

Abstract
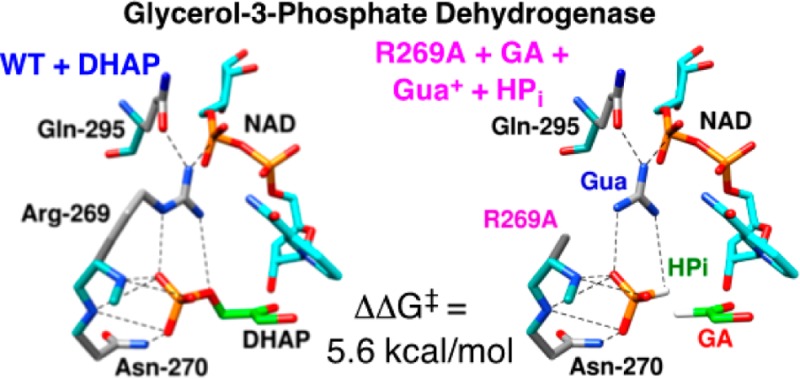
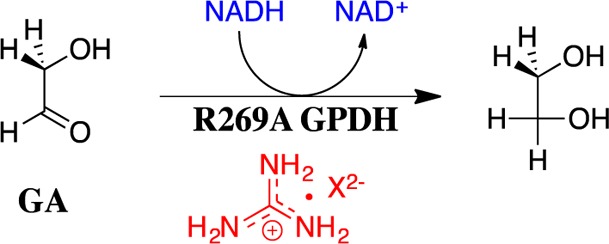
The stabilization of the transition state for hlGPDH-catalyzed reduction of DHAP due to the action of the phosphodianion of DHAP and the cationic side chain of R269 is between 12.4 and 17 kcal/mol. The R269A mutation of glycerol-3-phosphate dehydrogenase (hlGPDH) results in a 9.1 kcal/mol destabilization of the transition state for enzyme-catalyzed reduction of dihydroxyacetone phosphate (DHAP) by NADH, and there is a 6.7 kcal/mol stabilization of this transition state by 1.0 M guanidine cation (Gua+) [J. Am. Chem. Soc.2015, 137, 5312–5315]. The R269A mutant shows no detectable activity toward reduction of glycolaldehyde (GA), or activation of this reaction by 30 mM HPO32–. We report the unprecedented self-assembly of R269A hlGPDH, dianions (X2– = FPO32–, HPO32–, or SO42–), Gua+ and GA into a functioning catalyst of the reduction of GA, and fourth-order reaction rate constants kcat/KGAKXKGua. The linear logarithmic correlation (slope = 1.0) between values of kcat/KGAKX for dianion activation of wildtype hlGPDH-catalyzed reduction of GA and kcat/KGAKXKGua shows that the electrostatic interaction between exogenous dianions and the side chain of R269 is not significantly perturbed by cutting hlGPDH into R269A and Gua+ pieces. The advantage for connection of hlGPDH (R269A mutant + Gua+) and substrate pieces (GA + HPi) pieces, (ΔGS‡)HPi+E+Gua = 5.6 kcal/mol, is nearly equal to the sum of the advantage to connection of the substrate pieces, (ΔGS)GA+HPi = 3.3 kcal/mol, for wildtype hlGPDH-catalyzed reaction of GA + HPi, and for connection of the enzyme pieces, (ΔGS‡)E+Gua = 2.4 kcal/mol, for Gua+ activation of the R269A hlGPDH-catalyzed reaction of DHAP.
Introduction
The term self-assembly is used in biology,1 supramolecular chemistry2 and nanotechnology3 to describe molecules that spontaneously combine to form defined structures, which carry out complex functions. Protein catalysts and their substrates may be deconstructed into two or more pieces by cleavage of covalent bonds, but there have been limited studies on the reassembly of these pieces into a catalytically active unit, because of the anticipated difficulties in overcoming the entropic barrier to reassembly. An important and underappreciated exception is the rescue of mutant enzymes by small molecule analogues of the excised amino acid side chain.4 We have developed a protocol for comparing the reactivity of whole enzyme and substrate with the reactivity of the enzyme and substrate in pieces,5 and have applied this protocol to mutants of triosephosphate isomerase (TIM, K12G mutant),5b orotidine 5′-monophosphate decarboxylase (OMPDC, R235A mutant),5c and glycerol-3-phosphate dehydrogenase (GPDH, R269A mutant).5a In each case, the side chain cation of the essential amino acid sits at a cleft on the protein surface and interacts with the phosphodianion through a strong ion pair, as shown in Figure 1A for OMPDC and Figure 1B for GPDH from human liver (hlGPDH). The efficient rescue of these mutant enzymes reflects the ease of transfer of the exogenous cation from water to the protein surface, and the high stability of the transition state cation-phosphodianion pair.5a
Figure 1.

A comparison of the protein surfaces from X-ray crystal structures of the following: (A) The complex between OMPDC from yeast and 6-hydroxyuridine 5′-monophosphate (PDB entry 1DQX). (B) The nonproductive ternary complex of dihydroxyacetone phosphate (DHAP) and NAD+ with hlGPDH (PDP entry 1WPQ). These structures show the loops that trap the ligand in a protein cage shaded red, and the guanidine side chains at the protein surface shaded black. The respective enzyme-bound ligands at the structures represented by A and B are buried in the protein, with the phosphodianion projecting toward the surface, and in a stable ion pair with the guanidine side chains of R235 (OMPDC) or R269 (hlGPDH). Reproduced from ref (5a). Copyright 2015 American Chemical Society.
We now extend earlier work, and report the results of a kinetic study to characterize the unprecedented self-assembly of R269A mutant hlGPDH, dianions (e.g., X2– = HPi), guanidine cation (Gua+) and glycolaldehyde (GA) into a functioning catalyst of the reduction of GA by NADH (Scheme 1), and show that this self-assembly is carried out with a penalty of only 5.6 kcal/mol compared with the reduction of the whole substrate DHAP catalyzed by the whole enzyme, wildtype hlGPDH. The self-assembly of an active enzyme from these component parts is driven by strong electrostatic interactions between the anionic transition state and cationic protein catalyst, which stabilize the transition state for wildtype hlGPDH-catalyzed reduction of DHAP by between 12.4 and 17 kcal/mol.
Scheme 1.
