

Abstract
To evaluate whether obese patients with a binge eating disorder (BED) have an altered metabolic and inflammatory profile related to their eating behaviors compared with non-BED obese.
A total of 115 White obese patients consecutively recruited underwent biochemical, anthropometrical evaluation, and a 75-g oral glucose tolerance test. Patients answered the Binge Eating Scale and were interviewed by a psychiatrist. The patients were subsequently divided into 2 groups according to diagnosis: non-BED obese (n = 85) and BED obese (n = 30). Structural equation modeling analysis was performed to elucidate the relation between eating behaviors and metabolic and inflammatory profile.
BED obese exhibited significantly higher percentages of altered eating behaviors, body mass index (P < 0.001), waist circumference (P < 0.01), fat mass (P < 0.001), and a lower lean mass (P < 0.001) when compared with non-BED obese. Binge eating disorder obese also had a worse metabolic and inflammatory profile, exhibiting significantly lower high-density lipoprotein cholesterol levels (P < 0.05), and higher levels of glycated hemoglobin (P < 0.01), uric acid (P < 0.05), erythrocyte sedimentation rate (P < 0.001), high-sensitive C-reactive protein (P < 0.01), and white blood cell counts (P < 0.01). Higher fasting insulin (P < 0.01) and higher insulin resistance (P < 0.01), assessed by homeostasis model assessment index and visceral adiposity index (P < 0.001), were observed among BED obese. All differences remained significant after adjusting for body mass index. No significant differences in fasting plasma glucose or 2-hour postchallenge plasma glucose were found. Structural equation modeling analysis confirmed the relation between the altered eating behaviors of BED and the metabolic and inflammatory profile.
Binge eating disorder obese exhibited an unfavorable metabolic and inflammatory profile, which is related to their characteristic eating habits.
INTRODUCTION
Obesity is the second cause of death in the world and has reached epidemic proportions in recent years.1 The World Health Organization estimates that there are over 1 billion overweight adults globally, 300 million of whom are obese.1 Obesity is a chronic disease and is associated with numerous comorbidities, including type 2 diabetes mellitus (T2DM), cardiovascular disease, hypertension, and dyslipidemia. Moreover, obesity is a risk factor for major causes of morbidity and mortality2,3 and increasing evidence shows that a binge eating disorder (BED) affects a subset of obese patients.4,5
Binge eating disorder is a psychiatric disorder characterized by recurrent episodes of binge eating (ie, eating large amounts of food) and is associated with a loss of control and significant distress in the absence of the regular compensatory weight-reducing behaviors commonly observed among patients with bulimia nervosa.4 The prevalence of BED in the normal adult population varies from 2% to 5%, but this proportion rises up to 50% among obese adults seeking weight reduction.6 European data indicate that the general prevalence of BED in Italy is approximately 1.12% (0.26% among men and 1.92% among women).7 Binge eating disorder is strongly linked with obesity and binge eating per se is linked with a high burden of metabolic risk factors in the general population.5,8 Studies on obese patients with BED suggest an increased risk for the metabolic syndrome both in adults and adolescents.9,10 Several studies showed a higher prevalence of BED in young and overweight patients with T2DM.11,12 Furthermore, BED is associated with poorer glycemic control and higher rates of diabetic complications in adolescents and adults with type 1 diabetes.13,14 Binge eating disorder and binging behaviors are associated with poorer response to weight loss therapy.5,9 A few studies, however, have assessed the metabolic profile of BED obese without T2DM and no data are available on the relation between different eating behaviors and the metabolic profile of these patients.
Growing evidence suggests that obesity is associated with a chronic inflammatory state. Indeed, a body of studies suggests the presence of an overall, low-grade inflammation in obesity, with increased levels of several circulating factors, such as high-sensitive C-reactive protein (hs-CRP), tumor necrosis factor-α, interleukin-6, and other biologic markers of inflammation.15 Conversely, a reduction in body weight is accompanied by a decrease or even a normalization of these inflammatory markers, which are known to have a causal relationship with obesity and its comorbidities, such as insulin resistance, T2DM, and cardiovascular risk.15 Low-grade inflammation is strongly linked with obesity15 but, to our best knowledge, no data are available on the possible differences regarding the inflammatory profile between obese patients with and without BED.
The aim of this study was to evaluate whether obese patients with BED and without T2DM have a different metabolic and inflammatory profile related to their eating behaviors compared with non-BED obese. We also wished to test the hypothesis that the characteristic eating patterns of BED obese lead to an altered metabolic and inflammatory profile when compared with non-BED obese.
