

Abstract
Myotonic dystrophy type 1 (DM1) is a genetic disorder in which dominant-active DM protein kinase (DMPK) transcripts accumulate in nuclear foci, leading to abnormal regulation of RNA processing. A leading approach to treat DM1 uses DMPK-targeting antisense oligonucleotides (ASOs) to reduce levels of toxic RNA. However, basal levels of DMPK protein are reduced by half in DM1 patients. This raises concern that intolerance for further DMPK loss may limit ASO therapy, especially since mice with Dmpk gene deletion reportedly show cardiac defects and skeletal myopathy. We re-examined cardiac and muscle function in mice with Dmpk gene deletion, and studied post-maturity knockdown using Dmpk-targeting ASOs in mice with heterozygous deletion. Contrary to previous reports, we found no effect of Dmpk gene deletion on cardiac or muscle function, when studied on two genetic backgrounds. In heterozygous knockouts, the administration of ASOs reduced Dmpk expression in cardiac and skeletal muscle by > 90%, yet survival, electrocardiogram intervals, cardiac ejection fraction and muscle strength remained normal. The imposition of cardiac stress by pressure overload, or muscle stress by myotonia, did not unmask a requirement for DMPK. Our results support the feasibility and safety of using ASOs for post-transcriptional silencing of DMPK in muscle and heart.
Introduction
Myotonic dystrophy type 1 (DM1) is an autosomal dominant disorder resulting from expansion of a CTG repeat in the 3’ untranslated region of DMPK (1). While DM1 produces a wide spectrum of clinical signs, the main determinants of function and survival arise from cardiac, skeletal muscle and CNS effects. In skeletal muscle, DM1 causes progressive weakness, muscle wasting and repetitive action potentials (myotonia), culminating in respiratory failure [reviewed in (2)]. In the heart DM1 causes disease of the cardiac conduction system (CCS) (3). Electrocardiograms (ECGs) show prolongation of the PR interval or QRS duration in up to 80% of patients (3–5). The CCS defects typically begin in the second to fourth decade and progress slowly over time, leading to increased risk of sudden death (5,6).
Transcripts from the mutant DMPK allele are retained in nuclear foci (7,8), causing a 50% reduction of DM kinase protein. While reduced DMPK protein may contribute to cardiac symptoms, as discussed below, the evidence suggests that DM1 mainly results from a deleterious gain-of-function of the mutant RNA. The expression of RNA with expanded CUG repeats impacts nuclear regulation of gene expression through direct interaction with RNA binding proteins, such as Muscleblind-like (MBNL) 1 and 2, that have high affinity for CUG repeats (9–11). The resulting sequestration of MBNL protein affects several aspects of RNA processing, including alternative splicing, 3’ end formation, and maturation of miRNA (12–14). Expanded CUG repeats also activate signalling pathways (15), stabilize CELF1 protein (16,17), and may lead to repeat-associated non-ATG-dependent (RAN) translation (18).
Antisense oligonucleotides (ASOs) are in clinical use for post-transcriptional silencing of gene expression (19). The classical mechanism for ASO knockdown involves RNase H1, a ubiquitous enzyme that makes an endonucleolytic cleavage in the RNA strand of an ASO:RNA heteroduplex (20). Were it not for the limited biodistribution of ASOs to striated muscle, this mechanism would seem ideally suited to DM1 because [1] mutant DMPK transcripts and RNase H1 are both localized to the nucleus (7,21,8); [2] ASO-directed cleavage activity is higher in the nucleus than in the cytoplasm (22); [3] elimination of RNA with expanded CUG repeats has been shown to restore MBNL activity (23); and [4] knockdown of mutant DMPK mRNA would not impact DM kinase expression, since nuclear mRNAs are not translated (24).
This limitation, however, is not insurmountable. While ASO uptake in heart and muscle is relatively low (25,26), causing failure of target knockdown in most studies [ref. (24) and citations therein], there are several strategies to overcome this barrier. For example, in some dystrophies there are sarcolemma defects that permit greater access of ASOs to muscle fibres (27). However, this seems unlikely in DM1 where the muscle membrane is relatively intact. Alternatively, there has been significant progress in developing ASO formulations or chemical modifications that promote delivery to cardiac and skeletal muscle [reviewed in (28)]. Finally, we found that maximizing the potency of unformulated ASOs, by extensive optimization of targeting sequence and incorporation of 2'-4'-constrained ethyl nucleotides (29), can produce 50% knockdown of wild-type DMPK in heart and 46–79% knockdown in muscle, using weekly subcutaneous injections in non-human primates (30).
These findings raise a second question, addressed in the current study, about the requirement of DM kinase for normal function of cardiac and skeletal muscle. Although the precise function and physiological substrates of DMPK are unknown, this kinase is expressed more highly in cardiac, skeletal, and smooth muscle. Mice with heterozygous Dmpk gene deletion exhibited abnormal cardiac conduction (31,32), and homozygous deletion also produced skeletal myopathy and muscle weakness (33), suggesting that [1] the conduction system is sensitive to DMPK dose; [2] partial loss of DM kinase may contribute to the cardiac features of DM1; and [3] further knockdown in DM1 patients may carry risks of aggravating cardiac phenotypes, skeletal myopathy, or both. While it is possible that ASOs may preferentially target the mutant DMPK transcripts, because they are held in the nucleus where RNase H1 is localized, the extent of this allelic selectivity, if any, is presently unknown. Furthermore, studies in wild-type mice and monkeys have clearly shown that high-dose ASO regimens are capable of targeting normal DMPK transcripts in muscle and heart. Therefore, we have reassessed the physiological consequences of DMPK loss by examining cardiac and muscle function in knockout mice and mice with ASO-induced Dmpk knockdown.
Results
Transcriptional and post-transcriptional silencing of Dmpk in hearts
We used mice with heterozygous Dmpk deletion (+/-) to model DMPK reductions in human DM1. To study the influence of genetic background, we bred Dmpk knockout mice (34) onto C57Bl6 and FVB inbred strains. To study the combined effects of constitutive and post-developmental knockdown, we treated adult Dmpk +/- mice with ASO 486178, a 16-nt gapmer that targets a conserved region in the human and mouse 3′ UTR [Supplementary Material, Fig. S1A, reference (30)]. ASO 486178 was administered by weekly subcutaneous injection of 50 mg/kg for 6 weeks, then twice monthly until sacrifice.
Consistent with previous reports (33,34), immunoblot analysis showed that DMPK protein was absent in hearts of homozygous knockout (-/-) mice. DMPK protein in +/- mice was 49% of wild-type (WT) controls (Fig. 1A) , showing no dosage compensation at this locus. On the FVB background, 6 weeks of ASO administration to Dmpk +/- mice reduced Dmpk mRNA by 82 ± 1% and DMPK protein by 92 ± 2% in cardiac muscle, compared to saline-injected +/- littermates (Fig. 1A and Supplementary Material, Fig. S1B). Similarly, cardiomyocytes isolated from ASO-treated mice showed an 89 ± 2% reduction of DMPK protein (Fig. 1C). The knockdown efficiency was somewhat lower on the C57Bl6 background. DMPK protein levels in ASO-treated Dmpk +/- C57Bl6 mice were 50 ± 4% of saline-injected +/- controls (Fig. 1B).
