

Abstract
Intergenic and intragenic enhancers found inside topologically associated regulatory domains (TADs) express noncoding RNAs, known as enhancer RNAs (eRNAs). Recent studies have indicated these eRNAs play a role in gene regulatory networks by controlling promoter and enhancer interactions and topology of higher-order chromatin structure. Misregulation of enhancer and promoter associated noncoding RNAs (ncRNAs) could stabilize deleterious secondary DNA structures, noncoding RNA associated DNA/RNA hybrid formation, and promote collisions of transcription complexes with replisomes. It is revealing that many chromosomal aberrations, some associated with malignancies, are present inside enhancer and/or promoter sequences. Here, we expand on current concepts to discuss enhancer RNAs and enhancer transcription, and how enhancer transcription influences genomic organization and integrity.
Keywords: enhancer transcription, enhancer RNA, RNA exosome, bidirectional transcription, long noncoding RNA
Introduction to long noncoding RNAs (lncRNAs)
With the advent of high-throughput RNA and DNA sequencing technologies, our understanding both of RNA and the process of DNA transcription that generates nascent RNA has advanced rapidly. Many different techniques have been developed that identify ncRNAs, their structure, and potential functions [1–5]. Thus, the RNA field has expanded beyond messenger, ribosomal, and transfer RNAs to encompass a panoply of ncRNAs including miRNAs (miRs), short nuclear RNAs (snRNAs), short nucleolar RNAs (snoRNAs), siRNAs, piwi-interacting RNAs (piRNAs), lncRNAs, and others. lncRNAs differ from the other above-mentioned classes by their cutoff length, defined to be >200 nucleotides [6,7]. lncRNAs are located both intergenically and intragenically, are transcribed by RNA polymerase II (polII), lack an open reading frame, are able to be post-transcriptionally capped, polyadenylated, or spliced, and sometimes are transcribed in bidirectional orientations relative to protein-coding genes (reviewed in [8–10]).
Enhancer Sequences and the eRNAs They Generate
The transcriptional machinery generating the panoply of RNAs has undergone scrutiny, and current discoveries have revealed robust lncRNA expression at enhancer sites of the mammalian genome [11–13]. Enhancers are demarcated by certain characteristics, including a high ratio of histone H3 lysine 4 monomethylation (H3K4me1) to trimethylation (H3K4me3), as well as the acetylation of histone H3 on lysine 27 (H3K27Ac). Additionally, enhancers can bind coactivators and acetyltransferases such as CREB binding protein[CK1] and p300 (reviewed in [14]). Chromatin signatures, namely the H3K4me1 to H3K4me3 ratio, have been used to characterize extragenic transcription sites targeted by RNA polII, and it has been found that 70% of extragenic RNA polII peaks are associated with genomic regions possessing the canonical chromatin signature of an enhancer [15]. RNAs transcribed from enhancer sequences are named eRNAs, and, based on their length, a significant fraction of them fall into the category of lncRNAs. However, there are differences between eRNAs and lncRNAs as the former, for example, are rarely spliced (5%) whereas 30% of lncRNAs are (reviewed in [14]). ‘Superenhancers’ are characterized by clusters of enhancers (tenfold longer than other enhancers) that are hyperacetylated and are actively transcribed ([16], concept reviewed in [17]). Superenhancers frequently are found in the neighborhood where a large number of active genes are being transcribed [16]. These superenhancer regions may generate large numbers of antisense RNAs and eRNAs [12]. Since the first identification of eRNAs as a subtype of lncRNAs, many questions have been raised regarding their function including what cellular role eRNAs play, how they are controlled, and how they exert their effects on their target genes (whether in cis through looping with their targets or in trans). Equally important is the question of how eRNAs exert their function without causing chromosomal instability brought about by secondary DNA structure formation, as in eRNA associated DNA/RNA hybrid formation. In this review we address some of these critical questions.
Why Are eRNAs Important?
Unlike many other noncoding RNAs, for example, miRNAs, rRNAs, tRNAs etc., whose functions have been ascribed directly to the RNA species itself, the role of the eRNA moiety is debated. eRNAs could be merely a passive byproduct of enhancer transcription or they may have additional functions such as the active recruitment of transcription factors. Although the passive role of eRNAs is generally accepted, recent discoveries have suggested the latter may also be operative. For example, it was shown that regulatory element RNAs associated with promoter and enhancer sequences are capable of recruiting the transcription factor Yin-Yang (YY)1 [18]. The authors named this concept of RNA-induced transcription factor (TF) recruitment ‘TF trapping’. Furthermore, consistent with previous observations [12,13], enhancer-associated regulatory eRNAs are sensitive to the activity of the RNA exosome complex. Knockdown of RNA exosome activity results in increased eRNA accumulation but decreased YY1 recruitment to enhancers. The RNA exosome facilitates transcriptional termination of RNA polII-associated ncRNAs [11,12,19] to release the 3′ end of eRNAs from the transcription complex, allowing the nascent eRNAs to associate with transacting TFs and localize them to cognate enhancer sequences. Therefore, this indicates a role for eRNA processing and/or degradation in regulating TF binding and recruitment to enhancers. The 3′-end processing activity of the RNA exosome may be important for trimming the eRNAs to appropriate sizes that support functionality; alternatively, the degradation activity may reduce the level of free eRNAs (located unbound to the chromatin) that otherwise titer the TFs away from the DNA associated functional form of eRNA [18]. These recent observations may explain the observation that there is widespread expression of enhancers that are placed close to neuronally regulated genes [20]. Whether eRNA action in trans is universal or only applicable to certain regions of the genome needs to be investigated in greater detail, and is discussed later. Future work will also highlight what types of genes or group of genes are regulated by eRNA expression or enhancer activity. From the limited data sets currently available, one can speculate that developmentally regulated fast acting genes, particularly those regulated in and around superenhancers, could require this additional property of transcriptional activation of enhancers [12,16,21,22]. However, exhaustive analysis of enhancer mediated regulation of gene expression at specific loci of various subtypes of genes (e.g., fast acting, constitutive, developmentally regulated, and clustered) can only be accomplished following in-depth identification of enhancer coordinates, transcriptome profiles, and regulatory element mapping.
