

Abstract
The short-rib polydactyly syndromes (SRPS) encompass a radiographically and genetically heterogeneous group of skeletal ciliopathies that are characterized by a long narrow chest, short extremities, and variable occurrence of polydactyly. Radiographic abnormalities include undermineralization of the calvarium, shortened and bowed appendicular bones, trident shaped acetabula and polydactyly. In a case of SRPS we identified compound heterozygosity for mutations in IFT52, which encodes a component of the anterograde intraflagellar transport complex. The IFT52 mutant cells synthesized a significantly reduced amount of IFT52 protein, leading to reduced synthesis of IFT74, IFT81, IFT88 and ARL13B, other key anterograde complex members. Ciliogenesis was also disrupted in the mutant cells, with a 60% reduction in the presence of cilia on mutant cells and loss of cilia length regulation for the cells with cilia. These data demonstrate that IFT52 is essential for anterograde complex integrity and for the biosynthesis and maintenance of cilia. The data identify a new locus for SRPS and show that IFT52 mutations result in a ciliopathy with primary effects on the skeleton.
Introduction
Ciliopathies comprise a spectrum of disorders resulting from the defects in the biosynthesis and/or function of primary cilia (1,2). A subset of the ciliopathies have primary abnormalities of the skeleton and these include Ellis-van Creveld syndrome (EVC; OMIM 225500), cranioectodermal dysplasia (CED, also known as Sensenbrenner syndrome; OMIM 218330), asphyxiating thoracic dystrophy (ATD, also known as Jeune syndrome; OMIM 208500 and others), and the perinatal lethal short rib-polydactyly syndromes (SRPS; OMIM 208500 and others) (3). The SRPS are genetically and radiographically heterogeneous but all have small chests with markedly short ribs, micromelic limbs, reduced bone mineralization, and many cases have polydactyly. Visceral abnormalities can include cystic kidneys and livers and structural abnormalities of the heart. All forms of SRPS are thought to be inherited in an autosomal recessive manner.
Many of the gene defects characterized in the skeletal ciliopathies affect the components of ciliary intraflagellar (IFT) transport (2). IFT is a highly evolutionarily conserved process that serves to transport structural and signalling components along the ciliary axoneme from the cytoplasm to the ciliary tip via anterograde transport and then back to the cytoplasm via retrograde transport (4,5). The IFT-B anterograde complex is powered by kinesin motors and consists of 9 core components (IFT22, IFT25, IFT27, IFT46, IFT52, IFT70, IFT74, IFT81, and IFT88) and several peripheral subunits (IFT20, IFT54, IFT57, IFT80, IFT172, and others) (5). The IFT-A retrograde complex uses dynein motors and consists of 6 primary components (IFT43, IFT121/WDR35, IFT122, IFT139/TTC21B, IFT140, and IFT144/WDR19) and other ancillary proteins. These complexes interact with ciliary cargos and the anterograde and retrograde motors to facilitate axonemal transport. In Caenorhabditis elegans mutants, IFT-B subunit mutations primarily block assembly of cilia while IFT-A subunit mutations have been shown to lead to structurally and/or functionally defective cilia (6).
To date mutations in 10 genes have been found in cases of SRPS. DYNC2H1 (7,8), which encodes a retrograde (IFT-A) motor, is the main locus, and mutations in six additional genes encoding retrograde transport components [DYNC2LI1 (9), WDR34 (10), WDR60 (11), WDR35 (12), IFT140 (13), and TCTEX1D2 (14)] have also been characterized. Mutations in the gene encoding the IFT-B complex member IFT80 (15), the NEK1 kinase gene (16) and the KIAA0586 (17) centrosomal protein gene, have also been found to be responsible for SRPS. Among the non-lethal skeletal ciliopathies, mutations have been characterized in retrograde transport IFT-A component genes WDR19/IFT144 (18), IFT122 (19), IFT43 (20), and TTC21B/IFT139 (21); IFT-B complex component IFT172 (22); and two genes [EVC (23), EVC2 (24)] that encode basal body proteins. Mutations in a number of these genes can cause phenotypes along a spectrum of skeletal severity.
In this study, we report a non-consanguineous family with two fetuses affected by SRPS. Exome sequencing revealed compound heterozygosity for two mutations in IFT52. The mutations resulted in dramatic reduction of the protein product in cultured cells from mutant fibroblasts and chondrocytes, and reduced levels of several IFT-B core proteins, suggesting that IFT52 is central to anterograde complex assembly. Consequently, there was an absence of ciliogenesis in a high proportion of the cultured cells, and a loss of length regulation in the cells that had cilia. These data demonstrate that IFT52 is essential for ciliogenesis and cilia architecture as well as for IFT-A complex stability, and that mutations in IFT52 results in a ciliopathy with primary effects on the skeleton.
