory Animals of Hubei Province (Wuhan, PR China). For each sex, the individual body weights were within ±20% of the average. The mice were maintained in a room conditioned at 22 ± 3 °C, a relative humidity of 50% ± 20%, and a 12 h light/dark cycle. In this study, the mice were handled in accordance with the guidelines and protocols approved by the Ethical Committee of the Faculty of Veterinary Medicine (Huazhong Agricultural University). Prior to initiation of dosing, mice were quarantined for 1 week to evaluate any signs of disease and weight gain. The mice received free access to fresh water and basic diet during the one week acclimatization period.
For the experiments, the mice were randomly divided into four groups (N = 10 per group), including a control group (received the basic diet without feed additives) and three treated groups (administrated the same diet supplemented with 25, 55 and 110 mg/kg MEQ). Mice were separated by sex and housed five per group in shoebox cages with hardwood shavings as bedding. Food and water were supplied ad libitum and the treatment period lasted for 11 months. Symptoms and/or mortality were observed and carefully recorded each day during the 11 month period. The use of animals in this study was in compliance with the NIH Publication “The Development of Science-Based Guidelines for Laboratory Animal Care”72.
Coefficients and preparation of liver
Following 11 months of MEQ administration, all mice were subjected to fasting overnight. The mice were then weighted and sacrificed after being anesthetized with diethyl ether. After weighing the body and livers, the coefficient of liver was calculated as the ratio of liver (wet weight, mg) to body weight (BW) (g). The livers were excised, rinsed in phosphate buffered saline (PBS), weighed and then quickly frozen at −70 °C.
Histopathological examination of liver
For pathological studies, all histopathological tests were performed using standard laboratory procedures. Half of livers were preserved in 10% neutral-buffered formalin. After fixation, the livers were embedded in paraffin blocks then sliced into 5 μm sections by microtome and placed onto glass slides for hematoxylin-eosin (HE) staining. Slides were observed under an optical microscope (Olympus BX 41, Japan) for morphological alterations.
Biochemical analysis
For biochemical analysis, serum aliquots were obtained by placing the blood samples in serum tubes at a temperature of 24 °C for approximately 30 min. After clotting, the blood tubes were centrifuged at 3000 rpm for 10 min using a Himac CR 21 G centrifuge (Hitachi, Tokyo, Japan). Supernatants were removed and stored at −20 °C for further analysis. Serum chemistry was assessed using a Synchron CX4 Clinical System (Beckman Coulter, Brea, CA USA) according to the manufacturer’s protocol (Beijing Leadman Biochemistry Technology Co. Ltd, Beijing, China). The serum activities of ALB, ALP, ALT and AST were used as biochemical markers of hepatic damage.
LC/MS-ITTOF analysis of MEQ and its metabolites in liver
The detection of mequindox and its metabolites in the liver was carried out using the hybrid IT/TOF mass spectrometer coupled with a high-performance liquid chromatography system (Shimadzu Corp., Kyoto, Japan). The liquid chromatography system (Shimadzu) was connected to a solvent delivery pump (LC-20AD), an autosampler (SIL-20AC), a DGU-20A3 degasser, a photodiode array detector (SPD-M20A), a communication base module (CBM-20A) and a column oven (CTO-20AC).
