

Abstract
Empowered by modern genotyping and large samples, population structure can be accurately described and quantified even when it only explains a fraction of a percent of total genetic variance. This is especially relevant and interesting for humans, where fine-scale population structure can both confound disease-mapping studies and reveal the history of migration and divergence that shaped our species’ diversity. Here we review notable recent advances in the detection, use, and understanding of population structure. Our work addresses multiple areas where substantial progress is being made: improved statistics and models for better capturing differentiation, admixture, and the spatial distribution of variation; computational speed-ups that allow methods to scale to modern data; and advances in haplotypic modeling that have wide ranging consequences for the analysis of population structure. We conclude by outlining four important open challenges: The limitations of discrete population models, uncertainty in individual origins, the incorporation of both fine-scale structure and ancient DNA in parametric models, and the development of efficient computational tools, particularly for haplotype-based methods.
If one assumes humans across the globe are a single randomly mating population, it would result in only a 5–15% average error when predicting the proportion of observed heterozygotes at a locus. This closeness to an idealized randomly mating population is one vestige of how little evolutionary time has passed since the common origin of all humans in Africa. The departure from random mating predictions due to population differentiation has a classic quantitative measure, called FST, which appropriately takes on values of 5–15% in global samples of human populations [1–3]. If one zooms in within continental regions of the globe, FST tends to be even lower, regularly taking values below 1%, a threshold which we use here informally to define “fine-scale structure”. A triumph of the large datasets available to contemporary population geneticists is that they allow fine-scale structure to be detected and dissected to reveal population relationships (Figure 1).
Figure 1. Four methods for assessing population structure using large-scale single-nucleotide polymorphism data.
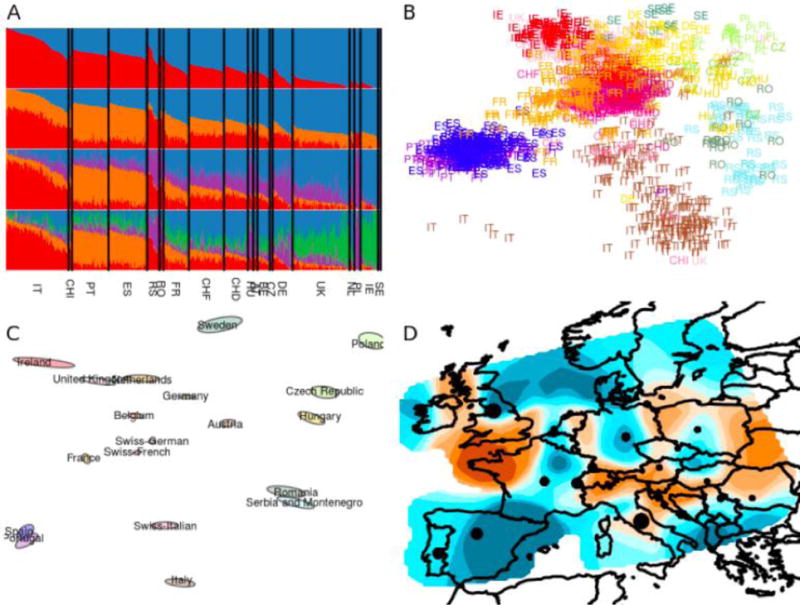
A) Ancestry proportion inference using ADMIXTURE. B) Principal components analysis. C) Plot of population in ‘geogenetic’ space using SpaceMix. D) Visualization of effective migration rates using EEMS (brown = low effective migration; blue = high effective migration). Each method is applied to the dataset analysed in [92] after filtering out populations with fewer than 10 individuals, where population identifiers are defined on the basis of grandparental ancestry. Structure is visible, even though FST-values average 0.004 between broad geographic regions in Europe [92].
The reason large datasets allow fine-scale structure to be dissected is a statistical phenomenon [4]. Subtle differences in average pairwise similarity become more apparent as the scale of a dataset increases (Figure 2). For principal component analysis (PCA), Patterson et al. [5] have argued that structure reveals itself much like a phase change in physics; namely if the product of the number of genetic markers (m) and individuals (n) is greater than 1/(FST2) then structure will be evident. The great fortune of human geneticists is that novel technologies have made it affordable to amass datasets large enough to characterize even very subtle population structure.
Figure 2. A large number of loci is required to reveal fine-scale population structure using PCA.