Figure 1.

DMPK knockdown in heart. (A) Heterozygous Dmpk knockout mice (+/-) on the FVB background received subcutaneous injection of saline or ASO 486178 (ASO) at 50mg/kg/wk (mpk/wk) for 1 month. Immunoblots for DMPK protein were performed on apical heart tissue and quantified, compared to wild-type (WT) and homozygous knockout (-/-) littermates (n = 3–4 per group). GAPDH was used as loading control. (B, C) Quantification of DMPK protein in (B) apical heart tissue of C57Bl6 +/- mice given saline or ASO for 1 month at 50mpk/wk (n = 3 per group); or (C) isolated cardiac myocytes of FVB +/- mice given saline or ASO at 50mpk/wk for 1 month then 50mpk biweekly for 2 months, compared to WT controls (n = 4–5 per group). (D) As in A, except that saline or ASO was started at 2 months of age and continued up to age 11–12 months, compared to age-matched WT and -/- littermates (n = 5–6 per group). (E) As in D, except on C57Bl6 background, (n = 4–5 per group). * = ANOVA P < 0.05 and T-test p ≤ 0.0084. † = T-test P < 0.05.
Dmpk silencing was sustained during long-term ASO administration. After 8–10 months of injections, cardiac Dmpk mRNA levels were reduced by 84 ± 3% and 68 ± 2% in FVB and C57Bl6 mice, respectively (Supplementary Material, Fig. S1C and D), and protein levels were reduced by 98 ± 2% and 78 ± 4% (Fig. 1D and E), as compared to saline-injected Dmpk +/- controls. Long-term administration of ASO 486178 had no effect on survival or body weight of Dmpk +/- mice (Supplementary Material, Fig. S2).
No effect of Dmpk silencing on cardiac conduction
Previous work showed that PR intervals are prolonged in mice with heterozygous deletion of Dmpk, suggesting that Dmpk is haploinsufficient in the CCS (31,32). We examined cardiac conduction in mice with constitutive and post-developmental Dmpk loss. Signal-averaged ECGs were performed under light inhalation anaesthesia at age 6 months, and repeated at age 10 months. We compared conduction intervals in wild-type, Dmpk -/-, saline-injected Dmpk +/- and ASO-injected Dmpk +/- mice. On both FVB and C57Bl6 backgrounds, we saw no effect of Dmpk gene deletion or ASO knockdown on heart rate, PR interval, or QRS duration (Fig. 2A–F). Analysis was continued for 18 months of age, and still no effects on ECG intervals were observed (Supplementary Material, Fig. S3).
Figure 2.

No alterations of ECG intervals with Dmpk loss. (A–C) Shown are surface ECG measurements of (A) heart rate; (B) PR interval; and (C) QRS duration in 6- and 10-month old FVB +/- mice given saline or ASO, with WT and -/- littermate controls (n = 9–11 per group; 6 males and 3–5 females per group). (D–F) Similar to A–C, except on C57Bl6 background (n = 11-12 per group; 5-6 males and 6 females per group). (G) Representative plot of heart rate from an implantable telemeter recording. Grey bars denote 15-min high- and low-activity segments chosen for signal averaging and conduction interval analysis. (H) Representative ECG traces from surface (top) and implantable telemetry (bottom). Scale bars are 120 msec. (I-K) Measurements from implantable telemetry of (I) heart rate; (J) PR interval; and (K) QRS duration in 11–12-month-old FVB Dmpk mice (n = 4–6 per group). ANOVA P-value is shown above each data set.
Previous work has shown that anaesthetic agents, administered by inhalation or parenteral injection, may affect cardiac electrophysiology in mice (35,36). To test the possibility that inhalation anaesthesia was masking subtle effects of Dmpk loss on cardiac conduction, we recorded ECGs using radiotelemetry. Recording devices were implanted in 11–12-month-old FVB mice. Signal-averaged ECG recordings were obtained from conscious unrestrained mice, beginning 8 days or more after device implantation. We analyzed ECGs during periods of high and low heart rate, reflecting normal diurnal fluctuations of mouse activity (Fig. 2G–H). Once again we found no effect of Dmpk gene ablation or antisense knockdown on heart rate, PR interval, or QRS duration, as compared to wild-type littermates (Fig. 2I–K).
No effect of Dmpk silencing on left ventricular (LV) contractile function
Although cardiac effects of DM1 fall mainly on the conduction system, up to 30% of patients eventually develop echocardiographic evidence of LV dysfunction (37). Studies have shown that Dmpk knockout mice do not develop histologic abnormalities in cardiac tissue (31,32), but detailed assessments of LV function have not been reported. We used echocardiography at ages 6 and 10 months to examine contractile function in relation to Dmpk expression level. On both FVB and C57Bl6 backgrounds there were no differences of ejection fraction, myocardial strain, LV mass, or other echocardiographic metrics among wild-type, Dmpk -/-, saline-injected Dmpk +/- and ASO-injected Dmpk +/- mice (Fig. 3A and Table 1). Additionally, heart weights were not altered by Dmpk silencing (Fig. 3B) and cardiac histology did not show fibrosis (Fig. 3C–F). The only between-group difference was that the estimated cardiac output was slightly increased in Dmpk -/- and ASO-treated +/- FVB mice at age 6 months (nominal P = 0.035, ANOVA). However, there was no significant difference at 10 months of age, and there were no parallel effects on the C57BL6 background.
Figure 3.