Results
Clinical description of the SRPS case
We studied a non-consanguineous family (International Skeletal Dysplasia Registry reference number R98-277) in which consecutive pregnancies resulted in prenatally identified fetuses with SRPS. Radiographic findings, taken at 20 and 23 weeks of gestation, respectively, were consistent between the two fetuses and showed undermineralized skulls, narrow thoraces with moderately shortened ribs and sharp angulations of some lower thoracic ribs, a flat appearance to the acetabular roofs, reverse campomelia of the humeri, mildly bent femurs, and no polydactyly (Figure 1A). These findings were consistent with a diagnosis of a form of SRPS. Histology of the cartilage growth plate from the second fetus (R98-277B) showed a very short and disorganized hypertrophic zone suggesting impairment of the transition from proliferating to hypertrophic chondrocytes, and an irregular margin at the transition between cartilage and bone (Figure 1B).
Figure 1.

Radiographic phenotype and growth plate histology in SRPS case R98-277B. (A) AP fetal radiograph illustrating the undermineralized skull, moderately short ribs, flat acetabular roof and shortened long bones with mild femoral/humeral bending. (B) Growth plate histology comparing the control (top) and SRPS case (bottom) demonstrating irregularity of the border between the hypertrophic zone (H) and bone (B) and the very short hypertrophic zone (the boxed area is expanded on the right). Scale bars are 100 µM.
Identification of mutations in IFT52 in a case of SRPS
To define the molecular basis of this SRPS case, exome sequencing was carried out as previously described (9) using DNA from case R98-277B under an approved human subjects protocol. Assuming recessive inheritance, the exome variants were filtered to focus on genes with homozygosity for a single variant or compound heterozygosity for two variants. All non-synonymous substitutions and variants affecting splice junction consensus sequences were annotated. The data were reviewed for variants in genes known to be associated with SRPS as well as other skeletal ciliopathies, and causative mutations at all of these loci were excluded. The data were then filtered against the variants listed by the Exome Sequencing Project (ESP) (25) and the Exome Aggregation Consortium (ExAC) (26) to remove all variants with population frequencies of 1% or more. In some instances genes could be excluded because both variants were inherited from the same parent and thus carried on the same allele. Eight genes were left as a result of the above filtering process (Supplementary Material, Table S1). Subsequently, variants in genes with a possible role in cilia were identified under the hypothesis that SRPS would result from a defect in cilia function. One gene, IFT52 at chromosome 20q12.13, was a particularly attractive candidate because the IFT52 protein is one of the IFT-B core components and partial loss of IFT52 function for the mouse orthologue resulted in multiple defects that arose during mouse embryogenesis, including left-right and ventral midline defects, abnormal craniofacial development, defective Hedgehog (Hh) signaling and polydactyly (27). These features were reminiscent of SRPS.
Compound heterozygosity for two variants was found and both were confirmed by Sanger sequence analysis (Figure 2). The first variant, which was inherited from the mother, was a single base deletion (c.878delT) in exon 10 that predicted a frameshift and premature stop codon (pLeu293AlafsX21), indicative of a null allele. The second variant was inherited from the father and was a missense change (c.595G > A) that implied a p.Ala199Thr substitution. Ala199 is evolutionarily conserved from humans to zebrafish (PhastCons = 1) (28) and the predicted consequence is deleterious (SIFT = 0.01) (29). Neither variant was found in public SNP databases, indicating that they are not common polymorphisms in the population.
Figure 2.

Mutations in IFT52. (A) The two mutations found are shown above a diagram of the domain structure of the IFT52 protein. Below the diagram, interactions between IFT52 and both IFT88 and IFT70 with the central region and IFT46 with the C-terminal domain are represented. (B) Segregation of the mutations in the family are shown on the pedigree with the paternally- and maternally-derived alleles indicated by red and green arrows, respectively.
Reduced IFT52 in SRPS fibroblasts
To define the consequence of the mutations at the RNA level, RT-PCR was performed using RNA extracted from cultured fibroblasts derived from case R98-277B and from normal controls, using primers that flank exon 7, which contained the missense mutation. Sequence analysis of the RT-PCR product revealed only sequences derived from the missense allele, c.595G > A (Figure 3A), suggesting that the transcript containing the frameshift variant was degraded through nonsense-mediated mRNA decay. Western blot analysis demonstrated significant reduction of IFT52 at the protein level in both fibroblasts and chondrocytes, whether the experiments were carried out in serum replete media or under reduced serum conditions used to promote ciliogenesis (Figure 3B). These data were consistent with instability and degradation of the protein product of the missense allele and an overall dramatic reduction of IFT52 in SRPS cells.