Experimental Section
Materials
Water was obtained from a Milli-Q Academic purification system. Q-Sepharose was purchased from GE Healthcare. Nicotinamide adenine dinucleotide reduced form (NADH, disodium salt), glycolaldehyde dimer, 2-(N-morpholino)ethanesulfonic acid sodium salt (MES, ≥ 99.5%), triethanolamine hydrochloride (≥99.5%) and d,l-dithiothreitol (DTT) were purchased from Sigma-Aldrich. Sodium phosphite (dibasic, pentahydrate) was purchased from Fluka. The water content was reduced to Na2HPO3·0.4H2O as previously described.6 Sodium fluorophosphate was a generous gift from Dr. Andrew Murkin. Sodium sulfate (anhydrous) was purchased from Mallinckrodt Chemicals. Guanidinium hydrochloride (electrophoresis grade, 99%), sodium hydroxide (1.0 N) and hydrochloric acid (1.0 N) were purchased from Fisher Scientific. All other chemicals were reagent grade or better and were used without further purification.
Solution pH was determined at 25 °C using an Orion Model 720A pH meter equipped with a Radiometer pHC4006–9 combination electrode that was standardized at pH 4.00, 7.00, and 10.00 at 25 °C. Stock solutions of NADH were prepared by dissolving the disodium form of the coenzyme in water and then stored at 4 °C. The concentration of NADH in these solutions was determined at 340 nm using the extinction coefficient of ε = 6200 M–1 cm–1. A stock solution of glycolaldehyde (200 mM monomer) was prepared by dissolving the dimer in water and storing the solution for at least 3 days at room temperature to allow for breakdown to the monomer.6,7 Stock solutions of guanidine hydrochloride, prepared by dissolving the salt in water and adjusting the pH to 7.5, were stored at room temperature. The concentration of the carbonyl form of GA is calculated from the total concentration of GA using an equilibrium constant Keq = [carbonyl]/[hydrate] = (6/94).8 MES and triethanolamine buffers were prepared by addition of 1 M NaOH or 1 M HCl and solid NaCl to give the desired acid/base ratio and final ionic strength.
The R269A mutant of hlGPDH was expressed and purified by published procedures.5a Concentrated solutions of R269A mutant hlGPDH (40 mg/mL) were dialyzed exhaustively against 20 mM triethanolamine buffer (pH 7.5) at 4 °C. The enzyme concentration was determined from the absorbance at 280 nm using the extinction coefficient of ε = 18 500 M–1 cm–1 and a subunit molecular mass of 37 500 Da.9 Stock solutions of hlGPDH were prepared by dilution with 20 mM triethanolamine (pH 7.5) that contains 10 mM DTT and 0.1 mg/mL BSA.
Kinetic Parameters for R269A hlGPDH-Catalyzed Reactions of GA in the Presence of Guanidinium Cation and Oxydianions
A Cary 300-Bio UV–vis spectrophotometer was used for initial velocity (vi) measurements. The drift in this instrument at 340 nm is <0.001 absorbance unit/hour. All enzyme assays were conducted at 25 °C, at a constant ionic strength of 0.12 maintained with NaCl and in a final volume of 1.0 mL. The initial velocity of hlGPDH-catalyzed reactions of NADH (≤5% substrate reaction) was calculated from the change in absorbance at 340 nm. Assay mixtures for R269A hlGPDH-catalyzed reduction of GA by NADH at pH 7.5 contained 10 mM triethanolamine (pH 7.5), 0.2 mM NADH, 1.8 or 3.6 mM of the carbonyl form of GA, from 0 to 30 mM guanidine hydrochloride and from 0 to 30 mM oxydianion at I = 0.12 (NaCl). The following enzyme concentrations were used in studies on activation by different dianions: [E] = 20 μm, HPO32–; [E] = 5–10 μm, FPO32–; [E] = 40 μm, SO42–. The initial velocity was determined over a 10–40 min reaction time. The pH was determined at the end of each reaction, and in no case was a significant (>0.01 unit) change observed in the starting pH.
Results
The R269A hlGPDH-catalyzed reduction of GA (3.6 mM carbonyl form of GA) by NADH (0.2 mM ≫ Km) in the absence of GPDH, in the presence of 20 μM GPDH, and in the presence of 20 μM GPDH and either 30 mM Gua+ or 30 mM phosphite dianion (HPi) was monitored at 340 nm for a total of 60 min. A marginally larger decrease in A340 was observed for the reaction in the presence of GPDH (ΔA340 ≈ 0.0102) than for the reaction in the absence of GPDH (ΔA340 ≈ 0.0060). This difference was not significantly affected by the addition of 30 mM Gua+ or 30 mM phosphite dianion (HPi). No attempt was made to rigorously demonstrate that the small difference between ΔA340 observed in the presence and absence of GPDH (ΔA340 ≈ 0.0042) represented the slow R269A hlGPDH-catalyzed reduction of GA. Instead, we use this difference to obtain an upper limit for reaction velocity. This sets an upper limit of 0.16 μM for the decrease in [NADH] for the 60 min R269A hlGPDH-catalyzed reduction of GA; and, the following upper limits for the rate constants for these hlGPDH-catalyzed reactions (Scheme 2): kcat/KGA ≤ 0.003 M–1 s–1 for R269A hlGPDH-catalyzed reduction of GA; kcat/KGAKHPi < 0.1 M–2 s–1 for activation of R269A hlGPDH-catalyzed reduction of GA by 30 mM HPi; and, kcat/KGAKGua < 0.1 M–2 s–1 for activation of R269A hlGPDH-catalyzed reduction of GA by 30 mM Gua+.
Scheme 2.

Figure 2 shows the effect of increasing concentrations of phosphite (HPi) (Figure 2A) fluorophosphate (FPi) (Figure 2B) and sulfate (SO42–) (Figure 2C) dianion on the observed second-order rate constant v/[E][GA] (M–1 s–1) for R269A hlGPDH-catalyzed reduction of the carbonyl form of GA by saturating 0.2 mM NADH at several different fixed concentrations of Gua+. The effect of increasing [HPi] on v/[E][GA] (M–1 s–1) for reactions activated by 30 mM or 15 mM Gua+ were examined at 60 mM (solid symbols), and at 30 mM (open symbols) total [carbonyl + hydrate] GA. These correspond to 3.6 mM and 1.8 mM, respectively, of the reactive carbonyl form of GA. In every case, the data obtained at the two concentrations of GA show a good fit to a single correlation line. This shows that reactions at total [GA] ≤ 60 mM are first-order in [GA], so that the apparent KGA for dissociation of GA from R269A mutant hlGPDH is ≫60 mM (Scheme 2).
Figure 2.