Possible Mechanism of eRNA Activity: cis versus trans
Much speculation of eRNA function has come from studying lncRNA-related mechanisms. The most basic concept in understanding eRNA function is whether they act in trans or in cis. Trans acting eRNAs generated following enhancer transcription are free to transition within the nuclear space and could affect a gene on another chromosome or upstream/downstream on the same chromosome (the trans model; see Panels B and C in Figure 1). An example of a lncRNA that likely functions in trans comes from studying HOTAIR (HOX [CK2]antisense intergenic RNA), a 2.2-kb lncRNA transcribed from the antisense strand of the HOXC cluster on human chromosome 12 [23]. Through knockdown experiments using siRNAs against HOTAIR, it was shown that diminution of HOTAIR levels had no significant effect on the HOXC genes[CK3] clustered around HOTAIR itself but rather led to transcriptional activation of HOXD genes (through the loss of H3K27me3 marks present at the HOXD locus) located on human chromosome 2. The obvious conclusion is that lncRNAs act in a trans fashion, although the situation is complicated by the ability of HOTAIR to act as a competitive endogenous RNA and to regulate the levels of miRNAs including miR-130a, miR-331-3p, and miR-124, so HOTAIR may have both direct and indirect methods of effecting its actions (reviewed in [24]).
Figure 1. [CK11]Models for Various Classes of Enhancers and Their Mechanism of Function.
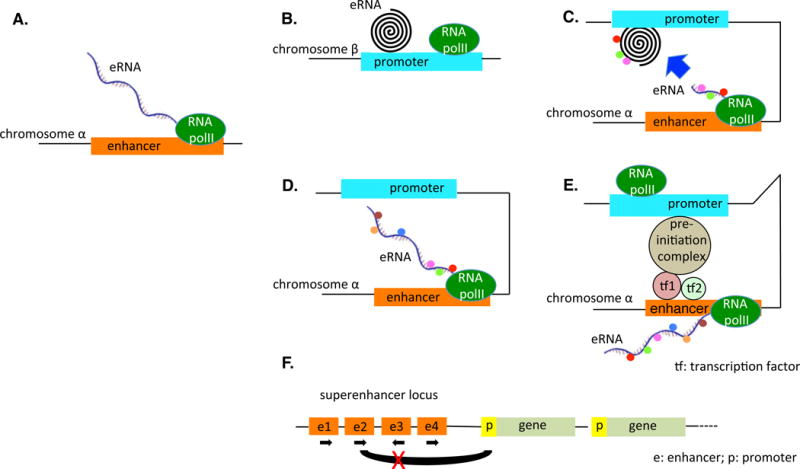
(A) eRNA is generated on chromosome α[CK12]. (B) eRNA acts in trans affecting a gene on a different chromosome (β). (C) eRNA acts in trans affecting a gene on the same chromosome. (D) eRNA acts in cis–trans. The transcript of the eRNA, bound by various proteins, binds to the target and influences expression. Alteration of the sequence of the transcript, thereby altering protein factor binding, helps to determine whether the sequence of the transcript itself or the act of transcription is critical. (E) cis model postulating that the eRNA transcript itself is not important; rather, the act of transcription of the enhancer allows the preinitiation complex to form a bridge between the enhancer region and the target region. (F) Superenhancer locus in which poorly understood interactions between various enhancers in various orientations may affect gene expression. The black loop between the second enhancer and the promoter of the first gene, for example, may function under circumstances but be blocked if the third enhancer, transcribed in the opposite orientation, is activated. Abbreviations: e, enhancer; eRNA, enhancer RNA; p, promoter; RNA polII, RNA polymerase II; tf, transcription factor.[CK13]
A variation on the trans acting model would be the cis–trans model, in which the ncRNA molecule itself, while being transcribed and thus still proximate to the promoter/enhancer from which it is being transcribed, brings that enhancer/promoter to the target sequence which could again be on a different chromosome or elsewhere on the same chromosome (see Panel D in Figure 1). In the cis–trans model, the eRNA moiety is a facilitator that brings two DNA segments together to interact. In an informative study published in 2008 that appears to correlate with a cis–trans model, it was suggested that ncRNAs (lncRNAs, as the transcripts were reported to be >200 nucleotides) are transcribed and serve as molecular ligands for RNA-binding proteins, which can be activated (or repressed) by the interaction with the ligand [25]. The ncRNAs are encoded in the genome close to the gene, which the RNA-binding protein represses (or activates). Thus, the ncRNA is transcribed, binds to the RNA-binding protein, and the RNA-binding protein then modulates the transcription of a gene close to the location of the ncRNA. This report was followed by a paper that likewise undertook a functional analysis of a long ncRNA adjacent to the Snai1 locus using reporter assays and demonstrated a role for this ncRNA in an RNA-dependent potentiation of neighboring gene expression [26]. Then, in 2011, it was shown that the lincRNA (long intergenic noncoding RNA), HOTTIP, found at the 5′ tip of the HOXA locus, coordinates the activation of several HOXA genes in vivo through chromosomal looping, whereby HOTTIP is brought into close proximity with its targets [27]. The HOTTIP RNA binds the adaptor protein WDR5[CK4] directly and targets the WDR5/MLL complex to the HOXA locus. It was noted that ectopic expression of HOTTIP RNA by retroviral transduction of lung fibroblasts, which do not express HOTTIP, failed to activate expression of distal HOXA genes. Ectopically expressed HOTTIP RNA, transcribed from retroviral insertion sites scattered randomly in the genome, may not be able to find 5′HOXA genes[CK5] whereas endogenous HOTTIP RNA is directly positioned near the 5′ HOXA genes by chromosomal looping, allowing interaction and control. Accordingly, HOTTIP would seem to behave as a cis–trans acting ncRNA. Finally, in the pure cis model, the ncRNA product itself is nonessential to any further biological effects; rather it is the act of transcription of the locus which opens the locus and brings the transcription preinitiation complex into alignment with the target (see Panel E in Figure 1). While it is conceivable that in the cis model the act of transcription and the opening of the locus could affect a target on another chromosome via interchromosomal interactions, this would require intricate and regulated coordination of chromosomal movement in the 3D space of the nucleus, along with overlaps of two different chromosomal territories [28–30]. Current models suggest that due to restriction of the chromosomal movements inside chromosomal territories, intrachromosomal interactions are more prevalent than interchromosomal interactions [28–30]. Thus, while the trans model of eRNA function is not at all impossible, it requires additional experimental evidence; the purely cis model would seem to be restricted to some enhancers that may not use the eRNAs they express for any particular purpose, so currently it is acceptable to say that the cis–trans model (Figure 1D) likely reflects the more prevalent and attractive mechanisms of enhancer function.