Figure 3.
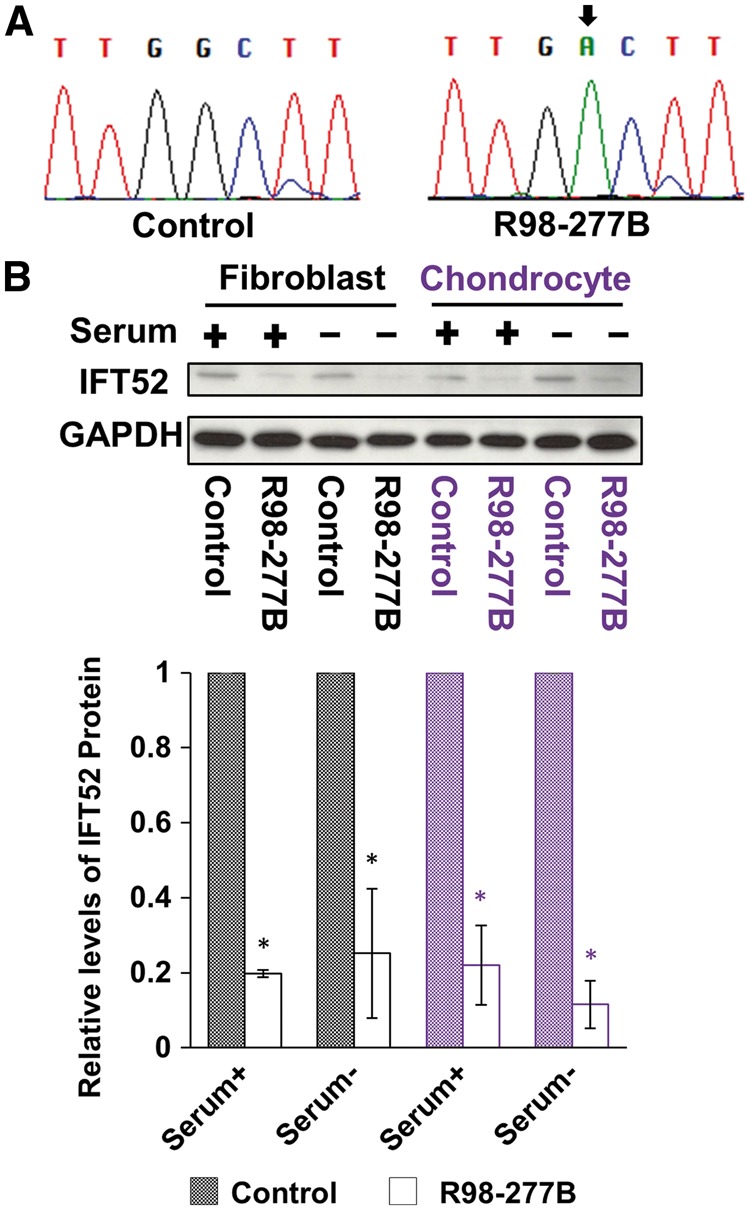
Consequences of IFT52 mutations. (A) Sequences of RT-PCR products derived from IFT52 mutant cells, demonstrating that only products of the missense allele with the c.595G > A mutation were found. The nucleotide change is indicated with a black arrow. (B) Immunoblotting revealed IFT52 protein reduction in the lysates of IFT52 mutant cultured fibroblasts and chondrocytes under both serum replete and serum restricted conditions as compared with controls. Below the Western blots, IFT52 abundance in SRPS cells was normalized to control cells, demonstrating a statistically significant reduction in IFT52 in SRPS cells (n = 3, *P < 0.05).
Destabilization of the IFT-B complex due to defects in IFT52
To determine the effect of defective IFT52 on other IFT-B components, Western blot analysis was used to quantify the expression of several IFT-B proteins by SRPS fibroblasts. Although the interactions among IFT proteins remain incompletely characterized, IFT52 has been shown to be a central part of the IFT-B core and directly interacts with IFT88 and IFT70 via its central domain and IFT46 and IFT74 via its C-terminal domain (30), as diagrammed in Figure 4A. IFT81 and IFT88 proteins were both reduced in SRPS cells in either the presence or absence of serum. In the presence of serum, neither control nor mutant cells synthesized detectable IFT74. However, under serum-free conditions, control cells synthesized IFT74 while synthesis was significantly reduced in SRPS cells (Figure 4B). Taken together, these observations suggest that abnormal IFT52 impairs anterograde IFT-B core assembly.
Figure 4.