Effect of inorganic dianions and Gua+ on R269A mutant hlGPDH-catalyzed reduction of GA by NADH, determined for reactions at pH 7.5, 25 °C, saturating [NADH] = 0.2 mM and I = 0.12 (NaCl). (A) The increase in v/[E][GA] (M–1 s–1), with increasing [HPO32–], for reactions in the presence of 30 mM (open symbols) or 60 mM (closed symbols) total GA [carbonyl + hydrate] and at different fixed concentrations of Gua+. The equilibrium constant for hydration of GA is Keq = [carbonyl]/[hydrate] = (6/94).8 Key: ([GA]carbonyl = 3.6 mM) (▼) 30 mM Gua+; (⧫), 25 mM Gua+; (■), 20 mM Gua+; (▲), 15 mM Gua+; (●), 10 mM Gua+; ([GA]carbonyl = 1.8 mM) (□), 30 mM Gua+; (◊), 15 mM Gua+. (B) The increase in v/[E][GA] (M–1 s–1), with increasing [FPO32–], for reactions at [GA]carbonyl = 3.6 mM and at different fixed concentrations of Gua+. Key: (▼) 30 mM Gua+; (⧫), 25 mM Gua+; (■), 20 mM Gua+; (▲), 15 mM Gua+; (●), 10 mM Gua+. (C) The increase in v/[E][GA] (M–1 s–1), with increasing [SO42–], for reactions at 3.6 mM GA and at different fixed concentrations of Gua+. Key: (▼) 30 mM Gua+; (⧫), 25 mM Gua+; (■), 20 mM Gua+; (▲), 15 mM Gua+; (●), 10 mM Gua+. Figure 3 shows the linear plots of the slopes of these linear correlations, (kcat/KGAKX)obs, against [Gua+].
The apparent third-order rate constants (kcat/KGAKX)obs (M–2 s–1) for dianion activation of R269A hlGPDH-catalyzed reduction of GA (carbonyl form), determined as the slopes of the linear correlations from Figure 2, are reported in Table 1. Figure 3 shows plots of (kcat/KGAKX)obs for dianion activation of R269A hlGPDH-catalyzed reduction of GA against the concentration of Gua+ activator. The slopes of these plots, reported in Table 1, are the fourth-order rate constants kcat/KGAKXKGua (M–3 s–1) for activation of R269A hlGPDH-catalyzed reduction of GA by the combined action of Gua+ and dianions X2– (Scheme 2).
Table 1. Kinetic Parameters for Activation of R269A hlGPDH-Catalyzed Reduction of GA (1.8 mM or 3.6 mM) by the Combined Action of a Dianion and Gua+a.
| activator |
kinetic parameter |
|||
|---|---|---|---|---|
| dianion | Gua+ (mM) | (kcat/KGAKX)obsb M–2 s–1 | kcat/KGAKXKGuac M–3 s–1 | |
| HPO32– | 10 | 3.4 ± 0.12d | 340 ± 10 | |
| 15 | 5.3 ± 0.19d | 5.2 ± 0.2e | ||
| 20 | 6.7 ± 0.15d | |||
| 25 | 8.4 ± 0.10d | |||
| 30 | 10.0 ± 0.13d | 10.2 ± 0.2e | ||
| FPO32– | 10 | 28.2 ± 0.6 | 2850 ± 50 | |
| 15 | 42.6 ± 0.7 | |||
| 20 | 54.8 ± 1.4 | |||
| 25 | 71.9 ± 1.5 | |||
| 30 | 85.5 ± 1.3 | |||
| SO42– | 10 | 0.257 ± 0.004 | 27 ± 1 | |
| 15 | 0.350 ± 0.002 | |||
| 20 | 0.494 ± 0.004 | |||
| 25 | 0.681 ± 0.007 | |||
| 30 | 0.884 ± 0.013 | |||
For reactions at pH 7.5, 25 °C, saturating [NADH] = 0.2 mM and I = 0.12 (NaCl).
Observed third-order rate constant for dianion activation, determined as the slope of the appropriate linear correlation shown in Figure 2.
Fourth-order rate constant kcat/KGAKXKGua (M–3 s–1) for activation, determined as the slope of linear correlations of (kcat/KGAKX)obs (M–2 s–1) against [Gua+] shown in Figure 3.
[GA]carbonyl = 3.6 mM.
[GA]carbonyl = 1.8 mM.
Figure 3.

Effect of increasing concentrations of Gua+ on the observed third-order rate constants (kcat/KGAKX)obs (M–2 s–1, Figure 2) for dianion activation of R269A mutant hlGPDH-catalyzed reduction of GA: solid symbols, [GA]carbonyl = 3.6 mM; open symbols, [GA]carbonyl = 1.8 mM. The slopes of these linear correlations are the fourth order rate constants kcat/KGAKXKGua reported in Table 1.
Discussion
This work combines two protocols from studies on the reactions of substrates in pieces and on enzymes in pieces.5a−5c,8,10 (1) The reactivity of hlGPDH in catalysis of the reaction of the substrate DHAP [(kcat/Km)WT] was compared with that for catalysis of the truncated substrate GA [(kcat/Km)GA] (Scheme 3) and for dianion activation of catalysis of GA ((kcat/Km)HPi/KX). These comparisons give changes in activation barriers (ΔΔG‡) that correspond to (a) the stabilization of the transition state for hlGPDH-catalyzed hydride transfer to DHAP by interaction with the substrate phosphodianion (ΔGT‡)Pi (eq 1) or, (b) the stabilization of the transition state for hlGPDH-catalyzed hydride transfer to GA by interaction with 1.0 M phosphite dianion (ΔGact)X (eq 2). The advantage obtained from connecting the pieces GA and HPi is calculated as (ΔGS‡)GA+X = −(ΔGT)Pi + (ΔGact‡)X (eq 3) where (ΔGS)GA+X is the connection energy as defined by W.P. Jencks.11 The relationships from eq 1–3 are illustrated by Scheme 4. hlGPDH is activated by a variety of inorganic dianions, which include FPO32–, HOPO32–, and SO42–. The values of (kcat/Km)X/KX and (ΔGact‡)X for dianion activation from earlier work are summarized in Table 2.10h We use the connection energy (ΔGS)GA+X = 3.3 kcal/mol for activation by HPO32– in our analyses, because phosphite dianion is the best steric and electronic dianion analogue for the phosphodianion of the whole substrate.
| 1 |
| 2 |
| 3 |
Scheme 3.