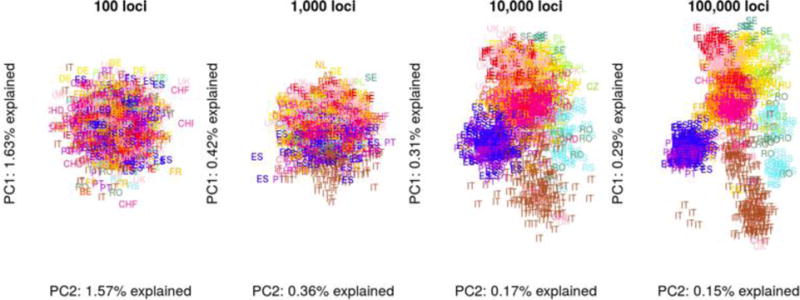
Four subsamples with an increasing number of loci were taken from the[93] dataset. Using 100 loci, Europe appears panmictic, whereas 1,000 loci are sufficient to establish a North-South cline. With 10,000 and 100,000 loci, fine-scale details are revealed.
Characterizations of fine-scale population structure are increasingly empowering the study of human population history, helping genetics play a role integral with linguistics, archaeology, and history in the study of the human past, as first envisioned by Cavalli-Sforza and colleagues [6]. Studies of population structure have also allowed the medical genetics community to build more robust genome-wide association studies for disease risk [7–9], enabled evolutionary geneticists to identify exceptional regions in the genome that have undergone local adaptation [10–12], and facilitated the individual-level ancestry assessment that is increasingly popular in personalized genomics [13], though not without criticism [14].
The most obvious advances in the study of fine-scale structure are the result of increased genome-wide single-nucleotide polymorphism (SNP) and sequencing data being collected from diverse regions across the world [15–17]. More profoundly, the recent availability of genome-wide data from archaeological samples of modern humans (“ancient DNA”, aDNA) is revolutionizing our understanding of the processes that generated present-day population structure [18]. As one stimulating example, aDNA studies suggest that levels of differentiation in Europe may have actually decreased during the late Neolithic period and Bronze Age, as at least three major proposed population lineages have intersected through time [17,19,20].
In this review, we highlight several of the most exciting advances in analysis methods in the past three years. Analysis methods are especially crucial when structure is fine-scale, and as we show, there has been extensive progress in numerous directions. To complete a picture of recent studies of human population structure, we recommend several other recent reviews [21–27]. We also highlight the Peopling of the British Isles Project [28] and two recent ancient DNA papers [19,20] as examples from the vanguard of fine-scale structure analysis using modern and ancient human data.
Developing and understanding metrics for population differentiation
While Wright’s FST has been a workhorse for describing population structure for decades, a novel set of f-statistics have become highly influential since their original publication [29,30] because of their utility in studying the recent admixture that is common in human history. In the past several years, the use of f-statistics has continued to develop; for example, Raghavan et al. [31] developed an ‘outgroup-f3’ statistic to provide a measure of similarity that is insensitive to population-specific drift, making it more interpretable than FST in many settings. The advent of sequencing data has also made possible a metric that is especially sensitive to recent population structure: the estimated time to the most recent common ancestor of shared doubleton variants [32]. Doubleton-based metrics are already showing their utility to detect fine-scale structure in several large sequencing studies [33–35].
Alongside the expansion of new metrics, there is an increasing understanding of the basic properties of existing ones. One area of concern is that FST has inconsistent values across different types of loci (e.g. microsatellite, SNP array, and sequence data differ by 0.05–0.07 in several meaningful human examples [36,37] also see [38]). As demonstrated by Jakobsson et al. [37], this discrepancy is largely explained by the frequency of the most frequent allele at a locus, which differs greatly between marker classes [37]. However, the statistical strategy used also has an impact, in particular when rare alleles are present [36]. Similar understandings need to be developed for f-statistics. In that vein, one of us [39] recently showed how f-statistics can be understood in general coalescent terms, with expectations that can be derived under arbitrary parametric population history models. Interestingly, the work revealed that the mean number of pairwise differences between populations is a better measure of differentiation than the outgroup-f3 statistic.
Refining and expanding models that handle human admixture
Recognizing that simple bifurcating trees are a poor model for human genetic diversity, several researchers have developed tree-building methods that take admixture and migration into account [29,40–42]. These methods typically build a guide tree and then add migration edges to represent recent admixture events. These methods represent a substantial advance and they are being used broadly; however, challenges remain with identifiability, exploring the space of all possible graphs, robustly selecting the number of migration edges, and exploiting tools from similar approaches developed independently in phylogenetics [43,44].
