


Abstract
While GWAS identify many disease-associated SNPs, using them to decipher disease mechanisms is hindered by the difficulty in mapping SNPs to genes. Most SNPs are in non-coding regions and it is often hard to identify the genes they implicate. To explore how far the SNP may be from the affected genes we used a pathway-based approach. We found that affected genes are often up to 2 Mbps away from the associated SNP, and are not necessarily the closest genes to the SNP. Existing approaches for mapping SNPs to genes leave many SNPs unmapped to genes and reveal only 86 significant phenotype-pathway associations for all known GWAS hits combined. Using the pathway-based approach we propose here allows mapping of virtually all SNPs to genes and reveals 435 statistically significant phenotype-pathway associations. In search for mechanisms that may explain the relationships between SNPs and distant genes, we found that SNPs that are mapped to distant genes have significantly more large insertions/deletions around them than other SNPs, suggesting that these SNPs may sometimes be markers for large insertions/deletions that may affect large genomic regions.
INTRODUCTION
Mapping SNPs to molecular process is crucial for understanding disease
A first step towards making molecular sense of GWAS is to map phenotype-associated SNPs to genes. While a SNP within a coding region is usually assumed to affect that gene, the majority of SNPs fall in non-coding regions and many of them are intergenic (1–3). This makes it difficult to determine which genes they affect (4) and, by extension, learn of their molecular contribution to the phenotype. Moreover, it has been shown that SNPs may occasionally affect distant genes (5), which makes the mapping of SNPs to genes even more challenging. Intergenic SNPs are often found in GWAS to be significantly associated with the studied phenotype (2). While linkage disequilibrium (LD) may help link some of these SNPs to nearby genes (2), many phenotype-associated SNPs are not in LD with any gene. It is a common practice to automatically map SNPs to the closest gene provided that it is close enough (see for example the Framingham heart study (6), which initiated many of the practices in GWAS), but there is no consensus as to the distance cutoff that should allow such mapping. The widely used SNP database dbSNP (7) uses an upstream cutoff of 2 Kbps and a downstream cutoff of 0.5 Kbps to map a SNP to a gene. A few studies have used larger cutoffs of up to 100 Kbps (8) and even 500 Kbps(9–12). But even these cutoffs leave many GWAS hits with no associations to any gene. Moreover, acknowledging that SNPs may affect distant genes (e.g. in the case of enhancers and repressors (9)) the common practice of mapping SNPs to the nearest gene may lead to false SNP-gene mapping.
Molecular pathways allow assessment of SNP-gene relationships
Results of GWAS have been increasingly used to associate phenotypes with pathways (13). This is based on the assumption that SNPs that are associated with the same phenotype are expected to affect the same biological processes and hence the same pathways. We recently introduced a framework that assesses associations between phenotypes and pathways based on SNPs and the genes to which they are mapped (14). Briefly, our framework determines whether the genes that are mapped to SNPs associated with a certain phenotype fall within the same pathway more than expected by chance. Here, we used this framework to statistically assess how far from the SNPs the causative genes may be. Specifically, for each phenotype, we tentatively assigned all associated SNPs to genes that are within a certain distance cutoff (e.g. 10 Kbps). We then checked whether these genes cluster into curated pathways more than expected by chance. Then, we repeated the same analysis, this time with a larger SNP-gene distance cutoff (e.g. 50 Kbps). In this second iteration, we map more SNPs to more genes (as more intergenic SNPs could be mapped to a gene). If these additional, more distant genes are mostly irrelevant for the phenotype, we expect that now the clustering of genes into pathways will be weaker than when considering only closer genes. However, if these more distant genes include many genes that affect the phenotype we expect that the clustering of these genes into pathways will be even stronger. We repeated this analysis for increasingly large distance cutoffs. Assigning SNPs to genes that are up to 200 Kbps away increased the number of significant phenotype-pathway associations. Beyond this distance the number started to diminish. However, the associations remained significant even when considering only genes that are up to 2 Mbps away from the SNP. Reviewing specific phenotypes, we found that in some cases associations to relevant molecular pathways were identified only once we allowed linking SNPs to very distant genes.
Possible association of SNPs and insertions/deletions (indels)
We compared the cases in which assigning SNPs to distant genes revealed more significant phenotype-pathway associations to the cases in which assigning SNPs to distant genes did not increase the number of associations. We found that there are significantly more indels in the chromosomal intervals between SNPs and genes that are associated through pathways compared to similar chromosomal segments between SNP and genes that are not associated. This suggests that in some cases, SNPs may be markers for large indels, which may affect large genomic regions and distant genes.
