

Abstract
The electrocardiographic QRS duration, a measure of ventricular depolarization and conduction, is associated with cardiovascular mortality. While single nucleotide polymorphisms (SNPs) associated with QRS duration have been identified at 22 loci in populations of European descent, the genetic architecture of QRS duration in non-European populations is largely unknown. We therefore performed a genome-wide association study (GWAS) meta-analysis of QRS duration in 13,031 African Americans from ten cohorts and a transethnic GWAS meta-analysis with additional results from populations of European descent. In the African American GWAS, a single genome-wide significant SNP association was identified (rs3922844, P = 4 × 10−14) in intron 16 of SCN5A, a voltage-gated cardiac sodium channel gene. The QRS-prolonging rs3922844 C allele was also associated with decreased SCN5A RNA expression in human atrial tissue (P = 1.1 × 10−4). High density genotyping revealed that the SCN5A association region in African Americans was confined to intron 16. Transethnic GWAS meta-analysis identified novel SNP associations on chromosome 18 in MYL12A (rs1662342, P = 4.9 × 10−8) and chromosome 1 near CD1E and SPTA1 (rs7547997, P = 7.9 × 10−9). The 22 QRS loci previously identified in populations of European descent were enriched for significant SNP associations with QRS duration in African Americans (P = 9.9 × 10−7), and index SNP associations in or near SCN5A, SCN10A, CDKN1A, NFIA, HAND1, TBX5 and SETBP1 replicated in African Americans. In summary, rs3922844 was associated with QRS duration and SCN5A expression, two novel QRS loci were identified using transethnic meta-analysis, and a significant proportion of QRS–SNP associations discovered in populations of European descent were transferable to African Americans when adequate power was achieved.
Introduction
The QRS duration, measured by the surface electrocardiogram (ECG), reflects depolarization and conduction of the electrical signal throughout the ventricular myocardium. Longer QRS duration is associated with an increased risk of cardiovascular (CV) mortality in Caucasians and all-cause mortality in African Americans from study populations unselected for specific CV disorders (1,2). Among patients hospitalized for heart failure with reduced left ventricular ejection fraction, prolonged QRS duration (≥ 120 milliseconds) is associated with post-discharge all-cause mortality (3). The QRS duration has been reported to be shorter in African Americans compared to Caucasians, even after taking into account cardiovascular risk factors and coronary heart disease (4–6).
Prior genome-wide association (GWA) studies of QRS duration performed in populations of European descent have identified 22 independent QRS loci (7,8). The genetic architecture of QRS duration among African Americans, by contrast, is largely unknown. Expanding GWA studies to populations of African descent holds the potential to refine association regions due to shorter-range linkage disequilibrium (LD) and to discover novel genetic associations given the presence of population-specific allele frequencies (9,10). We therefore performed a GWAS meta-analysis of QRS duration in a total of 13,031 African Americans from 10 cohort studies in order to: (1) fine-map the QRS association regions previously identified among those of European descent; (2) discover novel QRS loci through a meta-analysis of results from African American cohorts as well as a transethnic meta-analysis; (3) determine whether SNP associations with QRS duration discovered in populations of European descent are transferable to African Americans.
Results
GWAS meta-analysis of QRS duration in African Americans
We conducted a meta-analysis of 2,955,816 autosomal SNPs in 13,031 African Americans from 10 GWA studies of QRS duration (Supplementary Materials, Tables S1 and S2), with little evidence of genomic inflation in the individual cohorts (Supplementary Materials, Figs S1–S10 and Table S1) or the meta-analysis (Supplementary Material, Fig. S11, λ = 1.027). The percentage of European genetic ancestry was similar across the cohorts (Supplementary Material, Table S2) and was not significantly associated with QRS duration (Supplementary Material, Table S3).
A single SNP, rs3922844 (MAF = 0.42), in intron 16 of the cardiac sodium channel gene SCN5A, was associated with QRS duration among African Americans at the genome-wide significance threshold (β ± SE = 0.94 ms ± 0.12 ms, P = 4×10−14) (Table 1, Fig. 1 and Supplementary Material, Fig. S12). Adjustment for local ancestry minimally altered the association (Table 1). We fine-mapped the SCN5A-SCN10A region surrounding this signal by examining SNPs genotyped using the MetaboChip array in ARIC and WHI PAGE participants and imputed in the WHI SHARE participants (11). In the meta-analysis of the three cohorts, a second SNP in intron 16 of SCN5A, rs12635898, was in high LD with rs3922844 (HapMap YRI: r2 = 0.93, Table 2) and was similarly associated with QRS duration (Table 2). Furthermore, LD was low (1000 Genomes AFR population r2 = 0.03) between rs3922844 and rs7626962, an SCN5A missense mutation (S1103Y) associated with cardiac conduction and arrhythmias that is common in African Americans but rare in populations of European descent (12–15).
Table 1.
Association results for rs3922844 with and without adjustment for local ancestry estimates
| Assay | Adjusted for global ancestryd |
Adjusted for local ancestryd,e |
|||||
|---|---|---|---|---|---|---|---|
| Study | (G/I)a | CAFb | n | β ± SEGC | PGC | β ± SEGC | PGC |
| WHI | I | 0.42 | 4012 | 0.86 ± 0.21 | 6x10−5 | 0.90 ± 0.23 | 3x10−5 |
| ARIC | I | 0.41 | 2372 | 0.61 ± 0.28 | 0.03 | 0.68 ± 0.29 | 0.02 |
| JHS | I | 0.40 | 1918 | 1.41 ± 0.32 | 1x10−5 | 1.40 ± 0.33 | 2x10−5 |
| MESA | I | 0.43 | 1554 | 1.02 ± 0.36 | 0.005 | 1.17 ± 0.38 | 0.002 |
| Health ABC | G | 0.43 | 993 | 0.98 ± 0.47 | 0.04 | 0.99 ± 0.50 | 0.05 |
| HANDLS | G | 0.41 | 945 | 0.96 ± 0.43 | 0.03 | 0.85 ± 0.45 | 0.06 |
| CHS | G | 0.44 | 621 | 0.84 ± 0.56 | 0.13 | ND | ND |
| CFS | I | 0.39 | 315 | 0.16 ± 0.80 | 0.84 | 0.21 ± 0.82 | 0.80 |
| BLSA | G | 0.44 | 153 | 3.27 ± 0.99 | 9x10−4 | ND | ND |
| BHS | G | 0.42 | 147 | −1.14 ± 1.27 | 0.37 | ND | ND |
| Meta-analysis | |||||||
| All studies | 0.42 | 12,877 | 0.94 ± 0.12 | 4x10−14 | – | – | |
| LA studiesc | 0.42 | 12,109 | 0.91 ± 0.13 | 3x10−13 | 0.95 ± 0.13 | 7x10−13 | |
aG = directly genotyped SNP, I = imputed SNP.
bCoded allele frequency. C and T are the coded and non-coded alleles for rs3922844, respectively. CAF for the meta-analysis is the weighted average across the 10 cohorts.
cStudies with local ancestry (LA) estimates: WHI, ARIC, JHS, MESA, Health ABC, HANDLS, and CFS.
dβ and SE expressed in units of milliseconds.
eND = not determined.
Figure 1.

Regional association plot at SCN5A/SCN10A locus. African American SNP association meta-analysis results are plotted in the top panel, and meta-analysis results from cohorts of European ancestry are plotted in the bottom panel. The AA index SNP (rs3922844) is designated by a red diamond in both panels. The LD (r2) shown is relative to the AA index SNP and is based on HapMap YRI in the top panel and HapMap CEU in the bottom panel. Gray circles are SNPs without HapMap LD data. The X-axis marks the chromosomal position. Recombination rates estimated from African Americans and HapMap CEU individuals are shown in the top and bottom panels, respectively. The dashed horizontal lines in the top and bottom panels mark the GWAS significance level in African Americans (2.5 × 10 − 8) and populations of European descent (5.0 × 10 − 8), respectively.
Table 2.
Association results from dense SNP genotyping study at the SCN5A locus
| Coded/Reference allele (CAF) | ARIC |
WHI PAGE |
WHI SHARE |
Meta-analysis | rs3922844 LD (r2)c | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Positiona | n | β ± SEb (P) | n | β ± SEb (P) | n | β ± SEb (P) | β ± SEb (P) | ||
| rs12635898 | 38,601,069 | A/C (0.30) | 2911 | 1.03 ± 0.34 (0.002) | 725 | 1.18 ± 0.52 (0.02) | 3283 | 1.04 ± 0.24 (2x10−5) | 1.05 ± 0.19 (1x10−8) | 0.93 |
aPosition based on NCBI reference sequence build 36.
bβ and SE expressed in units of milliseconds.
cLD estimates between rs12635898 and rs3922844 based on rs12635898 genotype data from unrelated YRI samples on MetaboChip arrays used in the PAGE study and rs3922844 genotype data from unrelated YRI samples downloaded from HapMap phase 2 data.
While the most significant SNP associations reported from previous GWA studies of QRS interval among other ethnic groups are similarly located on chromosome 3 at the SCN5A/SCN10A locus, the SNP association region is broad, spanning approximately 300 kb, and multiple independent signals have been identified (Fig. 1) (7,8,16). By contrast, the genome-wide association among African Americans points to a single SNP (Fig. 1). The rs3922844-QRS association discovered in African American cohorts replicated in cohorts of European ancestry (P = 2×10−13) (Table 3), but this variant was not in high LD with other SCN5A or SCN10A index SNPs among European ancestry individuals (Table 3).