A more longstanding approach to study admixture is the use of global [45] and local ancestry models [46], and both have been advanced recently by computational speed-ups. When genome-wide SNP data first became available, difficulties in applying the now classic Bayesian method for admixture inference (STRUCTURE [45]) occasioned the development of fast maximum-likelihood based approaches [47,48]. The past two years have seen the return of Bayesian approaches with two new fast approaches that use variational approximations [49,50]. Impressively, teraSTRUCTURE [50] handles samples containing millions of individuals. Local ancestry approaches have recently been improved by the development of fast algorithms that leverage rare variants within ancestries [51] and wavelet techniques [52]. Unfortunately, distinguishing local ancestries among weakly differentiated source populations remains difficult. For these cases, alternative approaches based on admixture linkage disequilibrium, which side-step local ancestry inference, have proven fruitful for detecting admixture occurring in the recent past (e.g. within the last three thousand years) and suggest that admixture is widespread in human populations [53–55].
Latent factor models, such as sparse factor analysis and PCA, are also applicable for inference of admixture [56,57]. For PCA, more efficient algorithms have been implemented, allowing application to hundreds of thousands of individuals with reasonable run-times [58,59]. Another useful development has been the expansion of Procrustes approaches (used to align PCA and geography data [60]) to allow merging of samples with low-coverage sequence data into PCAs from heavily genotyped reference panels [61,62]. Novel factor models are also being developed that may have advantages for assessing admixed samples and controlling structure in association studies [63].
Putting geography into studies of population structure
Geographic information has been under-utilized in the study of fine-scale structure despite its central importance in the process of generating structure. Several recent approaches make progress by using Wishart distribution to model genetic similarity as a function of spatial distance. In one approach, Bradburd et al. [64] use a covariance model to visualize samples in a “geogenetic space”. In homogenous isolation-by-distance scenarios, the geogenetic space should mirror the geographic space. Deviations from homogeneity will result in adjusted placement of populations in geogenetic space. For example, barriers to migration result in larger geogenetic distances between populations. In some respects the methods should behave like PCA on spatial data [65], but with less of the sensitivity to uneven sample sizes that is typical of PCA [57,65]. Discrete admixture events can be accounted for by adding admixture links between sources and targets, in a manner similar to how recent tree-based methods add migration edges to model admixture (see above).
A second approach using the Wishart distribution is the EEMS method [66] which uses observed pairwise genetic similarities to estimate a map or “surface” of effective migration rates. The inferred migration rates are “effective” in that they reflect migration proportions scaled by effective population sizes under an equilibrium model. As few empirical systems (and especially humans) are at equilibrium, the surfaces should best be interpreted as a tool for visualizing patterns of genetic differentiation relative to geographic distance. In closely related work, Hanks and Hooten [67] independently develop a Wishart framework much like that in EEMS that can test whether a particular environmental variable is predictive of migration rates. Two additional exciting advances, not using the Wishart distribution, are a method (localdiff [68]) that uses locally computed FST values to visualize barriers, and an approach that models FST as a function of the bearing between two populations to study anisotropic patterns of spatial differentiation [69].
Extracting signatures of fine-scale structure in haplotype data
A very active and promising arena of research is in the development of haplotype-based methods for studying fine-scale structure. Local ancestry models (see above) have long been the only haplotype-based approach used to study structure, and haplotypes can be more informative for assignment [70,71], but a bevy of methods are being developed that leverage haplotypes in novel ways.
Methods using long shared haplotypes (also known as tracts of identity-by-descent, IBD) are particularly well-suited for the characterization and interpretation of fine-scale population structure [72]. As an example of their power, Ralph and Coop [73] showed that IBD patterns in Europe can reveal more subtle structure than simple pairwise SNP similarity. More recently Baharian et al [74] use the spatial distribution of long shared haplotypes to estimate dispersal rates in the context of the African-American history. These methods have exceptional promise, though interpretations can be complicated when shorter tracts are considered [75]. A related approach to IBD patterns is embodied in the fineSTRUCTURE model [76,77] which uses long shared haplotypes detected through the use of the Li and Stephens haplotype-copying model [78] and then processes them through a downstream analysis that includes mixture modeling of copying profiles. This model underlies the striking structure revealed in the Peopling of the British Isles project [28].
A second approach has been to focus on full chromosomal haplotype data using approximate coalescent models. Using the Sequential Markov Coalescent (SMC) approximation, it is now possible to study population divergence using small numbers of genomes [79,80]. As one example, Schiffels and Durbin suggest a novel non-parametric approach based on inference of cross-coalescent rates through time [80,81]. In related work, a recent approach for sampling coalescent genealogies genome-wide [82] is a remarkable achievement but has yet to be adapted to explicitly study population structure.
A drawback of most haplotype-based methods is that they are computationally very expensive. A major breakthrough is the development of fast and scalable algorithms for representing haplotype data in ways that make haplotype similarity evident and easy to query. One such algorithm, the Positional Burrows-Wheeler Transform (pBWT, [83]) is revolutionary in this regard, and a new extension [84] is also exciting as it links the Li and Stephens haplotype-copying model to the pBWT framework and provides approaches that can handle unphased data.