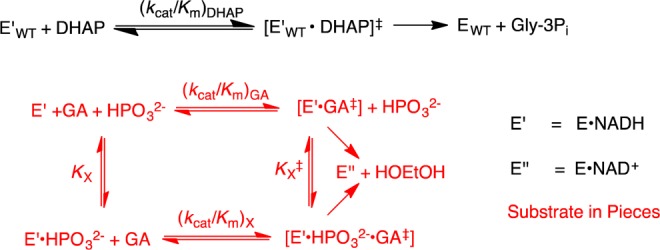
Scheme 4.

Table 2. Kinetic Parameters and Derived Gibbs Free Energy Terms for Reactions of the Substrate and Enzyme Pieces Catalyzed by TIM, OMPDC and hlGPDHa.
| enzyme | piece | (kcat/Km) (M–1 s–1)b | piece + activator(s) kcat/KmKX (M–2 s–1) or kcat/KmKXKcation (M–3 s–1)c | activationd | ΔG‡acte kcal/mol | EM (M) or (M)2f | (ΔGS‡)GA(EO)+X or (ΔGS)E+cation kcal/molg | |
|---|---|---|---|---|---|---|---|---|
| TIM (kcat/Km = 2.2 × 108 M–1 s–1)h | [1-13C]-GAq | 0.062 | GA + HPO32– | 2700 | 43500 | –6.3 | 81000 | 6.7 |
| (ΔGT‡)Pi = 13.0 kcal/moli | K12Gj | 300 | K12G + NH4+ | 3800 | 13 | –1.5 | 44000 | 6.3 |
| (ΔGT‡)K12G = 8.0 kcal/molj | K12G | 300 | K12G + CH3NH3+ | 20000 | 70 | –2.5 | 8300 | 5.3 |
| K12G | 300 | K12G + CH3CH2NH3+ | 100000 | 330 | –3.4 | 1700 | 4.4 | |
| K12G | 300 | K12G + HPO32– + CH3CH2NH3+ | not detected | |||||
| OMPDC (kcat/Km = 1.1 × 107 M–1 s–1)k | EOl | 0.026 | EO + HPO32– | 11700 | 4.5 × 105 | –7.7 | 940 | 4.1 |
| (ΔGT‡)Pi = 11.7 kcal/moll | R235Am | 610 | R235A + Gua+ | 69000 | 110 | –2.6 | 160 | 3.0 |
| (ΔGT‡)R235A = 5.8 kcal/molm | ||||||||
| hlGPDH (kcat/Km = 4.6 × 106 M–1 s–1)n | GAo | 0.05 | HPO32– | 16000 | 3.2 × 105 | –7.5 | 290 | 3.3 |
| GA | 0.05 | FPO32– | 75000 | 1.5 × 106 | –8.4 | |||
| GA | 0.05 | SO42– | 1100 | 2.2 × 104 | –5.9 | |||
| (ΔGT‡)Pi = 10.8 kcal/molo | R269Ap | 1.0 | R269A + Gua+ | 80000 | 80000 | –6.7 | 60 | 2.4 |
| (ΔGT‡)R269A = 9.1 kcal/molp | R269Ar | 1.0 | R269A+HPO32–+Gua+ | 340 [M–3 s–1] | 340 | 13500 [M2] | 5.6s | |
| R269Ar | 1.0 | R269A+FPO32– + Gua+ | 2850 | 2850 | ||||
| R269Ar | 1.0 | R269A+SO42– + Gua+ | 27 | 27 | ||||
For reactions at 25 °C.
Kinetic parameter for the wildtype-enzyme catalyzed reaction of the phosphodianion-truncated substrate piece, or for the truncated mutant-enzyme piece.
Kinetic parameter for activation of the substrate or enzyme piece by the designated activators. The third-order rate constants were determined for reactions of the given enzyme or substrate pieces, and the fourth-order rate constants are for combined activation by enzyme and substrate pieces.
The ratio of rate constants for the respective activated and unactivated enzyme-catalyzed reactions.
The Gibbs free energy for binding of the activator to the transition state complex for the unactivated reaction, calculated from the ratio in the previous column (eq 2 or 5).
The effective molarity for reaction of the enzyme or substrate piece, calculated from data in this Table using eq 3 or 6.13
The apparent energetic advantage for connecting the enzyme ((ΔGS‡)E+cation, eq 6) or substrate ((ΔGS)GA(EO)+X, eq 3) pieces.
Reactions at pH 7.0 [ref (12)].
Ref (10b).
Ref (5b).
Reactions at pH 7.1.
Ref (10h).
Ref (5c).
For reactions at pH 7.5.
Ref (10i).
Ref (5a).
Ref (10a).
This work.
(2) The reactivity of wildtype hlGPDH in catalysis of the reaction of DHAP, (kcat/Km)WT, was compared with that for R269A hlGPDH, (kcat/Km)R269A, and with the reactivity of the R269A mutant activated by 1.0 M Gua+, [(kcat/Km)Gua/KGua] (Scheme 5). These comparisons give differences in activation barriers (ΔΔG‡) that correspond to (a) the stabilization of the transition state for hlGPDH-catalyzed reduction of DHAP by interaction with the R269 side chain, (ΔGT‡)R269 (eq 4), or (b) the stabilization of the transition state for R269A hlGPDH-catalyzed reduction of DHAP by interaction with 1.0 M Gua+, (ΔGact)Gua (eq 5). The advantage obtained from connecting the enzyme pieces R269A hlGPDH and Gua+ is calculated as (ΔGS‡)E+Gua = (ΔGT)R269A + (ΔGact‡)Gua (eq 6). The relationships from eq 4–6 are illustrated by Scheme 6.
| 4 |
| 5 |
| 6 |
Scheme 5.
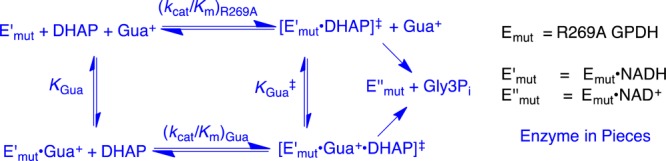
Scheme 6.

Energetics for the Self-Assembly of Substrate and Enzyme Pieces
The R269A mutant of hlGPDH shows no detectable activity toward catalysis of reduction of the truncated substrate GA by NADH (kcat/KGA ≤ 0.003 M–1 s–1) or for reduction of GA in the presence of 30 mM HPi or Gua+. However, R269A hlGPDH is activated for catalysis of reduction of GA by the combined binding of HPi or Gua+. Activation is also observed by FPO32– or SO42– in combination with Gua+ (Figure 3).