A total of 0.1 g of the liver sample was homogenized with 4.5 mL of 40 °C distilled water at the speed of 10,000 × g for 3 min in a model omni mixer homogenizer 17106 (OMNI International, Waterbury, CT, USA). Then 0.5 mL trichloroacetic acid was added at a final concentration of 10%. After vigorous shaking, the homogenate was centrifuged at 10,000 × g for 15 min to collect the supernatant. The mixed reagent [dichloromethane: acetonitrile (2:1, v/v)] was used to extract mequindox and its metabolites twice. Three milliliters of the mixed reagent was then added to the supernatant and vortex-mixed for 5 min. After vigorous shaking, the solution was centrifuged at 10,000 × g for 15 min. The lower liquid from the two extractions was merged and dried using N2 in a 40 °C water bath. The residue was reconstituted in 5 mL of 5% methanol. The reconstitution fluid was applied to a methanol (3 mL) and water (3 mL) pre-washed HLB 3cc cartridge (Waters Corporation, Milford, MA, USA). The reconstitution fluid was then sequentially washed with 3 mL of 5% methanol in water and 5 mL of methanol. The liver extracts were eluted into plastic tubes and evaporated to dryness under a nitrogen stream at 45 °C. Following drying, the residue was dissolved in 500 μL of LC-MS/MS mobile phase solution [acetonitrile: 0.1% formic acid (1:9, v/v)] and passed through a 0.22 μm filter membrane. The mixture (200 μL) was prepared for LC/MS-ITTOF analysis. Mequindox and its metabolites were identified based on a previous report14.
Oxidative stress assay
Assays of MDA, T-SOD, GSH and PCC levels in liver were performed using commercial kits. 8-OHdG was assayed by using a commercial ELISA kit. Data were analyzed according to the manufacturer’s instructions. Protein concentration in liver tissue was measured using BCA protein assay kit.
Expression of oxidative stress cytokines
The level of mRNA expression of oxidative stress-related genes, including p38, JNK, Nrf-2, Keap1, NF-кB, HO-1, GCLC, NQO-1, were determined by real-time quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). Total RNA was isolated from the liver homogenates using the Trizol reagent according to the manufacturer’s instructions. One microgram of RNA was reverse transcribed to cDNA using the ReverTra AceTM First Strand cDNA Synthesis Kit (Promega, USA). Synthesized cDNA was used for quantitative real-time PCR (Bio-Rad, USA) by SYBR® Premix Ex TaqTM RT-PCR kit (Takara, CodeDRR041 A, Japan).
Mouse specific primers were designed using Primer Express Software according to the software guidelines (Table 1). Each 25 μL reaction mixture consisted of 12.5 μL SYBR® Premix Ex TaqTM, 1.0 μL of each primer (10 μm), 2.0 μL of cDNA, and 8.5 μL Rnase Free H2O. For p38, JNK, HO-1, GCLC, the cycling conditions were as follows: step 1, 30 s at 95 °C; step 2, 45 cycles at 95 °C for 5 s, 55 °C for 30 s; step 3, dissociation stage. For Nrf-2, Keap1, NF-кB, NQO-1, the cycling conditions were as follows: step 1, 30 s at 95 °C; step 2, 45 cycles at 95 °C for 5 s, 60 °C for 30 s; step 3, dissociation stage. The housekeeping gene β-Actin was used as an internal calibrator reference gene for expression profiling of oxidative stress-related genes in this study.
Table 1. PCR Primers Used in the Gene Expression Analysis.
| Gene name | Description | Sequence (5′-3′) | Length (bps) |
|---|---|---|---|
| β-actin | mβ-actin - F | CTGTCCCTGTATGCCTCTG | 221 |
| mβ-actin - R | TTGATGTCACGCACGATT | ||
| P38 | mP38 - F | GGAGAAGATGCTCGTTTTGGA | 211 |
| mP38 - R | TTGGTCAAGGGGTGGTGG | ||
| JNK | mJNK - F | TCTCCAGCACCCATACATCAA | 150 |
| mJNK - R | TCCTCCAAATCCATTACCTCC | ||
| Nrf-2 | mNrf-2 - F | TCCTATGCGTGAATCCCAAT | 103 |
| mNrf-2 - R | GCGGCTTGAATGTTTGTCTT | ||
| Keap1 | mKeap1 - F | GATCGGCTGCACTGAACTG | 106 |
| mKeap1 - R | GGACTCGCAGCGTACGTT | ||
| NF-κB | mNF-κB - F | GGTGGAGGCATGTTCGGTA | 142 |
| mNF-κB - R | TGACCCCTGCGTTGGATT | ||
| NQO1 | mNQO1 - F | TTCTGTGGCTTCCAGGTCTT | 104 |
| mNQO1 - R | TCCAGACGTTTCTTCCATCC | ||
| GCLC | mGCLC - F | ATGTGGACACCCGATGCAGTATT | 200 |
| mGCLC - R | GTCTTGCTTGTAGTCAGGATGGTTT | ||
| HO-1 | mHO-1 - F | GACAGAAGAGGCTAAGACCGC | 213 |
| mHO-1 - R | TGGAGGAGCGGTGTCTGG |
Note: The primers were manufactured by Nanjing Genescript Co. Ltd. (Nanjing, PR China). P38, p38 MAP Kinase; JNK, c-Jun N-terminal protein kinase; Nrf-2, NF-E2-related factor 2; Keap1, Kelch-like ECH-associated protein1; NF-κB, nuclear factor κB; NQO1, NAD(P) H: quinoneoxidoreductase; HO-1, heme oxygenase 1; GCLC, catalytic subunit of glutamate-cysteine ligase.