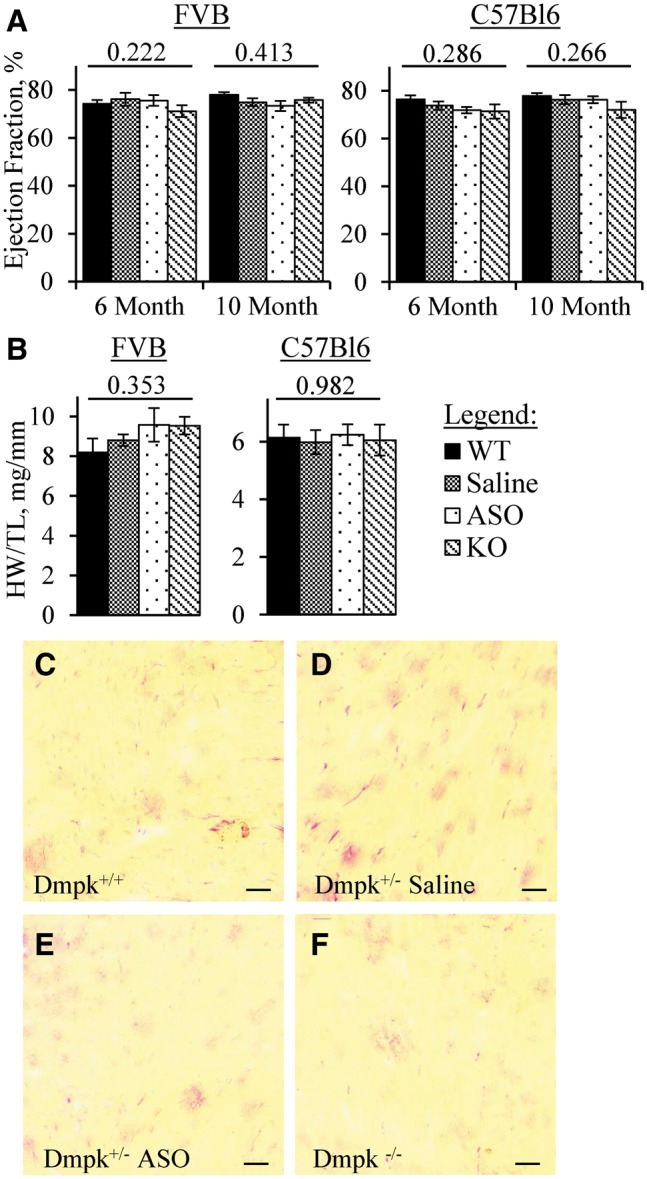
Dmpk silencing does not induce systolic dysfunction or cardiac fibrosis. (A) Ejection fraction determined by echocardiography in 6- and 10-month-old FVB Dmpk mice (n = 9–11 per group; 6 males and 3–5 females per group) and in C57Bl6 Dmpk mice (n = 11–12 per group; 5–6 males and 6 females per group). (B) Heart weight normalized to tibia length (HW/TL) in 11–12-month-old FVB Dmpk mice (n = 5-6 per group) and in 10–11-month-old C57Bl6 Dmpk mice (n = 4–5 per group). ANOVA P-Value above each data set. (C-F) Representative images of heart cryosections stained with picrosirius red, from 18-month old FVB WT, saline-injected Dmpk+/-, ASO-injected Dmpk+/-, and Dmpk -/- littermates, respectively. Low picrosirius staining in all sections indicates a lack of pathological fibrosis. Scale bar is 50 µm.
Table 1.
Echocardiographic function in Dmpk mice.
| Heart Rate (bpm) | Ejection Fraction (%) | Cardiac Output (mL/min) | LV Mass (mg) | Short Axis Strain (%) | Long Axis Strain (%) | |
|---|---|---|---|---|---|---|
| FVB/n 6 Month-Old | ||||||
| WT | 545 ±10 | 78.0 ±1.0 | 21.6 ±1.1 | 65.1 ±4.2 | 39.8 ±1.0 | 37.2 ±1.9 |
| Saline | 539 ±7 | 74.9 ±1.7 | 21.7 ±1.1 | 74.2 ±5.3 | 42.4 ±1.4 | 38.1 ±2.0 |
| ASO | 555 ±8 | 73.3 ±2.1 | 25.9 ±1.3 | 75.2 ±5.3 | 40.3 ±1.1 | 37.2 ±2.1 |
| KO | 544 ±8 | 75.7 ±1.0 | 24.8 ±1.4 | 78.4 ±4.2 | 40.3 ±1.5 | 37.7 ±1.5 |
| ANOVA P-value | 0.589 | 0.221 | 0.035 | 0.208 | 0.482 | 0.982 |
| FVB/n 10 Month-Old | ||||||
| WT | 557 ±7 | 74.2 ±1.6 | 22.9 ±2.0 | 86.2 ±5.5 | 39.3 ±1.4 | 36.6 ±1.3 |
| Saline | 563 ±10 | 76.1 ±2.7 | 21.8 ±1.4 | 83.1 ±5.9 | 41.7 ±1.8 | 37.5 ±1.1 |
| ASO | 557 ±7 | 75.6 ±2.2 | 24.0 ±1.1 | 84.1 ±5.6 | 40.2 ±1.2 | 36.4 ±1.6 |
| KO | 554 ±8 | 71.1 ±2.5 | 24.9 ±1.3 | 89.8 ±5.7 | 40.0 ±1.1 | 38.0 ±1.4 |
| ANOVA P-value | 0.878 | 0.413 | 0.431 | 0.841 | 0.699 | 0.829 |
| C57Bl6 6 Month-Old | ||||||
| WT | 565 ±8 | 76.3 ±1.7 | 19.2 ±0.9 | 51.9 ±2.5 | 42.2 ±0.8 | 40.6 ±1.1 |
| Saline | 571 ±7 | 73.8 ±1.7 | 20.4 ±1.2 | 51.2 ±3.1 | 42.5 ±1.3 | 39.6 ±1.3 |
| ASO | 570 ±11 | 71.9 ±1.3 | 19.2 ±1.2 | 48.6 ±2.4 | 41.3 ±0.9 | 37.4 ±1.3 |
| KO | 561 ±14 | 71.3 ±2.9 | 18.0 ±1.1 | 50.5 ±3.1 | 40.7 ±1.2 | 38.3 ±1.4 |
| ANOVA P-value | 0.865 | 0.286 | 0.503 | 0.917 | 0.626 | 0.179 |
| C57Bl6 10 Month-Old | ||||||
| WT | 565 ±9 | 77.8 ±1.3 | 20.4 ±1.3 | 57.6 ±3.2 | 42.6 ±0.9 | 38.5 ±1.3 |
| Saline | 584 ±9 | 76.3 ±1.9 | 20.8 ±1.4 | 56.1 ±3.5 | 43.5 ±1.5 | 38.1 ±1.3 |
| ASO | 580 ±12 | 76.2 ±1.5 | 21.9 ±1.0 | 52.8 ±2.6 | 43.7 ±1.0 | 36.5 ±1.4 |
| KO | 563 ±14 | 72.0 ±3.4 | 19.7 ±1.7 | 55.1 ±4.1 | 38.8 ±2.1 | 34.6 ±1.4 |
| ANOVA P-value | 0.455 | 0.266 | 0.699 | 0.779 | 0.068 | 0.168 |
Minor role of Dmpk in the cardiac response to pressure overload stress
Having found no cardiac requirement for DM kinase in sedentary mice, we postulated that Dmpk may have a role in cardiac compensation to overload stress. Pressure overload was imposed by placing a constricting band on the transverse aorta. This procedure often induces heart failure in mice within 8-10 weeks. The frequency of decompensated heart failure depends on strain background and severity of the aortic stenosis (38,39). We studied transverse aortic constriction (TAC) using FVB mice, a strain that previously displayed robust compensation to pressure overload (39). ASO 486178 or saline injections were initiated in Dmpk +/- and -/- mice at 2 months of age. One month later we determined baseline ejection fraction and then applied the TAC band. Age-matched WT littermates were also subjected to the TAC procedure. The post-operative mortality was low and similar across experimental groups (data not shown). The estimated trans-stenotic pressure gradient, as assessed by pulsed-wave Doppler, was around 120 mmHg, and similar across groups (Supplementary Material, Fig. S4A). Cardiac function was monitored by serial echocardiography (Fig. 4A). All experimental groups demonstrated a similar amount of left ventricular hypertrophy (Fig. 4B). At 3 weeks following TAC, the Dmpk -/- and ASO-treated +/- mice showed slight reduction of ejection fraction, as compared to WT controls (mean ejection fractions of 70% ± 1, 59% ± 2 and 62% ± 1; for WT, ASO-injected Dmpk +/- and Dmpk -/-, respectively, P = 0.0009, ANOVA). At 5-6 weeks following TAC the mean ejection fraction in ASO- and saline-treated Dmpk +/- mice was slightly reduced compared to WT controls (mean ejection fractions of 70% ± 1, 56% ± 1, 62% ± 1; for WT, ASO-injected Dmpk+/-, saline-injected Dmpk +/-, respectively, P = 0.0006, ANOVA). However, at 8 weeks following TAC banding there were no differences of ejection fraction between groups (P = 0.056, ANOVA) (Fig. 4A). Also, there was no difference of mean ejection fraction in ASO- versus saline-injected Dmpk +/- mice at any time point. Importantly, the frequency of decompensated heart failure, defined by the ratio of lung to body weight (LW/BW > 8 mg/g), was similar across groups (Fig. 4C). The mean heart and lung weights, normalized to tibia length, were also similar across groups (Supplementary Material, Fig. S4B and C).