Table 1 reports kcat/KGAKXKGua = 340 M–3 s–1 for the fourth order rate constant for activation of R269A hlGPDH for catalysis of reduction of GA by the combined action of X2– = HPi and Gua+ (Scheme 7). This represents a 340-fold rate enhancement, at a standard state of 1.0 M substrate and activator, compared with R269A mutant-enzyme catalyzed reaction of DHAP (kcat/Km = 1.0 M–1 s–1). The value of (kcat/Km)WT = 4.6 × 106 M–1 s–1 for wildtype hlGPDH-catalyzed reduction DHAP shows that the connection of the enzyme and substrate pieces to give the whole enzyme and substrate results in a 13 500-fold rate enhancement, which corresponds to a connection energy of (ΔGS‡)X+E+Gua = 5.6 kcal/mol (eq 7). Table 2 summarizes the values of kcat/KGAKXKGua for activation of hlGPDH-catalyzed reactions of GA by the combined action of dianions HPO32–, FPO32– or SO42– and Gua+.
| 7 |
Scheme 7.

TIM and GPDH both recover a part of the activity of the whole phosphorylated substrate when catalyzing the reaction of truncated substrate and phosphite dianion pieces, and a part of the reactivity of the whole enzyme when the mutant (R269A hlGPDH or K12G TIM) is combined with the truncated cation piece (Table 2). However, only R269A hlGPDH binds both substrate (GA + HPi) and enzyme (Gua+) pieces to form a robust active enzyme. By contrast, no activity is observed for K12G TIM in the presence of the corresponding substrate and enzyme (GA + HPi + EtNH3+) pieces.12 This difference in the tendency of the respective pieces for hlGPDH and TIM to assemble into an active catalyst is reflected by the large difference between the sum of the connection energies for the hlGPDH substrate and enzyme pieces ((ΔGS‡)GA+X + (ΔGS)E+Gua) = 3.3 + 2.4 = 5.7 kcal/mol, which favors the reaction of the pieces, and the larger sum for TIM ((ΔGS‡)GA+X + (ΔGS)E+RNH3) = 6.7 + 4.4 = 11.1 kcal/mol (Table 2). The intermediate value of ((ΔGS‡)GA+X + (ΔGS)E+Gua) = 4.1 + 3.0 = 7.1 kcal/mol determined for reactions catalyzed by wildtype and R235A mutant OMPDC predicts that if activation of R235A mutant OMPDC catalyzed decarboxylation of 1-(β-d-erythrofuranosyl)orotic acid (EO) by the combined action of HP1 and Gua+ is observed, then this activation will be weaker than for hlGPDH.
Capturing the Reactivity of the Whole in the Two Pieces
The substrate phosphodianion and exogenous phosphite dianion each strongly activate TIM for catalysis of the isomerization reaction; and, the effect of connecting the dianion to the carbon acid substrate is mainly to reduce the change in entropy associated with the binding of the whole substrate compared to pieces.14 The following observations for hlGPDH show, likewise, that the binding interactions of whole ligand or whole enzyme are captured in the complexes to the corresponding pieces, and that the effect of the covalent connection of the pieces is mainly to reduce the unfavorable loss in entropy associated with the binding of two or three ligands, compared to a single ligand.
-
(1)
The total connection energy for the reaction of the collection of enzyme and substrate pieces, (ΔGS‡)HPi+E+Gua = 5.6 kcal/mol (eq 7), is essentially equal to the sum of the connection energies (ΔGS)GA+HPi = 3.3 kcal/mol for the wildtype hlGPDH-catalyzed reactions of the substrate pieces GA + HPi and (ΔGS‡)E = 2.4 kcal/mol for the reaction of whole substrate catalyzed by the R269A + Gua+ enzyme pieces (Table 2). This observation of similar catalytic advantages from connection of the pieces at the ER269A·Gua+·GA·HPi quaternary complex (Figure 4D) and at the corresponding ER269A·Gua+·DHAP (Figure 4C) and EWT·GA·HPi ternary complexes (Figure 4B) is consistent with the conclusion that the respective transition states formed in the reaction of these different complexes are stabilized by essentially the same interactions with the protein catalyst. The observation of similar catalytic activity of the whole enzyme toward whole substrate, and of these collections of enzyme and substrate pieces, after correction of the nearly constant effects of attachment of the pieces, provides strong evidence that all of these reactions proceed through similarly structured Michaelis complexes, as illustrated by Figure 4.
-
(2)
Figure 5 shows the linear logarithmic correlation, with slope of 1.0, between fourth-order rate constants kcat/KGAKXKGua for activation of R269A hlGPDH-catalyzed reduction of GA by the combined action of dianions X2– and Gua+ and third order rate constants kcat/KGAKX for activation of wildtype hlGPDH-catalyzed reduction of GA by the same dianions. This correlation requires a similar stabilization of the transition states for these reactions by electrostatic interactions between the different exogenous dianions, and either the cationic side chain of wildtype hlGPDH or exogenous guanidinium cation at R269A hlGPDH. In other words, these electrostatic interactions are not significantly perturbed by cutting wildtype hlGPDH into the R269A hlGPDH and Gua+ pieces, as represented in Figures 4B and 4D.
Figure 4.
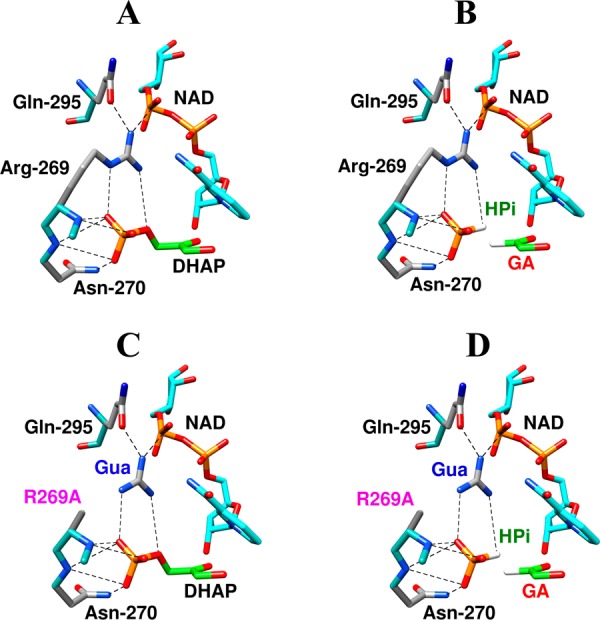
(A) Representations of the X-ray crystal structure (PDB entry 1WPQ) of the nonproductive ternary Michaelis complex between wildtype hlGPDH, DHAP and NAD+. (B–D) Representations, generated in silico from Figure 4A by deletion of the relevant covalent linkage(s) and maintaining a fixed position for the remaining atoms, of the following hypothetical Michaelis complexes: (B) wildtype hlGPDH, GA and HPi, (C) R269A hlGPDH, DHAP and Gua+, (D) R269A hlGPDH, GA, HPi and Gua+.