METHODS
Study Participants
A total of 115 White obese patients without T2DM were consecutively recruited in this cross-sectional study at the Department of Medical and Surgical Sciences of the University “Magna Graecia” of Catanzaro (Italy) from December 2013 to December 2014. Patients were enrolled according to the following eligibility criteria: aged between 20 and 65 years, body mass index (BMI) >30 kg/m2 and the ability to answer a self-reporting questionnaire. Exclusion criteria were as follows: pregnancy or having recently given birth, previous diagnosis of diabetes mellitus, known inflammatory disease, a history of malignant disease or pathologies, or drugs able to modify glucose metabolism.
After 12-hour fasting, a venous blood sample was drawn for laboratory determinations and a 75 g oral glucose tolerance test was performed with 0, 30, 60, 90, and 120 minutes of sampling for plasma glucose and insulin for each patient. Glucose tolerance status was defined on the basis of body mass index according to the American Diabetes Association criteria.16
Patients underwent anthropometrical evaluation, wearing light indoor clothing, and no shoes, with a standing height to the nearest 0.1 cm and a body weight to the nearest 0.1 kg at 8.00 am. Height and weight were measured using a portable stadiometer (Seca 220, GmbH & Co., Hamburg, Germany) and a balance scale (Seca 761, GmbH & Co., Hamburg, Germany); then, their BMI (kg/m2) was calculated. In addition, waist and hip circumference were measured, and body composition was estimated by bioelectrical impedance. Blood pressure was measured using a calibrated manual sphygmomanometer and a stethoscope on the brachial artery at the antecubital area of the left elbow with patients supine after 5 minutes of rest.
The Hospital Ethical Committee (Comitato Etico Azienda Ospedaliera “Mater Domini”) approved protocol in September 2013, and written informed consent was obtained from all patients. All the investigations were performed in accordance with the principles of the Declaration of Helsinki.
Psychiatric Evaluation
Patients answered the Binge Eating Scale (BES).17 Binge Eating Scale is an easy to administer test with adequate internal consistency and validity. It has been widely used in research either to measure binge eating severity in the nonpurge binge eating population or to determine whether potential research patients meet the inclusion criteria for binge eating. Binge Eating Scale is made up of 16 items describing the behavioral manifestations, feelings, and cognitions associated with binge eating. Each item consists of 4 statements that reflect a range of severity from which patients choose the 1 that best describes their perceptions and feelings about their eating behavior. A total BES score <17 indicates unlikely BED, a 17 to 27 score possible BED and values >27 probable BED.
Psychiatric researchers with adequate training in the field of eating disorders interviewed each patient a week later. The interviewers examined each patient by means of the Structured Clinical Interview for Diagnostic and Statistic Manual of Mental Disorders IV (DSM-IV) Axis I Disorders18 and the Binge Eating Disorder-Clinical Interview19 to respectively assess/confirm the diagnosis of BED and to deepen understanding of their eating behaviors (ie, night eating, postdinner eating, social eating, sweet eating, emotional eating, grazing, craving for carbohydrates, and hyperphagia) and exercise habits.
The clinical interview through the Structured Clinical Interview for DSM-IV Axis I Disorders confirmed the diagnosis of BED for all the obese patients with BES scores ≥17. No BED diagnosis was confirmed among patients with BES < 17.
According to the diagnosis of the BED, the patients were divided into 2 groups: non-BED obese patients (n = 85) and BED-obese patients (n = 30).
Laboratory Determinations
Plasma glucose, total and high-density lipoprotein (HDL) cholesterol, triglycerides, and uric acid concentrations were measured by enzymatic methods (Roche Diagnostics, Mannheim, Germany). Glycated hemoglobin (HbA1c) was measured with high-performance liquid chromatography using a National Glycohemoglobin Standardization Program certified automated analyzer and IFCC (Adams HA-8160 HbA1C analyzer, Menarini, Italy; normal reference range, 4.3%–5.9%).