Figure 4.

Effect of Dmpk silencing on cardiac response to pressure overload stress. FVB +/- (Het) and -/- (KO) mice were given saline or ASO at 50 mpk/wk starting at 2 months of age. Baseline measurements of (A) ejection fraction and (B) estimated LV mass were taken at age 3 months, along with age-matched WT and uninjected KO controls. Transverse aortic constriction (TAC) banding was performed after baseline measurements. Ejection fraction and LV mass were monitored at 3, 5–6, and 8 weeks following TAC banding (n = 17–19 for WT, Het+Saline, and Het+ASO at baseline to 5-6 weeks. n = 8–11 for KO, KO+Saline, and KO+ASO and for all groups at 8 weeks.) ANOVA P-value as shown above each data set. * = t-test P < 0.003, ** = t-test P < 0.00005. (C) Lung weight normalized to body weight (LW/BW mg/g) in FVB Dmpk mice 9 weeks post-TAC, with age-matched non-TAC controls. Dotted line denotes threshold for decompensated heart failure (LW/BW > 8mg/g). Fisher’s Exact Test showed no significant increase of lung weight in any group compared to WT controls.
No effect of Dmpk silencing on limb muscle strength
Previous studies of homozygous Dmpk knockout mice showed 40% reduction of force generation in sternomastoid muscle at 7-11 months of age (33). We examined muscle strength in homozygous Dmpk knockout mice and heterozygous mice under treatment with ASO 486178. Four weeks after initiating ASO injections, the level of Dmpk mRNA in quadriceps muscle of Dmpk +/- mice was decreased by 91 ± 1%, as compared to saline-injected +/- controls (Supplementary Material, Fig. S5A, FVB background). These results were consistent with our previous finding that knockdown efficiency in muscle is greater than in the heart (30). The Dmpk knockdown greater than 90% was sustained in muscle during long-term dosing on both FVB and C57Bl6 backgrounds (Supplementary Material, Fig. S5B and C). Notably, the finger flexor (grip) muscles are among the earliest and most-severely-affected muscles in human DM1. In contrast, in vivo grip strength in 8-9 month old ASO- and saline-injected Dmpk +/- mice was similar to Dmpk -/- and wild-type littermates (Fig. 5A). We also examined the ex-vivo contractile function of the extensor digitorum longus (EDL), a hindlimb muscle. At ages 10-11 months, there were no differences in peak tetanic specific force or force-frequency response in Dmpk -/-, saline-injected Dmpk +/-, ASO-injected Dmpk +/-, or wild-type mice (Fig. 5B and C, C57Bl6 background). At 18 months we found no dystrophic changes of limb muscle histology in Dmpk -/-, saline-injected Dmpk +/-, or ASO-injected Dmpk +/- mice (Fig. 5D–G).
Figure 5.
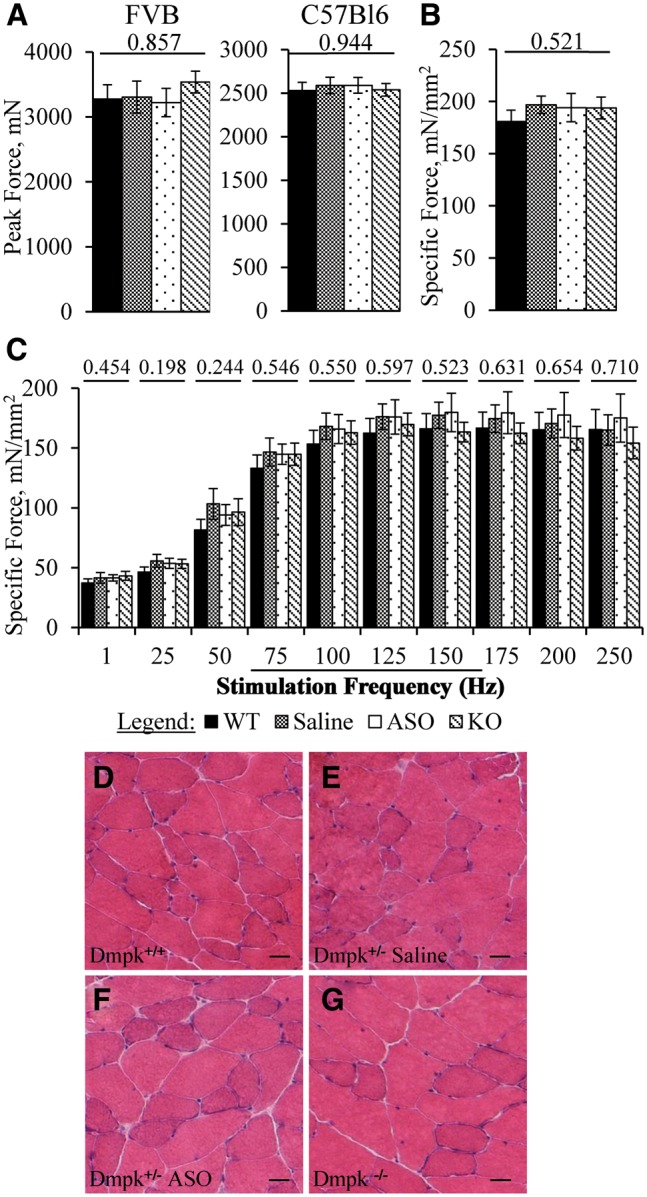
Dmpk silencing does not affect force generation or induce dystrophic changes in limb muscle. (A) Four-limb grip strength measurements of peak force from 8–9-month-old FVB Dmpk mice (n = 8–12), and 8–9-month-old C57Bl6 Dmpk mice (n = 11–12). ASO injections were initiated at age 2 months. (B) Ex vivo measurements of isometric, specific tetanic force and (C) force-frequency response of extensor digitorum longus (EDL) muscles from 10 to 11-month-old C57Bl6 Dmpk mice (n = 3–4 mice, or 5–8 muscles). ANOVA P-value above each data set. (D-G) Representative images of H&E from quadriceps muscle in 18-month old FVB WT, saline-injected Dmpk+/-, ASO-injected Dmpk+/-, and Dmpk -/- littermates, respectively. Scale bar is 25 µm.