Open challenges
Despite all the methodological progress, many fundamental challenges exist for fine-scale population structure studies. Many of these challenges stem from the difficulty of encapsulating the complexity of human population structure with mathematical models.
On the one hand, many models assume a small number of discrete, temporally continuous populations as the units of analysis. This is only an approximation to the structure of human populations on the ground, and there is a strong trade-off between the ease-of-analysis provided by using a small number of discrete populations and the error induced due to model violations. On the other hand, other models are not sufficiently parametric. Many available methods focus on summarizing the observed population structure in a form that facilitates interpretation, but without explicitly modeling the historical processes that shaped these patterns.
As a symptom of this problem, we currently lack a widely accepted generative model for human fine-scale data. Put another way, we do not have a clear simulation protocol to produce “realistic” human data with fine-scale structure. This hinders applications such as testing new methods and evaluating evolutionary models of disease variants in human populations. As an example, consider how recent aDNA studies have made clear that fine-scale structure in Europe is partly driven by temporally dynamic admixture patterns between 3 ancestral populations [17, 19, 20]; it is unclear from existing publications what pairing of simulation protocol and parameters would generate an in silico whole-genome dataset that replicates the basic features of European fine-scale structure.
A related persistent challenge is that choices must be made regarding assigning individual origins based on sampling location, individual birthplace, or an origin based on parental or grandparental ancestry. Given the ubiquity of human movement and admixture, the choice can complicate interpretations and/or result in samples being omitted when origins are unclear (e.g. grandparents of differing origins). The problem also arises when assigning aDNA samples into analysis units when they might vary by location, cultural context, and sampling time. One must always interpret results with the location assignment procedure in mind. Ideally, approaches can be developed that more explicitly model the uncertainty in this stage of analysis.
Another practical challenge with fine-scale population structure is that analysts must be especially cautious regarding model deviations such as ascertainment bias, heterogeneous linkage disequilibrium (LD) patterns, and complex mutational processes. For example, even after basic LD filtering, PCs can be affected by large blocks of SNPs with complex patterns of LD, such as those that arise due to structural variants like the polymorphic 4Mb Chr 8p23 inversion in Europe (e.g. PC2 in the study of [85]). Such patterns likely pollute the results of various methods that assume marker independence as well as haplotype-based methods that do not model complex LD patterns due to structural variants. One precaution is to inspect the PC loadings and repeat the PCA after removing any genomic regions with exceptionally high loadings. An example of a mutagenic process potentially influencing haplotype-based approaches is the error-prone DNA-Polymerase Zeta, which may introduce dinucleotide and other aberrant mutation patterns [86] that bias analyses when handled as distinct mutations.
A final precaution, and one of broader societal relevance, is that a viewer can become misled about the depth of population structure when casually inspecting visualizations using methods such as PCA, ADMIXTURE, EEMS or fineSTRUCTURE. For example, untrained eyes may overinterpret population clusters in a PCA plot as a signature of deep, absolute levels of differentiation with relevance for phenotypic differentiation. This is an ironic inverse of what Edwards harshly termed Lewontin’s fallacy [4], and what we might instead call Lewontin’s nightmare. To prevent these misinterpretations, first we encourage practitioners to make absolute metrics of differentiation clear to audiences (e.g. FST, PCA proportion of variance explained). Weak levels of differentiation, as measured by FST, imply that neutral quantitative traits will be weakly differentiated as well [3,87]. Second, visually displaying the geographic distribution of a manageable number of random markers from a dataset can be helpful for students and broader audiences to gain a direct sense of levels of population structure. Several resources make this feasible for human genetic datasets (GGV browser [88], ALFRED [89,90] and HGDP Selection Browser [91]).
Without any question, the study of fine-scale structure has been an exciting frontier of contemporary population genetics, with extensive progress and continued promise. As this work continues, we will begin to more fully understand the processes that shape fine-scale structure in humans, and have a more full perspective on human origins. Of broader relevance, this progress also provides guidance for studying other species with highly dynamic population histories, and many of the methods reviewed here are useful for applications outside of humans.
Acknowledgments
J.N. would like to acknowledge NIH support (R01HG00708, GM108805, 1U01 CA198933-01), and B.P. would like to acknowledge a Swiss NSF Postdoctoral Fellowship. We would also like to thank the members of the Novembre lab as well as Choongwon Jeong, Sohini Ramachandran, and Noah Rosenberg for helpful comments and discussions. The POPRES data used in the figures was accessed via dbGAP study accession phs000145.