Properties of Enhancers That Influence Genomic Integrity
As shown in Figure 1D, in the cis–trans model, the eRNA must find the target with which it interacts. However, these interactions occur in the context of various properties of enhancers that can potentially influence genome organization and stability. Below, we sketch some of these properties.
In wild-type cells, ncRNAs are synthesized at normal levels but are rapidly turned over by the RNase activity of the RNA exosome complex [12], making their investigation difficult; when the RNA exosome has compromised functionality or is deleted completely, the half-lives of the cell ncRNAs, including transcription start site (TSS)-RNAs at coding (protein coding) genes, intragenic antisense RNAs (as-RNAs), and noncoding (enhancers and lncRNAs) loci are extended. TSS-RNAs are ncRNAs >500 nucleotides in length, expressed divergently from the TSS of coding genes, and have their own cognate transcription start sites [11,31]. Transcription units with cognate transcription start sites, separated by 50 base pairs or more, that are directed in opposite directions on the two strands of a DNA locus, are referred to as divergent transcripts (Figure 2B, Key Figure). Locations that express these divergent transcripts may be more susceptible to activation-induced cytidine deaminease (AID) breaks and therefore break and rejoin other chromosomal areas also expressing divergent transcripts more easily, as we detail below. Accordingly, locations for genomic (in)stability and interaction are intimately related with divergent transcription.
Figure 2. (Key Figure). Divergent Transcription at Enhancer Sequence.
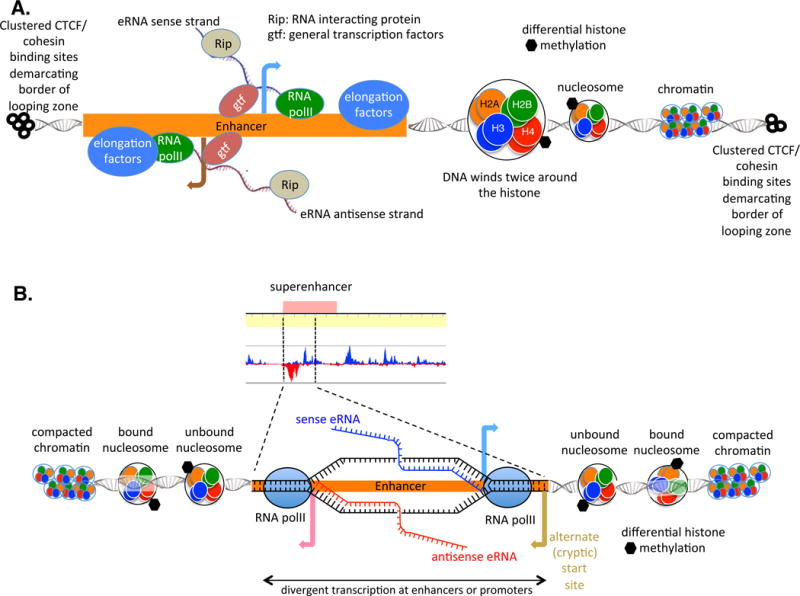
(A) Sense and antisense eRNA transcripts generated from an enhancer element. Transcripts may loop around and affect gene regulation by binding to promoters, as modeled in Figure 1. Note the presence of the clustered CTCF- and cohesin-binding sites, which demarcate the TAD. Looping between enhancers and promoters is believed to occur within a TAD but not between TADs. Looping occurs in open chromatin so that as distance from the enhancer increases, histones are compacted into nucleosomes and then further compacted into heterochromatin. Superenhancers are defined by altered histone methylation/acetylation. (B) In a superenhancer (series of enhancers in close proximity to one another causing high enhancer density), transcription of sense and antisense eRNAs in opposite orientations (by divergent enhancers) may mark the site for the cellular machinery as a location to which looping occurs. The antisense transcript may stall due to various hindrances, causing ‘flap-back’ of the nascent RNA and generating RNA–DNA structures called R loops. R loops attract AID whose activity causes alterations in the DNA and may allow looping to other areas (AID not shown in figure). Alternate (or cryptic) start sites may likewise generate transcripts (transcript not portrayed in figure). Abbreviations: AID, activation-induced deaminase; CTCF, [CK14]; eRNA, enhancer RNA; gtf, general transcription factor; Rip, RNA-interacting protein; RNA polII; RNA polymerase II; TAD, topologically associated domain.
In B cells, eRNAs or TSS-RNAs are transcribed at locations where enhancer and/or promoter-associated somatic mutation often occurs [11]. Likewise, as-RNAs are located within the bodies of genes in the B cell genome, observed to be translocation hotspots caused by somatic mutations [11]. Many intragenic and intergenic enhancer sequences are found at genomic coordinates that are close to or are overlapping with TSS-RNA or asRNA expression regions in the B cell genome. Although correlative, these observations point to the possibility of enhancer transcription promoting genomic instability via somatic mutagenesis and ensuing chromosomal translocations. Basically, then, the exosome knockout cells, because their ncRNAs are not as rapidly degraded, allow the identification of sites of divergent transcription (from which sense and anti-sense ncRNAs are expressed), and these sites of divergent transcription (at which eRNAs are located) (i) may overlap with the locations to which the B cell genome mutator AID is attracted [11,32–36] and (ii) may mark target points for genomic instability through mechanisms not yet completely understood but possibly including (i) unpacking of chromatin due to negative supercoiling created behind the RNA polII complexes; (ii) as-RNAs affecting chromatin modifications that enforce RNA polII stalling in the sense direction; and (iii) generation of single-stranded DNA structures (R loops) that are associated with RNA polII termination/stalling. A list of diseases proposed to be caused by enhancer/enhancer–cluster/superenhancer misregulation is highlighted in Table 1 and a discussion of hotspot characteristics that may contribute to their status as targets for mutation and recombination continues below.