Muscle fibres in DM1 are subjected to excessive calcium release due to involuntary runs of high frequency action potentials (myotonic discharges). As DMPK is implicated in calcium homeostasis (40) we postulated that Dmpk deficiency may predispose to muscle deterioration under stress of severe myotonia. To test this possibility we crossed Dmpk knockout mice with chloride channel 1 (Clcn1) null mice that display recessive generalized myotonia (adr mice (41), FVB background). The adr mice have severe myotonia by age 3 weeks that limits survival beyond 12 weeks (42). However, when examined at age 8-10 weeks, the hindlimb muscles did not show dystrophic pathology in Dmpk -/-•Clcn1-/- double knockout mice, as compared to littermates with single Clcn1-/- knockout (Supplementary Material, Fig. S6).
Discussion
The enzyme responsible for ASO-directed RNA cleavage, RNase H1, is predominantly nuclear (20,21). This localization may explain the observations that nuclear-retained RNAs, including CUG-expanded transcripts, seem particularly sensitive to ASO knockdown (23,43), and that rates of ASO cleavage in the nucleus are higher than in the cytoplasm (22). While these findings raise the possibility that ASOs may preferentially target mutant versus wild-type DMPK mRNA, no studies have directly compared their relative susceptibilities. Similarly, while other methods for allele-selective knockdown may exist, their utility for DM1 is uncertain. For example, in dominant disorders the most common approach is to design ASOs or siRNAs targeting sequence variants that distinguish mutant from wild-type transcripts (44,45). However, the application of this approach to DM1 would be limited because there are few exonic polymorphisms in DMPK and none have a minor allele frequency above 15%. Furthermore, targeting a polymorphism would limit the pool of candidate ASOs, thereby undermining efforts to optimize drug potency. Alternatively, viral-expressed antisense RNAs (46) or drugs that selectively inhibit transcription of expanded repeats (47) may provide other avenues for allele-selective silencing. However, these await further characterization at the level of delivery, safety and efficacy.
Since ASOs that are carefully optimized are fully capable of targeting wild-type DMPK in heart and muscle (30), we returned to the question of whether reduced translation of DMPK may contribute to symptoms of DM1, and whether additional knockdown may further compromise cardiac rhythm or muscle function. The latter is important because abnormal cardiac rhythm is an overriding concern in drug development. We expected to define a safety threshold for DM kinase reduction, and hopefully to establish useful physiological markers of DMPK insufficiency. We focused on the CCS because sudden death remains an important clinical problem in DM1 (5,6,48) and because previous work had indicated that the CCS is sensitive to changes of Dmpk expression (31,32).
Contrary to expectations, the major finding of our study was that constitutive or acquired DMPK loss had no effect on functional measures of cardiac or skeletal muscle in sedentary mice. Our key finding was that ECG recordings remained normal despite ASO administration for up to 16 months, producing sustained Dmpk reduction to levels that were nearly undetectable. The only exception was that cardiac output was slightly increased in FVB (but not C57Bl6) mice with Dmpk silencing. The mechanism for this effect is unknown, but vascular changes leading to reduced systemic resistance are possible. It is also evident that absence of DMPK was compatible with complex physiological adaptations to cardiac pressure overload. However, cardiac ejection fraction after TAC banding was slightly lower with Dmpk knockdown, suggesting that DM kinase has a minor role in the cardiac stress response.
Differences between our results and previous studies may relate to technical factors, strain background, or knockout alleles. In terms of technical factors, the previous studies were carried out before it was widely recognized that conditions for anaesthesia have major effects on mouse ECGs. The previous studies used injectable agents (ketamine and pentobarbital) and deep anesthesia (judging from reported heart rates), and procedures to maintain body temperature were not reported (31,32). Subsequently, it became clear that injectable anaesthetics and low body temperatures have major effects on ECG recordings in mice (35,36). We avoided these confounders by using light inhalation anaesthesia, maintaining body temperature and eventually by dispensing with anaesthesia altogether, using radiotelemetry recordings. We also used signal averaging to improve the precision of the ECG waveform analysis.
Alternatively, it is possible that differences of strain background or environmental conditions may have played a role. To minimize intersubject variance, we studied congenic knockout mice on two different genetic backgrounds (FVB and C57Bl6), whereas previous studies were performed on a mixed 129/C57Bl6 background.
A third possibility, which we tend to favour, is that cardiac conduction defects in knockout mice may arise through effects on flanking genes, rather than loss of DMPK itself. The conduction defects that were previously reported were observed in one founder line of Dmpk knockout mice (33). However, we had access to the other founder line (34), whose cardiac phenotype was not previously reported. Remarkably, the breakpoints for creating the Dmpk deletion were identical in both knockout lines. In both cases the targeted allele has replacement of the proximal Dmpk promoter, transcription start site and first 7 of 15 coding exons, eliminating all kinase homology domains (33,34). Genetic and expression analysis confirmed correct targeting and absence of Dmpk mRNA and protein for both founder lines (33,34). However, the selection cassettes for gene replacement were different, and remain in situ. This difference is potentially relevant because abundant evidence has shown that selection cassettes may interfere with expression of flanking genes [reviewed in (49)]. In this regard, it is noteworthy that gene density at the Dmpk locus is high, and that the 3‘end of Dmpk overlaps with regulatory elements for the flanking gene, Six5, which encodes a transcription factor expressed in cardiac cells (50). Studies have shown that heterozygous Six5 knockout mice develop adult-onset defects of the CCS (51). Although Six5 knockouts mainly exhibited changes in the infrahissian segments of the CCS, this observation nevertheless raises the possibility that cardiac conduction defects in one Dmpk knockout line may result from effects on one or more flanking genes, rather than Dmpk itself. Although beyond the scope of the current study, such a mechanism may have relevance to DM1 patients, since the expanded CTG repeat also exerts epigenetic effects on flanking genes (52–54). However, whichever explanation may apply, our study has clearly shown that DMPK is not required to maintain normal cardiac conduction on two genetic backgrounds.
Our study has several limitations. Although DMPK is highly conserved in humans and mice (86% identity), species differences in the structure and function of the CCS (55) may limit the extrapolation of our study to humans. However, it is noteworthy that ASOs produced 50% DMPK knockdown in cardiac tissue of cynomolgus monkeys, without affecting ECG intervals (30). Second, we did not perform intracardiac recordings or optical mapping of ventricular conduction. However, the rationale for such analysis was not compelling because we observed normal atrioventricular conduction by ECG, and the previous intracardiac recordings showed normal ventricular conduction in Dmpk knockout mice (56). Third, we assessed contractile function in limb muscle, leaving open that possibility that DMPK has a greater role in diaphragm, cranial, or trunk muscles.
Our study used an aggressive high-dose ASO regimen to produce an extreme target reduction within weeks (< 10% of wild-type) and then sustain it for up to 16 months. In contrast, the extent of DMPK knockdown required to obtain a clinical benefit in DM1 is unknown. In this regard, it is noteworthy that a 50% expression level of Mbnl1, the major Mbnl protein in skeletal muscle, is sufficient to sustain normal muscle function and splicing regulation in mice (13). Also, muscle function is preserved in pre-manifest DM1, despite the presence of conspicuous MBNL sequestration and corresponding changes of splicing regulation (57). Furthermore, the induction of severe myopathy in mice by Mbnl ablation required combinatorial deletion of 3 or more Mbnl alleles (i.e., Mbnl1-/- plus Mbnl2 +/-) (58). Finally, in mouse models we observed a reversal of DM1 phenotypes with partial knockdown of toxic RNA and incomplete release of Mbnl1 protein (23). Taken together, these findings suggest that our study produced greater Dmpk silencing than would be necessary to mitigate the RNA toxicity in DM1 patients, yet cardiac and muscle function remained normal. Accordingly, our results lessen concern that DMPK-targeting ASOs may produce significant cardiac or muscle toxicity in DM1 patients.
