


Abstract
Histone post-translational modifications, and specific combinations they create, mediate a wide range of nuclear events. However, the mechanistic bases for recognition of these combinations have not been elucidated. Here, we characterize crosstalk between H3T3 and H3T6 phosphorylation, occurring in mitosis, and H3K4me3, a mark associated with active transcription. We detail the molecular mechanisms by which H3T3ph/K4me3/T6ph switches mediate activities of H3K4me3-binding proteins, including those containing plant homeodomain (PHD) and double Tudor reader domains. Our results derived from nuclear magnetic resonance chemical shift perturbation analysis, orthogonal binding assays and cell fluorescence microscopy studies reveal a strong anti-correlation between histone H3T3/T6 phosphorylation and retention of PHD finger proteins in chromatin during mitosis. Together, our findings uncover the mechanistic rules of chromatin engagement for H3K4me3-specific readers during cell division.
INTRODUCTION
Chromatin undergoes extensive reorganization throughout the cell cycle, transitioning from a relatively relaxed state in interphase to a highly condensed state in mitosis and returning back to the interphase state after cell division is completed. Cell cycle progression is highly regulated and correlates with alterations in the structure and dynamics of the nucleosome, the core building block of chromatin. Nucleosomal histone proteins are subject to post-translational modifications (PTMs), including acetylation and methylation of lysine, methylation of arginine, and phosphorylation of serine and threonine. Over 500 histone PTMs have been identified by mass spectrometry analysis, and a number of these modifications, or epigenetic marks, have been shown to modulate chromatin structure and cell cycle specific processes (1).
Methylation of histone H3 at lysine 4 is one of the canonical PTMs (2–4). The trimethylated species (H3K4me3), which is found primarily in active promoters, is implicated in transcriptional regulation (5). Methylation of lysine residues in histones in general, and H3K4me3 in particular, is considered to be a stable mark compared to other PTMs (6,7). The H3K4me3 level remains fairly high and persists throughout the cell cycle owing to coordinating activities of K4-specific methyltransferases, such as the KMT2/SET1 family, and demethylases, such as the KDM5 family, though gene-specific alterations during mitosis have also been observed. A set of epigenetic readers, including a plant homeodomain (PHD) finger, a double tudor domain (DTD) and a double chromodomain (DCD), recognizes the H3K4me3 mark by enclosing K4me3 in an aromatic cage-like binding pocket (8). Preservation of H3K4me3 allows cells to recruit transcription complexes and resume normal gene expression patterns soon after mitosis completion via binding of epigenetic readers, present in these complexes, to H3K4me3-enriched promoters.
In contrast to lysine methylation, phosphorylation of serine and threonine residues in histone H3 fluctuates substantially throughout the cell cycle, rising sharply when cells enter mitosis (9). Phosphorylation of H3T3 (H3T3ph) is generated, or written, by the kinase Haspin early in prophase and is gradually erased by the PP1γ/Repo-Man phosphatase complex in anaphase (10,11). The H3T3ph mark recruits Survivin, a subunit of the chromosomal passenger complex (CPC) to chromatin, which is essential for proper chromosome segregation and modulation of the Aurora B kinase activity (12–15). The protein kinase CβI phosphorylates H3T6 during androgen receptor-dependent gene activation (16), however, a phosphatase responsible for the removal of this mark has not yet been identified.
The close proximity of the T3ph, K4me3 and T6ph residues in histone H3 suggests functional and regulatory interplay between the phosphorylation and methylation modifications that can influence the binding ability of readers of these PTMs. In 2003, Fischle et al. introduced the concept of binary phospho/methyl switches and proposed a role of these switches in the release of readers from chromatin (17). Since then, a substantial body of work has been carried out in support of this concept, which has provided a notable example of H3S10 phosphorylation promoting the dissociation of H3K9me3-binding heterochromatin protein 1 (HP1) from mitotic chromatin (18,19). Co-occurrence of H3T3ph/K4me, H3K9me/S10ph, H3K27me/S28ph and other phospho/methyl modifications has been confirmed experimentally, and combinatorial PTM-specific antibody studies reveal that K4me3 and T3ph can be present on the same histone H3 tail (20–22).
The inhibitory effect of H3T3 phosphorylation on the interaction with H3K4me3 has been reported for DCD of the ATP-dependent chromodomain helicase DNA-binding protein 1 (CHD1) (23) and the PHD finger of the transcription initiation factor TFIID subunit 3 (TAF3) (24). In vivo studies demonstrate that overexpression of Haspin leads to repression of TAF3-mediated gene transcription and a decreased association of the TFIID complex with chromatin, whereas this complex is retained at mitotic chromosomes as a result of Haspin knockdown (24). On-bead histone peptide library screening and peptide microarrays show a considerable attenuation in recognition of H3K4me3 by DTD of the demethylase KDM4A and by the PHD fingers of mixed lineage leukemia 5 (MLL5), death inducer obliterator 3 (DIDO3), inhibitor of growth 2 (ING2) and recombination activating gene 2 (RAG2) when the adjacent H3T3 is phosphorylated, whereas the effect of phosphorylation of H3T6 varies (25–27). The differential reduction in binding activities of these readers suggests that phospho/methyl switches provide a fine-tuned (possibly phase-specific) regulation of the H3K4me3-recognizing proteins, the mechanistic basis of which has not been well understood.
In this work, we detail the structural and molecular mechanisms by which the H3T3ph/K4me3/T6ph switches mediate activities of the H3K4me3-binding PHD and DTD readers. Our results derived from nuclear magnetic resonance (NMR) chemical shift perturbation (CSP) analysis, fluorescence binding assays, pull-downs, peptide microarrays and cell fluorescence microscopy studies reveal a strong anti-correlation between histone H3T3/T6 phosphorylation and the ability of these readers to recognize H3K4me3 or PHD finger-containing proteins to retain in chromatin during mitosis. Our data demonstrate clear mechanistic rules that drive chromatin engagement for H3K4me3-specific readers.