Table 1.
| Name of disease | Genetic defect | Identification of superenhancer | Disease alleviation | Refs |
|---|---|---|---|---|
| JIA | Superenhancer in JIA patient synovial fluid derived CD4+ memory–effector T cells | H3K27Ac ChIP | JQ1 inhibits superenhancer: reduced disease associated gene expression | 80 |
| Multiple myeloma | Translocation of 3′ IgH superenhancer to MYC gene | 18-fold more mediator, 16-fold more BRD4, higher H3K27 binding | Treatment with JQ1: loss of BRD4 at superenhancers and transcription elongation defects | 78 |
| Acute lymphoblastic leukemia | Introduction of binding motifs for MYB transcription factor upstream of TAL1 oncogene creating a super-enhancer | H3K27Ac ChIP | CRISPR-Cas9 deletion of enhancer region causes complete abrogation of TAL-1 expression | 79 |
| Diffuse large B cell lymphoma | Enhancer-associated factor BRD4 binds to POU2AF1, BCL6, PAX5, IRF8 genes; all essential for B cell fate determination, germinal center formation. | BRD4 and confirmed with H3K27Ac | JQ1 abolishes BRD4 binding | 22 |
| Cornelia de Lange syndrome | Defective NIPBL fails to facilitate looping of enhancers to promoters | 82 | ||
| HD | Downregulation of genes controlled by superenhancers in HD striatum | H3K27Ac | 77 | |
| RA | Half of RA risk genes in CD4+ T cells link to superenhancers | H3K27Ac | Treatment with Janus kinase inhibitor tofacitinib impacts superenhancer–gene interaction | 81 |
Abbreviations: BRD4, [CK17]; ChIP, chromatin immunoprecipitation; HD, Huntington’s disease; JIA, juvenile idiopathic arthritis; NIPBL,; RA, rheumatoid arthritis.
Divergent ncRNA Transcription and Genomic Instability at Enhancer Sites
Transcription of both TSS- and as-RNAs may stall frequently owing to various hindrances: high cytosine density in the DNA being transcribed (especially in CG rich promoters), association of various transcription factors with the DNA sequence (Figure 2A), negative supercoiling of the DNA (following RNA polymerases divergently traveling on DNA; Figure 2B), and single-stranded DNA nicks at promoters [37]. The negative supercoiling tension imparted on the DNA due to the divergently transcribing RNA polymerase can create nucleosome-depleted regions that have higher potential to accumulate single-stranded DNA structures. Single-stranded DNA is subject to mutation by AID, and the actions of AID may cause double-stranded breaks and thereby, ultimately, translocations between chromosomes. As an example, a specific area in the BCL6 gene was identified as a translocation hotspot [38]. The original report had demonstrated that translocations occur within intron 1 of the BCL6 gene, but it was unclear why the breaks occur within an ~2-kb region of the 11-kb length of intron 1 [39]. The breakpoint area is part of a superenhancer as defined by histone 3 lysine 27 acetylation (H3K27ac) density and there is a lncRNA in this 2-kb region; its boundaries match precisely the boundaries of the BCL6 translocation zone. Transcription of the lncRNA is in the opposite orientation to that of the BCL6 gene, leading to the consideration that divergent transcription may be implicated in defining translocation fragile zones. Divergent transcription results in single-stranded DNA, which is necessary for AID action, and it had been shown previously that AID actively deaminates cytidine targets in this particular breakpoint region [38,40,41]. This report in the literature connects divergent transcription, AID activity, lncRNA expression, superenhancer location, and genomic breakpoints.
Protein Binding That Mark Enhancers
It is known that intrachromosomal loops exist in DNA, which bring into close proximity disparate elements of the DNA ([42–44], reviewed in [45]), and are a prominent mechanism of action for ncRNAs. Two protein complexes are now universally accepted to be involved in DNA looping and genome organization; the components of the mediator complex and the CTCF [CK6]proteins. The Mediator complex [46] that binds at enhancer and promoter sites (Figure 3) facilitates the looping process between activating ncRNAs (ncRNA-a) and the targets of the ncRNA-as. ncRNA-as facilitate activation of neighboring genes. Loss of the Mediator complex diminishes looping between the ncRNA-a and its target [47]. The Mediator complex and CTCF are discussed in greater depth in Box 1, and extensive information regarding the mechanism of chromosome looping can be found in the following articles and reviews ([48–55]). The proper recruitment of CTCF, mediator proteins, and others may play a vital role in organizing the genome and preventing genomic instability.
Figure 3. High Resolution of Genome Structure and Looping.
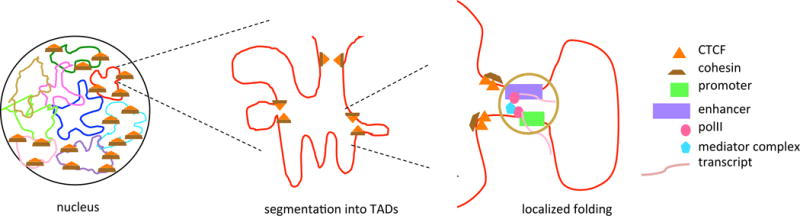
(A) Schematic of a mammalian nucleus with nine chromosomes indicated. Five of the chromosomes have TADs demarcated by CTCF/cohesin protein aggregates. (B) Magnified view of a TAD demarcated by CTCF/cohesin boundary marks. Note that subloops occur within the TAD – potentially bringing together a number of loops so that different promoters and enhancers interact with distant DNA sites concurrently (not indicated in figure) – but looping between TADs is thought not to occur except in states of disease. (C) Magnified view of the extrusion complex in B. Note the mediator complex interacting with the polymerases. Abbreviations: CTCF, [CK15]; TAD, topologically associated domain.