IFT complex B is unstable in the IFT52 mutant. (A) Schematic representation of the interactions among IFT-B core complex members and ARL13B based on published studies. The corresponding structural domains of IFT52 are indicated with brackets below the figure. (B) Whole-cell lysates from SRPS case and control fibroblasts were analyzed by Western blot using antibodies to IFT-B core proteins and ARL13B. The levels of these proteins in IFT52 mutant cells were markedly decreased, especially upon the stimulation of ciliogenesis in reduced serum media (n = 3, *P < 0.05).
To further explore the extent to which, apart from the core components, IFT52 abnormalities affected the IFT-B complex, the protein level of ARL13B, a membrane-associated small GTPase critical for the ciliary axoneme structure (31), was determined. The C. elegans orthologue of ARL13B has been shown to interact with IFT74 and IFT46 in C. elegans (32) (Figure 4A). Similar to the IFT-B core components, there was decreased ARL13B protein in the SRPS fibroblasts compared with control (Figure 4B). These data are thus consistent with the hypothesis that IFT52 is essential for the maintenance of the IFT-B core and the overall IFT-B complex.
IFT52 is required for the anterograde transport of IFT88
To examine the effect on IFT anterograde transport upon mutation of IFT52, the reduced amount of IFT88 observed by Western blot in the cultured cells was localized using immunofluorescence staining. In ciliated control fibroblasts we observed IFT88 at both the basal bodies and tips of the cilia (Figure 5). By contrast, in ciliated SRPS cells IFT88 was indistinguishable from background fluorescence and therefore could not be reliably associated with cilia (Figure 4B and 5). These data are consistent with a defect in anterograde transport in IFT52 mutant SRPS cells.
Figure 5.

IFT88 distribution in cilia. Immunofluorescence microscopy of SRPS and control ciliated cells stained with mouse anti-acetylated α-tubulin (red) and rabbit anti-IFT88 (green). The IFT88 signal was enriched at the basal bodies (single arrowhead) and tips of cilia (double arrowheads) in controls while in SRPS cells it could not be detected above background. Boxed regions are expanded at the bottom and separated into single signal panels with anti-acetylated α-tubulin (red) and anti-IFT88 (green). Scale bars represent 20 μm in the upper panels and 1 μm in the insets.
Abnormal cilia abundance and morphology with loss of IFT52
To assess the effect of the IFT52 mutations on ciliogenesis, cilia abundance and morphology were characterized in the SRPS fibroblasts and controls after 48 h of serum starvation. Cilia were co-immunostained with antibodies to ARL13B and acetylated α-tubulin (Figure 6A). Concordant with other studies describing the consequences of dysfunction of IFT-B complex components (22,33), the SRPS cells had significantly fewer ciliated fibroblasts compared with the control (SRPS mean 40.2% ciliated cells vs. control mean 79.2%, student’s t-test P-value= 0.0002, Figure 6B). Among the ciliated SRPS cells, the average length of cilia was shorter than in controls (SRPS vs. control mean length: 2.89 μm vs. 3.51μm, student’s t-test P-value: 6.312e-05). Moreover, among the ∼150 ciliated cells measured in the SRPS cells, there was loss of length regulation, with mutant cells having a more diverse length distribution, with an increased number of both highly shortened and elongated cilia relative to control cells (Figure 6C). These observations indicate that both ciliogenesis and cilia length regulation are defective in the IFT52 mutant cells.
Figure 6.

Cilia abundance and ciliary length. (A) Ciliary axonemes and basal bodies of SRPS fibroblasts were stained using mouse monoclonal anti-acetylated α-tubulin (green) and rabbit anti-ARL13B (red) antibodies, respectively. Representative cells with either elongated or shortened cilia as compared with control are shown. Scale bars represent 10 μm. (B) The fraction of ciliated cells in the SRPS case (40.2%) was significantly decreased compared with the control sample (79.2%). Cilia were enumerated from 324 patient cells and 287 control cells observed in four randomly sampled regions; *P = 0.0002. (C) Average cilia length in the SRPS case (2.89 μm) was significantly shorter than cilia length in control cells (3.51 μm), **P = 6.312e-05, and there was also a wider distribution of cilia lengths among SRPS cells. The cilia length data were calculated from 155 ciliated SRPS cells and 159 ciliated control cells.
Discussions
Through identification of IFT52 mutations in a case of SRPS, this study demonstrates the central role that IFT52 has in the stability of the IFT-B complex and the dramatic effect on skeletogenesis that results from its absence. SRPS was the result of compound heterozygosity for a missense mutation and a single base deletion leading to a frameshift. The frameshift mutation led to loss of the transcript from that allele, while the missense change destabilized IFT52 in cultured cells. The substitution implied by the missense mutation, p.Ala199Thr, was located within the N-terminal ‘GIFT’ [for GldG, intraflagellar transport (IFT)] domain. While the specific function of this domain is unknown, its sequence is highly conserved from archaea to eukarya (34).