Following amplification, a melting curve analysis was employed to verify the authenticity of the amplified product based on its specific melting temperature (Tm). The threshold cycle for the gene of interest and housekeeping gene, and the difference between their Ct values (ΔCt), were calculated. Relative quantitative analyses of gene expression were calculated using the 2−ΔΔCt data analysis method in accordance with the previous literature21,29,73,74. The housekeeping gene β-Actin was employed for compression as a control.
Statistical analysis
All results are expressed as mean ± SD. Statistical analysis was examined using the SPSS 15.0 software. Group differences were assessed by one-way analysis of variance followed by the least significance difference (LSD) test. p < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Liu, Q. et al. Toxic metabolites, MAPK and Nrf2/Keap1 signaling pathways involved in oxidative toxicity in mice liver after chronic exposure to Mequindox. Sci. Rep. 7, 41854; doi: 10.1038/srep41854 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
This work was supported by Natural Science Foundation of China (grant no. 31272614 and 31502115), Grants from 2016 National Risk Assessment of Quality and Safety of Livestock and Poultry Products (GJFP2016008), Research on the detection standard of veterinary drug residue (2662015PY021) and the Long-term development plan UHK.
Footnotes
The authors declare no competing financial interests.
Author Contributions Z.H.Y. conceived the idea; X.W. analyzed and discussed data; Q.Y.L. analyzed and discussed data and wrote the paper; Z.X.L. performed and revised the experiments; A.X.H. performed the experiments; S.Y.X., Q.H.W., I.A. and M.H.D. revised the paper. All the authors discussed the results and contributed to the final manuscript.
References
- Cheng G. et al. Systematic and Molecular Basis of the Antibacterial Action of Quinoxaline 1,4-Di-N-Oxides against Escherichia coli. Plos one 10, e0136450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. Y. et al. Further investigations into the genotoxicity of quinoxaline-di-N-oxides and their primary metabolites. Food Chem Toxicol 93, 145–157 (2016). [DOI] [PubMed] [Google Scholar]
- Vicente E. et al. Selective activity against Mycobacteriumtuberculosis of new quinoxaline 1,4-di-N-oxides. Bioorgan Med Chem 17, 385–389 (2009). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Two generation reproduction and teratogenicity studies of feeding cyadox in Wistar rats. Food Chem Toxicol 49, 1068–1079 (2011). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Genotoxic risk of quinocetone and its possible mechanism in in-vitro studies. Toxicol Res, doi: 10.1039/C1035TX00341E (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. et al. Deoxidation rates play a critical role in DNA damage mediated by important synthetic drugs, quinoxaline 1,4-dioxides. Chem Res Toxicol 28, 470–481 (2015). [DOI] [PubMed] [Google Scholar]
- Wu Y. et al. Development of a high-performance liquid chromatography method for the simultaneous quantification of quinoxaline-2-carboxylic acid and methyl-3-quinoxaline-2-carboxylic acid in animal tissues. J Chromatogr A 1146, 1–7 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang K. et al. Investigation of quinocetone-induced mitochondrial damage and apoptosis in HepG2 cells and compared with its metabolites. Environ Toxicol Pha 39, 555–567 (2015). [DOI] [PubMed] [Google Scholar]