Box 1. Roles of the Mediator Complex/Cohesin and CTCF in Loop Formation.
Mediator and cohesin physically and functionally connect the enhancers and core promoters of active genes in murine embryonic stem cells through the formation of large loops [71] (Figure 3); in different cell types mediator and cohesin bind to different promoters and enhancers, giving rise to different loops. The Mediator complex is a large, multiunit complex that functions as a transcriptional coactivator, interacting with general transcription factors as well as RNA polII. The flexibility of Mediator (and the ability to add and remove subunits from the complex) makes it well suited to bring together enhancers and promoters, although the question remains: what marks on the chromatin flag that particular area as one in which mediator/cohesin should bind[CK10] and which factors generate and erase those marks, facilitating new interactions? While protein factors must be involved in the selection of looping borders, the sequence of the DNA itself is the ultimate arbiter of interaction location selection. Mammalian CTCF-binding sites play a role in genomic topology and enhancer/promoter function [72] (Figure 3). CTCF is an important trans-acting factor that confers enhancer blocking insulator activity. By using CRISPR/Cas9 manipulation to alter CTCF-binding sites, using protocadherin and β-globin as model genes, inversion of CTCF-binding sites was shown to reconfigure the chromatin loops between distal enhancers and target promoters, altering gene-expression patterns. Both CTCF and cohesin are enriched at the borders of TADs, which may demarcate looping structures [73] (a generalized schematic is shown in Figure 3). TADs are megabase scale domains within which interactions between elements of chromatin are thought to occur; there are insulator elements at the potential ends of the TADs, which constrain interactions so that they occur within the TADs, not between them. TAD demarcations appear largely to be conserved across species [43]; rare human limb alterations, replicated in mice, arise when interactions occur across TADs, as opposed to within them [74]. Likewise, depletion of cohesin subunits by RNAi leads to the loss of long-range physical contacts (reviewed in [75]). The stability of cohesin and CTCF-mediated loops within TADs is unclear: are they stable or transient structures? Can an eRNA interact promiscuously, such that it loops with an upstream DNA at one time and temporally subsequently works with a (different) downstream DNA, or are loops stable elements? It has been shown that some CTCF elements are constrained at the level of DNA sequence and binding and others show more flexibility [76]. Accordingly, CTCF binding sites and histone modifications may affect looping possibilities and choices within the chromatin.
Formation at Enhancers of Secondary DNA Structures and Collisions with Replisomes
Expression of eRNAs at various enhancer sites or in superenhancer clusters may unwind DNA and potentially create single-stranded DNA that exist as R loops (or other structures such as G quartets [56]). These R loops contribute to genomic instability (Figure 2B). R loops are three-stranded DNA–RNA hybrid structures in which the nascent RNA strand binds to the DNA after the two nucleic acid strands exit the RNA polymerase holoenzyme, displacing a tract of single-stranded DNA. Miniature R loops are formed during DNA replication on the lagging strand, but these are physiological and not sources of genomic instability. When unwinding the DNA double helix at sites of transcription, negative supercoils arise behind and positive supercoils ahead of the moving RNA polymerase [57]. These conformations can destabilize the DNA, as demonstrated by the observation that highly transcribed genes have increased recombination and mutation rates compared to loci with lesser transcriptional activity (reviewed in [58]). The transcription and replication machineries themselves may play roles in the resulting mutational outcomes ([12], reviewed in [59]) overlapping the genomic coordinates where R-loop structures form. R loops associated with class switch recombination (CSR) when the antibody shuffles its isotype from the default μ class to a downstream class such as δ, γ, ε, or α, allowing different effector molecules to be recruited to the antibody – at G-rich switch sequences are physiological and stabilized due to G–G dimers formed on the same strands of DNA. The cell uses various methods to recognize and eliminate the R loops, including the use of transcription-coupled RNA-processing events associated with RNA degradation via RNA exosome complex or Xrn1, or splicing [60],[61],[12][CK7] and use of the activity of RNase H1 and RNase H2 enzymes [41].
Transcription-coupled negative supercoiling of the DNA template along with associated R-loop formation – when not rapidly resolved – can act as obstructions for the advancing replication fork (if cells transition into the S phase of the cell cycle). The R-loop structure may also be responsible for stalling the transcriptionally active RNA polymerase on the DNA template. Studies have postulated that such a transcription complex–replication complex collision can lead to DNA double-strand breaks, particularly at fragile sites in the genome [62–64]. IgH switch sequences are a special case, as it is here that transcription coupled R loops may be responsible for the formation of origin of replication sites that have a role in synapsis of DNA double-strand breaks [65]. Thus, in many different ways, transcription directionality of RNA polII (including at enhancer sites) at DNA polymerase complex associated sites in the genome may play a role in DNA double-strand break formation and genomic integrity regulation.