IFT52 is essential for maintaining the IFT-B complex, as the IFT52 mutations led to the reduction of the levels of multiple IFT-B proteins, as originally defined in Chlamydomonas (35). Accordingly, in our study, there was a dramatic reduction of IFT74, IFT81, IFT88, and ARL13B, all members of the IFT-B anterograde transport complex. During IFT-B complex assembly, IFT74 and IFT81 bind to each other and form a subcomplex with IFT27, IFT25 and IFT22 and then attach to a second subcomplex composed of IFT52, IFT88, IFT70 and IFT46, interacting directly with the C-terminal domains of IFT52 and IFT46 (30). These studies are concordant with recent high-resolution crystal structures detailing interactions between IFT70 and IFT52 as well as between IFT52 and IFT46 (30). Destabilization of the IFT-B core in SRPS is thus consistent with the central role of IFT52 in connecting the IFT-B core.
The role of IFT52 in the assembly of the IFT-B complex has also been observed in other species with defects in IFT52, which also result in defects in ciliogenesis in these organisms. In C. elegans, the cilia of the osm-6/ift52 mutant lacks ciliary axonemal microtubules beyond the transition zone (36). In the Chlamydomonas mutant of the IFT52 orthologue Bld1, the IFT-B complex is disrupted and flagella are completely depleted (35). Despite being the a component of the same complex, in Chlamydomonas the ift52 mutant has much more pronounced effects on IFT-B complex stability than ift88 and ift46 mutants, again reflecting the central organizational role of IFT52. Similarly, knockdown of ift52 in zebrafish resulted in fish with an abnormally curved body axis and pronounced ciliary defects characterized in nasal epithelia (37). Finally, hypomorphic mouse Ift52 mutants exhibited skeletal abnormalities, craniofacial defects and polydactyly as well as tight mesencephalic flexure, left-right and ventral midline defects (27). The recent structural studies of IFT-B core complex assembly provides a molecular rationale for the various ciliary phenotypes observed among these species, showing that the role IFT52 has in linking IFT88, IFT70, and IFT46 to the rest of the IFT-B core complex and indicating that defects in IFT52 lead to loss of these interactions and degradation of the other complex proteins. Thus IFT52 function appears to be evolutionarily conserved from Chlamydomonas to humans.
IFT52 is also required for localization of the IFT-B complex proteins. Immunoelectron microscopy studies have revealed that IFT52 clusters along the transition fibres and functions as a docking site for complex B proteins attaching to the transition fibres of the basal bodies (35,38). In Chlamydomonas, the complex B proteins are restricted to the proximal ends of basal bodies in the absence of IFT52 (35). Consistent with these observations, we observed loss of IFT88 signal in the cilia of SRPS fibroblasts, revealing a similar disruption of anterograde transport with the loss of IFT52.
Identification of IFT52 as a locus for SRPS underscores the importance of both anterograde and retrograde transport in cilia function in the skeleton, a finding bolstered by the recent characterization of an IFT52 mutation in a related but milder skeletal phenotype (39). Still unanswered is why defects in some ciliary proteins have such a pronounced effect on skeletal development. As the hierarchies of assembly of the IFT-B and IFT-A complexes are uncovered by studying cells from SRPS cases with mutations at the known loci, and as mutations at new loci are discovered through the study of additional SRPS cases, how such defects so severely and selectively affect the skeleton is expected to become apparent.
Materials and Methods
Exome sequencing and variant filtering
Genomic DNA was isolated from cultured fibroblasts derived from case R98-277B and from peripheral blood of both parents using standard procedures. The study was carried out under an approved University of California at Los Angeles human subjects’ protocol and written informed consent was obtained from the parents. Library construction and exome sequencing of DNA from the proband was carried out at the University of Washington Center for Mendelian Genomics. The exome-sequencing library was prepared with the NimbleGen SeqCap EZ Exome Library v2.0 kit and sequenced on the Illumina GAIIx platform. Reads were mapped with Novoalign and variants called using the Genome Analysis Toolkit following their Best Practices recommendations (40,41). Variants were filtered against public databases and annotated as previously described (9). The variants in IFT52 identified in case R98-277B were confirmed by Sanger sequence analysis of amplified DNA from the case and both parents. Primer sequences used were: IFT52-exon 7, F: 5’-CTTACAGGGCTC TCACCTTTGT-3’; R: 5’-GCAGGAAGACCAGTGAGAAAAC-3’; and IFT52-exon 10, F: 5’-CTTGCTCTTGCTGTGCTAAAAG-3’; R: 5’-CAG CC CAGTTTCTTTGTTGAC-3’.