Plasma insulin concentration was determined by a chemiluminescence-based assay (IMMULITE, Siemens Healthcare, Italy). High-sensitivity C reactive protein levels were measured by an automated instrument (CardioPhase hsCRP, Milan, Italy). The erythrocyte sedimentation rate (ESR) was measured automatically by the stopped-flow technique in a capillary microphotometer (Alifax Test 1 System Polverara, Italy). All others metabolites were measured by standard methods. Insulin resistance was estimated by the homeostasis model assessment (HOMA-IR) index, calculated from the fasting glucose, and insulin concentrations according to the following formula (fasting insulin × fasting glucose)/22.5.20 The visceral adiposity index (VAI) was calculated using the formulas proposed by Amato et al21 for men, VAI = [white blood cell (WC)/36.58 + (1.89 × BMI)] × (tryglicerides (TG)/0.81) × (1.52/HDL) and for women, VAI = [WC/39.68 + (1.88 × BMI)] × (TG/1.03) × (1.31/HDL).
Statistical Analysis
Data were analyzed using the Statistical Package for the Social Science, version 21.0 (SPSS Inc., Chicago, IL). Variables with skewed distribution, including triglyceride, hsCRP, ESR, WBC, and fasting insulin were natural log transformed for statistical analyses. Continuous data are expressed as means ± SD. Categorical variables were compared by χ2 test. Anthropometric and metabolic differences between groups were tested after adjusting for age, sex, and BMI using a general linear model. A P value <0.05 was considered statistically significant. Cohen's effect sizes (ES) were calculated for all significant findings, with values (negative or positive) of 0.2, 0.6, 1.2, and >1.2 indicating trivial, small, moderate, and large ES, respectively.22
The structural equation modeling using maximum likelihood estimation was conducted by means of SPSS/AMOS 21.0 to test for the validity of the hypothesized model. Structural equation modeling is a method that allows the simultaneous estimation of all relationships between observed (manifest or unmeasured) and unobserved (or latent) variables of a model. It is graphically represented with observed variables enclosed by rectangles and latent variables by circles. An assumed causal path between 2 variables is shown by a directed edge (single-headed arrow). Path coefficients on the edges are partial standardized regression coefficients, which measure the effect of 1 variable on another, while controlling for all other variables prior in the model; all coefficients show a positive association, but those with a minus sign indicate a negative association. Model fit was evaluated using the comparative fit index (CFI), the Tucker–Lewis incremental fit index (TLI), and root mean square error of approximation (RMSEA). The magnitudes of these indices were evaluated according to the recommendations of Hu and Bentler.23 For CFI and TLI, values of 0.90 and above were considered adequate, whereas values of 0.95 or above were considered very good; for RMSEA values of 0.08 and below were considered adequate and 0.05 or less very good. The ratio of χ2 and degree of freedom was also evaluated, considering that values below 3.0 are good and those below 2.0 are very good.