Materials and Methods
Animal Care and Antisense Dosing
Mice were housed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Dmpk knockout mice were obtained from Be Wieringa (34), and backcrossed to FVB/n and C57Bl6/n background, guided by genomic SNP markers and confirmed to be >99.7% congenic (Charles River, Troy, NY). The adr-mto2J mice, having a frame-shift deletion in exon 18 of Clcn1 (42), were obtained from Jackson Laboratories (Bar Harbor, MA) and backcrossed seven generations on to the FVB/n background.
Antisense Oligonucleotide (ASO) Treatment
ASO treatment was initiated at 2 months of age by subcutaneous injection in the interscapular region. ISIS 486178, a 16-mer gapmer targeting human and murine DMPK, was previously described (30). The injection frequency was 50mg/kg/wk for the first 6 weeks, then 50mg/kg bi-weekly.
Western Blotting
Apical heart tissue or cardiac myocytes were homogenized in buffer containing 50mM Tris-HCl, 150mM NaCl, 1% IGEPAL CA630, 0.25% sodium deoxycholate, at pH 7.4. 5mM EDTA and Halt protease and phosphatase inhibitor cocktail (Thermo-Fisher Scientific, Waltham, MA). Lysates were cleared by centrifugation at 10,000g for 20 min at 4 °C. The supernatants were diluted in loading buffer (LPS buffer, Li-cor, Lincoln, NE), heated to 95 °C for 5 min, separated on 10% polyacrylamide gels (Novex, Thermo-Fisher), and transferred onto PVDF membranes. Affinity-purified polyclonal rabbit anti-DMPK (1:200, Thermo-Fisher Scientific, Waltham, MA) and monoclonal mouse anti-GAPDH antibody (1:10,000, Thermo-Fisher Scientific) were used to probe membranes. Secondary anti-rabbit and anti-mouse IgG antibodies were used at 1:5000 for fluorescent detection (Odyssey, Li-cor, Lincoln, NE). Bands were quantified by densitometry using image J (NIH).
Cardiac Myocyte Isolation
Isolation was performed as previously described (59).
qRT-PCR
RNA was isolated from apical heart tissue or quadriceps muscle using Tri-Reagent. Measurement of Dmpk mRNA (Thermo-Fisher, Mm00446270_m1) was performed as previously described (60). GAPDH was used for normalization (Thermo-Fisher, Mm99999915_g1).
Surface Electrocardiography (ECG)
Anaesthesia was induced using 3% isoflurane and then maintained at 1.5-2%, delivered by nosecone. Body temperature was maintained using a heated water pad. Subcutaneous leads (Powerlab, AD Instruments, Sydney, Australia) were inserted in a lead II configuration. ECGs were recorded for approximately 5 min. Several minutes of stable recording were analyzed offline using ECG analysis software, as previously described (61). In brief, the software identifies all R peaks as a reference point to align each sinus cycle. RR, PR and QRS intervals were measured in the averaged waveform.
Implantable Electrocardiography
ETA-F10 telemeters (Data Science International (DSI), St. Paul, MN) were implanted as previously described (62). Leads were sutured to the body wall in a lead II configuration. Mice were allowed to recover for at least 8 days. ECGs were recorded in a quiet, isolated room over a 24-hour period using Ponemah software (DSI). Heart rates were then examined to assess circadian cycling. About 15-min recordings from intervals with sustained high or low heart rates were analyzed by signal averaging, and conduction intervals were determined.
Surface Echocardiography
Mice were examined under isoflurane anaesthesia as described above. Following hair removal, 2D-guided M-mode echocardiography was performed at the mid-papillary level (Visualsonics Vevo 2100) to measure left ventricular posterior and anterior wall thickness and internal diameter during systole and diastole. These values were then used to calculate left ventricular volume, ejection fraction, cardiac output and mass. Additionally, 2D echoes were obtained and speckle tracking analysis (Vevostrain) was performed to measure myocardial strain.
Grip Strength
Grip strength was determined as described previously (63). In brief, mice were pulled at a constant velocity by the tail across a metal wire grate attached to a force transducer (Dillon GTX). Peak force was recorded from three trials, and the highest peak force was chosen from each mouse for analysis.
Ex vivo Muscle Contraction
Ex vivo muscle strength was measured using a horizontal muscle contraction system (Aurora Scientific, Inc. Aurora Scientific, ON, Canada, Model 809B) equipped with a 300C-LR dual mode force transducer and a 701C stimulator, as described previously (64). In brief, extensor digitorum longus (EDL) muscles were isolated from anaesthetized mice (100mg/kg ketamine, 10mg/kg xylazine and 2mg/kg acepromazine), and suture was tied at both the proximal and distal tendons to mount the muscle between two platinum electrodes in a bath of oxygenated Tyrode solution. Muscle optimal length (Lo) and stimulus intensity were determined using a series of 1 Hz stimulations. Muscles were equilibrated with three 500 ms, 150 Hz tetanic stimulations separated by 1-min intervals. The final stimulation was used to determine tetanic specific force. The force-frequency protocol was a single twitch or 500 ms stimulation at the indicated frequencies. Muscle force was recorded using Dynamic Muscle Control software (Aurora Scientific, Inc. v5.415) and analyzed using Dynamic Muscle Analysis software (Aurora Scientific, Inc. v5.200). Muscle cross-sectional area and specific force were calculated as described previously (64).
Transverse Aortic Constriction (TAC) Banding
TAC banding was performed as previously described (65). Pulsed-wave Doppler measurements were taken 3 weeks post-TAC to determine the peak velocity of flow at the aortic stenosis, using the modified Bernoulli equation to calculate stenosis pressure gradient (38). Serial echocardiographic measurements were performed at 3, 5–6 and 8 weeks post-procedure.
Histological Analysis
Cryosections of quadriceps, gastrocnemius, and heart were cut at 8µm. Haematoxylin and eosin on skeletal muscle sections, and picrosirius red stain on heart sections, were performed as previously described (66,67).
Study Approval
All mouse procedures and treatments were reviewed and approved by the institutional Animal Resource committee at the University of Rochester.
Statistical Analysis
Data are expressed as mean ± standard error of the mean. Statistical significance was determined by one-way analysis of variance (ANOVA). Multiple comparisons were performed using the Bonferroni-corrected t-test. A Log-rank test was performed for survival analysis in Supplementary Material, Fig. S2. Fisher’s Exact test was used to compare groups in Fig. 4C.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
The authors would like to acknowledge Deanne Mickelsen, Sarah Leistman and Linda Richardson (University of Rochester) for technical support.