Reverse transcriptase PCR
RNA from cultured fibroblasts established from SRPS case R98-277B and a control fibroblast cell line was extracted using TRIZOL (Invitrogen) according to the manufacturer’s protocol. First strand cDNA was synthesized using SuperScript III First-Strand Synthesis SuperMix (Invitrogen), and a 208 bp fragment containing exon 7 of IFT52, was amplified using primer sequences F: 5’-CTGGGATCATTGATGAGGAAAG-3’; R: 5’-ATGTG ACATGAACCAAGCACTG-3’.
Western blot analysis
Cultured fibroblasts and chondrocytes from the case and controls were grown in DMEM supplemented with 10% fetal bovine serum (FBS). Cells were washed with PBS and incubated with 0.5% FBS containing media for 48 h in order to promote ciliogenesis. Cells were lysed with 1X RIPA Buffer supplemented with 1 mM PMSF and a protease inhibitor cocktail (Roche) at 4 °C for 30 min and extracted proteins were recovered in the supernatant after centrifugation at 16,000g for 10 min. 60–100 μg of lysate was used for each sample to be separated by 10% SDS-PAGE, and immunoblotting used the following primary antibodies: GAPDH (1:1000, Cell Signaling Technology: 2118), rabbit anti-IFT52 (1:1000, Protein Tech: 17534-1-AP), ARL13B (1:1000, Protein Tech: 17711-1-AP), IFT81 (1:1000, Protein Tech: 11744-1-AP), IFT74 (1:1000, Thermo Fisher: PA5-21746), IFT88 (1:1000, Abcam: ab184566). Horseradish peroxidase (HRP)-conjugated secondary antibodies were diluted at 1:1000 and incubated with the membranes for 1 h at room temperature. Each experiment was carried out three times and ImagJ (42) was used to scan the blots in order to quantify the amount of protein detected. Protein levels in SRP cells were first normalized to the loading control and then to the corresponding normalized control cell proteins. The Student’s t-test was used to determine statistical significance.
Immunofluorescence
Cultured fibroblasts from the case and control were seeded into 16-well plates at a density of 2.5× 104 cells per well and allowed to grow overnight. Cells were then serum starved for 48 h and fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were permeabilized using 0.1% triton-X-100 in PBS for 5 min at room temperature. Cells were blocked with 5% goat serum (Invitrogen) for 1 h at room temperature and then incubated with primary antibodies overnight at 4˚C. After washing with PBS, cells were incubated with Alexa Fluor 488/568- conjugated secondary antibodies (1:400; Invitrogen and Jackson ImmunoResearch) in PBS with 5% goat serum for 1 h at room temperature. The nuclei were stained with DAPI (Invitrogen) after the secondary antibody incubation (1:1000 dilution in 5% goat serum for 10 min at room temperature). Cells were extensively washed with PBS between each step.
Cilia abundance and ciliary length measurements
Ciliary axonemes and basal bodies were stained overnight at 4 °C using mouse anti-acetylated α-tubulin (1:2000; 6-11B-1; Sigma-Aldrich T6793), and rabbit anti-ARL13B (1:150; Proteintech 17711-1-AP) antibodies, respectively. Images were recorded using a Zeiss LSM780 microscope (×63 magnification, Carl Zeiss) and Axio Imager with Apotome (×20 magnification, Carl Zeiss). Mean numbers of ciliated cells were calculated from 324 SRPS cells and 287 control cells from four individual regions randomly selected from the plate. Cilia lengths were measured for 155 ciliated SRPS cells and 159 ciliated control cells randomly selected from the same plate. Data from SRPS and control cells were compared using the Protected Least Significant Difference (PLSD) of Fischer, with significance determined using the Student’s t-test.
Supplementary Material
Supplementary Material is available at HMG online
Acknowledgements
We thank the families for the generosity of their participation.
Conflict of Interest statement. None declared.
Funding
This study was supported in part by grants from the National Institutes of Health (NIDCR grant RO1 DE01957 and NIAMS grants R01 AR062651 and RO1 AR066124). S.P.T. was supported in part by NIH Training Grant in Genomic Analysis and Interpretation award T32 HG002536. Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW CMG) which is funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute Grant 1U54 HG006493. We also thank the March of Dimes and the Joseph Drown Foundation for their support of the International Skeletal Dysplasia Registry. We are grateful to the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA, supported with funding from NIH-NCRR shared resources grant (CJX1-443835-WS-29646), and NSF Major Research Instrumentation grant (CHE-0722519), for their assistance. The research was also supported by NIH/National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR000124.