RNA Polymerase Collisions during Convergent Transcription at Superenhancer Sites
Recent studies have highlighted that bidirectional transcription could create convergent transcription between two divergent transcription start sites, causing genomic instability. At some superenhancer sequences, owing to the close spacing between divergent enhancers, convergent transcription may occur between two RNA polII molecules initiating from divergent TSSs, as the RNA polII molecules enter the elongation phase (Figure 4A). Furthermore, that the collision of two RNA polymerases transcribing in a convergent manner may generate genomic instability has been considered. It has been postulated that the termination of enhancer RNA transcription through RNA exosome-dependent mechanisms prevents such instability [12]. It was also recently shown that the small nuclear RNA 7SK snRNA prevents convergent transcription-mediated DNA damage at enhancer sequences that are occupied by the bromodomain containing enhancer-binding protein Brd4 [66]. AID-induced chromosomal translocations and genomic instability at regions of the genome that have bidirectional transcription – potentially due to head-to-head transcription of TSS-associated and/or eRNA-associated transcription complexes around the promoters or genes inside gene bodies – have also been shown [11,38,67]. For example, in the first intron of the c-Myc oncogene, transcription complexes transcribing in a head-to-head orientation can be seen [11], and these regions of the myc gene contain translocation hotspots that are associated with the onset of Burkitt lymphoma in humans [68,69]. The mechanism of such instability is due either to the physical collision of the two RNA polymerases [67,70], and/or to the generation of nucleosome-free single-stranded DNA caused by the negative supercoiling present in the two passing RNA polII complexes, positioned in a divergent configuration during the process of transcription. These mechanistic possibilities are detailed in [40,41]. To this end, it has been shown that some of the bidirectionally transcribed regions of the B cell genome, where genomic instability occurs (including putative intragenic enhancers), have nucleosome depletion and accumulation of single-stranded DNA conformations ([11]).
Figure 4. Clustering of Sense–Antisense-Expressing Genes around Superenhancers.
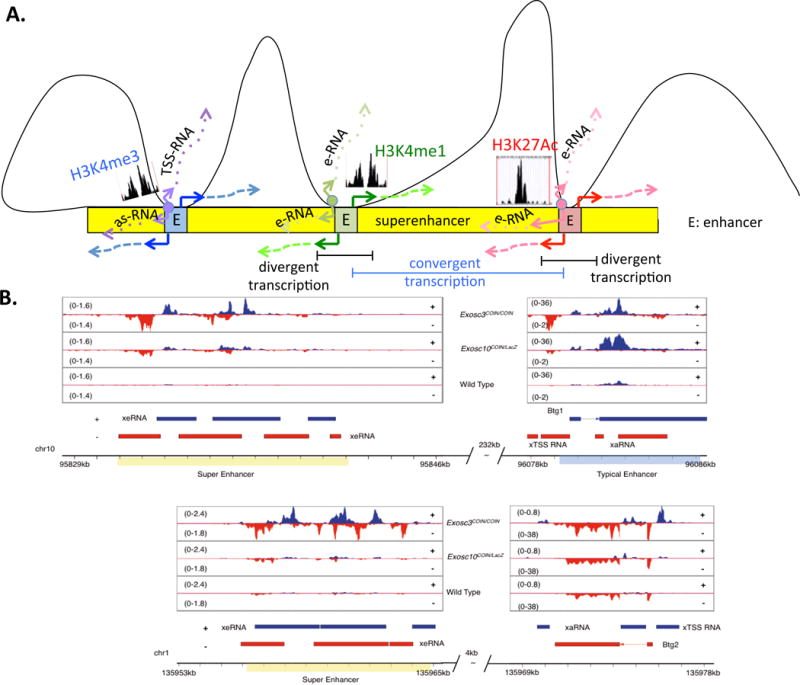
(A) A schematic showing (from left) a promoter (enriched for H3K4me3 marks and expressing TSS- and as-RNAs), (middle) an enhancer (enriched for H3K4me1 marks and expressing bidirectional enhancer RNAs), and (right) a superenhancer (enriched for H3K27Ac marks and expressing bidirectional enhancer RNAs) interacting with three separate enhancer loci within a superenhancer locus (blue, green, and red loci within the yellow superenhancer). Bidirectional transcription marks each of the three loci, potentially marking them as candidates for interaction with the superenhancer locus. Divergent transcripts are transcribed in opposite orientations (highlighted in black bar) but two closely-spaced divergent transcripts may form a situation of convergent transcription (highlighted in blue bar). (B) IGV[CK16] reads from cultured splenocytes illustrate that synthesis of as-RNAs may permit functional engagement with superenhancer elements to form higher-order chromosomal structures. (Top) A superenhancer–enhancer (overlapping the Btg1 gene) pair separated by a distance of 232 kb, expressing exosome-linked se-RNAs and TSS-RNAs, respectively. (Bottom) A superenhancer–enhancer (overlapping the Btg2 gene) pair separated by a distance of 4 kb, expressing exosome-linked se-RNAs and as-RNAs, respectively. Unshown: in examples where the enhancer–superenhancer are separated by distances >310 kb, the enhancer is less likely to express exosome-linked TSS-RNAs. Adapted from [12]. Abbreviations: as-RNA, antisense RNA; E, enhancer; eRNA, enhancer RNA; se-RNA, superenhancer RNA; TSS-RNA, transcription start site RNA.
Clustering of Sense–Antisense RNA Expressing Genes around Superenhancers
Finally, a less understood phenomenon is the clustering of sense/antisense RNA-expressing loci within a distance of ~300 kb surrounding superenhancers in B cells. Sense/antisense RNA-expressing promoters, enhancers, and superenhancers tend to cluster in developing cells like B cells [12]. A schematic of clustering or regulatory elements near an SE cluster[CK8] is shown in Figure 4A. As mentioned earlier, sense/antisense RNAs are potentially responsible for genomic instability. These sense/antisense RNAs inside and surrounding the SE clusters could be restricted in quantity by the activity of the RNA-processing complex, RNA exosome [12,62]. The reduction in RNA levels could be due to two reasons, the ncRNA degradation activity of the RNA exosome complex and/or the ability of RNA exosome to promote termination of antisense and sense ncRNAs expressed at regulatory sequences such as enhancers [11,19]. As an example, both the Btg1 and Btg2 genes (B cell translocation gene, Btg) that are often found to have chromosomal translocations in chronic lymphocytic leukemia (CLL) have high expression of antisense RNAs (visible following deletion of RNA exosome activity; Figure 4B) and placed close to superenhancer clusters (232 kb for Btg1 and 4 kb for Btg2) that show robust sense and antisense RNAs. For now, the mechanism of translocation can be ascribed to the presence of eRNA transcription at these loci only in a correlative manner, but future studies may be able to relate clustering of sense/antisense RNA expressing loci around superenhancer clusters with a new type of fragile site in the mammalian genome.