Figure 5.
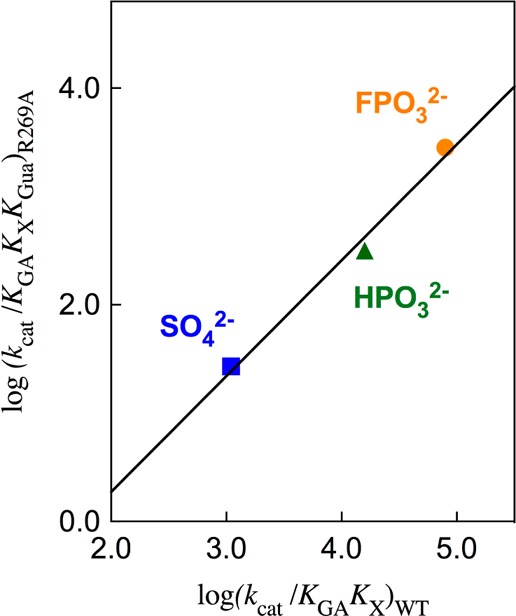
Linear logarithmic correlation, with slope of 1.0, between the fourth order rate constants kcat/KGAKXKGua for activation of R269A hlGPDH-catalyzed reduction of GA by the combined action of dianions X2– and Gua+ and the third order rate constants kcat/KGAKX for activation of wildtype hlGPDH-catalyzed reduction of GA by the same dianions.
Range of Connection Energies
Table 2 shows a variety of connection energies (ΔGS‡)GA (EO)+X for reactions of substrate pieces catalyzed by TIM, OMPDC, and hlGPDH. The connection energy reflects, first of all, the entropic advantage to the binding and reaction of a single whole ligand, compared to essentially the same ligand that has been cut into pieces (Figures 4A and 4B).11 However, it is difficult or impossible to rationalize variations in connection energies from Table 2 by consideration of entropic effects alone. For example, TIM and hlGPDH catalyze reactions of the same GA + HPi pieces and similar GAP and DHAP substrates. These enzymes might therefore be expected to show similar differences in the entropic price to the efficient binding and reaction of these substrates and pieces. By contrast, there is a large difference between the values of (ΔGS)GA+HPi = 6.7 and 3.3 kcal/mol determined, respectively, for TIM and hlGPDH.
The explanation for the small connection energies observed for hlGPDH is of particular interest, because this allows for efficient self-assembly of the enzyme and substrate pieces into a reactive complex. We propose that the large connection energy (ΔGS‡)GA+HPi = 6.7 kcal/mol for TIM reflects mainly the entropic advantage to the reaction of triosephosphates, and that the whole substrate and pieces show essentially the same binding interactions with the catalyst. By contrast we propose that the smaller connection energy (ΔGS)GA+HPi = 3.3 kcal/mol for hlGPDH includes a similar large (≈ 7 kcal/mol) entropic contribution, that is offset by the intrinsically tighter binding interactions to the pieces compared with whole substrate.
There is a larger total contribution of phosphodianion binding energy to catalysis by TIM (13.0 kcal/mol, Table 2) compared with hlGPDH (10.8 kcal/mol). This reflects the limited total substrate binding energy available to be utilized for catalysis of the reactions of triosephosphate substrates for TIM, and the imperative that TIM make the best possible use of these binding interactions. By comparison, there is a weaker imperative to optimize the utilization of the binding interactions of the phosphodianion of DHAP in catalysis by hlGPDH, because the binding energy from the large NADH cofactor is alone sufficient to obtain nearly the entire rate acceleration. This is the case for reactions catalyzed by alcohol dehydrogenase.15 We propose that hlGPDH fails to optimize the binding interactions for the whole substrate DHAP, and that there is a small, but significant, preference for binding the substrate in pieces. This may reflect the independent, unhindered, movement of the GA + HPi pieces to tightly bound conformations not accessible to the whole substrate DHAP, where such motion may be restricted by the covalent connection. Such preferential binding of the pieces would result in a reduction in the connection energy (ΔGS‡)GA+HPi.
Table 2 shows systematic variations in the connection energy (ΔGS‡)E+cation for the reactions of enzyme pieces that reflect, mainly, variations in the stabilization of the complex to the cationic piece by interactions with the protein catalyst. For example, the 1.9 kcal/mol decrease in the connection energy (ΔGS)E+RNH3 for rescue of K12G TIM from (ΔGS‡)E+HNH3 = 6.3 kcal/mol (R = H) to (ΔGS)E+EtNH3 = 4.4 kcal/mol (R = Et, Table 2) reflects increasing stabilization of the bound ammonium cation by interactions at a hydrophobic protein cleft.5b,16 The value of (ΔGS‡)E+HNH3 = 6.3 kcal/mol for the minimal activator NH4+ was proposed to represent the approximate entropic advantage of covalent connection of enzyme pieces.5b This is similar to (ΔGS)GA+HPi = 6.7 kcal/mol, which was estimated above as the approximate entropic advantage for connection of the substrate pieces for TIM. We likewise propose that the small connection energy of (ΔGS‡)E = 2.4 kcal/mol for R269A hlGPDH is due to stabilization of the complex between the cation and mutant enzyme by interactions with the amide side chain of Gln295 and the cofactor pyrophosphate (Figure 4).
The binding interactions to the alkyl groups of RNH3+ to TIM, which we propose are expressed as decreases in (ΔGS‡)E+RNH3, are not sufficiently strong to give detectable saturation of K12G TIM by 80 mM RNH3+.5b Similarly, there is no detectable saturation or hlGPDH by 80 mM Gua+. These results emphasize the difference between the readily measurable intrinsic binding energy associated with formation of complexes of these activating cations to the enzyme-bound transition state and the observed ligand binding energy, which is too small to measure in these experiments.
Importance of Enzyme–Substrate Ion-Pairs
The upper limit for the contribution of the substrate phosphodianion and the R269 guanidine side chain to the enzymatic rate acceleration is <19.9 kcal/mol, the sum of the contributions of the phosphodianion (10.8 kcal/mol, Scheme 4) and the side chain cation (9.1 kcal/mol) to the rate acceleration. This includes a direct interaction between these ions, which is counted twice in the sum of the contributions, and estimated as the 2.8 kcal/mol effect of the R269A mutation on the stability of the Michaelis complex to DHAP.5a The total stabilization of the transition state for hlGPDH-catalyzed hydride transfer by interactions with these ionic groups is therefore ≤ (19.9 – 2.8) ≤ 17.1 kcal/mol.