- Carta A., Corona P. & Loriga M. Quinoxaline 1,4-dioxide: a versatile scaffold endowed with manifold activities. Curr Med Chem 12, 2259–2272 (2005). [DOI] [PubMed] [Google Scholar]
- Chen Q. et al. Investigation of the genotoxicity of quinocetone, carbadox and olaquindox in vitro using Vero cells. Food Chem Toxicol 47, 328–334 (2009). [DOI] [PubMed] [Google Scholar]
- Ihsan A. et al. Acute and subchronic toxicological evaluation of Mequindox in Wistar rats. Regul Toxicol Phar 57, 307–314 (2010). [DOI] [PubMed] [Google Scholar]
- Ihsan A. et al. Long-term mequindox treatment induced endocrine and reproductive toxicity via oxidative stress in male Wistar rats. Toxicol Appl Pharm 252, 281–288 (2011). [DOI] [PubMed] [Google Scholar]
- Ihsan A. et al. Genotoxicity of quinocetone, cyadox and olaquindox in vitro and in vivo. Food Chem Toxicol 59, 207–214 (2013). [DOI] [PubMed] [Google Scholar]
- Huang L. et al. Metabolism, Distribution, and Elimination of Mequindox in Pigs, Chickens, and Rats. J Agri Food Chem 63, 9839–9849 (2015). [DOI] [PubMed] [Google Scholar]
- Cheng G. et al. Quinoxaline 1,4-di-N-Oxides: Biological Activities and Mechanisms of Actions. Front Pharmacol 7, 64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q. et al. Characterization of carbadox-induced mutagenesis using a shuttle vector PSP189 in mammalian cells. Mutat Res 638, 11–16 (2008). [DOI] [PubMed] [Google Scholar]
- Hao L., Chen Q. & Xiao X. 2006. Molecular mechanism of mutagenesis induced by olaquindox using a shuttle vector pSP189/mammalian cell system. Mutat Res 599, 21–25 (2006). [DOI] [PubMed] [Google Scholar]
- Ganley B., Chowdhury G., Bhansali J., Daniels J. S. & Gates K. S. Redox-activated, hypoxia-selective DNA cleavage by quinoxaline 1,4-di-N-oxide. Bioorgan Med Chem 9, 2395–2401 (2001). [DOI] [PubMed] [Google Scholar]
- Zhang K. et al. Cytotoxicity and genotoxicity of 1,4-bisdesoxyquinocetone, 3-methyl-quinoxaline-2-carboxylic acid (MQCA) in human hepatocytes. Res Vet Sci 93, 1393–1401 (2012). [DOI] [PubMed] [Google Scholar]
- Ihsan A. et al. Genotoxicity evaluation of Mequindox in different short-term tests. Food Chem Toxicol 51, 330–336 (2013). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Metabolites and JAK/STAT pathway were involved in the liver and spleen damage in male Wistar rats fed with mequindox. Toxicology 280, 126–134 (2011). [DOI] [PubMed] [Google Scholar]
- Azqueta A. et al. A quinoxaline 1,4-di-N-oxide derivative induces DNA oxidative damage not attenuated by vitamin C and E treatment. Chemico-biol Interact 168, 95–105 (2007). [DOI] [PubMed] [Google Scholar]
- Chowdhury G., Kotandeniya D., Daniels J. S., Barnes C. L. & Gates K. S. Enzyme-activated, hypoxia-selective DNA damage by 3-amino-2 quinoxalinecarbonitrile 1,4-di-N-oxide. Chem Res Toxicol 17, 1399–1405 (2004). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Fumonisins: oxidative stress-mediated toxicity and metabolism in vivo and in vitro. Arch Toxicol, doi: 10.1007/s00204-00015-01604-00208 (2015). [DOI] [PubMed] [Google Scholar]
- Liu J. et al. Mequindox induced cellular DNA damage via generation of reactive oxygen species. Mutat Res 741, 70–75 (2012). [DOI] [PubMed] [Google Scholar]