MATERIALS AND METHODS
Data
Phenotype-SNP associations were extracted from GWAS data in the NHGRI GWAS catalog (15). It contains manually curated entries of published GWAS, in which SNPs were associated with diseases, phenotypes, and traits. Unless otherwise stated we used the version from www.ebi.ac.uk/gwas on 9 September 2015. Gene symbols were taken from Genenames (16), while the genomic locations of SNPs and genes were taken from the UCSC genome browser (17). Biological pathways and their associated genes were taken from the KEGG pathway database (release 53) (18), and from ConsensusPathDB (CPDB) (19). From CPDB we took only KEGG pathways. Genomic indels were taken from DGV (20), a database of genomic structure variants (SV). These were used for the analysis of whether more indels fall in phenotype-associated SNP-gene regions. For the analysis of indels around SG regions (see below) we used GWAS Catalog version downloaded from www.genome.gov/gwastudies on 11/2011 with merged GWAS entries of the same phenotype as in (14).
Association of phenotypes to pathways
We define a SNP as a ‘phenotype-associated SNP’ if it is associated with a phenotype in the NHGRI GWAS catalog (see (15)). To determine whether a pathway is significantly associated with a phenotype we assess whether the genes of that phenotype fall within that pathway significantly more than expected by chance. The next paragraph describes the background model on which we determine the number of genes that are expected to cluster into a pathway by chance.
Assessing significance of phenotype-pathway associations
Assessment of the significance of the association between a phenotype and a pathway in a given distance cutoff x (e.g. 10 Kbps, 200 Kbps), hereby referred to as ‘cutoff’, is done as in (14), with a slight variation regarding the background model. Briefly, for each phenotype, with s SNPs associated with it according to GWAS, the number of phenotype-associated genes, denoted g, was recorded. For a distance cutoff of x, g is number of genes that are less than x bps from any of the s SNPs. We also recorded how many of these g genes fall into the same pathway. SNPs from GWAS have more genes in their vicinity compared to all SNPs (Figure 1), to account for that the expected number by chance was assessed by repeatedly picking s random SNPs from GWAS, mapping them to the genes that are less than x bps away and recording how many of these genes fall into the same pathway (note that in (14) genes were chosen randomly, and not SNPs). For each phenotype-pathway pair, this was repeated 1000 times. A phenotype is said to be significantly associated with a pathway with P-value <0.001, if <0.001 of these random resamplings resulted in an equal or greater number of genes which clustered into the pathway.
Figure 1.

Distribution of SNPs according to their proximity to the nearest gene. Bars depict percentage of SNPs that have a gene within a certain distance from them. Blue bars represent all known SNPs, the red bars represent only SNPs that were found by GWAS to be associated with phenotypes. The X-axis represents distance from the SNPs the Y-axis represents the percentage of SNPs that have a gene within that distance from them.
The random model should test whether genes that are close to phenotype-associated SNPs cluster into pathways more than expected by chance. However, it should take into account that neighboring genes on the chromosome might cluster into the same pathway regardless of the SNPs. We define a ‘segment’ as a stretch of contiguous base pairs encompassing one or more SNPs and the DNA around them up to a given distance cutoff. For example, for a phenotype with three associated SNPs in the following chromosomal locations: 9000, 35,000 and 40,000, on chromosome 3, using a distance cutoff of 10 Kbps, we should extend a segment 10 Kbps in each direction around each of these SNPs. In practice, we will end up with two segments, the first at 0–19,000, and the second at 25,000–50,000. Note that given the proximity of the first SNP to the end of the chromosome the effective size of the segment around it is 1.9 Kbps and not 2 Kbps. Given the proximity of the other two SNPs to each other their segments partially overlap to yield one joint segment of 2.5 Kbps rather than two separate segments of 2 Kbps each. Thus, these three SNPs highlight two chromosomal segments, one of 1.9 Kbps and one of 2.5 Kbps. To generate a random model we now select two segments of the same size as the two segments around the SNPs. To avoid biases, we restrict our selection to segments that surround reported SNPs. In particular, we first randomly chose two segments that are centered by a SNP, one that is 1.9 Kbps long and one that is 2.5 Kbps long. Next, in order to account for the original number of SNPs, the second segment, originally containing two SNPs, was divided by two arbitrary ‘SNPs’ distributed equally along the segment such that when applying the cutoff their combined segment will span 2.5 Kbps. As described in (14), the framework accounts for multiple testing. Briefly, let n be a number of phenotypes with associated SNPs that were found using the resampling procedure above to be significantly associated with pathways. Since the P-value for each phenotype was evaluated separately, one needs to assess the P-value of the overall result for all phenotypes. To this end, for each phenotype from n, a pseudo phenotype was created by randomly picking segments, as described above, corresponding in number and length to the original phenotype segments (note that in (14) pseudo phenotypes were made by randomly choosing genes, not segments). Then, the resampling procedure above is repeated for each of these n sets, to determine whether this pseudo phenotype turns out to pass the significance assessment described above. The number of ‘significant’ phenotype-pathway associations for each of the pseudo phenotypes is recorded. This is repeated 100 times, to yield a P-value for obtaining a certain number of significant phenotype-pathway associations for all phenotypes. The red bars/line in Figure 2 represent the median of these resampling procedures. Error bars on the red bars/line represent standard deviations.