Concluding Remarks and Future Directions
We have now discussed extensively the role of eRNAs in both normal and pathophysiological instances of genomic (in)stability. However, both practical and conceptual questions remain (See Outstanding Questions). On the practical side is one question relating to the cis versus cis/trans (or both) debate: whether the transcribed noncoding moiety itself is important or whether the act of transcription of the locus is critical to regulatory function of the ncRNA. To shed light on this issue may well involve isolation (and thereby identification) of factors bound to the ncRNA; point mutation of the critical residues of the lncRNA to prevent factor binding; and observation of changes in biological processes as the mechanism of lncRNA function is dissected in an individualized context. If it is the act of transcription that is important, then the point mutations on the ncRNA-expressing locus may not matter (assuming DNA mutations do not alter RNA polII recruitment and transcription rates at the lncRNA locus). If it is the ncRNA transcript itself that matters, decorated by proteins that interact with the target, then point mutations may be fatal to the biological readout. On the more conceptual side, a number of questions loom. We find three to be of particular interest. First, do breaks in enhancers (specifically those enhancers associated with TSS-RNAs) occur frequently? Transcriptional directionality at enhancer sites that cause DNA nicks on both strands theoretically should be repaired rapidly to prevent genomic instability. Do they break frequently, are they repaired rapidly, and if so, which pathways are responsible for their repair?
Outstanding Questions Box.
Enhancer RNA (eRNAs) may fall into a different category of biological regulator than other trans-acting non-coding RNAs, as the importance of looping within TADs and its role in disease and development comes into sharper focus. As highlighted in our review, the following subjects require deeper investigation:
-
□
Is the product of the eRNA itself (whether it acts in cis or in trans) critical for eRNA function, or is it the act of transcription–whereby the chromatin is exposed and can interact with other chromatin–that is at the heart of the role of the eRNA? eRNAs acting in cis do not lend themselves to fruitful overexpression studies and new techniques will be required.
-
□
How do RNA processing factors such as RNA exosome and the enhancer binding proteins such as the mediator complex affect eRNAs, their stability, their choice of interacting location in the genome, and their eventual degradation?
-
□
How does transcription coupled R-loop formation positively facilitate enhancer function? What are the cellular factors that prevent genomic instability at active enhancer sequences?
-
□
Is divergent transcription of enhancers important for their gene regulatory function?
Second, mutations inside genes that lead to loss of function or gain of function proteins are well characterized as contributing to disease onset. In a parallel question, do enhancer mutations affect physiology and disease, due to misregulation of gene expression that they control through their influence on promoter activity? Or, is the level of control even more complicated and complex, as mutations have heretofore unexplored effects on the loss of fine regulation of chromosomal organization inside loops/TADs?
Finally, and perhaps most importantly, what are the regulatory mechanisms such that enhancers and the associated cascades of gene expression they control differ for various cell types and during different stages of development? How are enhancers (and cluster of enhancers, and superenhancers) turned on and off, and how are mutational mistakes controlled as they do so?
Trends Box.
enhancer RNAs (eRNAs) may function to regulate promoter-enhancer interactions within Topologically Associated Domains.
Enhancer regions are targets of DNA altering events (mutations, transloations, etc.) associated with various diseases.
Divergently transcribed enhancers are particularly susceptible to genomic instability.
Enhancer instability could be due to R-loop or G-quartet formation.
Acknowledgments
Research in the Basu Laboratory is supported by NIH grant 1R01AI099195, and funding from Leukemia and Lymphoma Society of America and Pershing Square Sohn Cancer Research Alliance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feng Y, et al. Methods for the study of long noncoding RNA in cancer cell signaling. Methods Mol Biol. 2014;1165:115–143. doi: 10.1007/978-1-4939-0856-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullokandov G, et al. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods. 2012;9:840–846. doi: 10.1038/nmeth.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hennessy E, O’Driscoll L. MicroRNA expression analysis: techniques suitable for studies of intercellular and extracellular microRNAs. Methods Mol Biol. 2011;784:99–107. doi: 10.1007/978-1-61779-289-2_7. [DOI] [PubMed] [Google Scholar]
- 4.Siomi MC. PIWI-Interacting RNAs : Methods And Protocols[CK9] [Google Scholar]
- 5.Carmichael GG. Regulatory Non-Coding RNAs : Methods and Protocols [Google Scholar]
- 6.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 7.Hung T, Chang HY. Long noncoding RNA in genome regulation: prospects and mechanisms. RNA Biol. 2010;7:582–585. doi: 10.4161/rna.7.5.13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci. 2012;13:528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antoniou D, et al. Recent advances in the involvement of long non-coding RNAs in neural stem cell biology and brain pathophysiology. Front Physiol. 2014;5:155. doi: 10.3389/fphys.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kung JT, et al. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–669. doi: 10.1534/genetics.112.146704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pefanis E, et al. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature. 2014;514:389–393. doi: 10.1038/nature13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pefanis E, et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161:774–789. doi: 10.1016/j.cell.2015.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, et al. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 15.De Santa F, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47:8–12. doi: 10.1038/ng.3167. [DOI] [PubMed] [Google Scholar]
- 18.Sigova AA, et al. Transcription factor trapping by RNA in gene regulatory elements. Science. 2015;350:978–981. doi: 10.1126/science.aad3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemay JF, et al. The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat Struct Mol Biol. 2014;21:919–926. doi: 10.1038/nsmb.2893. [DOI] [PubMed] [Google Scholar]