MATERIALS AND METHODS
Cloning and protein purification
The DIDO PHD finger construct (aa 260–325) was cloned from full-length cDNA (Open Biosystems) into pDEST15 using Gateway® cloning technology. The ING1 PHD finger construct (aa 200–279) was cloned into pGex6P1 as described previously (28). The PHF8 PHD finger construct (aa 37–106) in a pGex6P1 vector was a kind gift from Xiaodong Cheng. Proteins were expressed in Rosetta2 (DE3) pLysS or BL21 (DE3) RIL in either Luria Broth or M19 minimal media supplemented with 50 μM ZnCl2 and 15NH4Cl. Cells were induced with 0.5 mM IPTG, grown for 16 h at 18°C and harvested by centrifugation at 6k rpm. Cells were lysed by sonication in lysis buffer containing 25 mM Tris pH 7.5 at 4°C, 150 mM NaCl, 3 mM dithiothreitol, 0.5% Triton X-100, 1 mM PMSF, 5 mM MgCl2, DNaseI and clarified by centrifugation at 15k rpm for 30 min. Proteins were purified on glutathione agarose beads (Pierce® cat# 16101) and the GST-tag cleaved using PreScission protease for at least overnight at 4°C. Cleaved proteins were concentrated into 25 mM Tris pH 7.5, 150 mM NaCl and 3 mM dithiothreitol. Unlabelled proteins were further purified by size exclusion chromatography using a HiPrep 16/60 Sephacryl S-100 column.
Nuclear magnetic resonance
NMR experiments were collected on a Varian INOVA 600 MHz spectrometer equipped with a cryogenic probe. CSP experiments were carried out at 298K using uniformly 15N-labelled PHD finger proteins. 1H, 15N HSQC spectra were recorded in the presence of increasing concentrations of histone H3 peptides (aa 1–12) carrying single, dual or triple modifications. Kd value was calculated by a nonlinear least-squares analysis in Kaleidagraph using the following equation:
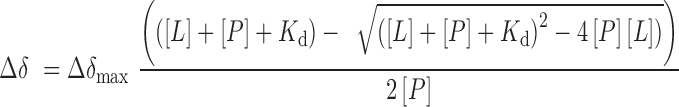 |
where [L] is the concentration of the peptide, [P] is the concentration of the protein, Δδ is the observed normalized chemical shift change and Δδmax is the normalized chemical shift change at saturation, calculated as
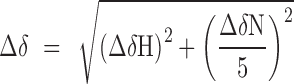 |
where δ is the chemical shift in p.p.m.
Fluorescence binding assay
Tryptophan fluorescence measurements were performed on a Fluoromax-3 spectrofluorometer at room temperature. The samples containing 1 or 2 μM PHD finger protein in 25 mM Tris pH 7.5, 150 mM NaCl, 3 mM DTT and increasing concentrations of histone H3 peptides were excited at 295 nm. Emission spectra were recorded from 320 to 380 nm with a 0.5 nm step size and a 0.5 s integration time, averaged over three scans. The Kd values were determined using a nonlinear least-squares analysis and the equation:
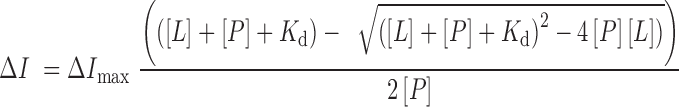 |
where [L] is the concentration of the peptide, [P] is the concentration of the protein, ΔI is the observed change of signal intensity and ΔImax is the difference in signal intensity of the free and bound states. The Kd values were averaged over three separate experiments, with error calculated as the standard deviation between runs.
Confocal scanning fluorescence microscopy
To analyse protein localization, RPE-1 cells were seeded on glass coverslips and grown in DMEM/F12 containing 10% FCS until 70% confluent. For immunofluorescence, cells were rinsed once in PBS, fixed in PBS containing 4% formaldehyde for 5 min and permeabilized for 5 min in PBS containing 0.5% Triton X-100. Blocking was achieved in PBS containing 1% bovine serum albumin (BSA) (1 h, room temperature). For detection of epitopes, cells were incubated with primary antibodies (1 h, room temperature), washed, incubated with secondary antibodies, washed again and mounted in Prolong Gold antifade (Life Technologies, Grand Island, NY). Primary antibodies against H3K4me3 (order number AB8580), H3T3ph (AB78351), H3T6ph (AB14102), PHF8 (AB84779) and DIDO (AB92868) were from Abcam (Cambridge, UK). The anti-ING1 (MAB5758) antibody was from R&D Systems (Minneapolis, MN). PHD proteins were detected with Alexa488-labelled secondary antibodies, and histones with Cy3-labelled secondary antibodies (Jackson Immunoresearch, West Grove, PA, USA). DNA was counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Confocal microscopy was performed using an IX81 microscope (Olympus, Tokio, Japan) with sequential excitation of fluorophores. Representative images corresponding to single confocal layers are shown in the figures. For Haspin inhibition, cells were treated for 24 h with 1 μM (2 μM in Figure 4C) CHR6494 (Tocris Bioscience, Bristol, UK) from a 500 μM stock in dimethylsulfoxide. Controls were treated with solvent only.
Figure 4.

Inhibition of histone H3T3 phosphorylation allows for re-association of the PHD finger-containing proteins with telophase chromatin. (A, B) Random cycling RPE-1 cells were incubated with 1 μM CHR6494 for 24 h and labelled as in Figure 3. Control cells were treated with DMSO solvent only. Histone modifications are depicted in green, DNA in blue and the PHD finger proteins in red. Pro- and telophases for the PHD finger proteins tested are shown in Supplementary Figure S5. (C) RPE-1 cells were incubated with 2 μM CHR6494 for 24 h and labelled as in (A). (D, E) RPE-1 cells were treated as under (A), and labelled with antibodies against DIDO (red) and either H3T6ph (green, D) or H3T3ph (green, E). DNA was counterstained with DAPI (blue). DIDO re-association with chromatin was observed in partially decondensed regions. Three right panels in (E) show magnification of the boxed area in the left panel. Scale bars, 5 μm.