Figure 2.

Associations based on mapping a SNP to genes at different intervals. (A) Number of pathways significantly associated with phenotypes if we map a SNP to all genes within a certain distance, in non-cumulative distance cutoffs (e.g. genes that are between 0–100 Kbps are not considered for the 100–200 Kbps interval, etc.). Red bars represent the number of associations expected by chance (median of 100 random resampling repetitions, see Methods). (B) Number of pathways significantly associated with phenotypes when for each distance cutoff genes are considered cumulatively (all genes between the SNPs and the distance cutoff are considered). Red line represents the number expected by chance, as above.
Assessing relationships between SNPs and insertions/deletions
Defining SNP-gene regions
We define a SNP-gene (SG) region as the chromosome area between a gene and a SNP to which it is assigned. We use this definition to explore whether more indels tend to occur inside phenotype-associated SG regions than non-associated SG regions.
Mapping indels to SG regions
To assess whether there is a relationship between SNPs and indels, we tested whether extracted indels from the DGV database reside in regions that are between phenotype-associated SNPs and linked genes (i.e. genes that contribute to a significant phenotype-pathway association). We defined two types of genomic regions that lie between a SNP and a gene (SG regions). A linked SG region lies between a phenotype-associated SNP and a gene that falls within a pathway significantly associated with that phenotype. In a non-linked SG region, the gene does not fall within a pathway that is significantly associated with the phenotype. Finally, non-SG regions are all regions that are not between a SNP and a gene. We compared the amount of indels found in these three types of genomic regions.
Note that we cross-referenced the locations of all deletions with the linked SG group, as well as the two other groups, in order to calculate the amount of deletions per group. Since the groups vary in size, i.e. the number of regions and their lengths are different for each group, we normalized the number of indels per nucleotide. For example, when considering SG regions that are 0.5–1 Mbps, we took all the linked SNP-gene pairs that are more than 0.5 Mbps but less than 1 Mbps apart. We then summed the length of all these regions in nucleotides. Then, we took all the known indels from DGV that fall within any of these regions and summed their cumulative length. Finally we divided the total length of the regions by the total length of the indels. The resulting number is the average different indels in which each nucleotide in the region appears. This was repeated for all region sizes and for all region types. Note, that currently, each position in the human genome appears, on average, in roughly 2 known indels.
In order to calculate significance for the amount of deletions in the group of linked SG regions, we employed random testing on each of our control groups. That is, we merged all the regions of a certain size, regardless of whether they come from linked SG regions or from a control. For each random run, two sets of 100 regions were randomly selected from the group and the amount of indels per nucleotide was calculated for each group, and the difference between the two groups was calculated. This was done 1000 times for each control group.
RESULTS
Most SNPs are outside genes, and are often very far from any gene
Of the SNPs curated in the UCSC genome database (17), 46% are inside a gene (i.e. downstream of the transcription start site (TSS)). Of the phenotype-associated SNPs listed in the NIH GWAS database (2), 59% are inside a gene. Figure 1 shows the cumulative fraction of SNPs according to their distance from the nearest gene. One third of phenotype-associated SNPs are more than 10 Kbps from the nearest gene and 15% of them are over 100 Kbps from the nearest gene. Bearing in mind that LD intervals are typically <10 Kbps long, Figure 1 indicates that over a third of SNPs that are associated with a phenotype are too distant to be mapped to any gene. Arguably, this suggests that very often SNPs reflect molecular effects that are more distant than currently assumed.