Table 3. SNP association transferability at the SCN5A/SCN10A locus
| European ancestry |
African ancestry |
rs3922844 LD (r2)d |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Index SNP/ annotation | Nearest gene | Chr (Position)h | Discovery population (Ref) | CA (Freq)a | β±SEGC (PGC)e | I2 f | Powerb | CA (Freq)a | β±SEGC (PGC)g | I2 f | Powerc | CEU | YRI | ASW |
| rs6801957/ | SCN10A | 3 | EA | T | 0.77 ± 0.07 | 45.3 | – | T | 0.54 ± 0.16 | 0.0 | 0.95 | 0.00 | 0.01 | ND |
| intron | (38,742,319) | (8) | (0.41) | (1x10−28) | (0.17) | (8x10−4) | ||||||||
| rs6795970/ | SCN10A | 3 | EA/Asian | A | 0.75 ± 0.07 | 44.8 | – | A | 0.44 ± 0.20 | 0.0 | 0.73 | 0.01 | 0.01 | 0.03 |
| missense | (38,741,679) | (7,16) | (0.40) | (5x10−27) | (0.10) | (0.03) | ||||||||
| rs9851724/ | SCN10A | 3 | EA | C | −0.66 ± 0.07 | 57.1 | – | C | −0.30 ± 0.16 | 7.2 | 0.82 | 0.00 | 0.01 | 0.01 |
| intergenic | (38,694,939) | (8) | (0.33) | (2x10−20) | (0.16) | (0.06) | ||||||||
| rs11710077/ | SCN5A | 3 | EA | T | −0.84 ± 0.09 | 23.8 | – | T | −0.87 ± 0.19 | 0.0 | 0.90 | 0.15 | 0.04 | 0.09 |
| intron | (38,632,903) | (8) | (0.21) | (6x10−22) | (0.11) | (7x10−6) | ||||||||
| rs11708996/ | SCN5A | 3 | EA | C | 0.79 ± 0.10 | 0.0 | – | C | 0.86 ± 0.80 | 0.0 | 0.29 | 0.10 | ND | ND |
| intron | (38,608,927) | (8) | (0.16) | (1x10−16) | (0.04) | (0.28) | ||||||||
| rs10865879/ | SCN5A | 3 | EA | C | 0.78 ± 0.08 | 53.6 | – | C | 0.46 ± 0.15 | 0.0 | 0.97 | 0.00 | 0.01 | ND |
| intergenic | (38,552,366) | (8) | (0.26) | (3x10−23) | (0.18) | (3x10−3) | ||||||||
| rs2051211/ | EXOG | 3 | EA | G | −0.44 ± 0.08 | 0.0 | – | G | −0.41 ± 0.15 | 0.0 | 0.38 | 0.20 | 0.00 | 0.05 |
| intron | (38,534,753) | (8) | (0.26) | (2x10−8) | (0.18) | (7x10−3) | ||||||||
| rs3922844/ | SCN5A | 3 | AA | C | 0.56 ± 0.08 | 23.7 | 0.99 | C | 0.94 ± 0.12 | 10.6 | – | – | – | – |
| intron | (38,599,257) | (0.69) | (2x10−13) | (0.42) | (4x10−14) | |||||||||
aCA = coded effect allele. Freq = frequency.
bPower to detect effect size reported in COGENT/CARe results using the coded effect allele frequency and trait variance as observed in European samples and keeping α = 0.002 adjusting for 22 SNPs.
cPower to detect effect size reported in European discovery population using the effect allele frequency and trait variance as observed in African-American samples and keeping α = 0.002 adjusting for 30 SNPs.
dLD between index SNP and rs3922844. ND indicates the SNP was not genotyped in the HapMap population or was monomorphic.
eIn non-African ancestry discovery rows, significance was based on the genome-wide discovery threshold and P-values ≤ 5.0x10 − 8 were bolded. In the African ancestry discovery row, significance was based on the replication threshold and P-values ≤ 0.002 were bolded. β and SE expressed in units of milliseconds.
fHeterogeneity I2 statistics bolded if the Cochran’s χ2 P-value ≤ 0.05.
gIn non-African ancestry discovery rows, significance was based on replication of the 30 previously identified SNPs and P-values ≤ 0.002 were bolded. In the African ancestry discovery row, significance was based on genome-wide discovery and P-values ≤ 2.5x10 − 8 were bolded. β and SE expressed in units of milliseconds.
hPosition based on NCBI reference sequence build 36.
SNP functional annotation and association with SCN5A expression in human cardiac tissue
Functional annotation indicated that rs3922844 and variants in LD (1000 Genomes AFR population r2 ≥ 0.2) overlapped and were near regulatory genomic features. These variants altered transcription factor binding motifs and overlapped with DNAse I hypersensitive sites (DHS) (Supplementary Material, Table S4). In multiple cell types, rs3922844 was located near a cluster of transcription factor binding events and a peak of Histone H3 Lysine 4 mono-methylation, a marker of putative enhancers (Supplementary Material, Fig. S13). Rs3922844 and variants in LD overlapped with DHS and enhancer histone marks in fetal heart tissue based on data from the Roadmap Epigenomics Mapping Consortium (Supplementary Materials, Table S4 and Fig. S14).
We next assessed the functional relevance of rs3922844 by examining its association with SCN5A RNA expression levels in human cardiac tissue. The rs3922844 C allele associated with longer QRS duration was also associated with lower SCN5A RNA expression in left atrial appendage samples from 289 individuals of European ancestry (β ± SE (units are RNA levels on log2 scale) = -0.11 ± 0.03, P = 1.6×10−3) and 40 individuals of African ancestry (β ± SE = -0.29 ± 0.11, P = 0.013), as well as in the meta-analysis across the two ethnic groups (β ± SE = -0.13 ± 0.03, P = 1.1×10−4, Fig. 2).
Figure 2.
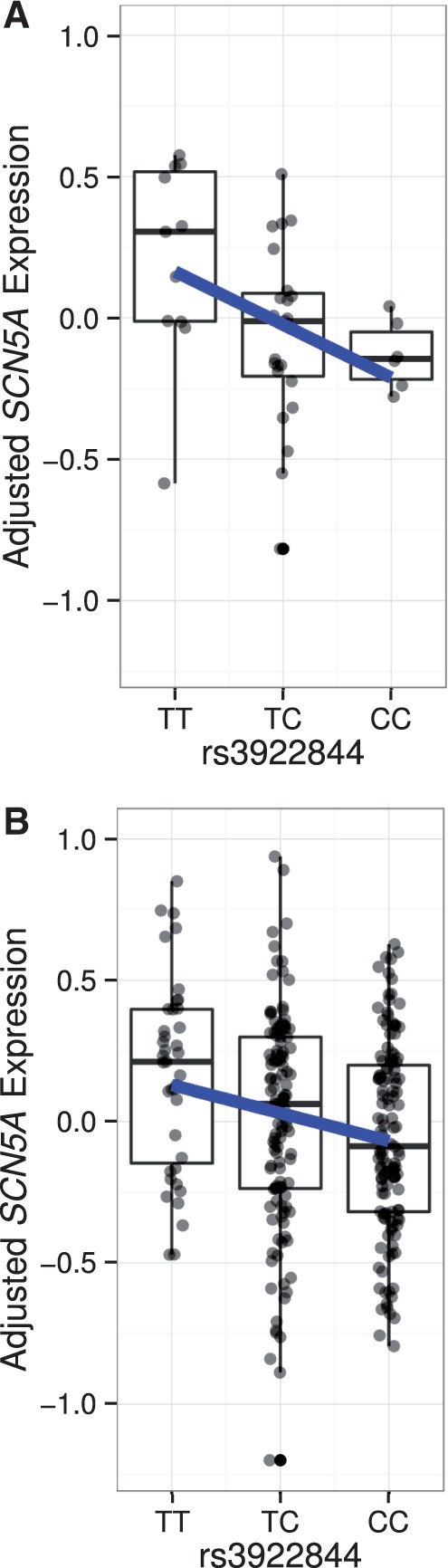
Association between rs3922844 genotype and SCN5A expression in human atrial tissue. Box plots display data from individuals of African ancestry (A) and European ancestry (B). Plotted along the Y-axis are RNA levels adjusted for covariates. The bottom and top of each box indicates the 25th and 75th percentiles, and the band within the box is the median. Whiskers extend to the most extreme value or the most extreme value within 1.5*interquartile range (IQR), whichever value is closer to the median. The fitted regression line is shown in blue.
Transethnic meta-analysis of QRS duration
QRS duration GWAS results from 13,031 African Americans (reported above) and from 40,407 European-ancestry individuals in the previously reported CHARGE analysis (8) were meta-analysed, with little evidence of genomic inflation (λ = 1.018, Supplementary Material, Fig. S15). In addition to the previously identified QRS loci (Supplementary Materials, Fig. S16 and Table S5), the transethnic meta-analysis identified SNPs at two novel loci associated with cardiac ventricular conduction: an intronic SNP (rs1662342, P = 4.9×10−8) in a myosin light chain regulatory gene, MYL12A, on chromosome 18 and an intergenic SNP (rs7547997, P = 7.9×10−9) near CD1E on chromosome 1 (Table 4, Figs 3 and 4). For both SNPs, the coded allele frequency was higher in African Americans than individuals of European ancestry (Table 4), indicating that the addition of African American participants may have provided greater gains in power for these two SNPs than the addition of an equivalent number of individuals of European ancestry.
Table 4.
Novel genome-wide significant SNP associations from transethnic meta-analysis
| European ancestry |
African ancestry |
Meta-analysis |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Positiona | Nearby Genes | SNP Annotation | CA (Freq)b | β ± SEGC (PGC)c | I2 d | CA (Freq)b | β ± SEGC (PGC)c | I2 d | CA (Freq)b | β ± SEGC (PGC)c | I2 d |
| rs1662342 | 18 | 3,245,301 | MYL12A | intron | A (0.14) | 0.53 ± 0.10 (3.1x10−7) | 34.7 | A (0.21) | 0.35 ± 0.16 (0.03) | 16.6 | A (0.16) | 0.47 ± 0.09 (4.9x10−8) | 0.0 |
| rs7547997 | 1 | 156,607,897 | CD1E, OR10T2, SPTA1 | intergenic | A (0.16) | 0.48 ± 0.10 (1.0x10−6) | 0.0 | A (0.59) | 0.38 ± 0.12 (1.5x10−3) | 18.8 | A (0.33) | 0.44 ± 0.08 (7.9x10−9) | 0.0 |
aPosition based on NCBI reference sequence build 36.
bCA = coded effect allele. Freq = frequency.
cβ and SE expressed in units of milliseconds.
dHeterogeneity I2 statistics bolded if the Cochran’s Χ2 P-value ≤ 0.05.
Figure 3.