Pull-down assays
GST-tagged reader domains were expressed in SoluBL21 cells (Amsbio) by induction with 1 mM IPTG at 16°C for 20 h. Cells were lysed by sonication in a buffer containing 50 mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 0.1 mM PMSF, 0.5 mg/ml lysozyme, and three units of Universal Nuclease (Pierce) and incubated with glutathione agarose beads (Pierce) for 2–3 h at 4°C. The beads were washed three times with preparation buffer (50 mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 10% glycerol, 1 μM ZnSO4). Proteins were eluted with preparation buffer supplemented with 10 mM reduced glutathione, dialyzed against 3 l of preparation buffer overnight at 4°C and concentrated by centrifugation. Streptavidin coated magnetic beads were pre-equilibrated in peptide binding buffer (50 mM Tris pH 8.0, 300 mM NaCl and 0.1% NP-40) and then incubated with 500 pmols of biotinylated histone peptides for 30 min at room temperature. All peptides were comprised of residues 1–20 of histone H3 and contained a lysine-linked biotin moiety at the C-terminus. Peptide coated beads were washed three times with peptide binding buffer and then incubated with 40 pmols of GST-tagged reader domains for 1.5 h at 4°C in the peptide binding buffer supplemented with 0.5% (w/v) BSA. Beads were washed three times with peptide binding buffer, and proteins were eluted by boiling samples for 10 min in 1xSDS loading buffer. Eluted proteins were resolved via SDS-PAGE and transferred to a PVDF membrane. The membrane was probed with an anti-GST antibody (Sigma) diluted 1:3500 in phosphate buffered saline supplemented with 0.1% (v/v) Tween-20 (PBST) and 5% (w/v) BSA. Following four washes with PBST, membranes were probed with an HRP conjugated anti-rabbit antibody (Sigma) diluted 1:10 000 in PBST + 5% BSA (w/v). Membranes were imaged on a BioRad Chemidoc in Chemiluminescence mode. The input represents 1% of the sample, and streptavidin beads without peptide served as the negative control.
Peptide microarrays
The peptide microarrays were generated and assayed as described previously (29) except that each sub-array contained two triplicate spots of each peptide. The array signal intensity was normalized to the signal intensity corresponding to the histone H3 peptide (residues 1–20) trimethylated at lysine 4 (H3K4me3).
RESULTS AND DISCUSSION
Phosphorylation of H3T3 abrogates binding of the DIDO PHD finger to H3K4me3
Structural analysis of the complexes between PHD fingers and histone H3K4me3 peptide reveals substantial differences in how H3T3 and H3T6 residues of the peptide are bound in the context of solvent accessibility of these threonine residues, hydrogen bonding contacts, electrostatic surface potential of the proteins and the rigidity of the binding pockets (Supplementary Figure S1a). To understand the effect of phosphorylation of the threonine residues, we selected the PHD fingers of DIDO, ING1 and PHF8, which differ significantly from each other from a structural point, for comparative studies (Supplementary Figure S1b). In the DIDO PHD-H3K4me3 complex, the H3T3 residue is buried in the binding pocket and is adjacent to a negatively charged residue of the rigid α-helix of the PHD finger. The H3T3 residue is more solvent accessible and is adjacent to a positively charged residue located in a flexible loop of the ING1 PHD finger. In the PHF8 complex, H3T3 is fully solvent exposed. In all three complexes the H3T6 residue is partially occluded.
To determine the impact of phosphorylation of H3T3 and H3T6 on the interaction of these PHD fingers, we purified 15N-labelled proteins and carried out 1H,15N heteronuclear single quantum coherence (HSQC) titration experiments (Figure 1). Addition of the H3K4me3 peptide (aa 1–12 of H3) induced large CSPs in the PHD fingers, indicating direct binding (left panels in Figure 1B, D and F). A gradual decrease in intensities of crosspeaks corresponding to the free states of the DIDO and PHF8 PHD fingers and a concomitant increase in intensities of crosspeaks corresponding to the bound states pointed to a slow exchange regime on the NMR time scale and tight interaction (Figure 1B and F). The precise values of dissociation constants (Kds) for the DIDO and PHF8 PHD fingers were measured by tryptophan fluorescence and found to be 1.1 μM and 2.5 μM, respectively (Figure 2A and B). Intermediate exchange regime observed upon addition of the H3K4me3 peptide to the ING1 PHD finger suggested a slightly weaker interaction, which was in agreement with previously determined binding affinity of 3.3 μM (28) (Figures 1D and 2A).
Figure 1.

Phosphorylation of histone H3T3 and H3T6 differentially affects binding of the PHD fingers to H3K4me3. (A, C, E) Surface representation of the DIDO, ING1 and PHF8 PHD fingers in complex with H3K4me3 peptide. Hydrogen bonds are shown as dotted red lines and water molecules are shown as blue spheres. PDB codes: 4L7X, 2QIC and 3KV4. (B, D, F) Superimposed 1H, 15N HSQC spectra of the indicated PHD fingers, collected upon titration with H3K4me3 (left), H3T3phK4me3 (middle) and H3K4me3T6ph (right) peptides (aa 1–12 of histone H3). Spectra are colour coded according to the protein:peptide molar ratio (inset). Crosspeaks are labelled for DIDO and ING1 but not for PHF8 as NMR assignments of PHF8 have not been performed.
Figure 2.

The molecular mechanism for the recognition of phosphorylated marks. (A) Binding affinities of the PHD fingers as measured by tryptophan fluorescence (or NMR for the PHF8 PHD-H3T3phK4me3 interaction). aTaken from (26,43). (B) Representative binding curves used to determine the Kd values by fluorescence and NMR. (C) Superimposed 1H,15N HSQC spectra of the ligand-free (black), H3K4me3-bound (orange), H3T3phK4me3-bound (blue) and H3K4me3T6ph-bound (green) ING1 PHD finger. (D) The histogram shows differences in normalized 1H,15N chemical shift changes in backbone amides of the ING1 PHD finger induced by H3T3phK4me3 versus H3K4me3 (blue), and by H3K4me3T6ph versus H3K4me3 (green). (E) Residues that exhibit unique CSPs (H3T3phK4me3 versus H3K4me3 and H3K4me3T6ph versus H3K4me) are coloured blue and green, respectively, on the surface of the ING1 PHD-H3K4me3 complex and labelled. PDB code: 2QIC.