References Cited
- 1.Lewontin RC. The Apportionment of Human Diversity. In: Dobzhansky T, Hecht MK, Steere WC, editors. Evolutionary Biology. Springer US; 1972. pp. 381–398. [Google Scholar]
- 2.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3**.Edge MD, Rosenberg NA. Implications of the apportionment of human genetic diversity for the apportionment of human phenotypic diversity. Stud Hist Philos Biol Biomed Sci. 2015;52:32–45. doi: 10.1016/j.shpsc.2014.12.005. A thoughtful review and analysis of a simple model to address the implications of population differentiation on neutral quantitative trait differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards AWF. Human genetic diversity: Lewontin’s fallacy. Bioessays. 2003;25:798–801. doi: 10.1002/bies.10315. [DOI] [PubMed] [Google Scholar]
- 5.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavalli-Sforza LL, Menozzi P, Piazza A. The history and geography of human genes. Princeton university press; 1994. [Google Scholar]
- 7.Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46:100–106. doi: 10.1038/ng.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno-Estrada A, Gignoux CR, Fernández-López JC, Zakharia F, Sikora M, Contreras AV, Acuña-Alonzo V, Sandoval K, Eng C, Romero-Hidalgo S, et al. Human genetics. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science. 2014;344:1280–1285. doi: 10.1126/science.1251688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corona E, Chen R, Sikora M, Morgan AA, Patel CJ, Ramesh A, Bustamante CD, Butte AJ. Analysis of the genetic basis of disease in the context of worldwide human relationships and migration. PLoS Genet. 2013;9:e1003447. doi: 10.1371/journal.pgen.1003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Novembre J, Di Rienzo A. Spatial patterns of variation due to natural selection in humans. Nat Rev Genet. 2009;10:745–755. doi: 10.1038/nrg2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frichot E, Schoville SD, Bouchard G, François O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol. 2013;30:1687–1699. doi: 10.1093/molbev/mst063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coop G, Witonsky D, Rienzo AD, Pritchard JK. Using Environmental Correlations to Identify Loci Underlying Local Adaptation. Genetics. 2010;185:1411–1423. doi: 10.1534/genetics.110.114819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shriver MD, Kittles RA. Genetic ancestry and the search for personalized genetic histories. Nat Rev Genet. 2004;5:611–618. doi: 10.1038/nrg1405. [DOI] [PubMed] [Google Scholar]
- 14.Weiss KM, Lambert BW. What type of person are you? Old-fashioned thinking even in modern science. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karakachoff M, Duforet-Frebourg N, Simonet F, Le Scouarnec S, Pellen N, Lecointe S, Charpentier E, Gros F, Cauchi S, Froguel P, et al. Fine-scale human genetic structure in Western France. Eur J Hum Genet. 2015;23:831–836. doi: 10.1038/ejhg.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yunusbayev B, Metspalu M, Metspalu E, Valeev A, Litvinov S, Valiev R, Akhmetova V, Balanovska E, Balanovsky O, Turdikulova S, et al. The Genetic Legacy of the Expansion of Turkic-Speaking Nomads across Eurasia. PLoS Genet. 2015;11:e1005068. doi: 10.1371/journal.pgen.1005068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, Kirsanow K, Sudmant PH, Schraiber JG, Castellano S, Lipson M. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–413. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pickrell JK, Reich D. Toward a new history and geography of human genes informed by ancient DNA. Trends Genet. 2014;30:377–389. doi: 10.1016/j.tig.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19*.Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B, Brandt G, Nordenfelt S, Harney E, Stewardson K, et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015;522:207–211. doi: 10.1038/nature14317. Along with Allentoft et al (2015), a large-scale analysis of Neolithic and Bronze Age ancient DNA samples from Europe and Asia to reveal evidence of major gene flow events from the Pontic-Caspian Steppe region into Europe within the past 5,000 years. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20*.Allentoft ME, Sikora M, Sjögren K-G, Rasmussen S, Rasmussen M, Stenderup J, Damgaard PB, Schroeder H, Ahlström T, Vinner L, et al. Population genomics of Bronze Age Eurasia. Nature. 2015;522:167–172. doi: 10.1038/nature14507. See entry for Haak et al (2015) [19] [DOI] [PubMed] [Google Scholar]
- 21.Schraiber JG, Akey JM. Methods and models for unravelling human evolutionary history. Nat Rev Genet. 2015;16:727–740. doi: 10.1038/nrg4005. [DOI] [PubMed] [Google Scholar]
- 22.Novembre J, Ramachandran S. Perspectives on human population structure at the cusp of the sequencing era. Annu Rev Genomics Hum Genet. 2011;12:245–274. doi: 10.1146/annurev-genom-090810-183123. [DOI] [PubMed] [Google Scholar]
- 23.Veeramah KR, Hammer MF. The impact of whole-genome sequencing on the reconstruction of human population history. Nat Rev Genet. 2014;15:149–162. doi: 10.1038/nrg3625. [DOI] [PubMed] [Google Scholar]