- 20.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qian J, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159:1524–1537. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapuy B, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rinn JL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhan A, Mandal SS. LncRNA HOTAIR: A master regulator of chromatin dynamics and cancer. Biochim Biophys Acta. 2015;1856:151–164. doi: 10.1016/j.bbcan.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orom UA, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dixon JR, et al. Chromatin domains: the unit of chromosome organization. Mol Cell. 2016;62:668–680. doi: 10.1016/j.molcel.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorkin DU, et al. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14:762–775. doi: 10.1016/j.stem.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun JB, et al. E3-ubiquitin ligase Nedd4 determines the fate of AID-associated RNA polymerase II in B cells. Gene Dev. 2013;27:1821–1833. doi: 10.1101/gad.210211.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavri R, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kodgire P, et al. Changes in RNA polymerase II progression influence somatic hypermutation of Ig-related genes by AID. J Exp Med. 2013;210:1481–1492. doi: 10.1084/jem.20121523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maul RW, et al. Topoisomerase I deficiency causes RNA polymerase II accumulation and increases AID abundance in immunoglobulin variable genes. DNA Repair (Amst) 2015;30:46–52. doi: 10.1016/j.dnarep.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwer B, et al. Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc Natl Acad Sci U S A. 2016;113:2258–2263. doi: 10.1073/pnas.1525564113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng S, et al. Non-coding RNA generated following lariat debranching mediates targeting of AID to DNA. Cell. 2015;161:762–773. doi: 10.1016/j.cell.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madabhushi R, et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Z, et al. Convergent BCL6 and lncRNA promoters demarcate the major breakpoint region for BCL6 translocations. Blood. 2015;126:1730–1731. doi: 10.1182/blood-2015-07-657999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Z, et al. BCL6 breaks occur at different AID sequence motifs in Ig-BCL6 and non-Ig-BCL6 rearrangements. Blood. 2013;121:4551–4554. doi: 10.1182/blood-2012-10-464958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pannunzio NR, Lieber MR. RNA polymerase collision versus DNA structural distortion: twists and turns can cause break failure. Mol Cell. 2016;62:327–334. doi: 10.1016/j.molcel.2016.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lieber MR. Mechanisms of human lymphoid chromosomal translocations. Nat Rev Cancer. 2016;16:387–398. doi: 10.1038/nrc.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang H, et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13:30–35. doi: 10.1016/j.stem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 43.Dixon JR, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cavalli G, Misteli T. Functional implications of genome topology. Nat Struct Mol Biol. 2013;20:290–299. doi: 10.1038/nsmb.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matharu N, Ahituv N. Minor loops in major folds: enhancer–promoter looping, chromatin restructuring, and their association with transcriptional regulation and disease. PLoS Genet. 2015;11:e1005640. doi: 10.1371/journal.pgen.1005640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lai F, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501. doi: 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fudenberg G, et al. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016;15:2038–2049. doi: 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hnisz D, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowley MJ, Corces VG. The three-dimensional genome: principles and roles of long-distance interactions. Curr Opin Cell Biol. 2016;40:8–14. doi: 10.1016/j.ceb.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lupianez DG, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene–enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dekker J, Misteli T. Long-range chromatin interactions. Csh Perspect Biol. 2015;7:a019356. doi: 10.1101/cshperspect.a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bouwman BAM, de Laat W. Getting the genome in shape: the formation of loops, domains and compartments. Genome Biol. 2015;16:154. doi: 10.1186/s13059-015-0730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merkenschlager M, Nora EP. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu Rev Genom Hum G. 2016;17:17–43. doi: 10.1146/annurev-genom-083115-022339. [DOI] [PubMed] [Google Scholar]
- 56.Bochman ML, et al. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kouzine F, et al. DNA topology and transcription. Nucleus. 2014;5:195–202. doi: 10.4161/nucl.28909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamperl S, Cimprich KA. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 2014;19:84–94. doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nature reviews. Genetics. 2012;13:204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grzechnik P, et al. Terminate and make a loop: regulation of transcriptional directionality. Trends Biochem Sci. 2014;39:319–327. doi: 10.1016/j.tibs.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan YA, et al. Mechanisms of genome instability induced by RNA-processing defects. Trends Genet. 2014;30:245–253. doi: 10.1016/j.tig.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garcia-Muse T, Aguilera A. Transcription–replication conflicts: how they occur and how they are resolved. Nat Rev Mol Cell Biol. 2016;17:553–563. doi: 10.1038/nrm.2016.88. [DOI] [PubMed] [Google Scholar]
- 63.Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- 64.Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nat Rev Genet. 2012;13:204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiedemann EM, et al. DNA replication origins in immunoglobulin switch regions regulate class switch recombination in an R loop-dependent manner. Cell Reports. doi: 10.1016/j.celrep.2016.11.041. (in press) [DOI] [PubMed] [Google Scholar]
- 66.Flynn RA, et al. 7SK-BAF axis controls pervasive transcription at enhancers. Nat Struct Mol Biol. 2016;23:231–238. doi: 10.1038/nsmb.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meng FL, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159:1538–1548. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lenoir GM, et al. Correlation between immunoglobulin light chain expression and variant translocation in Burkitt’s lymphoma. Nature. 1982;298:474–476. doi: 10.1038/298474a0. [DOI] [PubMed] [Google Scholar]
- 69.Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20:5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 70.Hobson DJ, et al. RNA polymerase II collision interrupts convergent transcription. Mol Cell. 2012;48:365–374. doi: 10.1016/j.molcel.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo Y, et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162:900–910. doi: 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lupianez DG, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vietri Rudan M, Hadjur S. Genetic tailors: CTCF and cohesin shape the genome during evolution. Trends Genet. 2015;31:651–660. doi: 10.1016/j.tig.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 76.Vietri Rudan M, et al. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015;10:1297–1309. doi: 10.1016/j.celrep.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Achour M, et al. Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington’s disease mice. Hum Mol Genet. 2015;24:3481–3496. doi: 10.1093/hmg/ddv099. [DOI] [PubMed] [Google Scholar]
- 78.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mansour MR, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peeters JG, et al. Inhibition of super-enhancer activity in autoinflammatory site-derived T cells reduces disease-associated gene expression. Cell Rep. 2015;12:1986–1996. doi: 10.1016/j.celrep.2015.08.046. [DOI] [PubMed] [Google Scholar]
- 81.Vahedi G, et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature. 2015;520:558–562. doi: 10.1038/nature14154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krantz ID, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]