- Huang X. J. et al. ROS mediated cytotoxicity of porcine adrenocortical cells induced by QdNOs derivatives in vitro. Chem-biol Interact 185, 227–234 (2010). [DOI] [PubMed] [Google Scholar]
- Zou J. et al. Olaquindox-induced genotoxicity and oxidative DNA damage in human hepatoma G2 (HepG2) cells. Mutat Res 676, 27–33 (2009). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. High risk of adrenal toxicity of N1-desoxy quinoxaline 1,4-dioxide derivatives and the protection of oligomeric proanthocyanidins (OPC) in the inhibition of the expression of aldosterone synthetase in H295R cells. Toxicol 341–343, 1–16 (2016). [DOI] [PubMed] [Google Scholar]
- Huang X. J. et al. Long-term dose-dependent response of Mequindox on aldosterone, corticosterone and five steroidogenic enzyme mRNAs in the adrenal of male rats. Toxicol Lett 191, 167–173 (2009). [DOI] [PubMed] [Google Scholar]
- Gong X., Ivanov V. N. & Hei T. K. 2,3,5,6-Tetramethylpyrazine (TMP) down-regulated arsenic-induced heme oxygenase-1 and ARS2 expression by inhibiting Nrf2, NF-kappaB, AP-1 and MAPK pathways in human proximal tubular cells. Arch Toxicol 90, 2187–200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. G. et al. Endosulfan induces COX-2 expression via NADPH oxidase and the ROS, MAPK, and Akt pathways. Arch Toxicol 89, 2039–2050 (2015). [DOI] [PubMed] [Google Scholar]
- Liao Y. C., Chen Y. F. & Lee T. C. Increased susceptibility of H-Ras(G12V)-transformed human urothelial cells to the genotoxic effects of sodium arsenite. Arch Toxicol 89, 1971–1979 (2015). [DOI] [PubMed] [Google Scholar]
- Romanov V., Whyard T. C., Waltzer W. C., Grollman A. P. & Rosenquist T. Aristolochic acid-induced apoptosis and G2 cell cycle arrest depends on ROS generation and MAP kinases activation. Arch Toxicol 89, 47–56 (2015). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. The critical role of oxidative stress in the toxicity and metabolism of quinoxaline 1,4-di-N-oxides in vitro and in vivo. Drug Metab Rev 48, 159–182 (2016). [DOI] [PubMed] [Google Scholar]
- Watanabe T., Sekine S., Naguro I., Sekine Y. & Ichijo H. Apoptosis Signal-regulating Kinase 1 (ASK1)-p38 Pathway- dependent Cytoplasmic Translocation of the Orphan Nuclear Receptor NR4A2 Is Required for Oxidative Stress-induced Necrosis. J Biol Chem 290, 10791–10803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ze Y. et al. Molecular mechanism of titanium dioxide nanoparticles-induced oxidative injury in the brain of mice. Chemosphere 92, 1183–1189 (2013). [DOI] [PubMed] [Google Scholar]
- Paradies G., Petrosillo G., Pistolese M. & Ruggiero F. M. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS lett 466, 323–326 (2000). [DOI] [PubMed] [Google Scholar]
- Kimura M. et al. Molecular basis distinguishing the DNA binding profile of Nrf2- Maf heterodimer from that of Maf homodimer. J Biol Chem 282, 33681–33690 (2007). [DOI] [PubMed] [Google Scholar]
- Kobayashi M. & Yamamoto M. Molecular mechanisms activating the Nrf2- Keap1 pathway of antioxidant gene regulation. Antioxid Redox Sign 7, 385–394 (2005). [DOI] [PubMed] [Google Scholar]
- Li W. & Kong A. N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mole Carcinogen 48, 91–104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedruzzi L. M., Stockler-Pinto M. B., Leite M. & Mafra D. Jr. Nrf2-keap1 system versus NF-kappaB: the good and the evil in chronic kidney disease? Biochimie 94, 2461–2466 (2012). [DOI] [PubMed] [Google Scholar]