Conflict of Interest statement. SKP and FCB are employed by Ionis Pharmaceuticals. CAT has received sponsored research support and consultation fees from Ionis Pharmaceuticals.
Funding
This work was supported by the National Institutes of Health [U54NS048843, U01NS072323]; a Howard Hughes Medical Institute ‘Med-into-Grad’ Fellowship to STC, and the Run America Foundation.
References
- 1.Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T., et al. (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell, 68, 799–808. [DOI] [PubMed] [Google Scholar]
- 2.Thornton C.A. (2014) Myotonic dystrophy. Neurol. Clin., 32, 705–719. viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips M.F., Harper P.S. (1997) Cardiac disease in myotonic dystrophy. Cardiovasc. Res., 33, 13–22. [DOI] [PubMed] [Google Scholar]
- 4.Groh W.J., Lowe M.R., Zipes D.P. (2002) Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J. Cardiovasc. Electrophysiol., 13, 444–448. [DOI] [PubMed] [Google Scholar]
- 5.Wahbi K., Meune C., Porcher R., Bécane H.M., Lazarus A., Laforêt P., Stojkovic T., Béhin A., Radvanyi-Hoffmann H., Eymard B., et al. (2012) Electrophysiological study with prophylactic pacing and survival in adults with myotonic dystrophy and conduction system disease. JAMA, 307, 1292–1301. [DOI] [PubMed] [Google Scholar]
- 6.Groh W.J., Groh M.R., Saha C., Kincaid J.C., Simmons Z., Ciafaloni E., Pourmand R., Otten R.F., Bhakta D., Nair G.V., et al. (2008) Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N. Engl. J. Med., 358, 2688–2697. [DOI] [PubMed] [Google Scholar]
- 7.Taneja K.L., McCurrach M., Schalling M., Housman D., Singer R.H. (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol., 128, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis B.M., McCurrach M.E., Taneja K.L., Singer R.H., Housman D.E. (1997) Expansion of a CUG trinucleotide repeat in the 3' untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. U S A, 94, 7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J., 19, 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mankodi A., Lin X., Blaxall B.C., Swanson M.S., Thornton C.A. (2005) Nuclear RNA foci in the heart in myotonic dystrophy. Circ. Res., 97, 1152–1155. [DOI] [PubMed] [Google Scholar]
- 11.Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S., Thornton C.A. (2006) Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet., 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- 12.Ho T.H., Charlet-B N., Poulos M.G., Singh G., Swanson M.S., Cooper T.A. (2004) Muscleblind proteins regulate alternative splicing. Embo J., 23, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. (2003) A muscleblind knockout model for myotonic dystrophy. Science, 302, 1978–1980. [DOI] [PubMed] [Google Scholar]
- 14.Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S., et al. (2012) Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins. Cell, 150, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batra R., Charizanis K., Manchanda M., Mohan A., Li M., Finn D.J., Goodwin M., Zhang C., Sobczak K., Thornton C.A., et al. (2014) Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell, 56, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timchenko L.T., Miller J.W., Timchenko N.A., DeVore D.R., Datar K.V., Lin L., Roberts R., Caskey C.T., Swanson M.S. (1996) Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res., 24, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philips A.V., Timchenko L.T., Cooper T.A. (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science, 280, 737–741. [DOI] [PubMed] [Google Scholar]
- 18.Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A., et al. (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U S A, 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raal F.J., Santos R.D., Blom D.J., Marais A.D., Charng M.J., Cromwell W.C., Lachmann R.H., Gaudet D., Tan J.L., Chasan-Taber S., et al. (2010) Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet, 375, 998–1006. [DOI] [PubMed] [Google Scholar]
- 20.Wu H., Lima W.F., Zhang H., Fan A., Sun H., Crooke S.T. (2004) Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem., 279, 17181–17189. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki Y., Holmes J.B., Cerritelli S.M., Sakhuja K., Minczuk M., Holt I.J., Crouch R.J. (2010) An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol. Cell. Biol., 30, 5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vickers T.A., Crooke S.T. (2015) The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res., 43, 8955–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F., Thornton C.A. (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature, 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halstead J.M., Lionnet T., Wilbertz J.H., Wippich F., Ephrussi A., Singer R.H., Chao J.A. (2015) Translation. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science, 347, 1367–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett C.F., Swayze E.E. (2010) RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol., 50, 259–293. [DOI] [PubMed] [Google Scholar]
- 26.Bennett C.F. (2008) ST, C. (ed.), In Antisense Drug Technology: Principles, Strategies and Applications. CRC Press, Boca Raton, pp. 273–304. [Google Scholar]
- 27.Heemskerk H., de Winter C., van Kuik P., Heuvelmans N., Sabatelli P., Rimessi P., Braghetta P., van Ommen G.J., de Kimpe S., Ferlini A., et al. (2010) Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther., 18, 1210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Järver P., Coursindel T., Andaloussi S.E., Godfrey C., Wood M.J., Gait M.J. (2012) Peptide-mediated Cell and In Vivo Delivery of Antisense Oligonucleotides and siRNA. Mol. Ther. Nucleic Acids, 1, e27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seth P.P., Siwkowski A., Allerson C.R., Vasquez G., Lee S., Prakash T.P., Wancewicz E.V., Witchell D., Swayze E.E. (2009) Short antisense oligonucleotides with novel 2'-4' conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J. Med. Chem., 52, 10–13. [DOI] [PubMed] [Google Scholar]
- 30.Pandey S.K., Wheeler T.M., Justice S.L., Kim A., Younis H., Gattis D., Jauvin D., Puymirat J., Swayze E.E., Freier S.M., et al. (2015) Identification and Characterization of Modified Antisense Oligonucleotides Targeting DMPK in Mice and Nonhuman Primates for the Treatment of Myotonic Dystrophy Type 1. J. Pharmacol. Exp. Ther., 355,329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berul C.I., Maguire C.T., Aronovitz M.J., Greenwood J., Miller C., Gehrmann J., Housman D., Mendelsohn M.E., Reddy S. (1999) DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Invest., 103, R1–R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berul C.I., Maguire C.T., Gehrmann J., Reddy S. (2000) Progressive atrioventricular conduction block in a mouse myotonic dystrophy model. J. Interv. Card. Electrophysiol., 4, 351–358. [DOI] [PubMed] [Google Scholar]
- 33.Reddy S., Smith D.B., Rich M.M., Leferovich J.M., Reilly P., Davis B.M., Tran K., Rayburn H., Bronson R., Cros D., et al. (1996) Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet., 13, 325–335. [DOI] [PubMed] [Google Scholar]
- 34.Jansen G., Groenen P.J., Bächner D., Jap P.H., Coerwinkel M., Oerlemans F., van den Broek W., Gohlsch B., Pette D., Plomp J.J., et al. (1996) Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet., 13, 316–324. [DOI] [PubMed] [Google Scholar]