- 24.Sousa V, Peischl S, Excoffier L. Impact of range expansions on current human genomic diversity. Curr Opin Genet Dev. 2014;29:22–30. doi: 10.1016/j.gde.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Scally A, Durbin R. Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet. 2012;13:745–753. doi: 10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
- 26.François O, Waits LP. Landscape Genetics. John Wiley & Sons, Ltd; 2015. Clustering and Assignment Methods in Landscape Genetics; pp. 114–128. [Google Scholar]
- 27.Barbujani G, Ghirotto S, Tassi F. Nine things to remember about human genome diversity. Tissue Antigens. 2013;82:155–164. doi: 10.1111/tan.12165. [DOI] [PubMed] [Google Scholar]
- 28**.Leslie S, Winney B, Hellenthal G, Davison D, Boumertit A, Day T, Hutnik K, Royrvik EC, Cunliffe B, Wellcome Trust Case Control Consortium 2 et al. The fine-scale genetic structure of the British population. Nature. 2015;519:309–314. doi: 10.1038/nature14230. An impressive revelation of fine-scale structure within the small geographic area of the British Isles using the fineSTRUCTURE method. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461:489–494. doi: 10.1038/nature08365. An important paper for its empirical analysis of a major admixture even in Indian population history and its development of f-statistics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30**.Patterson NJ, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. Ancient Admixture in Human History. Genetics. 2012;192(3):1065–93. doi: 10.1534/genetics.112.145037. A comprehensive presentation of the influential f-statistics with applications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raghavan M, Skoglund P, Graf KE, Metspalu M, Albrechtsen A, Moltke I, Rasmussen S, Stafford TW, Jr, Orlando L, Metspalu E, et al. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature. 2014;505:87–91. doi: 10.1038/nature12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Mathieson I, McVean G. Demography and the age of rare variants. PLoS Genet. 2014;10:e1004528. doi: 10.1371/journal.pgen.1004528. An innovative paper developing the idea of using shared sequence similarity around doubleton variants to assess recent coalescent times, demographic history, and structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818–825. doi: 10.1038/ng.3021. [DOI] [PubMed] [Google Scholar]
- 34.UK10K Consortium. Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JRB, Xu C, Futema M, et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sidore C, Busonero F, Maschio A, Porcu E, Naitza S, Zoledziewska M, Mulas A, Pistis G, Steri M, Danjou F, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015;47:1272–1281. doi: 10.1038/ng.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhatia G, Patterson N, Sankararaman S, Price AL. Estimating and interpreting FST: the impact of rare variants. Genome Res. 2013;23:1514–1521. doi: 10.1101/gr.154831.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37*.Jakobsson M, Edge MD, Rosenberg NA. The relationship between F(ST) and the frequency of the most frequent allele. Genetics. 2013;193:515–528. doi: 10.1534/genetics.112.144758. A careful analysis of bounds on FST that can help explain differences in values found across different marker types. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jost L. GST and its relatives do not measure differentiation. Mol Ecol. 2008;17:4015–4026. doi: 10.1111/j.1365-294x.2008.03887.x. [DOI] [PubMed] [Google Scholar]
- 39*.Peter BM. Admixture, Population Structure, and F-Statistics. Genetics. 2016;202:1485–1501. doi: 10.1534/genetics.115.183913. A synthetic review relating f-statistics to coalescent times and mathematical concepts from phylogenetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pickrell JK, Pritchard JK. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet. 2012;8:e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipson M, Loh P-R, Levin A, Reich D, Patterson N, Berger B. Efficient Moment-Based Inference of Admixture Parameters and Sources of Gene Flow. Mol Biol Evol. 2013;30:1788–1802. doi: 10.1093/molbev/mst099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42*.Kamm JA, Terhorst J, Song YS. Efficient computation of the joint sample frequency spectra for multiple populations. arXiv:1503.01133 [math, q-bio] 2015 doi: 10.1080/10618600.2016.1159212. Develops an efficient framework to calculate likelihoods under arbitrary population trees with admixture. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huson DH, Rupp R, Scornavacca C. Phylogenetic networks: concepts, algorithms and applications. Cambridge University Press; Cambridge UK: 2010. [Google Scholar]