We analyzed 1420 phenotypes and all their associated SNPs in GWAS. A pathway and a phenotype are said to be associated with each other if the genes mapped to the phenotype-associated SNPs were significantly (P-value<0.001, resampling) enriched in that pathway. Figure 2 shows the number of pathways that are significantly associated with phenotypes, for genes at different distances from the SNPs for all 1420 phenotypes. Figure 2A shows the number of associations for non-cumulative intervals of 100 Kbps. That is, the first bar from the left presents the number of significant phenotype-pathway associations found when assigning each SNP to all the genes that are less than 100 Kbps from it. The second bar represents the number of significant phenotype-pathway associations found when ignoring any gene that is less than 100 Kbps away, and assigning SNPs to all the genes that are more than 100 Kbps, but <200 Kbps away. For each interval, we also assessed the number of associations expected by chance, based on the number of SNPs in the interval and the number of KEGG pathways in CPDB. The difference between the observed number of phenotype-pathway associations (blue bars) and the number of associations expected by chance (red bars) is greatest for the genes that are between 100 and 200 Kbps away from the SNPs. That is, the genes with the strongest effect on the phenotype are close to the SNP but not necessarily very close. The number of phenotype-pathway associations decreases as we get further away from SNPs, suggesting that in these intervals there are fewer genes that affect the phenotype, but it remains significantly greater than what we expect by chance even when we consider only DNA segments that are >1 Mbps away from the SNPs. The number of phenotype-pathway associations reaches what we expect by chance only at a distance of 2.1 Mbps.
Figure 2B shows the number of significant phenotype-pathway associations that could be identified when assigning cumulatively all the genes in increasingly long distance cutoffs from the SNPs. That is, the first point from the left represents the number of significant phenotype-pathway associations found when assigning a SNP to a gene only if it is inside the gene (0 bp). The fifth point, however, represents the number of significant phenotype-pathway associations found when adding all the genes that are between 100 and 200 Kbps from the SNPs to the genes that are between 0 bp and 100 Kbps from the SNPs. If we map a gene to a SNP only if the SNP falls inside that gene (i.e. downstream of the TSS), we found a mere 86 significant phenotype-pathway associations for all phenotype-associated SNPs in GWAS. When we mapped all genes that are <10 Kbps from a SNP to that SNP (including those that are inside the gene), there are 161 significant phenotype-pathway associations. As we extend the considered distance interval, the number continues to grow until it reaches 435 associations at a distance of 200 Kbps. Beyond this distance the number begins to decline and reaches that of the random model at around 1000 Kbps, suggesting that at this distance we introduce more noise (genes that are not related to the phenotype) than signal (genes that account for the phenotype). In the cumulative analysis (Figure 2B) the number expected by chance increases at greater distances but in the non-cumulative analysis (Figure 2A) it stays the same (as the interval size remains constant). This explains why the real data and the random model merge closer to the SNPs in Figure 2B than in Figure 2A. As we move away from the SNP, fewer genes in an interval are relevant to the phenotypes. These smaller numbers are visible against the random model of the non-cumulative analysis, but not on the background of the larger number of genes in the cumulative analysis.
Identifying relevant pathways only at large distances
Some significant phenotype-pathway associations can be revealed only when distant SNP-gene assignments are considered. A few examples are presented in Table 1. For instance, 14 SNPs are reported in the GWAS catalog to be associated with multiple myeloma (hyperdiploidy). As shown in Table 1, when mapping these SNPs to genes that are <10 Kbps away, the genes did not significantly cluster into any pathway. Increasing the distance to 400 Kbps still did not identify any significant phenotype-pathway associations. However, when assigning SNPs to genes that are up to 500 Kbps away from SNPs, two pathways emerged as being significantly associated with that phenotype, a pathway named ‘Pathways in cancer’ and ‘Melanoma’, both are arguably biologically related to the phenotype. Similarly, many SNPs were found to be associated with eye color but looking at nearby genes didn't lead to associations with any pathways. The relevance of this phenotype to the pathway ‘melanogenesis’ is revealed only when we map those SNPs to genes that are up to 100 Kbps away.
Table 1. Association between phenotypes and pathways at incrementing distance cutoffs.