References
- 1.Badano J.L., Mitsuma N., Beales P.L., Katsanis N. (2006) The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet., 7, 125–148. [DOI] [PubMed] [Google Scholar]
- 2.Waters A.M., Beales P.L. (2011) Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol., 26, 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber C., Cormier-Daire V. (2012) Ciliary disorder of the skeleton. Am. J. Med. Genet. C Semin. Med. Genet., 160C, 165–174. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa H., Marshall W.F. (2011) Ciliogenesis: building the cell's antenna. Nat. Rev. Mol. Cell Bio., 12, 222–234. [DOI] [PubMed] [Google Scholar]
- 5.Taschner M., Bhogaraju S., Lorentzen E. (2012) Architecture and function of IFT complex proteins in ciliogenesis. Differentiation, 83, S12–S22.22118932 [Google Scholar]
- 6.Pan X.Y., Ou G.S., Civelekoglu-Scholey G., Blacque O.E., Endres N.F., Tao L., Mogilner A., Leroux M.R., Vale R.D., Scholey J.M. (2006) Mechanism of transport of IFT particles in C-elegans cilia by the concerted action of kinesin-II and OSM-3 motors. J. Cell Biol., 174, 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merrill A.E., Merriman B., Farrington-Rock C., Camacho N., Sebald E.T., Funari V.A., Schibler M.J., Firestein M.H., Cohn Z.A., Priore M.A., et al. (2009) Ciliary Abnormalities Due to Defects in the Retrograde Transport Protein DYNC2H1 in Short-Rib Polydactyly Syndrome. Am. J. Hum. Genet., 84, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dagoneau N., Goulet M., Genevieve D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L., et al. (2009) DYNC2H1 Mutations Cause Asphyxiating Thoracic Dystrophy and Short Rib-Polydactyly Syndrome, Type III. Am. J. Hum. Genet., 84, 706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor S.P., Dantas T.J., Duran I., Wu S.L., Lachman R.S., Nelson S.F., Cohn D.H., Vallee R.B., Krakow D., Geno U.W.C.M. (2015) Mutations in DYNC2LI1 disrupt cilia function and cause short rib polydactyly syndrome. Nat. Commun., 6,7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber C., Wu S.L., Kim A.S., Sigaudy S., Sarukhanov A., Serre V., Baujat G., Sang K.H.L.Q., Rimoin D.L., Cohn D.H., et al. (2013) WDR34 Mutations that Cause Short-Rib Polydactyly Syndrome Type III/Severe Asphyxiating Thoracic Dysplasia Reveal a Role for the NF-kappa B Pathway in Cilia. Am. J. Hum. Genet., 93, 926–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McInerney-Leo A.M., Schmidts M., Cortes C.R., Leo P.J., Gener B., Courtney A.D., Gardiner B., Harris J.A., Lu Y.P., Marshall M., et al. (2013) Short-Rib Polydactyly and Jeune Syndromes Are Caused by Mutations in WDR60. Am. J. Hum. Genet., 93, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mill P., Lockhart P.J., Fitzpatrick E., Mountford H.S., Hall E.A., Reijns M.A.M., Keighren M., Bahlo M., Bromhead C.J., Budd P., et al. (2011) Human and Mouse Mutations in WDR35 Cause Short-Rib Polydactyly Syndromes Due to Abnormal Ciliogenesis. Am. J. Hum. Genet., 88, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A.M., Gerber S., Delphin N., Bigot K., et al. (2012) Mainzer-Saldino Syndrome Is a Ciliopathy Caused by IFT140 Mutations. Am. J. Hum. Genet., 90, 864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gholkar A.A., Senese S., Lo Y.C., Capri J., Deardorff W.J., Dharmarajan H., Contreras E., Hodara E., Whitelegge J.P., Jackson P.K., et al. (2015) Tctex1d2 associates with short-rib polydactyly syndrome proteins and is required for ciliogenesis. Cell Cycle, 14, 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavalcanti D.P., Huber C., Sang K.H.L., Baujat G., Collins F., Delezoide A.L., Dagoneau N., Le Merrer M., Martinovic J., Mello M.F.S., et al. (2011) Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J. Med. Genet., 48, 88–92. [DOI] [PubMed] [Google Scholar]
- 16.Thiel C., Kessler K., Giessl A., Dimmler A., Shalev S.A., von der Haar S., Zenker M., Zahnleiter D., Stoss H., Beinder E., et al. (2011) NEK1 Mutations Cause Short-Rib Polydactyly Syndrome Type Majewski. Am. J. Hum. Genet., 88, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alby C., Piquand K., Huber C., Megarbane A., Ichkou A., Legendre M., Pelluard F., Encha-Ravazi F., Abi-Tayeh G., Bessieres B., et al. (2015) Mutations in KIAA0586 cause lethal ciliopathies ranging from a hydrolethalus phenotype to short-rib polydactyly syndrome. Am. J. Hum. Genet., 97, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbo M., Filhol E., Bole-Feysot C., et al. (2011) Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A Gene WDR19. Am. J. Hum. Genet., 89, 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A., et al. (2010) Cranioectodermal dysplasia, sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet., 86, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arts H.H., Bongers E.M.H.F., Mans D.A., van Beersum S.E.C., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A.M., Schuurs-Hoeijmakers J.H.M., de Leeuw N., et al. (2011) C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet., 48, 390–395. [DOI] [PubMed] [Google Scholar]
- 21.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., et al. (2011) TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet., 43, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halbritter J., Bizet A.A., Schmidts M., Porath J.D., Braun D.A., Gee H.Y., McInerney-Leo A.M., Krug P., Filhol E., Davis E.E., et al. (2013) Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet., 93, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz-Perez V.L., Ide S.E., Strom T.M., Lorenz B., Wilson D., Woods K., King L., Francomano C., Freisinger P., Spranger S., et al. (2000) Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet., 24, 283–286. [DOI] [PubMed] [Google Scholar]
- 24.Galdzicka M., Patnala S., Hirshman M.G., Cai J.F., Nitowsky H., Egeland J.A., Ginns E.I. (2002) A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Mol. Genet. Metab., 77, 291–295. [DOI] [PubMed] [Google Scholar]
- 25.Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/), date last accessed March 2015. in press.