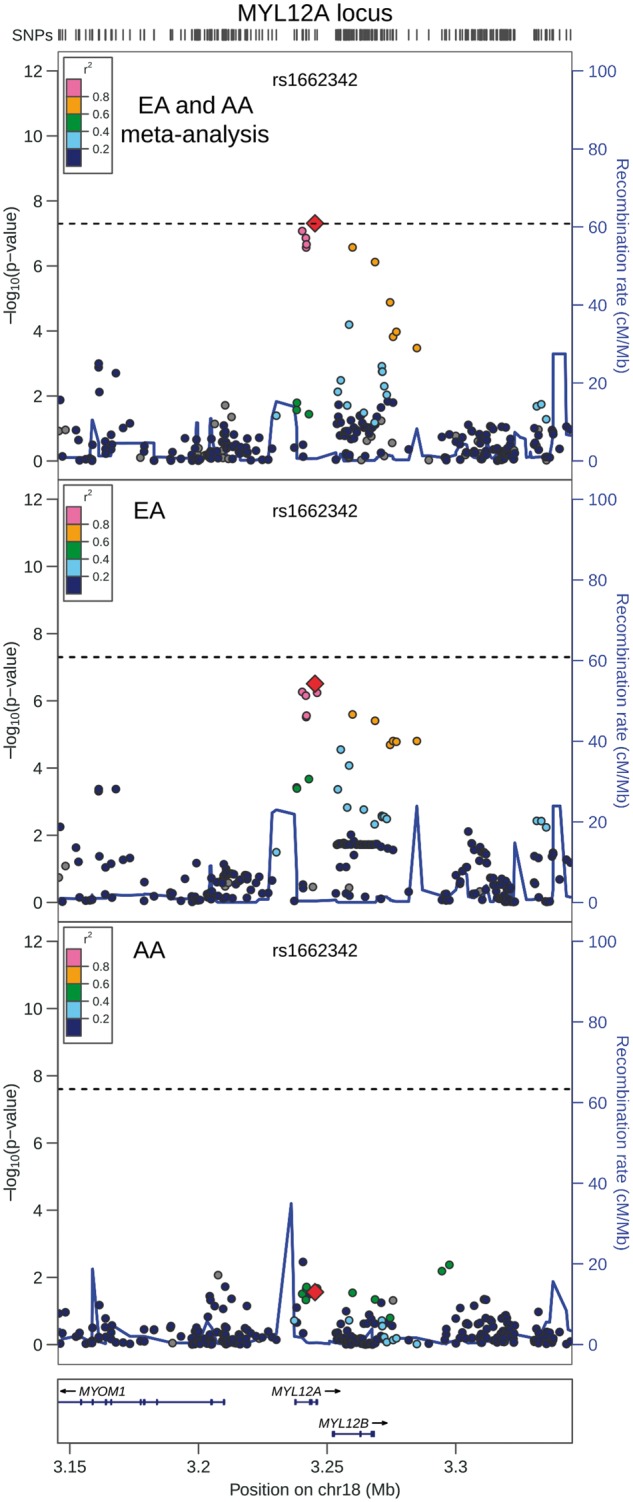
Regional association plot of transethnic meta-analysis results at MYL12A locus. Transethnic meta-analysis SNP association results are plotted in the top panel, meta-analysis results from cohorts of European ancestry are plotted in the middle panel, and African American SNP association meta-analysis results are plotted in the bottom panel. The transethnic index SNP (rs1662342) is designated by a red diamond in all panels. The LD (r2) shown is relative to the index SNP and is based on HapMap CEU in the top and middle panels and is based on HapMap YRI in the bottom panel. Gray circles are SNPs without HapMap LD data. The X-axis marks the chromosomal position. Recombination rates averaged across HapMap populations, in HapMap CEU individuals, and in African Americans are shown in the top panel, middle panel, and bottom panel, respectively. The dashed horizontal line in the top and middle panels marks the GWAS significance level for European ancestry (5.0 × 10 − 8) and in the bottom panel marks the GWAS significance level for African Americans (2.5 × 10 − 8).
Figure 4.
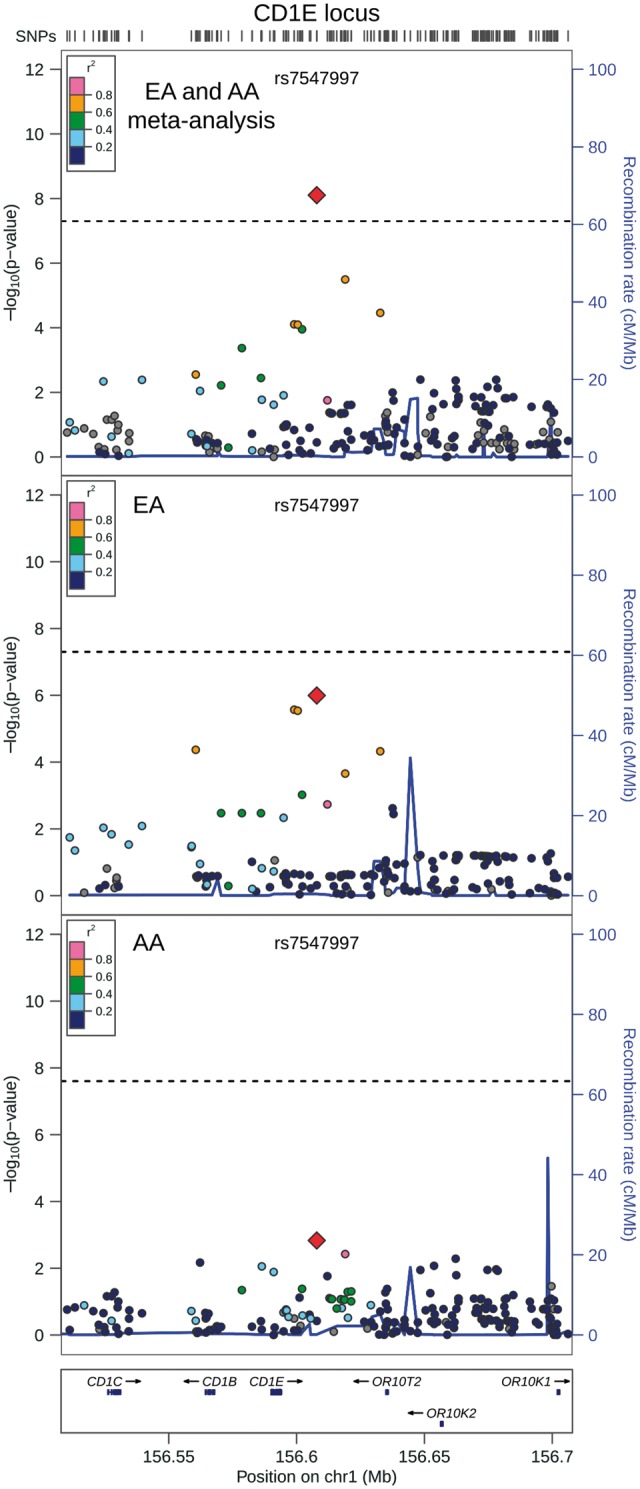
Regional association plot of transethnic meta-analysis results at chromosome 1q23.1. Transethnic meta-analysis SNP association results are plotted in the top panel, meta-analysis results from cohorts of European ancestry are plotted in the middle panel, and African American SNP association meta-analysis results are plotted in the bottom panel. The transethnic index SNP (rs7547997) is designated by a red diamond in all panels. The LD (r2) shown is relative to the index SNP and is based on HapMap CEU in the top and middle panels and is based on HapMap YRI in the bottom panel. Gray circles are SNPs without HapMap LD data. The X-axis marks the chromosomal position. Recombination rates averaged across HapMap populations, in HapMap CEU individuals, and in African Americans are shown in the top panel, middle panel, and bottom panel, respectively. The dashed horizontal line in the top and middle panels marks the GWAS significance level for European ancestry (5.0 × 10−8) and in the bottom panel marks the GWAS significance level for African Americans (2.5 × 10−8).
We examined whether rs1662342 or rs7547997 were eQTLs in human left atrial appendage tissue. While the rs1662342 A allele was nominally associated with higher MYL12A RNA levels in the meta-analysis of results from 289 individuals of European ancestry and 40 individuals of African ancestry (β ± SE = 0.08 ± 0.04, P = 0.03), the association was not significant after correction for multiple testing. Gene expression for only two genes (CD1C and CD1E) was detected within 250 kb upstream and downstream of rs7547997, and this SNP was not significantly associated with expression of either of these genes (P > 0.05).
Gene set enrichment and transferability of QRS-associated loci and SNPs
A gene set enrichment analysis (GSEA) revealed that genes identified from the 22 European-descent QRS loci were enriched for significant SNP associations in the African American QRS meta-analysis results, suggesting the transferability of QRS associations at the gene-set level between the two population groups. Gene-based P-values for 9 of the 22 QRS loci were in the top 95th percentile of all gene scores genome-wide in African Americans, whereas only one would be expected by chance, and the 22 QRS loci were significantly enriched for significant QRS-SNP associations in African Americans compared with randomly sampled gene sets (GSEA empirical P = 9.9×10−7). Importantly, the direction of the association was the same for all index SNPs at the 22 previously identified QRS loci in both ethnic groups, further supporting the transferability of associations between those of European and African ancestry (Tables 3 and 5, Supplementary Material, Table S6).
Table 5.
SNP replication results with adequate power from GWAS in African Americans
| European ancestry |
African ancestry |
LD (r2)d |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Index SNP/ annotation | Nearby gene | Chr (Position)h | Discovery population (Ref) | CA (Freq)a | β±SEGC (PGC)e | I2 f | Powerb | CA (Freq)a | β±SEGC (PGC)g | I2 f | Powerc | CEU | YRI | ASW |
| rs9470361/ | CDKN1A | 6 | EA (8) | A | 0.87 ± 0.08 | 14.6 | – | A | 0.43 ± 0.13 | 17.1 | 0.99 | 0.95 | 0.91 | ND |
| intergenic | (36,731,357) | (0.25) | (3x10−27) | (0.31) | (1x10−3) | |||||||||
| rs1321311/ | CDKN1A | 6 | EA (7) | A | 0.94 ± 0.15 | ND | – | A | 0.40 ± 0.12 | 0.0 | 0.99 | 0.95 | 0.70 | ND |
| intergenic | (36,730,878) | (0.21) | (3x10−10) | (0.39) | (1x10−3) | |||||||||
| rs7756236/ | CDKN1A | 6 | AA | G | 0.83 ± 0.08 | 34.3 | 0.99 | G | 0.53 ± 0.13 | 3.0 | – | – | – | – |
| intergenic | (36,735,031) | (0.26) | (2x10−26) | (0.30) | (4x10−5) | |||||||||
| rs11153730/ | PLN | 6 | EA (8) | C | 0.59 ± 0.07 | 5.3 | – | C | 0.32 ± 0.13 | 0.0 | 0.91 | 0.92 | 0.27 | ND |
| intergenic | (118,774,215) | (0.49) | (1x10−18) | (0.29) | (0.02) | |||||||||
| rs4307206/ | PLN | 6 | AA | A | 0.54 ± 0.07 | 14.4 | 0.99 | A | 0.52 ± 0.15 | 0.0 | – | – | – | – |
| intron | (118,920,013) | (0.45) | (5x10−16) | (0.21) | (4x10−4) | |||||||||
| rs9436640/ | NFIA | 1 | EA (8) | G | −0.59 ± 0.07 | 51.2 | – | G | −0.51 ± 0.12 | 0.0 | 0.95 | 0.54 | 0.53 | 0.69 |
| intron | (61,646,265) | (0.46) | (5x10−18) | (0.37) | (2x10−5) | |||||||||
| rs2207791/ | NFIA | 1 | AA | G | −0.59 ± 0.07 | 44.5 | 0.99 | G | −0.62 ± 0.13 | 6.7 | – | – | – | – |
| intron | (61,667,490) | (0.48) | (1x10−17) | (0.31) | (1x10−6) | |||||||||
| rs13165478/ | HAND1 | 5 | EA (8) | A | −0.55 ± 0.07 | 64.6 | – | A | −0.45 ± 0.14 | 0.0 | 0.93 | 1.00 | 1.00 | ND |
| intergenic | (153,849,233) | (0.36) | (7x10−14) | (0.53) | (1x10−3) | |||||||||
| rs13185595/ | HAND1 | 5 | AA | G | 0.56 ± 0.07 | 64.8 | 0.99 | G | 0.44 ± 0.13 | 0.0 | – | – | – | – |
| intergenic | (153,852,363) | (0.63) | (9x10−14) | (0.47) | (5x10−4) | |||||||||
| rs883079/ | TBX5 | 12 | EA (8) | G | 0.49 ± 0.08 | 8.3 | – | G | 0.60 ± 0.13 | 0.0 | 0.76 | 0.90 | 0.92 | 0.75 |
| 3’ UTR | (113,277,623) | (0.25) | (1x10−10) | (0.33) | (1x10−6) | |||||||||
| rs3825214/ | TBX5 | 12 | EA (7) | G | 1.06 ± 0.15 | ND | – | G | 0.49 ± 0.14 | 0.0 | 0.99 | 0.71 | 0.36 | 0.65 |
| intron | (113,279,826) | (0.22) | (3x10−13) | (0.25) | (3x10−4) | |||||||||
| rs7312625/ | TBX5 | 12 | AA | G | 0.46 ± 0.08 | 15.9 | 0.99 | G | 0.66 ± 0.13 | 0.0 | – | – | – | – |
| intron | (113,284,357) | (0.28) | (1x10−9) | (0.29) | (7x10−7) | |||||||||
| rs7342028/ | VTI1A | 10 | EA (8) | T | 0.48 ± 0.08 | 0.0 | – | T | 0.18 ± 0.12 | 0.0 | 0.81 | 1.0 | 0.44 | 0.46 |
| intron | (114,469,252) | (0.27) | (5x10−10) | (0.54) | (0.12) | |||||||||
| rs7907361/ | VTI1A | 10 | AA | G | −0.47 ± 0.08 | 0.0 | 0.96 | G | −0.39 ± 0.12 | 23.8 | – | – | – | – |
| intron | (114,458,127) | (0.74) | (1x10−9) | (0.47) | (9x10−4) | |||||||||
| rs4687718/ | TKT | 3 | EA (8) | A | −0.63 ± 0.11 | 0.0 | – | A | −0.06 ± 0.12 | 0.0 | 0.98 | ND | 0.05 | 0.02 |
| intron | (53,257,343) | (0.14) | (6x10−9) | (0.53) | (0.64) | |||||||||
| rs9311496/ | TKT | 3 | AA | A | 0.46 ± 4.49 | 0.0 | 0.00 | A | −0.48 ± 0.26 | 0.0 | – | – | – | – |
| intron | (53,347,118) | (0.001) | (0.92) | (0.06) | (0.06) | |||||||||
| rs17391905/ | CDKN2C | 1 | EA (8) | G | −1.35 ± 0.23 | 4.0 | – | G | −0.12 ± 0.26 | 0.0 | 0.99 | 0.13 | 0.02 | ND |
| intergenic | (51,318,728) | (0.05) | (9x10−9) | (0.07) | (0.65) | |||||||||
| rs11205809/ | CDKN2C | 1 | AA | A | 0.11 ± 0.10 | 6.6 | 0.90 | A | 0.42 ± 0.12 | 0.0 | – | – | – | – |
| intergenic | (51,273,357) | (0.84) | (0.25) | (0.48) | (8x10−4) | |||||||||
aCA = coded effect allele. Freq = frequency.