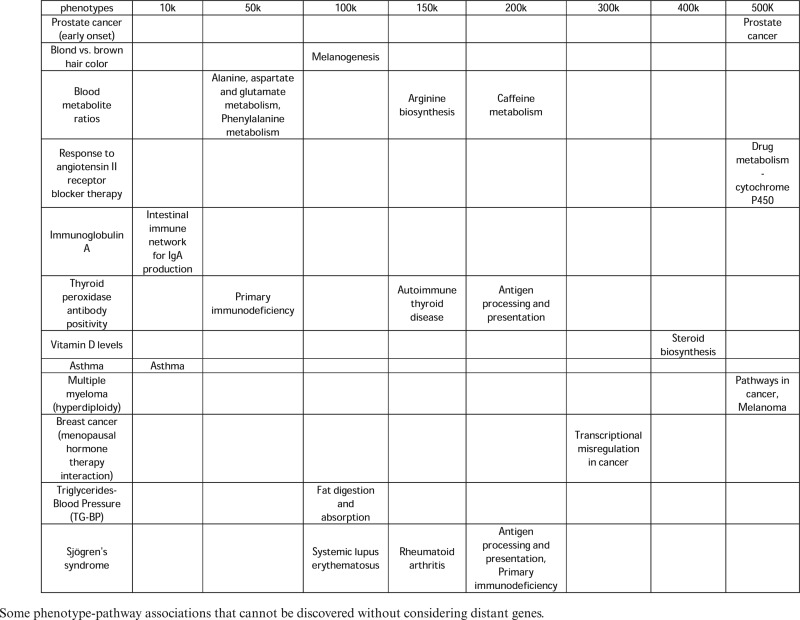
|
Comparing SNPs that are linked to distant genes to SNPs that are not
Figure 3 compares the indels around SNPs that are mapped to distant genes and indels around SNPs that are not mapped to distant genes. Briefly, a phenotype-associated SNP and a gene are said to be linked if (i) the gene belongs to a KEGG pathway, and (ii) the gene is less than x bp away from the SNP such that the phenotype and the pathway are significantly associated for a distance cutoff that is ≤x bp. The chromosomal region between the SNP and the gene is then referred to as a ‘linked SNP-gene (SG) region’. Similarly, we defined a chromosomal segment as a ‘non-linked SG region’ if it is between a SNP and gene that are not significantly linked by a pathway (Methods). We collected all known indels from the Database of Genomic Variants (20), which lists indels observed in healthy human subjects. We then binned all SNP-gene pairs according to the physical distance between the SNP and the gene (in Mbps). For each such DNA segment, we counted the number of different known indels. For SNP-gene pairs that are less than 10 Kbps apart, there was no difference between linked SG region and non-linked SG regions. The difference in the number of indels appears at a distance of 50 Kbps, grows until 100 Kbps, where it starts to decline, and disappears again for SNP-gene pairs that are 2 Mbps apart (P-value < 0.001 up to a distance of 300 Kbps, and P-value ≤0.01 up to a distance of 1 Mbps).
Figure 3.

Average number of indels between SNPs and genes at different distances. Each point depicts the normalized average number of known indels between a SNPs and genes at a certain distance. The blue points represent indels between SNPs and genes that are linked to each other through a significance association with a pathway. Red points represent SNPs and genes that are not mapped to each other. For example, SNPs that are linked by pathways to genes that are 150 Kbps away have, on average, 3.5 known indels for every nucleotide in that distance. SNPs that have genes 150 Kbps away, but these genes are not associated to them by pathways, have on average only 2.1 indels per nucleotide.
As additional controls, we also analyzed chromosomal regions that do not contain known SNP or genes (non-SNP-gene regions), as well as all chromosomal regions that do not encompass linked SG regions (that is, they cover non-linked SNP-gene regions and non-SNP-gene regions). As shown in Table 2, in both of these additional controls, the average number of indels was very similar regardless of the length of the region we tested. The average number of known indels per nucleotide in linked SG regions varies between 2.28, for SNPs that are very close the gene, and 3.69 for SNPs that are linked to genes that are 100 Kbps away, a range of 1.41. For the three other types of regions we considered, the number of indels was never above 2.78 indels per nucleotide, and the maximum range was 0.41 (Table 2).
Table 2. Average number of indels per base-pair in different types of chromosomal regions.