The doubly modified histone H3T3phK4me3 peptide did not cause CSPs in the DIDO PHD finger (Figure 1B, middle panel), implying that the presence of phosphorylated H3T3 completely abrogates the association with H3K4me3. H3T3 is buried in a narrow cleft, flanked by Trp291 and Glu308 of the DIDO PHD finger, and is involved in an intricate network of intermolecular and intrapeptide hydrogen bonds (Figure 1A). The hydroxyl group of T3 is hydrogen bonded to Glu308, the side chain of which protrudes over T3, making this threonine essentially solvent inaccessible in the complex. Furthermore, the side chain of T3 forms a water-mediated hydrogen bond with the side chain amide of H3Q5. Phosphorylation of T3 would not only eliminate these hydrogen bonds but also result in a steric clash. Finally, addition of the negatively charged phosphate group to H3T3 is likely precluded due to electrostatic repulsion with the negatively charged carboxylic moiety of Glu308. On the other hand, the DIDO PHD finger is less sensitive to phosphorylation of H3T6, which caused a ∼5-fold decrease in H3K4me3-binding affinity (Kd = 5.5 μM for H3K4me3T6ph peptide) (Figure 2A). Most likely, two hydrogen bonds involving the hydroxyl group of T6 and the backbone amide of Phe281 and the backbone carbonyl of Asn279 would be disrupted by phosphorylation, leading to a weaker interaction (Figure 1A).
H3T3ph and H3T6ph differentially impact binding of the ING1 and PHF8 PHD fingers to H3K4me3
In contrast to DIDO, phosphorylation of H3T3 and H3T6 affected interaction of the ING1 PHD finger with H3K4me3 to the same extent. Binding affinity of the ING1 PHD finger was reduced ∼7- and ∼5-fold when T3 and T6, respectively, were phosphorylated as measured by tryptophan fluorescence (Figure 2A), and NMR titration experiments further corroborated these results (Figure 1D). Both H3T3phK4me3 and H3K4me3T6ph peptides induced CSPs in the ING1 PHD finger in fast exchange regime, indicative of a reduced association with the doubly modified peptides. The lower sensitivity of this PHD finger to T3ph, as compared to DIDO, is possibly because the side chain of T3 in the ING1 PHD-H3K4me3 complex is more solvent exposed and is not involved in the formation of hydrogen bonds. In place of acidic Glu308 in DIDO, which is located in a rigid α-helix essential for structural stability of the DIDO PHD finger, ING1 contains a basic Lys246 residue in close proximity to T3 (Figure 1C). The positively charged side chain of Lys246 may form favourable electrostatic contacts with the phosphate group of T3. Additionally, Lys246 is found in a flexible loop that may alleviate steric clashes with T3ph by swaying away. The modest inhibitory effect of H3T3ph was comparable to the inhibitory effect of H3T6ph, which is likely the result of disruption of a pair of hydrogen bonds formed between the hydroxyl group of T6 and Ser219 and Gly221 of the PHD finger in the ING1 PHD-H3K4me3 complex.
Although the side chain of T3 is fully solvent accessible in the PHF8 PHD-H3K4me3 complex, phosphorylation of H3T3 affected binding of this protein to H3K4me3 to a great extent. Titration of H3T3phK4me3 peptide to the PHF8 PHD finger produced resonance changes in the intermediate-to-fast exchange regime, which indicated that the H3T3ph mark substantially weakened the binding (Figure 1F, middle panel). Plotting normalized CSPs versus peptide concentration yielded a Kd of 174 μM that is much higher than the Kd for the interaction with H3K4me3. In the PHF8 PHD-H3K4me3 complex, H3T3 is not involved in intermolecular contacts, however, it forms intra-peptide hydrogen bonds, linking the side chain amide of Q5 and the backbone carbonyl group of H3A1 through a water-mediated hydrogen bond (Figure 1E). This stabilization of the peptide conformation could be essential for strong interaction. T3ph could also induce a conformational change in the histone H3 tail by interacting with the adjacent positively charged K4me3 and locking H3 in a conformation that impairs binding of the readers. Furthermore, similarly to DIDO and in contrast to ING1, the PHD finger of PHF8 contains a rigid α-helix next to the H3T3 binding pocket. Therefore, the inflexible nature of the H3K4me3 binding site can play an important role in preventing phosphorylation of H3T3. Unlike H3T3ph, the H3T6ph mark had a less pronounced effect on binding of the PHF8 PHD finger to H3K4me3, reducing the interaction by 2-fold (Figure 2A).
Mechanistic principles for H3T3ph- and H3T6ph-mediated inhibition
Comparison of CSPs observed in the PHD fingers of ING1 or PHF8 upon binding of H3T3phK4me3, H3K4me3T6ph and H3K4me3 peptides and of DIDO upon binding of H3K4me3T6ph and H3K4me3 peptides revealed overall similar patterns, implying that these peptides occupy the same binding site in each protein (Figure 1B, D and F, and Supplementary Figure S2). However, the magnitude of perturbations varied for several amide resonances, indicating a change in local chemical environment of these amides due to formation of the complexes with each peptide. In addition, a few crosspeaks shifted in different directions upon binding of the H3K4me3T6ph peptide. For example, in the ING1 PHD finger, the amide resonances of Gly221, Glu222, Ile224 and Cys226 were uniquely perturbed by H3T3phK4me3 and the amide resonances of Val218, Ser219, Tyr220, Gly221 and Glu222 were uniquely perturbed by H3K4me3T6ph as compared to CSPs caused by the unphosphorylated peptide (Figure 2C and D). Mapping the uniquely perturbed residues onto the structure of the H3K4me3-bound ING1 PHD finger reveals well-defined shallow pockets where phosphorylated H3T3 and H3T6 are bound (Figure 2E). The Ser219 and Gly221 residues that form hydrogen bonds with the hydroxyl moiety of T6 in the ING1 PHD-H3K4me3 complex were most sensitive to the presence of the phosphate group on T6.
Interestingly, the changes in direction of CSPs were observed in all three PHD fingers and only due to binding of H3K4me3T6ph (indicated by dotted ovals in Figure 1B, D and F). It has been shown that the PHD fingers form critical contacts with the A1-R2-T3-K4me3 fragment of the histone tail in the PHD-H3K4me3 complexes (8), which may explain why this domain is more sensitive to phosphorylation of T3 and is less sensitive to phosphorylation of T6. Phosphorylation of H3T6 may not affect binding of the N-terminal H3A1-R2-T3 residues while disturbing priming of the histone residues in close vicinity to T6, which would result in a binding mode slightly different from that of seeing for the unphosphorylated H3K4me3 peptide.