bPower to detect effect size reported in COGENT/CARe results using the effect allele frequency and trait variance as observed in European samples and keeping α = 0.002 adjusting for 22 SNPs.
cPower to detect effect size reported in European discovery population using the effect allele frequency and trait variance as observed in African-American samples and keeping α = 0.002 adjusting for 30 SNPs.
dLD between AA and EA index SNPs. ND indicates the SNP was not genotyped in the HapMap population or was monomorphic.
eIn non-African ancestry discovery rows, significance was based on the genome-wide discovery threshold and P-values ≤ 5.0x10 − 8 were bolded. In African ancestry discovery rows, significance was based on the replication threshold and P-values ≤ 0.002 were bolded. β and SE expressed in units of milliseconds.
fHeterogeneity I2 statistics bolded if the Cochran’s Χ2 P-value ≤ 0.05.
gIn non-African ancestry discovery rows, significance was based on replication of the 30 previously identified SNPs and P-values ≤ 0.002 were bolded. In African ancestry discovery rows, significance was based on discovery within the 22 loci and P-values ≤ 1.4×10 − 5 were bolded. β and SE expressed in units of milliseconds.
hPosition based on NCBI reference sequence build 36.
SNP association transferability at SCN5A/SCN10A locus
The most significant SNP associations reported from previous GWA studies of QRS interval among other ethnic groups are similarly located on chromosome 3 at the SCN5A/SCN10A locus, and multiple independent signals have been identified (Fig. 1) (7,8,16). The coded allele frequencies for all SCN5A/SCN10A European-ancestry index SNPs were lower among African Americans than individuals of European ancestry, which along with the smaller sample size among the African Americans we examined, reduces power to replicate (Table 3). Of the 4 independent European descent QRS index SNPs with adequate power (≥80%) to replicate among African Americans, 2 were significant, including the top SNP-QRS association among individuals of European ancestry, rs6801957 (P = 8×10−4 among African Americans, Table 3).
The absence of genome-wide significant QRS associations among SNPs within SCN10A in African Americans does not appear to be due to a difference in the MAF distribution of SCN10A SNPs between populations of European and African descent (Supplementary Material, Table S7). Compared to populations of European descent (Supplementary Material, Fig. S17), LD was reduced and haplotypes were shorter in the SCN10A region in populations of African descent (Supplementary Materials, Figs S18 and S19), which could impact the ability for assayed SNPs to tag potential non-genotyped causal variants.
SNP association transferability at the remaining known QRS loci
At the remaining 21 QRS loci (7,8), there was adequate power (≥80%) to replicate European-ancestry index SNPs at 8 loci, and index SNPs at 4 of these 8 loci replicated in African Americans at the multiple-testing corrected significance threshold of P = 0.002 (CDKN1A, NFIA, HAND1 and TBX5, Table 5, binomial test P = 4×10−4). Of the 13 loci where there was not adequate power to replicate, a European-ancestry index SNP at one locus replicated in African Americans (SETBP1, Supplementary Material, Table S6). At the more liberal replication significance threshold of 0.05, European-ancestry index SNPs at 5 of the 8 loci with adequate power replicated (binomial test P = 2×10−5), and index SNPs at 7 of the 13 loci without adequate power replicated (binomial test P = 1×10−6) (Table 5 and Supplementary Material, Table S6).
We next expanded our characterization of each of the 22 European-ancestry QRS loci to identify the most significant SNP association with QRS duration among African Americans (African-American index SNPs) and to determine the LD between European and African-ancestry index SNPs. Other than SCN5A, two other loci (TBX5 and NFIA) contained African-ancestry index SNPs that passed the significance threshold for discovery (P ≤ 1.4×10 − 5) within the 22 QRS loci (Table 5). The African-ancestry index SNP associations in TBX5 (rs7312625) and NFIA (rs2207791) replicated at the genome-wide significance level in populations of European descent (Table 5). For both loci, our results did not provide evidence for allelic heterogeneity, as the African-ancestry index SNPs were in moderate LD with the European-ancestry index SNPs (HapMap ASW r2 ≥ 0.65, HapMap CEU r2 ≥ 0.5) (Table 5, Supplementary Materials, Fig. S20 and S21).
Fine-mapping intervals based on Bayes factors and the resulting posterior probabilities generated from a transethnic meta-analysis with MANTRA (17) were constructed at the 22 QRS loci previously identified in populations of European descent and the two new QRS loci reported here. At each locus, the 95% credible set (CS) defines the genomic boundary that contains the smallest set of SNPs accounting for 95% of the posterior probability (Supplementary Material, Table S8). Other than the loci at which the 95% CS contained a single SNP, the largest percentage decrease from the genome-wide significant interval discovered in European populations to the 95% CS in the transethnic meta-analysis was at the TBX5 locus (Supplementary Material, Fig. S20).
Discussion
Our genetic association study of cardiac ventricular conduction among African Americans identified variants in a putative transcriptional regulatory region within intron 16 of the cardiac sodium channel SCN5A that were associated with QRS duration and SCN5A RNA levels. Furthermore, two novel loci associated with cardiac ventricular depolarization and conduction were identified through a transethnic GWAS meta-analysis. Finally, our study demonstrated the transferability of QRS-SNP associations between populations of European and African-descent by both gene-set enrichment analysis as well as direct evaluation of top QRS-SNP associations.
Our GWAS meta-analysis with additional fine-mapping identified two SNPs in high LD (rs3922844 and rs12635898, HapMap YRI: r2 = 0.93) in intron 16 of the cardiac sodium channel SCN5A gene associated with QRS duration among African Americans. SCN5A encodes the pore-forming α subunit of the cardiac voltage-gated sodium channel Nav1.5, and opening of the Nav1.5 channel drives rapid membrane depolarization during the cardiac action potential (18). Common and rare SCN5A genetic variants have been associated with cardiac depolarization, conduction, and repolarization (7,8,19). The most significantly associated SNP identified in this study (rs3922844) has been previously shown to associate with atrioventricular conduction (PR interval) among African Americans that included a subset of the same cohorts as this study (20,21), similar to other SNPs at this locus where variants that prolong PR interval also prolong QRS duration.
Functional annotation indicated that rs3922844 and other intronic variants in LD overlapped with a putative intronic transcriptional regulatory region. While functional intronic enhancers have not been identified in SCN5A, transcriptional enhancers are commonly found within intronic regions (22). Consistent with rs3922844’s overlap with putative transcriptional regulatory features, we found that rs3922844 was associated with SCN5A expression in human atrial tissue. The rs3922844 C allele associated with longer QRS duration was also associated with lower SCN5A RNA expression in human cardiac tissue, supporting the hypothesis that fewer available Nav1.5 channels would lead to subtly slower depolarization and conduction in cardiac tissue. It is intriguing that both genome-wide significant SNPs were located within intron 16 of SCN5A, which is immediately adjacent to an alternative splicing event that skips exons 17 and 18, resulting in the production of the non-functional Nav1.5b isoform that contains a partial deletion of the sixth transmembrane spanning segment of the DII domain and a deletion of a large segment of the intracellular DII-DIII linker (18). While the array-based expression data in our eQTL study did not enable our examination of specific transcript isoforms, future studies could investigate this potential molecular consequence that could lead to an increased production of non-functional transcripts that would further reduce the number of functional Nav1.5 voltage-gated sodium channels.
Using a transethnic meta-analysis approach, two novel QRS loci were identified: one in the gene MYL12A on chromosome 18 (rs1662342) and one in an intergenic region on chromosome one (rs7547997) near a cluster of five CD1 genes. For both SNPs, the higher allele frequency in African Americans combined with the 13,031 additional sample size increased power to find associations in the transethnic meta-analysis.
While the association between intronic SNP rs1662342 and MYL12A gene expression did not pass multiple test correction, the nominal significance of the association provided suggestive evidence that rs1662342 could be associated with MYL12A gene expression. MYL12A encodes the myosin regulatory light chain that binds to a variety of myosin heavy chain IIs (MHC IIs) in multiple cell types (23). MYL12A is expressed in the heart and cardiac myocytes in humans (24). Knockdown of MYL12A in mouse fibroblasts resulted in a significant reduction in cellular contractility, a disruption of cellular structure and morphology, and a decrease in non-muscle MHC II expression (23). The most significant SNP associations with RR interval in populations of European descent were located in MYH6, the α-heavy chain subunit of cardiac myosin (7,25), and the associations replicated in African Americans (26). Our results suggest that genetic variation in a different component of myosin, myosin regulatory light chain, may play a role in QRS duration.