- 26.Lek M., Karczewski K., Minikel E., Samocha K., Banks E., Fennell T., O'Donnell-Luria A., Ware J., Hill A., Cummings B. et al. (2015) Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv, doi: http://dx.doi.org/10.1101/030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu A.M., Wang B.L., Niswander L.A. (2005) Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development, 132, 3103–3111. [DOI] [PubMed] [Google Scholar]
- 28.Felsenstein J., Churchill G.A. (1996) A hidden Markov Model approach to variation among sites in rate of evolution. Mol. Biol. Evol., 13, 93–104. [DOI] [PubMed] [Google Scholar]
- 29.Kumar P., Henikoff S., Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1082. [DOI] [PubMed] [Google Scholar]
- 30.Taschner M., Kotsis F., Braeuer P., Kuehn E.W., Lorentzen E. (2014) Crystal structures of IFT70/52 and IFT52/46 provide insight into intraflagellar transport B core complex assembly. J. Cell Biol., 207, 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantagrel V., Silhavy J.L., Bielas S.L., Swistun D., Marsh S.E., Bertrand J.Y., Audollent S., Attie-Bitach T., Holden K.R., Dobyns W.B., et al. (2008) Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet., 83, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cevik S., Sanders A.A.W.M., Van Wijk E., Boldt K., Clarke L., van Reeuwijk J., Hori Y., Horn N., Hetterschijt L., Wdowicz A., et al. (2013) Active Transport and Diffusion Barriers Restrict Joubert Syndrome-Associated ARL13B/ARL-13 to an Inv-like Ciliary Membrane Subdomain. Plos Genet., 9, e1003977. doi: 10.1371/journal.pgen.1003977 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrault I., Halbritter J., Porath J.D., Gerard X., Braun D.A., Gee H.Y., Fathy H.M., Saunier S., Cormier-Daire V., Thomas S., et al. (2015) IFT81, encoding an IFT-B core protein, as a very rare cause of a ciliopathy phenotype. J. Med. Genet., 52, 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beatson S., Ponting C.P. (2004) GIFT domains: linking eukaryotic intraflagellar transport and glycosylation to bacterial gliding. Trends Biochem. Sci., 29, 396–399. [DOI] [PubMed] [Google Scholar]
- 35.Richey E.A., Qin H.M. (2012) Dissecting the sequential assembly and localization of intraflagellar transport particle complex B in Chlamydomonas. Plos One, 7, 7(8):e43118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perkins L.A., Hedgecock E.M., Thomson J.N., Culotti J.G. (1986) Mutant sensory cilia in the nematode Caenorhabditis Elegans. Dev. Biol., 117, 456–487. [DOI] [PubMed] [Google Scholar]
- 37.Tsujikawa M., Malicki J. (2004) Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron, 42, 703–716. [DOI] [PubMed] [Google Scholar]
- 38.Deane J.A., Cole D.G., Seeley E.S., Diener D.R., Rosenbaum J.L. (2001) Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr. Biol., 11, 1586–1590. [DOI] [PubMed] [Google Scholar]
- 39.Girisha K.M., Shukla A., Trujillano D., Bhavani G.S., Hebbar M., Kadavigere R., Rolfs A. (2016) A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet, 90, 536–539. [DOI] [PubMed] [Google Scholar]
- 40.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics, 43, 11.10.1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome. Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider C.A., Rasband W.S., Eliceiri K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]