RESULTS
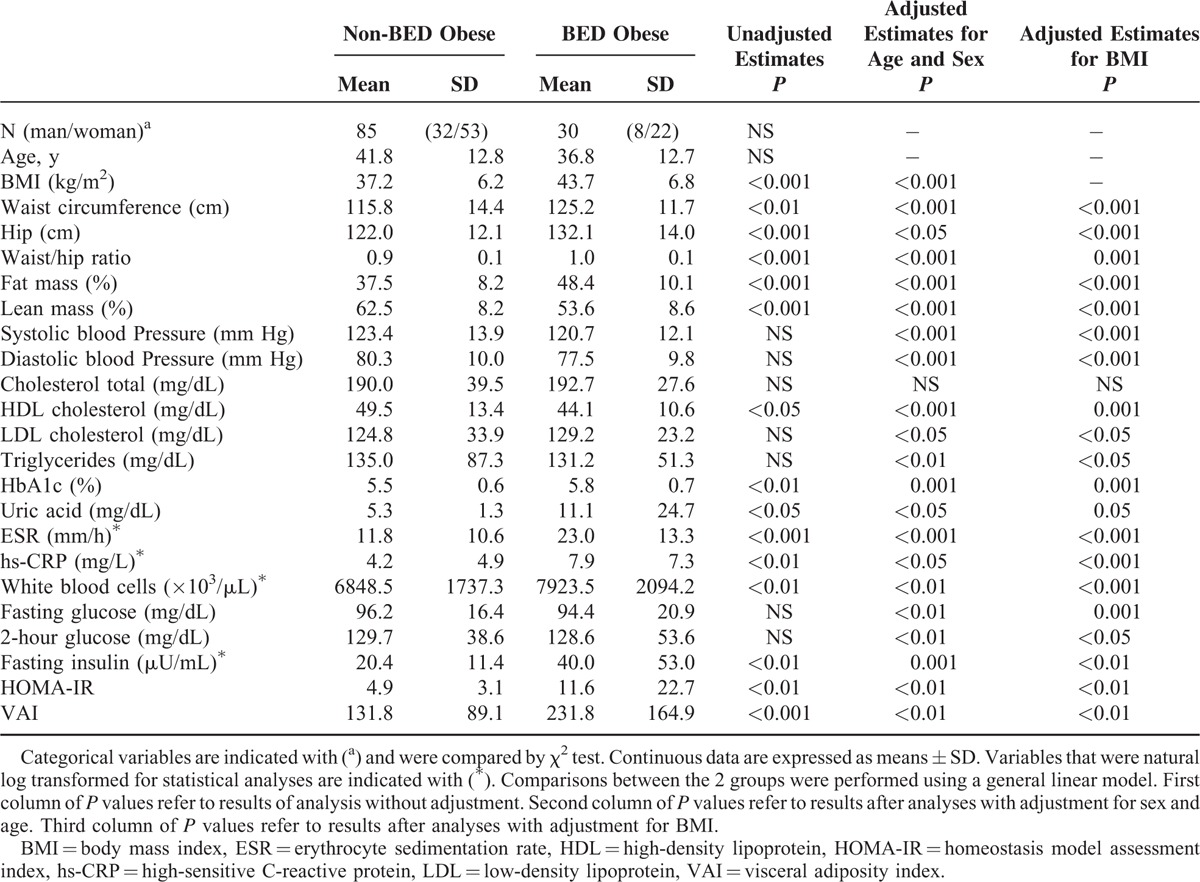
Table 1 shows the anthropometrical characteristics and laboratory findings for the 2 study groups. There were no differences in age and sex between BED and non-BED obese. Binge eating disorder obese exhibited significantly higher BMI, waist circumference, hip circumference, waist/hip ratio, and fat mass, and a lower lean mass as compared with non-BED obese; these differences remained significant after adjusting for BMI (Table 1). There were no differences between the groups in blood pressure, total cholesterol, low-density lipoprotein cholesterol, triglycerides. Binge eating disorder obese had a worse metabolic and inflammatory profile, exhibiting significantly lower HDL cholesterol and higher levels of HbA1c, uric acid, ESR, hs-CRP, and WBC and all these differences remained significant after adjusting for BMI (Table 1). Binge eating disorder obese also exhibited significantly higher levels of fasting plasma insulin and higher degree of insulin resistance as assessed by HOMA-IR index and VAI compared with non-BED obese; similarly, differences remained significant after adjusting for BMI. No significant differences were found in fasting glucose and 2-hour postload plasma glucose. Blood pressure, low-density lipoprotein cholesterol, triglycerides, fasting glucose, and 2-hour postload plasma glucose were statistically significant after adjusting for BMI.
TABLE 1.
Comparison of Anthropometrical and Laboratory Characteristics Between Groups
Regarding eating habits, BED obese revealed significantly higher percentages of grazing, emotional eating, night eating, sweet eating, and craving for carbohydrates than non-BED obese (Figure 1).
FIGURE 1.
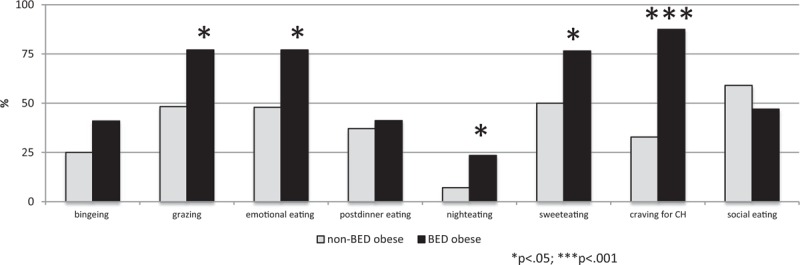
Eating behaviors in binge eating disorder and non-binge eating disorder obese patients.
Structural equation modeling was conducted to test the hypothesis that altered eating behaviors affect anthropometrical, metabolic, and inflammatory variables. The model achieved goodness-of-fit: χ2 = 69.755; P = 0.261; ratio of χ2 and degree of freedom = 1.107; CFI = 0.980; TLI = 0.970; and RMSEA = 0.032. In our model, altered eating behaviors (ie, night eating, sweet eating, craving for carbohydrates, emotional eating, and grazing) showed an indirect effect on waist circumference mediated by the diagnosis of BED and body weight; waist circumference had an indirect effect on HbA1c mediated by insulin resistance assessed either by HOMA-IR or VAI; and VAI had a direct effect on the inflammatory indexes (ie, ESR, hs-CRP, and uric acid; Figure 2).