- Lee S. E. et al. Induction of heme oxygenase-1 inhibits cell death in crotonaldehyde-stimulated HepG2 cells via the PKC-delta-p38-Nrf2 pathway. Plos one 7, e41676 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. M., Huang S. M., Liu C. L. & Hu M. L. Apo-8′-lycopenal induces expression of HO-1 and NQO-1 via the ERK/p38-Nrf2-ARE pathway in human HepG2 cells. J Agr Food Chem 60, 1576–1585 (2012). [DOI] [PubMed] [Google Scholar]
- Zhao D. X. et al. Reactive oxygen species-dependent JNK downregulated olaquindox-induced autophagy in HepG2 cells. J Appl Toxicol 35, 709–716 (2015). [DOI] [PubMed] [Google Scholar]
- Zhao W. X. et al. Olaquindox-induced apoptosis is suppressed through p38 MAPK and ROS-mediated JNK pathways in HepG2 cells. Cell Biol Toxicol 29, 229–238 (2013). [DOI] [PubMed] [Google Scholar]
- Yang W. et al. Quinocetone triggers oxidative stress and induces cytotoxicity and genotoxicity in human peripheral lymphocytes of both genders. J Sci Food Agr 93, 1317–1325 (2013). [DOI] [PubMed] [Google Scholar]
- Yu M. et al. Nrf2/ARE is the potential pathway to protect Sprague-Dawley rats against oxidative stress induced by quinocetone. Regul Toxicol Pharm 66, 279–285 (2013). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Genomic and proteomic analysis of the inhibition of synthesis and secretion of aldosterone hormone induced by quinocetone in NCI-H295R cells. Toxicology 350–352, 1–14 (2016). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. A chronic toxicity study of cyadox in Wistar rats. Regul. Toxicol. Phar. 59, 324–333 (2011). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Acute and sub-chronic oral toxicological evaluations of quinocetone in Wistar rats. Regul Toxicol Phar 58, 421–427 (2010). [DOI] [PubMed] [Google Scholar]
- Wang X. et al. Mechanism of adrenocortical toxicity induced by quinocetone and its bidesoxy quinocetone metabolite in porcine adrenocortical cells in vitro. Food Chem Toxicol 84, 115–124 (2015). [DOI] [PubMed] [Google Scholar]
- Zhang K. et al. Identification of oxidative stress and responsive genes of HepG2 cells exposed to quinocetone, and compared with its metabolites. Cell Biol Toxicol 30, 313–329 (2014). [DOI] [PubMed] [Google Scholar]
- Dai C., Tang S., Li D., Zhao K. & Xiao X. Curcumin attenuates quinocetone-induced oxidative stress and genotoxicity in human hepatocyte L02 cells. Toxicol Mech Method 25, 340–346 (2015). [DOI] [PubMed] [Google Scholar]
- Li Z. et al. Research on olaquindox induced endoplasmic reticulum stress related apoptosis on nephrotoxicity. J Hygi Res 44, 444–450 (2015). [PubMed] [Google Scholar]
- Zhang C. M. et al. TNFR1/TNF-alpha and mitochondria interrelated signaling pathway mediates quinocetone-induced apoptosis in HepG2 cells. Food Chem Toxicol 62, 825–838 (2013). [DOI] [PubMed] [Google Scholar]
- Zhang T. et al. c-Myc influences olaquindox-induced apoptosis in human hepatoma G2 cells. Mol Cell Biochem 354, 253–261 (2011). [DOI] [PubMed] [Google Scholar]
- Zou J. et al. Olaquindox induces apoptosis through the mitochondrial pathway in HepG2 cells. Toxicol 285, 104–113 (2011). [DOI] [PubMed] [Google Scholar]