DIDO, ING1 and PHF8 are excluded from H3K4me3-containing chromatin during mitosis
Reduced affinity of the PHD fingers for H3K4me3 in the presence of T3ph or T6ph suggested a role for the H3T3ph/K4me3/T6ph phospho/methyl switches in mitosis. To examine the effect of phosphorylation on binding of the PHD fingers to H3K4me3 in a cellular context, random cycling RPE-1 cells were labelled with antibodies against ING1, DIDO or PHF8, and studied by confocal fluorescence microscopy. Antibodies against H3K4me3, H3T3ph and H3T6ph were used to identify mitotic phases. All three PHD finger-containing proteins co-localized with H3K4me3 during interphase, with some PHF8 also appearing in the cellular periphery (Figure 3 and Supplementary Figure S3). Even though the H3K4me3 mark persisted throughout mitosis, the three proteins dissociated from chromatin in prophase and remained detached from chromatin until telophase. Some small differences in the pattern of localization for DIDO were due to its association with microtubules (30). Dissociation from chromatin coincided with the appearance of H3T3ph and H3T6ph, which was very robust from prophase until metaphase; in the second half of mitosis, from anaphase onward, phosphorylation slowly decreased but remained discernible until late telophase. In agreement with the in vitro biochemical results discussed above, the cell data also suggested that T3ph and T6ph might have a role in the disengagement of the PHD finger proteins from chromatin in vivo.
Figure 3.

Exclusion of the PHD finger-containing proteins from mitotic chromatin coincides with phosphorylation of histone H3T3 and H3T6. Random cycling RPE-1 cells were labelled for ING1 and counterstained for H3K4me3, H3T3ph and H3T6ph. Histone modifications, DNA and ING1 are green, blue and red, respectively. Scale bars, 5 μm. Mitotic phases for DIDO and PHF8 are given in Supplementary Figure S3.
Although H3T3ph affected binding of the DIDO, ING1 and PHF8 PHD fingers to H3K4me3 to a various extent in vitro, all three full-length proteins were efficiently expelled from mitotic chromatin, and we did not detect apparent differences in their expulsion rates. These results indicate that even a 7-fold reduction in binding affinity of the ING1 PHD finger for H3K4me3 upon H3T3 phosphorylation is sufficient to dismiss the protein in vivo. The dissociation of the PHD finger proteins from chromatin can also be promoted if T3 and T6 are phosphorylated on the same histone tail, because in vitro, binding of the ING1 PHD finger to the triple PTM-containing peptide, H3T3phK4me3T6ph, was markedly reduced (Supplementary Figure S4). We also cannot exclude the possibility that other yet-identified processes associated with H3T3 phosphorylation may condense chromatin promoting the rapid exclusion of the PHD finger proteins.
Inhibition of H3T3ph increases the re-association of DIDO and ING1 with chromatin
The H3T3ph mark is produced by the mitotic kinase Haspin (10). To investigate whether reduction in H3T3ph levels could lead to retention of the PHD finger proteins at chromatin, we treated RPE-1 cells with 1 μM of the CHR6494 Haspin inhibitor (31) and visualized DIDO and ING1 localization using confocal fluorescence microscopy 24 h post-treatment (Figure 4A, B and Supplementary Figure S5). Although Haspin inhibition resulted in pronounced reduction of H3T3ph in prophase, DIDO and ING1 were mainly excluded from chromatin and only a portion of these proteins remained attached to chromatin in the inhibitor-treated cells. Retention is more clearly visible in cells treated with a higher inhibitor concentration (2 μM); these cells are unable to complete mitosis but instead show co-localization of DIDO and ING1 on poorly condensed H3K4me-positive chromatin (Figure 4C and Supplementary Figure S6).
Strikingly, we observed an earlier re-association of DIDO and ING1 with chromatin at the end of cell division (in telophase) in the inhibitor-treated cells as compared to untreated cells (Figure 4A, B and Supplementary Figure S5). These data substantiated the importance of H3T3 phosphorylation in mediating the repulsion of DIDO and ING1 from chromatin. The H3T6ph mark underwent minor suppression in the inhibitor-treated cells, implying that this PTM does not contribute significantly to the observed effect; however, we cannot rule out the possibility that mitotic chromatin would retain the PHD finger proteins to a higher degree if phosphorylation of both H3T3 and H3T6 is blocked (Figure 4D). Since the kinases that mediate T6 phosphorylation in mitosis remain to be identified, testing this hypothesis by small molecule inhibition is unfeasible at the present time.
Notably, wherever we observed re-association of the PHD finger proteins with chromatin in telophase, this was in the regions characterized by partial decondensation and absence of residual T3ph or T6ph (Figure 4E and data not shown). As shown in Figure 4E, chromatin underwent local structural rearrangements rather than a uniform decondensation in telophase, and DIDO localized primarily to more open decondensed regions. In agreement with the idea that chromatin condensation and PHD finger protein binding to H3K4me3 are mutually exclusive, the combined action of complete dephosphorylation and increased protein concentration can also stimulate further decondensation. These data suggest that phosphorylation of H3T3/T6 not only correlates with initial chromatin condensation in prophase but might also be important for the maintenance of a condensed state until the end of mitosis.
A variable degree anti-correlation effect is observed for other H3 readers
To establish whether the effect of the H3T3ph/K4me3/T6ph switching is conserved and regulates binding activities of other proteins known to associate with methylated histone H3 tail, we examined an extended set of H3K4me3-readers by pull-down assays, microarrays and NMR (Figure 5). This includes the PHD fingers of BPTF, DIDO, ING1, PHF8, Rag2 and Taf3, the DTD module of KDM4A, and the DCD module of CHD1, as well as a H3T3ph-reader, the BIR domain of Survivin. As shown in Figure 5A, binding of all PHD fingers to H3K4me3 was considerably compromised by phosphorylation of T3. Phosphorylation of T6 had a lesser effect, although it notably reduced binding of the Rag2 PHD finger. In full agreement with published data, symmetrical dimethylation of R2 augmented the Rag2 interaction, whereas asymmetrical dimethylation of R2 diminished binding of the Taf3 PHD finger (32,33). The peptide microarrays further corroborated the pull-down results, showing a strong inhibitory effect of H3T3ph for each PHD finger tested in this study, whereas the inhibitory effect of H3T6ph was milder (Figure 5B and Supplementary Figure S7).