The candidate gene for the second novel QRS locus is not as obvious, as rs7547997 was not associated with gene expression of nearby genes in our eQTL study. SNP rs7547997 is located within an intergenic region on chromosome one near a cluster of five CD1 genes, which mediate lipid and glycolipid antigen presentation to T cells (27) and near a cluster of 15 olfactory receptor genes (28). SPTA1, which encodes α-spectrin, is located 240 kb from rs7547997, but measures of SPTA1 RNA did not pass quality filters in our eQTL study and rs7547997’s association with SPTA1 gene expression could not be determined. Spectrin, a tetramer composed of α-β dimers, acts as an actin crosslinking and molecular scaffold protein that regulates cell shape and membrane protein localization (29). Spectrin binds ankyrin-G, and ankyrin-G is required for Nav1.5 membrane targeting and excitability (29). The E1053K SCN5A variant disrupts the ankyrin-G/Nav1.5 binding interaction and results in Brugada Syndrome (30). In mouse cardiomyocytes, β-spectrin co-localizes with ankyrin-G and Nav1.5 (31). β-spectrin also targets CaMKII to Nav1.5 where CaMKII regulates Nav1.5 by phosphorylation (31). While SPTA1 might be an attractive candidate gene, further studies are needed to identify causal gene(s) and variant(s) in the novel QRS association region identified on chromosome one.
Our transethnic meta-analysis results provide evidence that a large proportion of SNP and loci associations with QRS duration are shared between populations of European and African descent. Gene-set enrichment analysis revealed that the 22 previously reported European-descent QRS-associated loci were enriched for significant QRS-SNP associations among African Americans compared with randomly selected gene sets matched for gene set characteristics such as gene size and LD properties, which provided evidence for replication at the gene-set level. Where the power for replication was adequate, half of the European-descent index SNPs were also associated with QRS duration among African Americans. Even among the SNPs where power was inadequate, the majority were at least nominally associated with QRS duration among African Americans. Furthermore, the direction of the association for all European-ancestry index SNPs was the same in both ethnic groups. While these results taken together provide evidence for the transferability of QRS genetic associations from one ethnic group to another, there are some exceptions. There are SNPs associated with QRS duration among those of European descent, for instance rs9851724 near SCN10A (Table 3) and rs4687718 in TKT (Table 5), where despite adequate power, no evidence for association was identified among African Americans.
In addition to replication sample size and population-specific allele frequencies, SNP associations could fail to replicate across continental ancestry groups for several reasons, including: (1) population-specific causal variants, (2) population-specific LD between assayed SNPs and causal variants, and (3) interactions between SNPs and population-specific non-genetic factors (9,32). In a systematic survey of SNPs identified through GWA studies (GWAS SNPs), the allele frequency and LD with nearby SNPs differed significantly between population groups for a number of GWAS SNPs, suggesting that at least some GWAS SNPs identified in European populations might not generalize to other populations (10). In addition, it has been posited that rare variants, which are more likely to be population-specific (33–36), can create synthetic associations with common variants identified in GWA studies, which would result in the lack of transferability of findings across populations (37). Others have argued that synthetic associations might exist, but they are unlikely to account for most GWAS results (38,39). While a direct test of the synthetic association hypothesis requires a comprehensive collection of rare and common variants, our GWAS in African Americans provides an opportunity to test a prediction from the synthetic association hypothesis that SNP associations would not generalize across populations.
Well-powered studies for a limited number of traits and diseases have provided evidence that a majority of GWAS SNPs discovered in populations of European descent generalize to multiple populations (40–43). For instance, a high proportion of SNP associations with blood lipids discovered in populations of European descent generalized to populations of non-European descent (44,45), but allelic heterogeneity was observed at some loci (45,46). A recent systematic examination of GWAS SNP replication across populations found that 45.8% of GWAS SNPs initially identified in populations of European descent replicated in East Asian populations, and the percentage increased to 76.5% when replication attempts were limited to those that achieved sufficient power (47). The same study found that only 7 of 73 (9.6%) SNP associations replicated in populations of African descent, and the replication percentage only increased to 20% among the 25 replication attempts that achieved sufficient power (47). In an analysis of five traits and diseases in the Population Architecture Using Genomics and Epidemiology (PAGE) study, a consortium of multi-ancestry population-based studies, a significant proportion of GWAS SNPs discovered in European populations replicated in populations of non-European descent (43). However, the effect estimates in non-European populations tended to be closer to the null, especially in African Americans (43). Our analysis of SNP associations with QRS duration is consistent with findings from other traits; namely, SNP associations generalize at most loci but not all. GWA studies of ECG traits other than QRS duration have been conducted in African Americans, and SNP association transferability has been found (13,20,21,26,48,49). In two large meta-analyses, the proportion of SNP associations that replicated in African Americans was 7 of 13 for RR interval and 10 of 22 for QT interval at α = 0.05 (26,48). While Dickson et al. predicted that synthetic associations would be inconsistent across populations (37), results from our study and others indicate that a majority of SNP associations do generalize across populations. Continuing to perform well-powered GWA studies in African Americans should reveal whether SNP associations for a variety of traits and conditions are transferable.
In conclusion, by conducting a GWAS meta-analysis of QRS duration in African Americans, we refined the SCN5A association region to a single intron, and the associated SNP rs3922844 was also associated with reduced SCN5A expression in human atrial tissue in individuals of African and European ancestry. Two novel genome-wide significant SNP associations in or near intriguing candidate genes (MYL12A and SPTA1) were identified using transethnic meta-analysis. In addition, the high proportion of QRS associations that were transferable between populations of European and African-descent indicated that, at many of the associated loci, common genetic variation shared across populations contributes to QRS duration.
Materials and Methods
Study samples and ECG recordings
The following ten cohorts with African American participants contributed to this study (in order of decreasing sample size): the Women’s Health Initiative (WHI), the Atherosclerosis Risk in Communities (ARIC) study, the Jackson Heart Study (JHS), the Multi-Ethnic Study of Atherosclerosis (MESA), the Health, Aging, and Body Composition Study (Health ABC), the Healthy Aging in Neighborhoods of Diversity across the Life Span study (HANDLS), the Cardiovascular Health Study (CHS), the Cleveland Family Study (CFS), the Baltimore Longitudinal Study on Aging (BLSA) study, and the Bogalusa Heart Study (BHS). Detailed descriptions of the study samples and ECG recording methods are provided in Supplementary Material, Text S1. The European-descent participants and cohorts that contributed to this analysis have been previously described (8). The study was approved by the Institutional Review Board at all participating institutions. All individuals included in this analysis provided written informed consent.
Genotyping and genotype imputation
Cohorts used Affymetrix or Illumina SNP genotyping arrays and applied quality control filters to samples and SNPs (Supplementary Material, Table S1). Participants unlikely to be of African descent based on principal component analysis were excluded from the analysis (Supplementary Material, Text S1). Genotype imputation was performed using MACH or Beagle software. Individual studies performed genotype imputation using reference haplotypes that consisted of either a mixture of phased haplotype data from HapMap 2 YRI and CEU in a 1:1 ratio or a combination of HapMap 2 YRI and CEU in a 1:1 ratio and HapMap 3 YRI, CEU, and ASW in a 1:1:1 ratio (Supplementary Material, Table S1). Detailed descriptions of genotyping methods, quality control steps, and imputation methods can be found in Supplementary Materials, Text S1 and Table S1.
Genome-wide association analysis among African Americans
Study participants were excluded from analysis based on the following criteria: missing covariates, younger than 18 years of age, atrial fibrillation on the ECG, history of heart failure or myocardial infarction, QRS duration ≥ 120 ms, Wolff-Parkinson-White pattern, pacemaker or defibrillator implant, or use of class I and III antiarrhythmic medications. Genetic association analysis was performed in each cohort using linear regression models with the following covariates: age, sex, study site (if multiple sites were present), BMI, height, and principal components derived from principal component analysis (PCA) of genotype data (50) (Supplementary Material, Text S1). The exclusion criteria and covariate adjustment we applied were the same as those applied in a previous GWAS of QRS duration in populations of European descent (8), which facilitated comparisons of results between the two studies. In addition to adjustment for global genetic ancestry estimates, the genome-wide significant SNP association was additionally adjusted for local genetic ancestry estimates (see below for local ancestry estimation details). GWA studies in ARIC, JHS, MESA, and CFS were performed as part of the Candidate gene Association Resource (CARe) using QRS standardized residuals adjusted for the covariates mentioned above (51,52). All cohorts except for CFS performed a GWAS using PLINK, R, MACH2QTL, or Merlin (Supplementary Materials, Table S1 Table and Text S1). The family-based CFS study performed a GWAS using linear mixed models to account for relatedness (52,53). A subcohort of JHS was family-based, but previous analyses determined that the use of methods accounting for family structure had minimal influence on effect estimates, so linear regression was used as in previous studies (52). Imputed allele dosages were modelled with an additive mode of inheritance. When available, results from directly genotyped SNP data were used in preference over those from imputed SNP data. Detailed descriptions of cohort-specific GWAS analytic methods can be found in S1 Text.
Prior to meta-analysis, GWAS results from each cohort were filtered to remove SNPs with minor allele frequency (MAF) < 0.01 or imputation quality scores < 0.3. Effect estimates and their standard errors estimated from cohorts that used QRS standardized residuals as the phenotype (ARIC, JHS, MESA, and CFS) were transformed back to units of milliseconds by multiplying by the study-specific standard deviation of the residuals. Cohort-specific GWAS results were combined using fixed effect meta-analysis with inverse variance weights as implemented in METAL (54). SNPs that were non-autosomal or were only present in a single study were excluded from analysis. Genomic control was applied to the results from each cohort prior to meta-analysis and to the results of the meta-analysis (double GC-correction) (Supplementary Material, Table S1) (55). To maintain an experiment-wide type I error rate of 0.05, a genome-wide significance threshold of 2.5×10−8 was pre-specified based on a Bonferroni correction for the 2 million independent common variants estimated to exist in the genomes of individuals of African ancestry (56). Heterogeneity across samples was assessed using Cochran’s Χ2 test of heterogeneity with 9 degrees of freedom (57) and the I2 statistic (58). A transethnic GWAS meta-analysis of QRS duration was performed using fixed-effect inverse variance weighted meta-analysis to combine the double GC-corrected African American GWAS meta-analysis results with the double GC-corrected European ancestry CHARGE GWAS meta-analysis results (8).