We speculate that 17 kcal/mol is larger than the true contribution of interactions between the enzyme cation and the substrate phosphodianion to transition state stabilization, because of cooperativity between development of these interactions. The magnitude of these cooperative stabilizing interactions, which are eliminated by truncation of either the substrate phosphodianion or the enzyme cation, may be estimated by comparing the sum of the effects of separate truncation of the phosphodianion and cation ((10.8 + 9.1 – 2.8) = 17.1 kcal/mol) with the total effect of the two truncations on kcat/Km = 4.6 × 106 M–1 s–1 for the reaction catalyzed by wildtype hlGPDH. A rate constant of 1.2 × 10–6 M–1 s–1 is predicted for a total 17 kcal/mol destabilization of the transition state for wildtype hlGPDH-catalyzed reduction of DHAP. By comparison, our protocol fail to detect R269A hlGPDH-catalyzed reduction of GA and set an upper limit of kcat/Km ≤ 0.003 M–1 s–1 for this reaction. This sets limits of 17 kcal/mol ≥ ΔΔG† ≥ 12.4 kcal/mol for the stabilization of the transition state for hlGPDH-catalyzed reduction of DHAP by interactions that involve the transition state, the protein catalyst, the enzyme cationic side chain and the substrate phosphodianon. These limits correspond to a rate acceleration of between 109- and 1012.5-fold.
Cooperativity in the expression of the electrostatic interactions of these ionic groups will cause the transition state stabilization determined by summing the effects of mutation of the enzyme [R269A mutation] and substrate [truncation of phosphodianion] to exceed the true stabilization obtained from these interactions. For example, if closure of the enzyme flexible loop (Figure 1B) over the substrate phosphodianion results in a change in the environment of the enzyme active site that enhances electrostatic stabilization of the transition state, then truncation of the phosphodianion will both weaken the driving force for enzyme-activating loop closure and weaken the transition state stabilization from ion pairing interactions to the cationic side chain of R269. These ion pairing interactions are also lost at R269A mutant of hlGPDH, so that this part of this side chain stabilization of the transition state will be included twice when summing the effects of the separate truncation of the phosphodianion and the cationic side chain.
Other Enzymes
Other enzymes that catalyze reactions of phosphodianion substrates have been examined to determine whether the large 109- and 1012.5-fold contribution of this binding determinate to the stabilization of the transition state for catalysis by GPDH is an exception, the rule, or if the contribution to catalysis varies systematically from enzyme to enzyme. The present results are not exceptional, but the data is too limited to establish rules.
Triosephosphate Isomerase
The total stabilization of the transition state for TIM-catalyzed isomerization by interactions with the substrate phosphodianion and K12 side chain is [13.0 + 8.0 – 2.3] = 18.7 kcal/mol, where the first two terms give the sum of the effects of separate truncation of these groups (Table 2), and the third is the estimated 2.3 kcal/mol effect of the K12G mutation on the stability of the Michaelis complex to DHAP,5a which is counted twice in the first sum. This sets an upper limit of 18.7 kcal/mol ≤ ΔΔG† for transition state stabilization that is greater than the upper limit of 17 kcal/mol ≤ ΔΔG† for hlGPDH.
Orotidine 5′-Monophosphate Decarboxylase
The placement of K12 and R269 at the active sites of TIM and hlGPDH, respectively, is substantially different from that of R235 at OMPDC. All three side chains interact with the substrate phosphodianion, but only the side-chain of OMPDC interacts exclusively with the dianion, because its 10 Å separation from the orotate ring precludes direct interaction with the ring at the decarboxylation transition state. Consequently, the K12G and R269A mutations result in significant decreases in the respective values of kcat/Km for catalysis of reaction of the truncated substrate GA,10h,12 but the R235A mutation has no significant effect on kcat/Km for decarboxylation of the truncated substrate 1-(β-d-erythrofuranosyl)orotic acid (EO) by wildtype OMPDC.10d,10e This absolute requirement for the presence of the substrate phosphodianion to observe stabilization of the decarboxylation transition state by interactions with the cationic side chain of R235 demonstrates a strong cooperativity in the development of these cation-dianion electrostatic interactions. The contribution of this ion-pair to the total transition state stabilization is therefore equal to the 5.8 kcal/mol effect of the R235A mutation on catalysis by OMPDC.
Conclusions and Speculation
GPDH has evolved to optimize transition state stabilization from interactions that involve the cationic side chain of R269 and the phosphodianion of substrate DHAP. The large (12.4–17.1) kcal/mol stabilization obtained from these interactions provides a telling example of the power of electrostatic interactions in promoting enzymic catalysis.17 The most interesting element in the architecture of GPDH is the flexible loop, which closes over the phosphodianion of DHAP18 and converts GPDH from a floppy unliganded structure to a tighter and rigid closed caged complex, which provides for optimal protein–ligand interactions.19 We propose the following imperatives for evolution of this type of enzyme architecture.20
-
(1)
GPDH, TIM and OMPDC exist in an open form that allows substrate access to the active site, and then undergo a change in enzyme conformation to a caged complex that provides for optimal protein ligand interactions.19
-
(2)
These conformational changes, which convert the open enzymes to their closed form, are sufficiently fast to support turnover numbers of as large as kcat = 8000 s–1 for TIM-catalyzed isomerization of GAP.12,21 Such rapid reorganization of enzymes to their catalytic conformation is possible for floppy proteins with the TIM barrel fold,22 that contain unstructured loops. By comparison, the caged complex between substrate diaminopimelate (DAP) and DAP-epimerase is formed by an “oyster shell-like” clamping that involves movement of relatively rigid fixed protein domains.23 The smaller kcat ≈ (70–80) s–1 for DAP epimerase24 compared with 8000 s–1 TIM12 is consistent with an intrinsically slower motion of these rigid domains compared with the unstructured protein loops at a TIM barrel.20
-
(3)
The closed form of GPDH is relatively rigid and presumably provides for optimal positioning of protein side chains next to the enzyme-bound ligand.17a The entropic cost of this conformational change, which restricts the motion of the flexible peptide backbone and catalytic side chains attached to this backbone, is unclear. This cost is recovered as an enhancement of stabilizing electrostatic interactions at the preorganized closed form of GPDH.19b,25
Our treatment emphasizes the large transition state stabilization obtained from electrostatic interactions at the rigid closed conformation of GPDH,17 and the similarity in the total stabilizing electrostatic interactions of the protein catalyst with either the bound whole substrate or bound substrate pieces (Figure 5).10b,14b We suggest that the covalent connection, which reduces the entropic cost of binding the whole substrate compared to the substrate pieces, plays little or no role in positioning the substrate at the enzyme active site.