Figure 5.

The phospho/methyl switching mechanism is conserved. (A) Peptide pulldown experiments comparing the binding of readers to the indicated H3 peptides (residues 1–20). R2me2s and R2me2a are symmetrically and asymmetrically, respectively, dimethylated arginine 2. All peptides were biotinylated at the C-terminus and were preincubated with streptavidin-coated beads prior to addition of reader domains. Beads without peptide served as the negative control. (B) Binding of readers to the indicated peptides was probed by peptide microarrays. Signal intensity was normalized to the signal intensity for binding to H3K4me3 peptide. (C) Superimposed 1H,15N HSQC spectra of DTD of KDM4A collected upon titration with indicated peptides. Spectra are colour coded according to the protein:peptide molar ratio (inset).
Like the PHD fingers, other readers of the H3K4me3 mark, DTD of KDM4A and DCD of CHD1, were less sensitive to the H3T6ph modification, however H3T3ph substantially decreased binding of DTD and abrogated binding of DCD to H3K4me3 (Figure 5A and C). The BIR domain of Survivin that prefers histone H3 tail phosphorylated at T3 was capable of tolerating K4 methylation to some extent (Figure 5A), supporting previously determined binding affinities of this domain for H3T3ph and H3T3phK4me3 (5 and 10 μM, respectively) (14). Together, these results indicate that readers of the histone H3 tail can differentially recognize the H3T3ph-K4me3-T6ph combination of PTMs, which can be a prerequisite for eliciting distinct biological responses.
CONCLUDING REMARKS
Histone PTMs, and the patterns they create, regulate a wide range of nuclear events. In this study, we assessed the impact of H3T3 and H3T6 phosphorylation, occurring in mitosis, on binding of effector proteins to H3K4me3—a mark associated with promoters of actively transcribed genes. We found that H3T3ph and H3T6ph correlate with chromatin condensation and anti-correlate with binding of the PHD finger-containing proteins to chromatin, even though H3K4me3 persists throughout mitosis. Our results suggest that phosphorylation, particularly of H3T3, disrupts the interaction of the PHD fingers with H3K4me3 in vitro and can play a role in repulsion of their host proteins from mitotic chromatin.
Trimethylation of H3K4 is a well-established modification found near transcription start sites (TSS) of active genes (5). To activate or maintain gene expression, H3K4me3 recruits transcriptional and chromatin-remodeling complexes harbouring H3K4me3-specific readers to TSS. H3T3ph can act as a repressive PTM, blocking binding of readers and preventing association of these complexes with chromatin.
Although transcription programs are mostly halted during cell division, they must be re-activated in daughter cells to maintain cellular identity. Preservation of H3K4me3 in mitotic chromatin (6) suggests that this modification may function, at least in part, as a bookmarking PTM (34–36), helping to maintain epigenetic memory during mitosis and faithfully restore gene expression patterns upon exit from mitosis. H3T3ph can prevent demethylation of H3K4me3 through hindering the association of H3K4me3-specific demethylases. Consequently, in addition to being a repressive PTM, H3T3ph can also play a role in passing on inheritable information about transcriptional and other chromatin-related programs from mother to daughter cells by shielding H3K4me3. Thus, the phospho/methyl switching may be one of the fundamental mechanisms used by the cell to preserve epigenetic information during cell division to facilitate re-activation of precise transcription programs in progeny cells.
Studies using combinatorial modification-specific antibodies have demonstrated the presence of T3ph and K4me3 on the same histone tail (21), however, how these marks are spatio-temporally controlled and what is the chain of sequential events leading to the formation of this dual PTM remains unclear. The H3T3ph mark, generated (or written) by the Haspin kinase, is removed by the PP1γ/Repo-Man phosphatase complex and read by the BIR domain of Survivin, with the latter being required for chromatin localization of CPC and subsequent phosphorylation of H3S10 (10–15) (Figure 6A). The in vitro kinase activity of Haspin on histone peptides is reduced by 28-, 2.6- and 1.2-fold when the adjacent H3K4 residue in the peptides is tri-, di- and monomethylated, respectively (37,38). The structure of the Haspin kinase domain in complex with the substrate H3 peptide helps to explain this strong selectivity (39). The side chain of unmodified K4 of the histone peptide is partially buried and it makes electrostatic and hydrogen bonding contacts with two aspartic acids of the protein (Figure 6B). Although there is enough space in the pocket to accommodate one or two methyl groups of methyllysine, priming of a trimethylated species would be less tolerable due to steric hindrance and loss of hydrogen bonds. While there is no information available on the impact of K4 methylation on the PP1γ/Repo-Man phosphatase complex activity, trimethylation of H3K4 appears to have a small effect on reading of this mark by Survivin, diminishing binding of its BIR domain to H3T3ph 2-fold (14). The side chain of histone K4 in the crystal structure of the BIR-H3T3ph complex is mostly solvent accessible and, therefore, could be methylated without compromising the interaction of Survivin with H3T3ph (13–15). Together, these reports suggest that K4 methylation does not prevent the recruitment of CPC to mitotic chromosomes.
Figure 6.

Crosstalk between histone H3T3ph and H3K4me3 PTMs. (A) A cartoon depicting writers, readers and erasers of H3T3ph and H3K4me3. (B) A close view of the histone-binding sites in the indicated complexes. PDB codes: 4OUC, 2W5Z, 3UIG, 4L7X and 2V1D. (C) A schematic showing potential transitions and interactions involving H3T3phK4me.