- Huang X. J. et al. Interactions of NADPH oxidase, renin-angiotensin-aldosterone system and reactive oxygen species in mequindox-mediated aldosterone secretion in Wistar rats. Toxicol Lett 198, 112–118 (2010). [DOI] [PubMed] [Google Scholar]
- Yu M. et al. Quinocetone-induced Nrf2/HO-1 pathway suppression aggravates hepatocyte damage of Sprague-Dawley rats. Food Chem Toxicol 69, 210–219 (2014). [DOI] [PubMed] [Google Scholar]
- Wang D. et al. Pu-erh black tea supplementation decreases quinocetone-induced ROS generation and oxidative DNA damage in Balb/c mice. Food Chem Toxicol 49, 477–484 (2011). [DOI] [PubMed] [Google Scholar]
- Zhao X. J. et al. Dynamic metabolic response of mice to acute mequindox exposure. J Proteome 10, Res, 5183–5190 (2011). [DOI] [PubMed] [Google Scholar]
- Liu Z. Y., Huang L. L., Chen D. M. & Yuan Z. H. Metabolism of mequindox in liver microsomes of rats, chicken and pigs. Rapid Commun Mass 24, 909–918 (2010). [DOI] [PubMed] [Google Scholar]
- Junnotula V., Sarkar U., Sinha S. & Gates K. S. Initiation of DNA strand cleavage by 1,2,4-benzotriazine 1,4-dioxide antitumor agents: mechanistic insight from studies of 3-methyl-1,2,4-benzotriazine 1,4-dioxide. J Am Chem Soc 131, 1015–1024 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole J. S. et al. Photochemical electron transfer reactions of tirapazamine. J Photoch Photobio 75, 339–345 (2002). [DOI] [PubMed] [Google Scholar]
- El-Khatib M., Geara F., Haddadin M. J. & Gali-Muhtasib H. Cell death by the quinoxaline dioxide DCQ in human colon cancer cells is enhanced under hypoxia and is independent of p53 and p21. Radiat Oncol, doi: 10.1186/1748-717X-5-107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. Y. & Lee T. H. Antioxidant enzymes as redox-based biomarkers: a brief review. BMB Rep 48, 200–208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J. et al. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol 36, 655–662 (2015). [DOI] [PubMed] [Google Scholar]
- Liu Z. Y. & Sun Z. L. The metabolism of carbadox, olaquindox, mequindox, quinocetone and cyadox: an overview. J Med Chem 9, 1017–1027 (2013). [DOI] [PubMed] [Google Scholar]
- Prasad S., Ravindran J. & Aggarwal B. B. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem 336, 25–37 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui S. et al. Renal injury and Nrf2 modulation in mouse kidney following chronic exposure to TiO(2) nanoparticles. J Agri Food Chem 61, 8959–8968 (2013). [DOI] [PubMed] [Google Scholar]
- Luo L. et al. Butylated hydroxyanisole induces distinct expression patterns of Nrf2 and detoxification enzymes in the liver and small intestine of C57BL/6 mice. Toxicol Appl Pharm 288, 339–348 (2015). [DOI] [PubMed] [Google Scholar]
- NRC. The Development of Science based Guidelines for Laboratory Animal Care, Proceedings of the November 2003 International Workshop. National Academy Press, Washington, DC (2004). [PubMed] [Google Scholar]
- Yang Y. et al. Olaquindox induces DNA damage via the lysosomal and mitochondrial pathway involving ROS production and p53 activation in HEK293 cells. Environ Toxicol Pha 40, 792–799 (2015). [DOI] [PubMed] [Google Scholar]
- Sánchez-Gómez F. J. et al. Detoxifying Enzymes at the Cross-Roads of Inflammation, Oxidative Stress, and Drug Hypersensitivity: Role of Glutathione Transferase P1-1 and Aldose Reductase. Front Pharmacol 7, 237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]