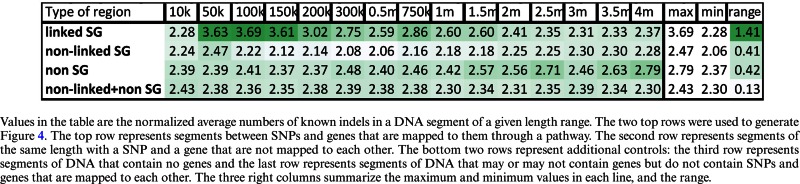
|
DISCUSSION
Determining statistically justified cutoff for mapping genes to SNPs
Figure 2 reveals that if we look at traditional distance cutoffs of <10 Kbps, we will identify relatively few associations between phenotypes and pathways based on SNPs. As mentioned above, it is common practice to assign SNPs to genes based on a distance cutoff. Studies use a variety of cutoffs, such as 2 Kbps (21), 5 Kbps (22–24), 20 Kbps (25–27), 100 Kbps (8,28). Some studies suggested to use a cutoff of 500 Kbps (9–12), since enhancers and repressors may be as distant as 500 Kbps from their genes (9). As shown in Figure 2A, assigning SNPs to distant genes, while ignoring nearby genes, yields clustering of genes near phenotype-associated-SNPs into pathways at a significantly higher rate than genes near randomly selected SNPs. The nearest 100 Kbps interval identified 416 associated pathways as opposed to 137.5 associations expected by chance. However, when we ignore the genes that allowed for these associations and consider only genes that are between 100 and 200 Kbps, the number of significant associations remains much higher than expected by chance. As we map to a SNP genes that are further away from it, the number of significant phenotype-pathway associations declines but remains higher than the number of phenotype-pathway associations expected by chance, until 1 Mbps where it merges with the number of associations expected by chance. These results suggest that SNPs are more likely to be relevant to closer genes, but that even very distant genes are affected by the variation.
The random model in this analysis had to be considered carefully. Since functionally related genes may be adjacent on the chromosomes neighboring genes may cluster into the same pathway more than random genes. To account for that, our random model was based on picking up random SNPs and considering DNA stretches of the same size around them. This way, we can make sure that the difference between the real data and the random model should be attributed to the effect of SNPs on distant genes and not to the fact that genes of the same pathway are likely to be near each other.
It is difficult to determine, statistically, what is the optimal distance for linking a SNP to genes. Such optimal cutoff would be a distance from the SNP beyond which the addition of more genes does not improve the identification of phenotype-pathway associations. The difficulty stems from the change in the random model. In Figure 2A each of the compared intervals has the same size, and thus approximately the same number of genes. Therefore, for each interval the number of associations expected by chance is very similar. In this analysis we still discover significant associations with pathways even when we consider genes that are 1.9 Mbps away from the SNPs. However, Figure 2B compares stretches of different sizes. Since a stretch of 1 Mbps has more genes than a stretch of 0.2 Mbps, the number of associations expected by chance is larger for longer intervals. Indeed, in Figure 2B, the expected number of random association increases with distance. Figure 4 shows the difference between the number of associations in the real data and the number expected by chance for the cumulative analysis. The number grows rapidly until 200 Kbps and drops rapidly for longer distance. These results suggest that linking SNPs to genes that are up to 750 Kbps away is justified statistically both by the cumulative and the by the non-cumulative analyses and is likely to reveal true functional connections. While our results show that causative genes are often found up to 2 Mbps away from the SNP, further analysis is required to define ways to tease these genes out for specific phenotypes and specific distances.
Figure 4.

Difference between observed and expected number of associated pathways. Subtracting the numbers represented by the red line in Figure 2B from the numbers represented by the blue line, reveals that until 200 Kbps away from the SNP, associations with pathways improves as we include more distant genes. From this distance on, adding of more distant genes decreases the number of significant associations.
Mapping SNPs to multiple genes, rather than one, is more revealing
While some studies try to offer tools that help in identifying a single gene within that interval (29,30), it is a common practice to map the SNP to more than one gene within a given cutoff (11,27,31,32). It has been argued that such gene-based approach can resolve some of the reproducibility challenges of the SNP-based approach and would capture more of the potential risk-conferring SNPs (33,34). As seen in Figure 2, assigning a SNP to all the genes in an interval identifies significant phenotype-pathway associations. This corroborates the suggestion that a paradigm of one-SNP-many-genes may be more useful in unraveling the molecular basis of the phenotype.
The effect of the genome 3D structure
While SNPs are commonly regarded as affecting nearby genes, effects on distant genes have been demonstrated. For example, in the case of enhancers (5,35), which may affect distant genes (36,37) or even genes on other chromosomes (38,39) because of the 3D organization of the genome (40,41). The 3D structure of the genome may be a major factor in the effect of SNPs on distant genes, and may explain many of the effects we observed in this study. The rapid accumulation of data regarding the 3D arrangement of the genome would probably allow, in the near future, for the mapping of SNPs to their 3D spatial neighborhood. However, with the amount of structural data available at the moment, this is not yet possible. Our results suggest that non-local effects are more common than previously assumed. We observe that this effect attenuates with distance. Indeed, one may expect that while 3D packing could bring any two points on the genome to close proximity, the probability of two points to be close in 3D decreases with their physical distance on the chromosome.