Conformational changes of GPDH and other enzymes must be sufficiently fast to support catalytic turnover. Proposals for a more profound influence of the dynamics of the enzyme conformational change on the enzymatic rate acceleration add one more layer of complexity to our understanding of enzyme catalysis. These proposals have staying power, in part because it is difficult to rigorously demonstrate the absence of a contribution of dynamics to catalysis. Furthermore, almost anything done to a protein changes its dynamics, so that an explanation for any experimental result is immediately at hand.14a,26 We therefore emphasize that the results reported in this paper are consistent with a model that considers the conformational change that connects the open and closed forms of GPDH, independent of the protein dynamics along the low free-energy pathway that connects these structures.10a,19b,25
Acknowledgments
This work was generously supported by the following grants from the US National Institutes of Health: GM116921 and GM039754.
The authors declare no competing financial interest.
References
- Lasker K.; Förster F.; Bohn S.; Walzthoeni T.; Villa E.; Unverdorben P.; Beck F.; Aebersold R.; Sali A.; Baumeister W. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 1380–1387. 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lehn J.-M. Angew. Chem., Int. Ed. Engl. 1990, 29, 1304–1319. 10.1002/anie.199013041. [DOI] [Google Scholar]; b Lawrence D. S.; Jiang T.; Levett M. Chem. Rev. 1995, 95, 2229–2260. 10.1021/cr00038a018. [DOI] [Google Scholar]
- Grzelczak M.; Vermant J.; Furst E. M.; Liz-Marzán L. M. ACS Nano 2010, 4, 3591–3605. 10.1021/nn100869j. [DOI] [PubMed] [Google Scholar]
- Peracchi A. Curr. Chem. Biol. 2008, 2, 32–49. 10.2174/187231308783334162. [DOI] [Google Scholar]
- a Reyes A. C.; Koudelka A. P.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2015, 137, 5312–5315. 10.1021/jacs.5b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Go M. K.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2010, 132, 13525–13532. 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Barnett S. A.; Amyes T. L.; Wood M. B.; Gerlt J. A.; Richard J. P. Biochemistry 2010, 49, 824–826. 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Olucha J.; Meneely K. M.; Lamb A. L. Biochemistry 2012, 51, 7525–7532. 10.1021/bi300472n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Biochemistry 2007, 46, 5841–5854. 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. Biochemistry 2008, 47, 4575–4582. 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Biochemistry 2007, 46, 5841–5854. 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud A.; Wilkins M. R.; Appel R. D.; Bairoch A. Proteomics Protocols Handbook 2005, 571–607. 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]; b Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. Nucleic Acids Res. 2003, 31, 3784–8. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Go M. K.; Amyes T. L.; Richard J. P. Biochemistry 2009, 48, 5769–5778. 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2014, 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. Biochemistry 2013, 52, 537–546. 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. Biochemistry 2013, 52, 7500–7511. 10.1021/bi401117y. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. Biochemistry 2012, 51, 4630–4632. 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2011, 133, 6545–6548. 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Amyes T. L.; Richard J. P.; Tait J. J. J. Am. Chem. Soc. 2005, 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]; h Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2015, 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Tsang W.-Y.; Amyes T. L.; Richard J. P. Biochemistry 2008, 47, 4575–4582. 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Spong K.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2013, 135, 18343–18346. 10.1021/ja4107513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4046–50. 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Koudelka A.; Amyes T. L.; Richard J. P. Biochemistry 2010, 49, 5377–5389. 10.1021/bi100538b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby A. J. Adv. Phys. Org. Chem. 1980, 17, 183–278. 10.1016/S0065-3160(08)60129-X. [DOI] [Google Scholar]
- a Zhai X.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2015, 137, 15185–15197. 10.1021/jacs.5b09328. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhai X.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2014, 136, 4145–4148. 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plapp B. V.; Charlier H. A. Jr.; Ramaswamy S. Arch. Biochem. Biophys. 2016, 591, 35–42. 10.1016/j.abb.2015.12.009. [DOI] [PubMed] [Google Scholar]
- Joseph-McCarthy D.; Lolis E.; Komives E. A.; Petsko G. A. Biochemistry 1994, 33, 2815–23. 10.1021/bi00176a010. [DOI] [PubMed] [Google Scholar]
- a Warshel A. J. Biol. Chem. 1998, 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]; b Warshel A.; Sharma P. K.; Kato M.; Parson W. W. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1647–1676. 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. Curr. Opin. Struct. Biol. 2010, 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wolfenden R. Mol. Cell. Biochem. 1974, 3, 207–11. 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]; b Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Zhai X.; Malabanan M. M. Bioorg. Chem. 2014, 57, 206–212. 10.1016/j.bioorg.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desamero R.; Rozovsky S.; Zhadin N.; McDermott A.; Callender R. Biochemistry 2003, 42, 2941–2951. 10.1021/bi026994i. [DOI] [PubMed] [Google Scholar]
- Bar-Even A.; Milo R.; Noor E.; Tawfik D. S. Biochemistry 2015, 54, 4969–4977. 10.1021/acs.biochem.5b00621. [DOI] [PubMed] [Google Scholar]
- Pillai B.; Cherney M. M.; Diaper C. M.; Sutherland A.; Blanchard J. S.; Vederas J. C.; James M. N. G. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8668–8673. 10.1073/pnas.0602537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman J. S.; Nichols J. S. J. Biol. Chem. 1984, 259, 8907–8914. [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Biochemistry 2013, 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Glowacki D. R.; Harvey J. N.; Mulholland A. J. Nat. Chem. 2012, 4, 169–176. 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]; b Antoniou D.; Schwartz S. D. J. Phys. Chem. B 2011, 115, 15147–15158. 10.1021/jp207876k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Francis K.; Kohen A. Curr. Opin. Chem. Biol. 2014, 21, 19–24. 10.1016/j.cbpa.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Frushicheva M. P.; Mills M. J. L.; Schopf P.; Singh M. K.; Prasad R. B.; Warshel A. Curr. Opin. Chem. Biol. 2014, 21, 56–62. 10.1016/j.cbpa.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]