- 44.Yu Y, Degnan JH, Nakhleh L. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 2012;8:e1002660. doi: 10.1371/journal.pgen.1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- 48.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raj A, Stephens M, Pritchard JK. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Datasets. Genetics. 2014;197:573–89. doi: 10.1534/genetics.114.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gopalan P, Hao W, Blei DM, Storey JD. Scaling probabilistic models of genetic variation to millions of humans [Internet] bioRxiv. 2015 doi: 10.1101/013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown R, Pasaniuc B. Enhanced methods for local ancestry assignment in sequenced admixed individuals. PLoS Comput Biol. 2014;10:e1003555. doi: 10.1371/journal.pcbi.1003555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanderson J, Sudoyo H, Karafet TM, Hammer MF, Cox MP. Reconstructing Past Admixture Processes from Local Genomic Ancestry Using Wavelet Transformation. Genetics. 2015;200:469–481. doi: 10.1534/genetics.115.176842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53**.Hellenthal G, Busby GBJ, Band G, Wilson JF, Capelli C, Falush D, Myers S. A Genetic Atlas of Human Admixture History. Science. 2014;343:747–751. doi: 10.1126/science.1243518. A large-scale application of methods that detect and characterize admixture without using local ancestry reconstruction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Busby GBJ, Hellenthal G, Montinaro F, Tofanelli S, Bulayeva K, Rudan I, Zemunik T, Hayward C, Toncheva D, Karachanak-Yankova S, et al. The Role of Recent Admixture in Forming the Contemporary West Eurasian Genomic Landscape. Curr Biol. 2015;25:2518–2526. doi: 10.1016/j.cub.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loh P-R, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, Berger B. Inferring admixture histories of human populations using linkage disequilibrium. Genetics. 2013;193:1233–1254. doi: 10.1534/genetics.112.147330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engelhardt BE, Stephens M. Analysis of population structure: a unifying framework and novel methods based on sparse factor analysis. PLoS Genet. 2010;6:e1001117. doi: 10.1371/journal.pgen.1001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McVean G. A genealogical interpretation of principal components analysis. PLoS Genet. 2009;5:e1000686. doi: 10.1371/journal.pgen.1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abraham G, Inouye M. Fast principal component analysis of large-scale genome-wide data. PLoS One. 2014;9:e93766. doi: 10.1371/journal.pone.0093766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galinsky KJ, Bhatia G, Loh P-R, Georgiev S, Mukherjee S, Patterson NJ, Price AL. Fast Principal-Component Analysis Reveals Convergent Evolution of ADH1B in Europe and East Asia. Am J Hum Genet. 2016;98:456–472. doi: 10.1016/j.ajhg.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang C, Szpiech ZA, Degnan JH, Jakobsson M, Pemberton TJ, Hardy JA, Singleton AB, Rosenberg NA. Comparing spatial maps of human population-genetic variation using Procrustes analysis. Stat Appl Genet Mol Biol. 2010;9 doi: 10.2202/1544-6115.1493. Article 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang C, Zhan X, Bragg-Gresham J, Kang HM, Stambolian D, Chew EY, Branham KE, Heckenlively J, FUSION Study. Fulton R, et al. Ancestry estimation and control of population stratification for sequence-based association studies. Nat Genet. 2014;46:409–415. doi: 10.1038/ng.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skoglund P, Malmström H, Raghavan M, Storå J, Hall P, Willerslev E, Gilbert MTP, Götherström A, Jakobsson M. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science. 2012;336:466–469. doi: 10.1126/science.1216304. [DOI] [PubMed] [Google Scholar]
- 63.Hao W, Song M, Storey JD. Probabilistic models of genetic variation in structured populations applied to global human studies. Bioinformatics. 2016;32:713–721. doi: 10.1093/bioinformatics/btv641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64**.Bradburd GS, Ralph PL, Coop GM. A Spatial Framework for Understanding Population Structure and Admixture. PLoS Genet. 2016;12:e1005703. doi: 10.1371/journal.pgen.1005703. Develops a unique and flexible modeling approach that assumes a base model of isolation-by-distance but allows for long-range admixture. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 2008;40:646–649. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66**.Petkova D, Novembre J, Stephens M. Visualizing spatial population structure with estimated effective migration surfaces. Nat Genet. 2016;48:94–100. doi: 10.1038/ng.3464. A novel approach that visualizes population structure using maps of effective migration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67**.Hanks EM, Hooten MB. Circuit Theory and Model-Based Inference for Landscape Connectivity. J Am Stat Assoc. 2013;108:22–33. A novel approach for assessing the contribution of landscape features to patterns of spatial differentiation. [Google Scholar]