The effect of indels
Another possible explanation for the relationships of SNPs and distant genes may be that SNPs are markers for large structural variations that may affect large chromosomal segments. If such a relationship exists, we should observe more indels between SNPs and their linked genes than in other genomic regions of similar size. The suggestion that SNPs may be markers to structural variations has been made before (42). Our results indicate this may be more frequent than previously assumed. The control group we selected, that of DNA stretches that lie between SNP and genes that are not significantly linked, is similar to our test group of significantly linked SNP-gene regions, and avoids a bias toward long intergenic regions, which may have unique traits (e.g. low complexity or enrichment of ALU or different packing). The non-linked SG group was tested as a whole, meaning that regions overlapping with the linked SG group were not removed. Therefore, indels residing in a region overlapping a linked SG region as well as a non-linked SG region were counted in both groups. Still, the difference between the two sets of genomic regions was significant.
We extracted all primary indels in the DGV database. The data from DGV suggests that, on average, any nucleotide in the genome appears in approximately two different observed indels. Many entries in the DGV database are partially overlapping. As such, it is possible for multiple deletions to encompass the same region on the chromosome. We calculated in how many different known indels each nucleotide appears, on average, for significantly linked SG and for non-significantly linked ones. Importantly, our results show that there are more deletions for SNP-gene links that are up to 2 Mbps apart (Figure 3). Beyond this distance we cannot see a difference between significantly linked SNP-gene pairs and random SNP-gene pairs. These results may suggest that in some cases SNPs are markers for structural variations. To validate this suggestion, further analysis is required. Specifically, full genome sequence of many thousands of genomes is may establish that specific deleterious alleles are associated with large indels. Current publically available genomes are not sufficient for such analysis.
The success of the approach proposed here for mapping SNPs to genes relies on the current knowledge of pathways. Unraveling more of the networks that underlie biological processes will increase both the number and the quality of known pathways. This will improve our ability to map SNPs to genes and phenotypes to molecular processes.
FUNDING
Microsoft research. Funding for open access charge: Microsoft research.
Conflict of interest statement. None declared.
REFERENCES
- 1.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A.. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicolae D.L., Wen X., Voight B.F., Cox N.J. Coverage and characteristics of the Affymetrix GeneChip Human Mapping 100K SNP set. PLoS Genet. 2006;2:e67–e67. doi: 10.1371/journal.pgen.0020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witte J.S. Genome-wide association studies and beyond. Annu. Rev. Public Health. 2010;31:9–20. doi: 10.1146/annurev.publhealth.012809.103723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visel A., Rubin E.M., Pennacchio L.A. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahmood S.S., Levy D., Vasan R.S., Wang T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet. 2014;383:999–1008. doi: 10.1016/S0140-6736(13)61752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sherry S.T. dbSNP: the NCBI database of genetic variation. Nucleic Acids Research. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoof N., Iles M.M., Bishop D.T., Newton-Bishop J.A., Barrett J.H., Genomel Consortium et al. Pathway-based analysis of a melanoma genome-wide association study: analysis of genes related to tumour-immunosuppression. PLoS ONE. 2012;6:e29451. doi: 10.1371/journal.pone.0029451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K., Li M., Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L., Zhang L., Zhao Y., Xu L., Shang Y., Wang Q., Li W., Wang H., Li X. Prioritizing risk pathways : a novel association approach to searching for disease pathways fusing SNPs and pathways. Bioinformatics. 2009;25:237–242. doi: 10.1093/bioinformatics/btn613. [DOI] [PubMed] [Google Scholar]
- 11.Luo L., Peng G., Zhu Y., Dong H., Amos C.I., Xiong M. Genome-wide gene and pathway analysis. Eur. J. Hum. Genet. 2010;18:1045–1053. doi: 10.1038/ejhg.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Y.-F., Li J., Chen Y., Zhang L.-S., Deng H.-W. A new permutation strategy of pathway-based approach for genome-wide association study. BMC bioinformatics. 2009;10:429–429. doi: 10.1186/1471-2105-10-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. 2010;11:843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 14.Brodie A., Tovia-Brodie O., Ofran Y. Large scale analysis of phenotype-pathway relationships based on GWAS results. PLoS One. 2014;9:e100887. doi: 10.1371/journal.pone.0100887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray K.A., Daugherty L.C., Gordon S.M., Seal R.L., Wright M.W., Bruford E.A. Genenames.org: the HGNC resources in 2013. Nucleic Acids Res. 2013;41:D545–D552. doi: 10.1093/nar/gks1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karolchik D., Hinrichs A.S., Kent W.J. The UCSC Genome Browser. Curr. Protoc. Bioinformatics. 2012 doi: 10.1002/0471250953.bi0104s40. Chapter 1, Unit 1.4. [DOI] [PubMed] [Google Scholar]