Global and local genetic ancestry was estimated in ARIC, JHS, MESA, and CFS cohorts using ANCESTRYMAP (59) and HAPMIX (60) as previously described (52). For the WHI cohort, global ancestry was estimated using Frappe (61) and local ancestry was estimated using SABRE (62). For the HANDLS and Health ABC cohorts, STRUCTURE (63) was used to estimate global ancestry, as previously described for the Health ABC cohort (64), and LAMP (65) was used to estimate local ancestry. Additional details of global and local ancestry estimation can be found in S1 Text. Linear regression was used to estimate the association between global genetic ancestry estimates and QRS duration.
Variant annotation was performed using HaploReg (66), which leveraged data from the 1000 Genomes Project (36), ENCODE (67), and the Roadmap Epigenomics Mapping Consortium (68).
Genome-wide SNP results from the GWAS in African Americans and the transethnic GWAS will be made available through the dbGaP CHARGE summary results site under dbGaP accession phs000930.
Gene set enrichment analysis
A gene set enrichment analysis (GSEA) was performed using MAGENTA version 2.4 (69) on the QC-filtered double GC-corrected African American GWAS meta-analysis results. Briefly, MAGENTA assigns a gene score based on the most significant SNP association in the gene region, corrects for potential gene score confounders (e.g., gene size and number of independent SNPs), and determines the proportion of gene scores in the gene set above a specified percentile cut-off (i.e., the leading edge fraction). The construction of gene scores from the minimum QRS-SNP association P-value for each gene region enabled a gene score to be significant in the presence of allelic heterogeneity, i.e., different SNPs that are the most significantly associated with QRS duration at each locus in populations of European and African descent. The significance of the gene set’s leading edge fraction was determined using an empirical null distribution of 10,000 leading edge fractions from randomly sampled gene sets matched to the user-defined gene set by gene score confounder characteristics. To enable MAGENTA to calculate the number of SNPs per gene for populations of African descent, we generated a genome-wide list of SNP positions after SNPs in high LD (pairwise genotypic correlation r2 ≥ 0.8) were removed by applying PLINK’s LD pruning function to HapMap phase 2 YRI genotype data using sliding windows of 50 SNPs moving by 5 SNP increments (70).
The candidate gene set was created using the gene closest to the index SNP from the GWAS of QRS duration previously conducted by the CHARGE consortium (8), the largest GWAS of QRS duration, and included: SCN5A, SCN10A, CDKN1A, PLN, NFIA, HAND1, TBX20, SIPA1L1, TBX5, TBX3, VTI1A, SETBP1, STRN, TKT, CRIM1, CDKN2C, PRKCA, IGFBP3, CASQ2, KLF12, LRIG1, DKK1, and GOSR2. SCN5A and SCN10A were individually included in our gene set because the previously published CHARGE GWAS of QRS duration determined that SNP associations in these adjacent genes were independent (8).
Locus-specific replication and regional association analysis
The 30 European ancestry index SNPs (EA index SNPs) from the 22 previously reported EA QRS loci that were examined for replication in African Americans were 3 SCN10A SNPs, 3 SCN5A SNPs, 1 EXOG SNP, 2 CDKN1A SNPs, 2 TBX5 SNPs, and a single SNP from each of the other 19 previously reported loci (7,8,16). To account for the 30 examined SNPs, a significance threshold of 0.002 was adopted. The following parameters were used in power calculations for the 30 SNP replication analysis: α = 0.002, African American sample size = 13,031, African American QRS duration mean ± SD = 89.23 ± 9.70 ms (weighted average and pooled SD across the 10 COGENT/CARe cohorts), the previously reported SNP effect size, and the SNP coded allele frequency in populations of African descent (weighted average across the 10 COGENT/CARe cohorts).
In the analysis of the genomic region surrounding each of the European-ancestry index SNPs, each region was defined as the genomic interval encompassing all SNPs with QRS association P-values ≤ 5×10−8 from the largest GWAS meta-analysis conducted in individuals of European ancestry (8), and then extending the genomic interval boundaries by 100 kb in both directions. To set the significance threshold, the number of independent SNPs in these 22 regions was determined from HapMap phase 2 YRI SNP genotype data using the same LD pruning procedure that was described for our GSEA method. For the 22 previously reported QRS loci, 3,526 independent SNPs were identified and a Bonferroni-based significance threshold of 1.4×10−5 was adopted.
At each of the 22 previously identified QRS loci, the most significant SNP association detected in African Americans (AA index SNP) was also examined in GWAS results from populations of European descent (8). Power to replicate African-ancestry index SNP associations in populations of European descent was calculated using the following parameters: α = 0.002, sample size of individuals with European ancestry = 40,407, QRS duration mean ± SD from individuals of European ancestry = 88.32 ± 10.13 ms (estimated from 2,845 CHS subjects of European ancestry after application of exclusion factors described above), African-ancestry index SNP effect size, and the previously reported SNP coded allele frequency in populations of European descent (8). All power calculations were performed using QUANTO (71).
Regional association plots were created using LocusZoom (72), customized to separately plot recombination rates estimated from African Americans (73) and HapMap CEU individuals. For Supplementary Materials, Figs S17–S19, LD plots were created using Haploview and LD blocks were estimated using 95% confidence bounds on D prime (74,75).
95% Credible set construction
Regions used to construct credible sets (CS) at the 22 QRS loci discovered in populations of European descent were defined as genomic intervals encompassing all SNPs with QRS association P-values ≤ 5×10 − 8 reported by Sotoodehnia et al. (8), and then extending those genomic intervals by 100 kb in both directions. At the two new QRS loci reported here, intervals 100 kb upstream and downstream of the transethnic index SNP were used as regions for CS construction. Transethnic meta-analysis of fixed-effect meta-analysis results from populations of European descent (8) and African Americans was performed using MANTRA (17). As previously described (76) and in the context of a transethnic meta-analysis (77), for n SNPs in a region, a Bayes factor (BFi) was estimated for each SNPi using MANTRA, and the posterior probability for SNPi is equal to
SNPs with high posterior probabilities are more likely to be associated with a trait than SNPs with low posterior probabilities. SNPs at each region were ranked by their posterior probabilities in decreasing order, and the 95% CS consisted of the smallest set of ranked SNPs for which the cumulative sum of their posterior probabilities reached 0.95. The genomic interval of a 95% CS was the range of the positions of the SNPs in the CS.
Metabochip analysis of SCN5A-SCN10A gene region
To more comprehensively evaluate genetic variation in the SCN5A-SCN10A genomic region, we examined SNP associations using the MetaboChip, a high-density custom Illumina iSelect array that includes SNPs from the 1000 Genomes Project (11,78). In the SCN5A-SCN10A region (NCBI build 36 positions 38,490,026 - 38,818,967), 654 MetaboChip SNPs were directly genotyped and passed QC filters (SNP and sample call rate > 90%, concordance among blind duplicates > 98%, HWE P-value > 0.001, MAF > 0.01) in ARIC and WHI-PAGE participants. MetaboChip SNPs were imputed in WHI-SHARE participants using genome-wide genotype data (Affymetrix 6.0 SNP array), as previously described (79).
Gene expression analysis in human atrial tissue
Human left atrial appendage and pulmonary vein trimming tissues were obtained with written informed consent from 289 European-ancestry and 40 African-ancestry patients undergoing cardiac surgery. Use of discarded surgical tissue was approved by the Institutional Review Board of the Cleveland Clinic. Total RNA was extracted using TRIzol. Genome-wide RNA levels were measured using Illumina HT12 v.3 and v.4 expression arrays. RNA expression levels were background corrected, log2-transformed, quantile normalized, and batch adjusted. Genome-wide SNPs were genotyped in these subjects using Illumina Hap550 and Hap610 arrays, and multidimensional scaling (MDS) was performed. SNP association with RNA levels was determined separately for each racial group using linear regression with SNPs coded as dosages and additive adjustment for sex, tissue location, MDS dimensions, and surrogate variables, which were included to reduce expression heterogeneity and improve power to detect eQTLs (80). Effect estimates were expressed on the log2-transformed RNA scale. Analysis was performed using expression probes detected in at least 10% of samples within 250 kb of the query SNP. Nine eQTLs were examined: rs3922844 and SCN5A, rs1662342 and probes in MYL12A, TGIF1 (2 probes), LPIN2 (2 probes), and MYOM1, and rs7547997 and probes in CD1C and CD1E, and an eQTL P-value < 0.006 (0.05/9) was deemed significant. SCN5A exonic locations are expressed relative to the longest isoform, NM_198056.2, which contains 28 exons.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
The authors wish to acknowledge the contributions of the involved research institutions, study investigators, field staff, and study participants of WHI, ARIC, JHS, MESA, Health ABC, HANDLS, CHS, CFS, BLSA, and BHS.
The authors would also like to acknowledge the generous sharing of QRS GWAS results from the CHARGE consortium. The following individuals are members of the CHARGE QRS Consortium: Nona Sotoodehnia, Aaron Isaacs, Paul I.W. de Bakker, Marcus Dörr, Christopher Newton-Cheh, Ilja M. Nolte, Pim van der Harst, Martina Müller, Mark Eijgelsheim, Alvaro Alonso, Andrew A. Hicks, Sandosh Padmanabhan, Caroline Hayward, Albert Vernon Smith, Ozren Polasek, Steven Giovannone, Jingyuan Fu, Jared W. Magnani, Kristin D. Marciante, Arne Pfeufer, Sina A. Gharib, Alexander Teumer, Man Li, Joshua C. Bis, Fernando Rivadeneira, Thor Aspelund, Anna Köttgen, Toby Johnson, Kenneth Rice, Mark P.S. Sie, Amanda Ying Wang, Norman Klopp, Christian Fuchsberger, Sarah H. Wild, Irene Mateo Leach, Karol Estrada, Uwe Völker, Alan F. Wright, Folkert W. Asselbergs, Jiaxiang Qu, Aravinda Chakravarti, Moritz F. Sinner, Jan A. Kors, Astrid Petersmann, Tamara B. Harris, Elsayed Z. Soliman, Patricia B. Munroe, Bruce M. Psaty, Ben A. Oostra, L. Adrienne Cupples, Siegfried Perz, Rudolf A. de Boer, André G. Uitterlinden, Henry Völzke, Timothy D. Spector, Fang-Yu Liu, Eric Boerwinkle, Anna F. Dominiczak, Jerome I. Rotter, Gé van Herpen, Daniel Levy, H.-Erich Wichmann, Wiek H. van Gilst, Jacqueline C.M. Witteman, Heyo K. Kroemer, W.H. Linda Kao, Susan R. Heckbert, Thomas Meitinger, Albert Hofman, Harry Campbell, Aaron R. Folsom, Dirk J. van Veldhuisen, Christine Schwienbacher, Christopher J. O’Donnell, Claudia Beu Volpato, Mark J. Caulfield, John M. Connell, Lenore Launer, Xiaowen Lu, Lude Franke, Rudolf S.N. Fehrmann, Gerard te Meerman, Harry J.M. Groen, Rinse K. Weersma, Leonard H. van den Berg, Cisca Wijmenga, Roel A. Ophoff, Gerjan Navis, Igor Rudan, Harold Snieder, James F. Wilson, Peter P. Pramstaller, David S. Siscovick, Thomas J. Wang, Vilmundur Gudnason, Cornelia M. van Duijn, Stephan B. Felix, Glenn I. Fishman, Yalda Jamshidi, Bruno H Ch Stricker, Nilesh J. Samani, Stefan Kääb, Dan E. Arking.