- 35.Appleton G.O., Li Y., Taffet G.E., Hartley C.J., Michael L.H., Entman M.L., Roberts R., Khoury D.S. (2004) Determinants of cardiac electrophysiological properties in mice. J. Interv. Card. Electrophysiol., 11, 5–14. [DOI] [PubMed] [Google Scholar]
- 36.Chaves A.A., Dech S.J., Nakayama T., Hamlin R.L., Bauer J.A., Carnes C.A. (2003) Age and anesthetic effects on murine electrocardiography. Life Sci., 72, 2401–2412. [DOI] [PubMed] [Google Scholar]
- 37.Bhakta D., Groh M.R., Shen C., Pascuzzi R.M., Groh W.J. (2010) Increased mortality with left ventricular systolic dysfunction and heart failure in adults with myotonic dystrophy type 1. Am. Heart J., 160, 1137–1141. [DOI] [PubMed] [Google Scholar]
- 38.Mohammed S.F., Storlie J.R., Oehler E.A., Bowen L.A., Korinek J., Lam C.S., Simari R.D., Burnett J.C., Redfield M.M. (2012) Variable phenotype in murine transverse aortic constriction. Cardiovasc. Pathol., 21, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao S., Ho D., Vatner D.E., Vatner S.F. (2011) Echocardiography in Mice. Curr. Protoc. Mouse Biol., 1, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benders A.A., Groenen P.J., Oerlemans F.T., Veerkamp J.H., Wieringa B. (1997) Myotonic dystrophy protein kinase is involved in the modulation of the Ca2+ homeostasis in skeletal muscle cells. J. Clin. Invest., 100, 1440–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinmeyer K., Klocke R., Ortland C., Gronemeier M., Jockusch H., Gründer S., Jentsch T.J. (1991) Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature, 354, 304–308. [DOI] [PubMed] [Google Scholar]
- 42.Lueck J.D., Lungu C., Mankodi A., Osborne R.J., Welle S.L., Dirksen R.T., Thornton C.A. (2007) Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am. J. Physiol. Cell Physiol., 292, C1291–C1297., [DOI] [PubMed] [Google Scholar]
- 43.Lennox K.A., Behlke M.A. (2016) Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res., 44, 863–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller V.M., Xia H., Marrs G.L., Gouvion C.M., Lee G., Davidson B.L., Paulson H.L. (2003) Allele-specific silencing of dominant disease genes. Proc. Natl. Acad. Sci. U S A, 100, 7195–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carroll J.B., Warby S.C., Southwell A.L., Doty C.N., Greenlee S., Skotte N., Hung G., Bennett C.F., Freier S.M., Hayden M.R. (2011) Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther., 19, 2178–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.François V., Klein A.F., Beley C., Jollet A., Lemercier C., Garcia L., Furling D. (2011) Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat. Struct. Mol. Biol., 18, 85–87. [DOI] [PubMed] [Google Scholar]
- 47.Coonrod L.A., Nakamori M., Wang W., Carrell S., Hilton C.L., Bodner M.J., Siboni R.B., Docter A.G., Haley M.M., Thornton C.A., et al. (2013) Reducing levels of toxic RNA with small molecules. ACS Chem. Biol., 8, 2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Die-Smulders C.E., Howeler C.J., Thijs C., Mirandolle J.F., Anten H.B., Smeets H.J., Chandler K.E., Geraedts J.P. (1998) Age and causes of death in adult-onset myotonic dystrophy. Brain, 121, 1557–1563. [DOI] [PubMed] [Google Scholar]
- 49.Müller U. (1999) Ten years of gene targeting: targeted mouse mutants, from vector design to phenotype analysis. Mech. Dev., 82, 3–21. [DOI] [PubMed] [Google Scholar]
- 50.Heath S.K., Carne S., Hoyle C., Johnson K.J., Wells D.J. (1997) Characterisation of expression of mDMAHP, a homeodomain-encoding gene at the murine DM locus. Hum. Mol. Genet., 6, 651–657. [DOI] [PubMed] [Google Scholar]
- 51.Wakimoto H., Maguire C.T., Sherwood M.C., Vargas M.M., Sarkar P.S., Han J., Reddy S., Berul C.I. (2002) Characterization of cardiac conduction system abnormalities in mice with targeted disruption of Six5 gene. J. Interv. Card. Electrophysiol., 7, 127–135. [DOI] [PubMed] [Google Scholar]
- 52.Thornton C.A., Wymer J.P., Simmons Z., McClain C., Moxley R.T. (1997) Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet., 16, 407–409. [DOI] [PubMed] [Google Scholar]
- 53.Klesert T.R., Otten A.D., Bird T.D., Tapscott S.J. (1997) Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat. Genet., 16, 402–406. [DOI] [PubMed] [Google Scholar]
- 54.López Castel A., Nakamori M., Tomé S., Chitayat D., Gourdon G., Thornton C.A., Pearson C.E. (2011) Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet., 20, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nikolaidou T., Aslanidi O.V., Zhang H., Efimov I.R. (2012) Structure-function relationship in the sinus and atrioventricular nodes. Pediatr. Cardiol., 33, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saba S., Vanderbrink B.A., Luciano B., Aronovitz M.J., Berul C.I., Reddy S., Housman D., Mendelsohn M.E., Estes N.A., Wang P.J. (1999) Localization of the sites of conduction abnormalities in a mouse model of myotonic dystrophy. J. Cardiovasc. Electrophysiol., 10, 1214–1220. [DOI] [PubMed] [Google Scholar]
- 57.Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T., et al. (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol., 74, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee K.Y., Li M., Manchanda M., Batra R., Charizanis K., Mohan A., Warren S.A., Chamberlain C.M., Finn D., Hong H., et al. (2013) Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med., 5, 1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roth G.M., Bader D.M., Pfaltzgraff E.R. (2014) Isolation and physiological analysis of mouse cardiomyocytes. J. Vis. Exp., e51109.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heid C.A., Stevens J., Livak K.J., Williams P.M. (1996) Real time quantitative PCR. Genome Res., 6, 986–994. [DOI] [PubMed] [Google Scholar]
- 61.Auerbach D.S., Jones J., Clawson B.C., Offord J., Lenk G.M., Ogiwara I., Yamakawa K., Meisler M.H., Parent J.M., Isom L.L. (2013) Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One, 8, e77843.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cesarovic N., Jirkof P., Rettich A., Arras M. (2011) Implantation of radiotelemetry transmitters yielding data on ECG, heart rate, core body temperature and activity in free-moving laboratory mice. J. Vis. Exp., 57, pii: 3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meyer O.A., Tilson H.A., Byrd W.C., Riley M.T. (1979) A method for the routine assessment of fore- and hindlimb grip strength of rats and mice. Neurobehav. Toxicol., 1, 233–236. [PubMed] [Google Scholar]
- 64.Hakim C.H., Li D., Duan D. (2011) Monitoring murine skeletal muscle function for muscle gene therapy. Methods in Mol. Biol., 709, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.deAlmeida A.C., van Oort R.J., Wehrens X.H. (2010) Transverse aortic constriction in mice. J. Vis. Exp., 38, pii: 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Junqueira L.C., Bignolas G., Brentani R.R. (1979) Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J., 11, 447–455. [DOI] [PubMed] [Google Scholar]
- 67.Wissowzky A. (1876) Ueber das Eosin als Reagenz auf Hamoglobin und die Bildung von Blutgetassen und Blutkorporchen bie Saugetier und Huhnerembryonen. Archiv Fur Mikroskopische Anatomie, 479–496. [Google Scholar]