- 18.Kanehisa M., Goto S., Furumichi M., Tanabe M., Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamburov A., Stelzl U., Lehrach H., Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41:D793–D800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macdonald J.R., Ziman R., Yuen R.K., Feuk L., Scherer S.W. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2013;42:D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chelala C., Khan A., Lemoine N.R. SNPnexus: a web database for functional annotation of newly discovered and public domain single nucleotide polymorphisms. Bioinformatics. 2009;25:655–661. doi: 10.1093/bioinformatics/btn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee P.H., Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36:D820–D824. doi: 10.1093/nar/gkm904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X., Wang L., Hu B., Guo M., Barnard J., Zhu X. Pathway-based analysis for genome-wide association studies using supervised principal components. Genetic Epidemiol. 2010;34:716–724. doi: 10.1002/gepi.20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torkamani A., Topol E.J., Schork N.J. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92:265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L.S., Hutter C.M., Potter J.D., Liu Y., Prentice R.L., Peters U., Hsu L. Insights into colon cancer etiology via a regularized approach to gene set analysis of GWAS Data. Am. Journal of Hum. Genet. 2012;86:860–871. doi: 10.1016/j.ajhg.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang M., Liang L., Xu M., Qureshi A.a., Han J. Pathway analysis for genome-wide association study of Basal cell carcinoma of the skin. PloS One. 2011;6:e22760. doi: 10.1371/journal.pone.0022760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weng L., Macciardi F., Subramanian A., Guffanti G., Potkin S.G., Yu Z., Xie X. SNP-based pathway enrichment analysis for genome-wide association studies. BMC Bioinformatics. 2011;12:99–99. doi: 10.1186/1471-2105-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang K., Zhang H., Kugathasan S., Annese V., Bradfield J.P., Russell R.K., Sleiman P.M., Imielinski M., Glessner J., Hou C., et al. Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn Disease. Am J Hum Genet. 2009;84:399–405. doi: 10.1016/j.ajhg.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehne B., Lewis C.M., Schlitt T. From SNPs to genes: disease association at the gene level. PLoS One. 2011;6:e20133. doi: 10.1371/journal.pone.0020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bakir-Gungor B., Sezerman O.U. A new methodology to associate SNPs with human diseases according to their Pathway Related Context. PLoS ONE. 2012;6:e26277. doi: 10.1371/journal.pone.0026277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holmans P., Green E.K., Pahwa J.S., Ferreira M.A., Purcell S.M., Sklar P., Wellcome Trust Case-Control C., Owen M.J., O'Donovan M.C., Craddock N. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraft P., Raychaudhuri S. Complex diseases, complex genes: keeping pathways on the right track. Epidemiology. 2009;20:508–511. doi: 10.1097/EDE.0b013e3181a93b98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neale B.M., Sham P.C. The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004;75:353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng G., Luo L., Siu H., Zhu Y., Hu P., Hong S., Zhao J., Zhou X., Reveille J.D., Jin L., et al. Gene and pathway-based second-wave analysis of genome-wide association studies. Eur J Hum Genet. 2010;18:111–117. doi: 10.1038/ejhg.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blackwood E.M. Going the Distance: A Current View of Enhancer Action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- 36.Visel A., Akiyama J.A., Shoukry M., Afzal V., Rubin E.M., Pennacchio L.A. Functional autonomy of distant-acting human enhancers. Genomics. 2009;93:509–513. doi: 10.1016/j.ygeno.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang Y.C., Zheng Q., Gregory B.D., Wang L.S. High-throughput identification of long-range regulatory elements and their target promoters in the human genome. Nucleic Acids Res. 2013;41:4835–4846. doi: 10.1093/nar/gkt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams A., Spilianakis C.G., Flavell R.A. Interchromosomal association and gene regulation in trans. Trends Genet. 2010;26:188–197. doi: 10.1016/j.tig.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ong C.T., Corces V.G. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrascheck M., Escher D., Mahmoudi T., Verrijzer C.P., Schaffner W., Barberis A. DNA looping induced by a transcriptional enhancer in vivo. Nucleic Acids Res. 2005;33:3743–3750. doi: 10.1093/nar/gki689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinds D.A., Kloek A.P., Jen M., Chen X., Frazer K.A. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]