Conflict of Interest statement. Bruce M. Psaty serves on the DSMB of a clinical trial funded by the manufacturer (Zoll LifeCor) and on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Anne M. Butler has received investigator-initiated support from Amgen and AstraZeneca for unrelated work.
Funding
This work was supported by the National Institutes of Health [R01 HL088456 to N.S., R01 HL116747 to N.S., R01 HL111089 to N.S., R01 HL091244 to N.S., K99 HL098458 to C.L.A., 5T32CA009330-30 to A.M.B., R01 ES017794 to E.A.W., P20MD006899 to S.G.B., and U24AG051129 to D.S.E.], the Laughlin Family [to N.S.], and the German Research Foundation [SCHNA 1149/3-1 to R.B.S.].
As part of the National Heart, Lung, and Blood Institute (NHLBI)-sponsored Candidate gene Association Resource (CARe) project, the ARIC, JHS, MESA, and CFS studies contributed parent study data, ancillary study data, and DNA samples through the Broad Institute of Harvard and MIT (N01-HC-65226) to create a genotype/phenotype database for wide dissemination to the biomedical research community.
Atherosclerosis Risk in Communities (ARIC): The ARIC study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. The authors thank the staff and participants of the ARIC study for their important contributions. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
Cleveland Family Study (CFS): Case Western Reserve University (NIH HL 46380, M01RR00080).
Jackson Heart Study (JHS): The Jackson Heart Study is supported by contracts HHSN268201300046C, HHSN268201300047C, HHSN268201300048C, HHSN268201300049C, HHSN268201300050C from the National Heart, Lung, and Blood Institute and the National Institute on Minority Health and Health Disparities.
Multi-Ethnic Study of Atherosclerosis (MESA): University of Washington (N01-HC-95159), Regents of the University of California (N01-HC-95160), Columbia University (N01-HC-95161), Johns Hopkins University (N01-HC-95162, N01-HC-95168), University of Minnesota (N01-HC95163), Northwestern University (N01-HC-95164), Wake Forest University (N01-HC-95165), University of Vermont (N01-HC-95166), New England Medical Center (N01-HC-95167), Harbor-UCLA Research and Education Institute (N01-HC-95169), Cedars-Sinai Medical Center (R01-HL-071205), and University of Virginia (subcontract to R01-HL-071205).
Health, Aging, and Body Composition Study (Health ABC): The Health ABC study was supported by NIA contracts N01AG62101, N01AG62103, and N01AG62106. The genome-wide association study was funded by NIA grant 1R01AG032098-01A1 to Wake Forest University Health Sciences and genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
Healthy Aging in Neighbourhoods of Diversity across the Life Span Study (HANDLS): The HANDLS study was in part supported by the intramural research program of the National Institute on Aging and the National Center for Minority Health and Health Disparities, National Institutes of Health. This research was supported by the Intramural Research Program of the NIH, National Institute on Aging and the National Center on Minority Health and Health Disparities (contract # Z01-AG000513 and human subjects protocol # 2009-149). Data analyses for the HANDLS study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Md. (http://biowulf.nih.gov).
Women’s Health Initiative (WHI): The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C. This manuscript was prepared in collaboration with investigators of the WHI, and has been reviewed and/or approved by the Women’s Health Initiative (WHI). WHI investigators are listed at https://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf. Funding for WHI SHARe genotyping was provided by NHLBI Contract N02-HL-64278.
Cardiovascular Health Study (CHS): This CHS research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, HHSN268200960009C; and NHLBI grants HL080295, HL087652, HL105756, HL103612, HL120393, and HL130114, HL085251 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org/. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Baltimore Longitudinal Study of Aging (BLSA): The BLSA was supported in part by the Intramural Research Program of the NIH, National Institute on Aging. A portion of that support was through a R&D contract with MedStar Research Institute.
Bogalusa Heart Study (BHS): BHS is supported by grants R01ES021724 from National Institute of Environmental Health Science and R01AG016592 from the National Institute on Aging. Analysis performed at STSI/TSRI was supported by U54 NS056883 and Scripps Genomic Medicine.
The Population Architecture Using Genomics and Epidemiology (PAGE) program is funded by the National Human Genome Research Institute (NHGRI), supported by U01HG007416 (CALiCo), U01HG007417 (ISMMS), U01HG007397 (MEC), U01HG007376 (WHI), and U01HG007419 (Coordinating Center). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The complete list of PAGE members can be found at http://www.pagestudy.org. Assistance with data management, data integration, data dissemination, genotype imputation, ancestry deconvolution, and general study coordination was provided by the PAGE Coordinating Center (U01HG007419). The PAGE consortium thanks the staff and participants of all PAGE studies for their important contributions.
Funding for the atrial tissue eQTL study was provided by NIH 5R01HL090620, NIH 5R01HL111314, Fondation Leducq CVD-07-03, European North American Atrial Fibrillation Research Alliance.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Badheka A.O., Singh V., Patel N.J., Deshmukh A., Shah N., Chothani A., Mehta K., Grover P., Savani G.T., Gupta S., et al. (2013) QRS duration on electrocardiography and cardiovascular mortality (from the National Health and Nutrition Examination Survey-III). Am. J. Cardiol, 112, 671–677. [DOI] [PubMed] [Google Scholar]
- 2.Mentz R.J., Greiner M.A., DeVore A.D., Dunlay S.M., Choudhary G., Ahmad T., Khazanie P., Randolph T.C., Griswold M.E., Eapen Z.J., et al. (2015) Ventricular conduction and long-term heart failure outcomes and mortality in African Americans: insights from the Jackson Heart Study. Circ. Heart Fail., 8, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang N.C., Maggioni A.P., Konstam M.A., Zannad F., Krasa H.B., Burnett J.C., Jr., Grinfeld L., Swedberg K., Udelson J.E., Cook T., et al. (2008) Clinical implications of QRS duration in patients hospitalized with worsening heart failure and reduced left ventricular ejection fraction. JAMA, 299, 2656–2666. [DOI] [PubMed] [Google Scholar]
- 4.Vitelli L.L., Crow R.S., Shahar E., Hutchinson R.G., Rautaharju P.M., Folsom A.R. (1998) Electrocardiographic findings in a healthy biracial population. Atherosclerosis Risk in Communities (ARIC) Study Investigators. Am. J. Cardiol., 81, 453–459. [DOI] [PubMed] [Google Scholar]
- 5.Rautaharju P.M., Prineas R.J., Kadish A., Larson J.C., Hsia J., Lund B. (2006) Normal standards for QT and QT subintervals derived from a large ethnically diverse population of women aged 50 to 79 years (the Women's Health Initiative [WHI]). Am. J. Cardiol., 97, 730–737. [DOI] [PubMed] [Google Scholar]
- 6.Walsh J.A., 3rd, Prineas R., Daviglus M.L., Ning H., Liu K., Lewis C.E., Sidney S., Schreiner P.J., Iribarren C., Lloyd-Jones D.M. (2010) Prevalence of electrocardiographic abnormalities in a middle-aged, biracial population: Coronary Artery Risk Development in Young Adults study. J. Electrocardiol., 43, 385 e381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holm H., Gudbjartsson D.F., Arnar D.O., Thorleifsson G., Thorgeirsson G., Stefansdottir H., Gudjonsson S.A., Jonasdottir A., Mathiesen E.B., Njolstad I., et al. (2010) Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet., 42, 117–122. [DOI] [PubMed] [Google Scholar]
- 8.Sotoodehnia N., Isaacs A., de Bakker P.I., Dorr M., Newton-Cheh C., Nolte I.M., van der Harst P., Muller M., Eijgelsheim M., Alonso A., et al. (2010) Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet., 42, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg N.A., Huang L., Jewett E.M., Szpiech Z.A., Jankovic I., Boehnke M. (2010) Genome-wide association studies in diverse populations. Nat. Rev. Genet., 11, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casto A.M., Feldman M.W. (2011) Genome-wide association study SNPs in the human genome diversity project populations: does selection affect unlinked SNPs with shared trait associations? PLoS Genet., 7, e1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matise T.C., Ambite J.L., Buyske S., Carlson C.S., Cole S.A., Crawford D.C., Haiman C.A., Heiss G., Kooperberg C., Marchand L.L., et al. (2011) The Next PAGE in understanding complex traits: design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) Study. Am. J. Epidemiol., 174, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Splawski I., Timothy K.W., Tateyama M., Clancy C.E., Malhotra A., Beggs A.H., Cappuccio F.P., Sagnella G.A., Kass R.S., Keating M.T. (2002) Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science, 297, 1333–1336. [DOI] [PubMed] [Google Scholar]
- 13.Jeff J.M., Brown-Gentry K., Buxbaum S.G., Sarpong D.F., Taylor H.A., George A.L., Jr., Roden D.M., Crawford D.C. (2011) SCN5A variation is associated with electrocardiographic traits in the Jackson Heart Study. Circ. Cardiovasc. Genet., 4, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magnani J.W., Brody J.A., Prins B.P., Arking D.E., Lin H., Yin X., Liu C.T., Morrison A.C., Zhang F., Spector T.D., et al. (2014) Sequencing of SCN5A identifies rare and common variants associated with cardiac conduction: Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. Circ. Cardiovasc. Genet., 7, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilkhanoff L., Arking D.E., Lemaitre R.N., Alonso A., Chen L.Y., Durda P., Hesselson S.E., Kerr K.F., Magnani J.W., Marcus G.M., et al. (2014) A common SCN5A variant is associated with PR interval and atrial fibrillation among African Americans. J. Cardiovasc. Electrophysiol., 25, 1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers J.C., Zhao J., Terracciano C.M., Bezzina C.R., Zhang W., Kaba R., Navaratnarajah M., Lotlikar A., Sehmi J.S., Kooner M.K., et al. (2010) Genetic variation in SCN10A influences cardiac conduction. Nat. Genet., 42, 149–152. [DOI] [PubMed] [Google Scholar]
- 17.Morris A.P. (2011) Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol., 35, 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroeter A., Walzik S., Blechschmidt S., Haufe V., Benndorf K., Zimmer T. (2010) Structure and function of splice variants of the cardiac voltage-gated sodium channel Na(v)1.5. J. Mol. Cell. Cardiol., 49, 16–24. [DOI] [PubMed] [Google Scholar]
- 19.Zimmer T., Surber R. (2008) SCN5A channelopathies–an update on mutations and mechanisms. Prog. Biophys. Mol. Biol., 98, 120–136. [DOI] [PubMed] [Google Scholar]
- 20.Smith J.G., Magnani J.W., Palmer C., Meng Y.A., Soliman E.Z., Musani S.K., Kerr K.F., Schnabel R.B., Lubitz S.A., Sotoodehnia N., et al. (2011) Genome-wide association studies of the PR interval in African Americans. PLoS Genet., 7, e1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler A.M., Yin X., Evans D.S., Nalls M.A., Smith E.N., Tanaka T., Li G., Buxbaum S.G., Whitsel E.A., Alonso A., et al. (2012) Novel loci associated with PR interval in a genome-wide association study of 10 African American cohorts. Circ. Cardiovasc. Genet., 5, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadhouders R., van den Heuvel A., Kolovos P., Jorna R., Leslie K., Grosveld F., Soler E. (2012) Transcription regulation by distal enhancers: who's in the loop? Transcription, 3, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park I., Han C., Jin S., Lee B., Choi H., Kwon J.T., Kim D., Kim J., Lifirsu E., Park W.J., et al. (2011) Myosin regulatory light chains are required to maintain the stability of myosin II and cellular integrity. Biochem. J., 434, 171–180. [DOI] [PubMed] [Google Scholar]