- 68.Duforet-Frebourg N, Blum MGB. Nonstationary Patterns of Isolation-by-Distance: Inferring measure of local genetic differentiation with Bayesian kriging. Evolution. 2014;68:1110–1123. doi: 10.1111/evo.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jay F, Sjödin P, Jakobsson M, Blum MGB. Anisotropic isolation by distance: the main orientations of human genetic differentiation. Mol Biol Evol. 2013;30:513–525. doi: 10.1093/molbev/mss259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gattepaille LM, Jakobsson M. Combining markers into haplotypes can improve population structure inference. Genetics. 2012;190:159–174. doi: 10.1534/genetics.111.131136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duforet-Frebourg N, Gattepaille LM, Blum MGB, Jakobsson M. HaploPOP: a software that improves population assignment by combining markers into haplotypes. BMC Bioinformatics. 2015;16:242. doi: 10.1186/s12859-015-0661-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palamara PF, Lencz T, Darvasi A, Pe’er I. Length distributions of identity by descent reveal fine-scale demographic history. Am J Hum Genet. 2012;91:809–822. doi: 10.1016/j.ajhg.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73*.Ralph P, Coop G. The Geography of Recent Genetic Ancestry across Europe. PLoS Biol. 2013;11:e1001555. doi: 10.1371/journal.pbio.1001555. An impressive demonstration of how long haplotype sharing can reveal finer structure and includes models for inferring coalescent time distributions from shared haplotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baharian S, Barakatt M, Gignoux CR, Shringarpure S, Errington J, Blot WJ, Bustamante CD, Kenny EE, Williams SM, Aldrich MC, et al. The Great Migration and African-American Genomic Diversity. PLoS Genet. 2016;12:e1006059. doi: 10.1371/journal.pgen.1006059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiang CWK, Ralph P, Novembre J. Conflation of Short Identity-by-Descent Segments Bias Their Inferred Length Distribution. G3. 2016;6:1287–1296. doi: 10.1534/g3.116.027581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76**.Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8:e1002453. doi: 10.1371/journal.pgen.1002453. Introduces an efficient framework to model very-recent population genetic structure and admixture based on the Li and Stephens haplotype copying model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lawson DJ, Falush D. Population identification using genetic data. Annu Rev Genomics Hum Genet. 2012;13:337–361. doi: 10.1146/annurev-genom-082410-101510. [DOI] [PubMed] [Google Scholar]
- 78.Li N, Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165:2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schiffels S, Durbin R. Inferring human population size and separation history from multiple genome sequences. Nat Genet. 2014;46:919–25. doi: 10.1038/ng.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harris K, Nielsen R. Inferring demographic history from a spectrum of shared haplotype lengths. PLoS Genet. 2013;9:e1003521. doi: 10.1371/journal.pgen.1003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rasmussen MD, Hubisz MJ, Gronau I, Siepel A. Genome-wide inference of ancestral recombination graphs. PLoS Genet. 2014;10:e1004342. doi: 10.1371/journal.pgen.1004342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83**.Durbin R. Efficient haplotype matching and storage using the positional Burrows–Wheeler transform (PBWT) Bioinformatics. 2014;30:1266–1272. doi: 10.1093/bioinformatics/btu014. Presents a conceptual breakthrough promising efficient algorithms for analysis of large-scale haplotype data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84*.Lunter G. Fast haplotype matching in very large cohorts using the Li and Stephens model. bioRxiv. 2016 doi: 10.1093/bioinformatics/bty735. Presents an algorithm for using the framework of the Positional Burrows Wheeler Transform to efficiently compute likelihoods under the Li & Stephens copying model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tian C, Plenge RM, Ransom M, Lee A, Villoslada P, Selmi C, Klareskog L, Pulver AE, Qi L, Gregersen PK, et al. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genet. 2008;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Harris K, Nielsen R. Error-prone polymerase activity causes multinucleotide mutations in humans. Genome Res. 2014;24:1445–1454. doi: 10.1101/gr.170696.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berg JJ, Coop G. A population genetic signal of polygenic adaptation. PLoS Genet. 2014;7:e1004412. doi: 10.1371/journal.pgen.1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marcus J, Novembre J. Visualizing the Geography of Genetic Variants. bioRxiv. 2016 doi: 10.1093/bioinformatics/btw643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rajeevan H, Soundararajan U, Kidd JR, Pakstis AJ, Kidd KK. ALFRED: an allele frequency resource for research and teaching. Nucleic Acids Res. 2012;40:D1010–5. doi: 10.1093/nar/gkr924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Osier MV, Cheung K-H, Kidd JR, Pakstis AJ, Miller PL, Kidd KK. ALFRED: An allele frequency database for anthropology. Am J Phys Anthropol. 2002;119:77–83. doi: 10.1002/ajpa.10094. [DOI] [PubMed] [Google Scholar]
- 91.Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]