Since Haspin activity is considerably weakened, though not abolished, by H3K4me3 and not by H3K4me2/me1, it remains to be determined whether the kinase activity is sufficient to generate H3T3phK4me3 through phosphorylating H3K4me3 directly, or H3K4me3 species has to be demethylated first and then phosphorylated to H3T3phK4me2/me1 and methylated to H3T3phK4me3 (Figure 6C). It is also unclear whether K4-specific lysine methyltransferases (KMTs) would efficiently methylate in the presence of T3ph. The crystal structure of the catalytic SET domain of MLL1 (one of the KMTs that generate H3K4me3) in complex with histone H3 peptide shows that the hydroxyl group of T3 is partially masked, which may account for a ∼5-fold decrease in methyltransferase activity of MLL1 for the histone peptide substrate phosphorylated at T3 (40) (Figure 6B). We do not yet know the effect of H3T3 phosphorylation on the catalytic activity of the KDM5 family of K4me2/me3-demethylases, however, H3T6 phosphorylation has been shown to inhibit activity of the H3K4me2-specific demethylase, LSD1 (also known as KDM1A) (16). In the KDM1A-H3K4M structure, the hydroxyl group of H3T3 is completely buried in a deep negatively charged binding pocket (41), thereby implying that phosphorylation of T3 is most likely prohibited. These observations support the role of H3T3 phosphorylation as a shielding PTM.
Our data demonstrate a robust anti-correlation between T3 phosphorylation and binding of PHD finger proteins to H3K4me3-containing mitotic chromatin. However, many reader-containing proteins are components of multisubunit enzymatic complexes, which can further modify the local chromatin landscape through adding or removing PTMs. For example, ING1 is a component of the Sin3A/HDAC deacetylase complex, whereas PHF8 is a H3K9me-specific KDM. This adds yet another layer of complexity to our efforts of establishing and generalizing epigenetic mechanisms. Furthermore, mechanisms other than those that are epigenetic-driven may contribute to preventing protein binding to mitotic chromatin. The distribution of ING1 and DIDO in dividing cells, concentrated on either side of the chromosome pool, suggests interaction with the mitotic spindle. A small pool of DIDO interacts with tubulin in interphase (30), and reducing the affinity for H3K4me3 by T3 phosphorylation would shift equilibrium towards these interactions. Additionally, it has been reported that PHF8 can dissociate from chromatin in prophase due to CDK1-mediated phosphorylation of PHF8 serine residues (42), hence modifiable residues in other PHD finger proteins could also contribute to their translocation from chromatin during mitosis.
Supplementary Material
Acknowledgments
We thank P. Pena and R. Hom for help with experiments and X. Cheng for kindly providing cDNA of PHF8.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01 GM106416 and GM100907 to T.G.K., GM110058 to B.D.S.]; K.V.W. was supported in part by Spanish Ministry of Economics and Competitiveness [SAF2013-42289-R]. J.G. is an NIH NRSA predoctoral fellow (F31 CA189487). C.M.G. is a CSIC predoctoral fellow (FPI BES-2014-068580) and was supported in part by Spanish Ministry of Economics and Competitiveness [SAF2013-42289-R]; UNC Lineberger Cancer Center Postdoctoral Fellowship Award (to S.A.S.). National Institute of General Medical Sciences [Award Number: ‘100907’, ‘106416’, ‘110058’]. Funding for open access charge: R01 GM106416.
Conflict of interest statement. None declared.
REFERENCES
- 1.Huang H., Lin S., Garcia B.A., Zhao Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015;115:2376–2418. doi: 10.1021/cr500491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black J.C., Van Rechem C., Whetstine J.R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wozniak G.G., Strahl B.D. Hitting the ‘mark’: interpreting lysine methylation in the context of active transcription. Biochim. Biophys. Acta. 2014;1839:1353–1361. doi: 10.1016/j.bbagrm.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Strahl B.D., Ohba R., Cook R.G., Allis C.D. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl Acad. Sci. U.S.A. 1999;96:14967–14972. doi: 10.1073/pnas.96.26.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruthenburg A.J., Allis C.D., Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 6.Kouskouti A., Talianidis I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 2005;24:347–357. doi: 10.1038/sj.emboj.7600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barth T.K., Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem. Sci. 2010;35:618–626. doi: 10.1016/j.tibs.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Musselman C.A., Kutateladze T.G. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawicka A., Seiser C. Histone H3 phosphorylation - a versatile chromatin modification for different occasions. Biochimie. 2012;94:2193–2201. doi: 10.1016/j.biochi.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai J., Sultan S., Taylor S.S., Higgins J.M. The kinase Haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005;19:472–488. doi: 10.1101/gad.1267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian J., Lesage B., Beullens M., Van Eynde A., Bollen M. PP1/Repo-man dephosphorylates mitotic histone H3 at T3 and regulates chromosomal aurora B targeting. Curr. Biol. 2011;21:766–773. doi: 10.1016/j.cub.2011.03.047. [DOI] [PubMed] [Google Scholar]
- 12.Kelly A.E., Ghenoiu C., Xue J.Z., Zierhut C., Kimura H., Funabiki H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science. 2010;330:235–239. doi: 10.1126/science.1189505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeyaprakash A.A., Basquin C., Jayachandran U., Conti E. Structural basis for the recognition of phosphorylated histone h3 by the survivin subunit of the chromosomal passenger complex. Structure. 2011;19:1625–1634. doi: 10.1016/j.str.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Du J., Kelly A.E., Funabiki H., Patel D.J. Structural basis for recognition of H3T3ph and Smac/DIABLO N-terminal peptides by human survivin. Structure. 2012;20:185–195. doi: 10.1016/j.str.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niedzialkowska E., Wang F., Porebski P.J., Minor W., Higgins J.M., Stukenberg P.T. Molecular basis for phosphospecific recognition of histone H3 tails by Survivin paralogs at inner centromeres. Mol. Biol. Cell. 2012;23:1457–1466. doi: 10.1091/mbc.E11-11-0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metzger E., Imhof A., Patel D., Kahl P., Hoffmeyer K., Friedrichs N., Muller J.M., Greschik H., Kirfel J., Ji S., et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010;464:792–796. doi: 10.1038/nature08839. [DOI] [PubMed] [Google Scholar]