FIGURE 2.
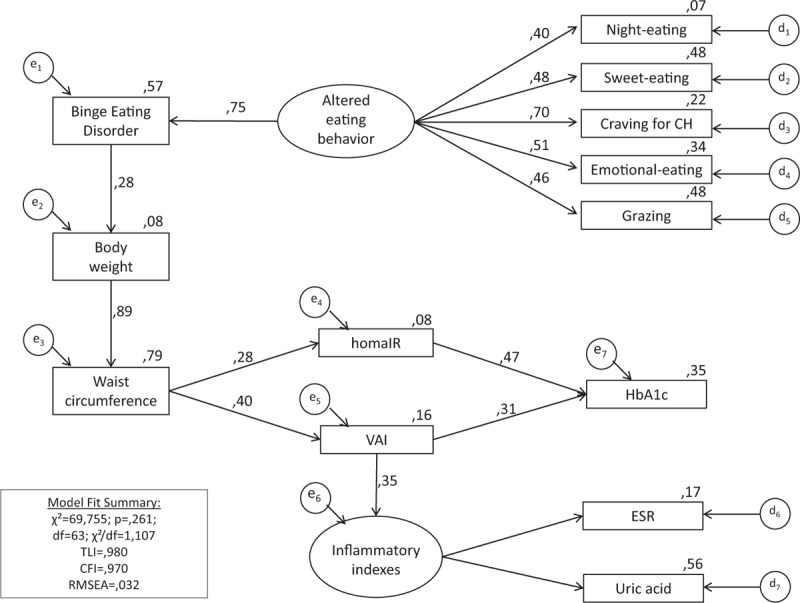
Path diagram showing the relation between eating behaviors and metabolic and inflammatory profile. Craving for BED = binge eating disorder, CFI = comparative fit index, CH = craving for carbohydrates, CMIN = χ2, df = degree of freedom, ESR = erythrocyte sedimentation rate; RMSEA = root mean square error of approximation, TLI = Tucker–Lewis Index, VAI = visceral adiposity index.
DISCUSSION
In this study, we showed an unfavorable metabolic and inflammatory profile in obese patients with BED as compared with non-BED obese.
Notably, we reported, for the first time that BED obese, exhibited a worse anthropometric and metabolic profile in relation to their different eating behaviors, as compared with non-BED obese. There are several potential mechanisms by which binge eating may cause or contribute to obesity. The characteristic eating patterns of BED individuals seem to be associated with specific metabolic abnormalities of obesity. For example, the ingestion of fewer but larger meals has adverse metabolic consequences than the ingestion of frequent, small meals, including increase in fasting glucose, insulin secretion, serum lipids, and deterioration of glucose tolerance.10,24 Eating rapidly results in elevated serum lipids, higher waist-hip circumference ratio, and liver steatosis.25 The mechanism(s) underlying the relationship between eating rate and fatty liver are not known. As reported by Kral et al,25 it is hypothesized that the enhance and rapid glucose absorption, mediated by a brisk insulin response via cephalic phase release, an incretin effect and rapid intestinal handling, causes fatty infiltration of the liver via glucose toxicity. Alone or in conjunction with rapid absorption of lipid, glucose and insulin increases may lead to insulin resistance and metabolic syndrome with central/visceral fat distribution and dyslipidemia. Besides, increased insulin levels induced by binging may contribute to increased hunger.26 In fact, BED patients eat significantly more than do non-BED obese during the stimulation of a binge eating episode.27 Despite the relation between specific metabolic alterations and these eating features of BED individuals (ie, speed and amount), little is known about the metabolic consequences of the other eating habits typically observed in BED individuals (eg, craving for carbohydrates, sweet eating, grazing, or emotional eating).
Herein, we provide evidence that BED obese showed a higher frequency of altered eating behaviors (ie, binging, grazing, emotional eating, sweet eating, and craving for carbohydrates) than non-BED obese.