- 24.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., et al. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U S A, 101, 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eijgelsheim M., Newton-Cheh C., Sotoodehnia N., de Bakker P.I., Muller M., Morrison A.C., Smith A.V., Isaacs A., Sanna S., Dorr M., et al. (2010) Genome-wide association analysis identifies multiple loci related to resting heart rate. Hum. Mol. Genet., 19, 3885–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deo R., Nalls M.A., Avery C.L., Smith J.G., Evans D.S., Keller M.F., Butler A.M., Buxbaum S.G., Li G., Miguel Quibrera P., et al. (2013) Common genetic variation near the connexin-43 gene is associated with resting heart rate in African Americans: a genome-wide association study of 13,372 participants. Heart Rhythm, 10, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen N.R., Garg S., Brenner M.B. (2009) Antigen Presentation by CD1 Lipids, T Cells, and NKT Cells in Microbial Immunity. Adv. Immunol., 102, 1–94. [DOI] [PubMed] [Google Scholar]
- 28.Malnic B., Godfrey P.A., Buck L.B. (2004) The human olfactory receptor gene family. Proc. Natl. Acad. Sci. U S Aa, 101, 2584–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith S., Curran J., Hund T.J., Mohler P.J. (2012) Defects in cytoskeletal signaling pathways, arrhythmia, and sudden cardiac death. Front. Physiol., 3, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohler P.J., Rivolta I., Napolitano C., LeMaillet G., Lambert S., Priori S.G., Bennett V. (2004) Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U S Aa, 101, 17533–17538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hund T.J., Koval O.M., Li J., Wright P.J., Qian L., Snyder J.S., Gudmundsson H., Kline C.F., Davidson N.P., Cardona N., et al. (2010) A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest., 120, 3508–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teo Y.Y., Small K.S., Kwiatkowski D.P. (2010) Methodological challenges of genome-wide association analysis in Africa. Nat. Rev. Genet., 11, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gravel S., Henn B.M., Gutenkunst R.N., Indap A.R., Marth G.T., Clark A.G., Yu F., Gibbs R.A., Bustamante C.D. (2011) Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. U S Aa, 108, 11983–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tennessen J.A., Bigham A.W., O'Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science, 337, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu W., O'Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., et al. (2013) Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature, 493, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. (2010) Rare variants create synthetic genome-wide associations. PLoS Biol., 8, e1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson C.A., Soranzo N., Zeggini E., Barrett J.C. (2011) Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biol., 9, e1000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wray N.R., Purcell S.M., Visscher P.M. (2011) Synthetic associations created by rare variants do not explain most GWAS results. PLoS Biol., 9, e1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waters K.M., Stram D.O., Hassanein M.T., Le Marchand L., Wilkens L.R., Maskarinec G., Monroe K.R., Kolonel L.N., Altshuler D., Henderson B.E., et al. (2010) Consistent association of type 2 diabetes risk variants found in europeans in diverse racial and ethnic groups. PLoS Genet., 6, pii: e1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haiman C.A., Chen G.K., Blot W.J., Strom S.S., Berndt S.I., Kittles R.A., Rybicki B.A., Isaacs W.B., Ingles S.A., Stanford J.L., et al. (2011) Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet., 7, e1001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen F., Chen G.K., Millikan R.C., John E.M., Ambrosone C.B., Bernstein L., Zheng W., Hu J.J., Ziegler R.G., Deming S.L., et al. (2011) Fine-mapping of breast cancer susceptibility loci characterizes genetic risk in African Americans. Hum. Mol. Genet., 20, 4491–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlson C.S., Matise T.C., North K.E., Haiman C.A., Fesinmeyer M.D., Buyske S., Schumacher F.R., Peters U., Franceschini N., Ritchie M.D., et al. (2013) Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLoS Biol., 11, e1001661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dumitrescu L., Carty C.L., Taylor K., Schumacher F.R., Hindorff L.A., Ambite J.L., Anderson G., Best L.G., Brown-Gentry K., Buzkova P., et al. (2011) Genetic determinants of lipid traits in diverse populations from the population architecture using genomics and epidemiology (PAGE) study. PLoS Genet., 7, e1002138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coram M.A., Duan Q., Hoffmann T.J., Thornton T., Knowles J.W., Johnson N.A., Ochs-Balcom H.M., Donlon T.A., Martin L.W., Eaton C.B., et al. (2013) Genome-wide characterization of shared and distinct genetic components that influence blood lipid levels in ethnically diverse human populations. Am. J. Hum. Genet., 92, 904–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Y., Waite L.L., Jackson A.U., Sheu W.H., Buyske S., Absher D., Arnett D.K., Boerwinkle E., Bonnycastle L.L., Carty C.L., et al. (2013) Trans-ethnic fine-mapping of lipid loci identifies population-specific signals and allelic heterogeneity that increases the trait variance explained. PLoS Genet., 9, e1003379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marigorta U.M., Navarro A. (2013) High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet., 9, e1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith J.G., Avery C.L., Evans D.S., Nalls M.A., Meng Y.A., Smith E.N., Palmer C., Tanaka T., Mehra R., Butler A.M., et al. (2012) Impact of ancestry and common genetic variants on QT interval in African Americans. Circ. Cardiovasc. Genet., 5, 647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeff J.M., Ritchie M.D., Denny J.C., Kho A.N., Ramirez A.H., Crosslin D., Armstrong L., Basford M.A., Wolf W.A., Pacheco J.A., et al. (2013) Generalization of variants identified by genome-wide association studies for electrocardiographic traits in African Americans. Ann. Hum. Genet., 77, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet., 38, 904–909. [DOI] [PubMed] [Google Scholar]
- 51.Musunuru K., Lettre G., Young T., Farlow D.N., Pirruccello J.P., Ejebe K.G., Keating B.J., Yang Q., Chen M.H., Lapchyk N., et al. (2010) Candidate gene association resource (CARe): design, methods, and proof of concept. Circ. Cardiovasc. Genet., 3, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lettre G., Palmer C.D., Young T., Ejebe K.G., Allayee H., Benjamin E.J., Bennett F., Bowden D.W., Chakravarti A., Dreisbach A., et al. (2011) Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet., 7, e1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen M.H., Yang Q. (2010) GWAF: an R package for genome-wide association analyses with family data. Bioinformatics, 26, 580–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willer C.J., Li Y., Abecasis G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Devlin B., Roeder K. (1999) Genomic control for association studies. Biometrics, 55, 997–1004. [DOI] [PubMed] [Google Scholar]
- 56.Pe'er I., Yelensky R., Altshuler D., Daly M.J. (2008) Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol., 32, 381–385. [DOI] [PubMed] [Google Scholar]
- 57.Whitehead A., Whitehead J. (1991) A general parametric approach to the meta-analysis of randomized clinical trials. Stat. Med., 10, 1665–1677. [DOI] [PubMed] [Google Scholar]
- 58.Higgins J.P., Thompson S.G. (2002) Quantifying heterogeneity in a meta-analysis. Stat. Med., 21, 1539–1558. [DOI] [PubMed] [Google Scholar]
- 59.Patterson N., Hattangadi N., Lane B., Lohmueller K.E., Hafler D.A., Oksenberg J.R., Hauser S.L., Smith M.W., O'Brien S.J., Altshuler D., et al. (2004) Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet., 74, 979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Price A.L., Tandon A., Patterson N., Barnes K.C., Rafaels N., Ruczinski I., Beaty T.H., Mathias R., Reich D., Myers S. (2009) Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet., 5, e1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang H., Peng J., Wang P., Risch N.J. (2005) Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol., 28, 289–301. [DOI] [PubMed] [Google Scholar]
- 62.Tang H., Coram M., Wang P., Zhu X., Risch N. (2006) Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet., 79, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pritchard J.K., Stephens M., Donnelly P. (2000) Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nalls M.A., Wilson J.G., Patterson N.J., Tandon A., Zmuda J.M., Huntsman S., Garcia M., Hu D., Li R., Beamer B.A., et al. (2008) Admixture mapping of white cell count: genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am. J. Hum. Genet., 82, 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pasaniuc B., Sankararaman S., Kimmel G., Halperin E. (2009) Inference of locus-specific ancestry in closely related populations. Bioinformatics, 25, i213–i221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ward L.D., Kellis M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R., et al. (2010) The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol., 28, 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segre A.V., Groop L., Mootha V.K., Daly M.J., Altshuler D. (2010) Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet., 6, e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gauderman W.J. (2002) Sample size calculations for matched case-control studies of gene-environment interaction. Stat. Med., 21, 35–50. [DOI] [PubMed] [Google Scholar]
- 72.Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R., Willer C.J. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics, 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hinch A.G., Tandon A., Patterson N., Song Y., Rohland N., Palmer C.D., Chen G.K., Wang K., Buxbaum S.G., Akylbekova E.L., et al. (2011) The landscape of recombination in African Americans. Nature, 476, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barrett J.C., Fry B., Maller J., Daly M.J. (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 21, 263–265. [DOI] [PubMed] [Google Scholar]
- 75.Gabriel S.B., Schaffner S.F., Nguyen H., Moore J.M., Roy J., Blumenstiel B., Higgins J., DeFelice M., Lochner A., Faggart M., et al. (2002) The structure of haplotype blocks in the human genome. Science, 296, 2225–2229. [DOI] [PubMed] [Google Scholar]
- 76.Wellcome Trust Case Control C., Maller J.B., McVean G., Byrnes J., Vukcevic D., Palin K., Su Z., Howson J.M., Auton A., Myers S., et al. (2012) Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet., 44, 1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Asimit J.L., Hatzikotoulas K., McCarthy M., Morris A.P., Zeggini E. (2016) Trans-ethnic study design approaches for fine-mapping. Eur. J. Hum. Genet., 24(9):1330–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.(2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu E.Y., Buyske S., Aragaki A.K., Peters U., Boerwinkle E., Carlson C., Carty C., Crawford D.C., Haessler J., Hindorff L.A., et al. (2012) Genotype imputation of Metabochip SNPs using a study-specific reference panel of ∼4,000 haplotypes in African Americans from the Women's Health Initiative. Genet. Epidemiol., 36, 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leek J.T., Storey J.D. (2007) Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet., 3, 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]