- 17.Fischle W., Wang Y., Jacobs S.A., Kim Y., Allis C.D., Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischle W., Tseng B.S., Dormann H.L., Ueberheide B.M., Garcia B.A., Shabanowitz J., Hunt D.F., Funabiki H., Allis C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 19.Hirota T., Lipp J.J., Toh B.H., Peters J.M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- 20.Garcia B.A., Barber C.M., Hake S.B., Ptak C., Turner F.B., Busby S.A., Shabanowitz J., Moran R.G., Allis C.D., Hunt D.F. Modifications of human histone H3 variants during mitosis. Biochemistry. 2005;44:13202–13213. doi: 10.1021/bi050906n. [DOI] [PubMed] [Google Scholar]
- 21.Markaki Y., Christogianni A., Politou A.S., Georgatos S.D. Phosphorylation of histone H3 at Thr3 is part of a combinatorial pattern that marks and configures mitotic chromatin. J. Cell Sci. 2009;122:2809–2819. doi: 10.1242/jcs.043810. [DOI] [PubMed] [Google Scholar]
- 22.Sawicka A., Seiser C. Sensing core histone phosphorylation - a matter of perfect timing. Biochim. Biophys. Acta. 2014;1839:711–718. doi: 10.1016/j.bbagrm.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flanagan J.F., Mi L.Z., Chruszcz M., Cymborowski M., Clines K.L., Kim Y., Minor W., Rastinejad F., Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 24.Varier R.A., Outchkourov N.S., de Graaf P., van Schaik F.M., Ensing H.J., Wang F., Higgins J.M., Kops G.J., Timmers H.T. A phospho/methyl switch at histone H3 regulates TFIID association with mitotic chromosomes. EMBO J. 2010;29:3967–3978. doi: 10.1038/emboj.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garske A.L., Oliver S.S., Wagner E.K., Musselman C.A., LeRoy G., Garcia B.A., Kutateladze T.G., Denu J.M. Combinatorial profiling of chromatin binding modules reveals multisite discrimination. Nat. Chem. Biol. 2010;6:283–290. doi: 10.1038/nchembio.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gatchalian J., Futterer A., Rothbart S.B., Tong Q., Rincon-Arano H., Sanchez de Diego A., Groudine M., Strahl B.D., Martinez A.C., van Wely K.H., et al. Dido3 PHD modulates cell differentiation and division. Cell Rep. 2013;4:148–158. doi: 10.1016/j.celrep.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ali M., Rincon-Arano H., Zhao W., Rothbart S.B., Tong Q., Parkhurst S.M., Strahl B.D., Deng L.W., Groudine M., Kutateladze T.G. Molecular basis for chromatin binding and regulation of MLL5. Proc. Natl Acad. Sci. U.S.A. 2013;110:11296–11301. doi: 10.1073/pnas.1310156110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peña P.V., Hom R.A., Hung T., Lin H., Kuo A.J., Wong R.P., Subach O.M., Champagne K.S., Zhao R., Verkhusha V.V., et al. Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J. Mol. Biol. 2008;380:303–312. doi: 10.1016/j.jmb.2008.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothbart S.B., Krajewski K., Nady N., Tempel W., Xue S., Badeaux A.I., Barsyte-Lovejoy D., Martinez J.Y., Bedford M.T., Fuchs S.M., et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012;19:1155–1160. doi: 10.1038/nsmb.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez de Diego A., Alonso Guerrero A., Martinez A.C., van Wely K.H. Dido3-dependent HDAC6 targeting controls cilium size. Nat. Commun. 2014;5:3500. doi: 10.1038/ncomms4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huertas D., Soler M., Moreto J., Villanueva A., Martinez A., Vidal A., Charlton M., Moffat D., Patel S., McDermott J., et al. Antitumor activity of a small-molecule inhibitor of the histone kinase Haspin. Oncogene. 2012;31:1408–1418. doi: 10.1038/onc.2011.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan C.C., Matthews A.G., Jin Y., Chen C.F., Chapman B.A., Ohsumi T.K., Glass K.C., Kutateladze T.G., Borowsky M.L., Struhl K., et al. Histone H3R2 symmetric dimethylation and histone H3K4 trimethylation are tightly correlated in eukaryotic genomes. Cell Rep. 2012;1:83–90. doi: 10.1016/j.celrep.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermeulen M., Mulder K.W., Denissov S., Pijnappel W.W., van Schaik F.M., Varier R.A., Baltissen M.P., Stunnenberg H.G., Mann M., Timmers H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 34.Zaidi S.K., Young D.W., Montecino M.A., Lian J.B., van Wijnen A.J., Stein J.L., Stein G.S. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat. Rev. Genet. 2010;11:583–589. doi: 10.1038/nrg2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao R., Nakamura T., Fu Y., Lazar Z., Spector D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011;13:1295–1304. doi: 10.1038/ncb2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blobel G.A., Kadauke S., Wang E., Lau A.W., Zuber J., Chou M.M., Vakoc C.R. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol. Cell. 2009;36:970–983. doi: 10.1016/j.molcel.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eswaran J., Patnaik D., Filippakopoulos P., Wang F., Stein R.L., Murray J.W., Higgins J.M., Knapp S. Structure and functional characterization of the atypical human kinase Haspin. Proc. Natl Acad. Sci. U.S.A. 2009;106:20198–20203. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han A., Lee K.H., Hyun S., Lee N.J., Lee S.J., Hwang H., Yu J. Methylation-mediated control of aurora kinase B and Haspin with epigenetically modified histone H3 N-terminal peptides. Bioorg. Med. Chem. 2011;19:2373–2377. doi: 10.1016/j.bmc.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 39.Maiolica A., de Medina-Redondo M., Schoof E.M., Chaikuad A., Villa F., Gatti M., Jeganathan S., Lou H.J., Novy K., Hauri S., et al. Modulation of the chromatin phosphoproteome by the Haspinprotein kinase. Mol. Cell. Proteomics. 2014;13:1724–1740. doi: 10.1074/mcp.M113.034819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Southall S.M., Wong P.S., Odho Z., Roe S.M., Wilson J.R. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol. Cell. 2009;33:181–191. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 41.Forneris F., Binda C., Adamo A., Battaglioli E., Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 42.Liu W., Tanasa B., Tyurina O.V., Zhou T.Y., Gassmann R., Liu W.T., Ohgi K.A., Benner C., Garcia-Bassets I., Aggarwal A.K., et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peña P.V., Davrazou F., Shi X., Walter K.L., Verkhusha V.V., Gozani O., Zhao R., Kutateladze T.G. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.