We did not observe a significant difference in binge eating frequency between BED and non-BED obese patients. The symptom “binge” does not directly imply with the diagnosis of BED. “Binge eating” is a behavior marked by consumption of a large amount of food within a short period of time with the sense of loss of control. But to fulfill the diagnosis of BED,4 the patient needs to have other symptoms, such as eating quickly, eating until feeling full, eating even if not hungry, eating alone for embarrassment and/or feeling disgusted, depressed, or guilty for overeating. So binge is a characteristic symptom but not an exclusive symptom of patients with BED. On the contrary, DSM-IV requires that binges occur at least 2 days a week for 6 months, so many obese patients could not satisfy the criteria of frequency while still binging. As binging becomes a distraction related to aversive emotional states, BED obese tend more frequently to eat in response to emotions.28 Our results are in agreement with previous data showing that emotional eating is associated with the preference for sweet foods and that BED obese crave carbohydrates more than fats, compared with non-BED obese.29 As previously reported, grazing, another dysfunctional eating behavior that consists of smaller, subjective episodes of overeating, was significantly more evident among BED obese as compared with non-BED obese. We postulate that the compensatory pattern, demonstrated in healthy lean patients, could be impaired in patients who are already obese and have chronic binge habits. These habits could be responsible, in association with obesity, for the worse metabolic profile observed in BED obese.30 Accordingly, we found that BED obese exhibited a significantly higher BMI, fat mass, fasting insulin levels, HOMA-IR index, and HbA1c when compared with non-BED obese. Moreover, among BED obese, we found an increased abdominal circumference, an indirect index of visceral adiposity and an increased VAI, an indicator of a dysfunction of adipose tissue.21,31,32 In obese patients, visceral adipose tissue accumulation has been associated with an increased production of free fatty acids, interleukin-6, tumor necrosis factor-α, hs-CRP, and a decreased production of adiponectin, each of which may contribute to insulin resistance. It is known that these cytokines and chemokines activate intracellular pathways that lead to the development of insulin resistance, increasing the risk of T2DM.33 A previously study demonstrated that women with BED had significantly reduced plasma levels of adiponectin.34 Accordingly, it is conceivable that increased visceral adiposity observed in BED obese may be responsible for increased insulin resistance, estimated by the HOMA-IR index.
Importantly, we observed that the BED obese showed elevated inflammatory markers, such as hs-CRP, ESR, and WBC counts. Several studies showed that chronic subclinical inflammation is associated with T2DM, cardiovascular disease, and patients at high risk of developing T2DM.35–39 Elevation in hs-CRP is considered a marker of cardiovascular risk that has also been correlated with insulin resistance.33 Increasing evidence has shown that the accumulation of lipids in adipose tissue and the expansion of fat mass determine the initiation of the obesity-induced inflammatory process through the production of proinflammatory cytokines and chemokines by the fat tissue.33
All these differences remained significant after adjusting for BMI. Our results, in line with Hudson et al,9 disagree with Abraham et al.8 A possible explanation is that the research by Hudson et al was drawn among obese patients, as in our case, whereas Abraham et al used a large population-based cohort comparing obese (most bingers) with overweight (most nonbingers).
Moreover, we found, that BED obese showed higher uric acid levels than non-BED obese did. Elevation in uric acid has been associated with obesity and insulin resistance, all risk factors for atherosclerosis, and T2DM.40 The prooxidant and proinflammatory effects of uric acid that interfere with glucose uptake may explain this association. Furthermore, hyperuricemia is frequently documented in patients with cardiovascular diseases and with subclinical organ damage.41
Structural equation modeling analysis helps to explain the relation between eating behaviors and metabolic impairment. Night eating, sweet eating, grazing, emotional eating, and craving for carbohydrates may lead to increased waist circumference, an index of visceral adiposity, and insulin resistance, which, in turn, may favor production of inflammatory molecules, and alteration in glucose metabolism. The mechanism(s) by which the eating behaviors are able to induce an alteration of metabolic profile and/or the inflammatory profile is not known. It might represent a direct effect of binge eating, perhaps because of the large amount of food ingested in typical eating binges.42 Also, rapid consumption of large amounts of food can increase oxidative and inflammatory stress,43,44 and inflammatory changes could represent an important causal pathway for developing metabolic alterations. Alternatively, because BED appears to be partially caused by genetic factors independent of obesity,45–47 it is possible that these or other underlying nongenetic factors might be responsible for these alterations.
The novelty of this article is that an eating disorder, namely BED, and the related altered eating behaviors, can help to explain an impaired metabolic and inflammatory profile in a group of obese patients that could have an increased cardiometabolic risk.
Strengths of this study are the exclusion of patients with T2DM or confounding disorders characterized by elevation in inflammatory molecules, inclusion of both sexes, the accurate anthropometric, metabolic, inflammatory and psychiatric characterization, and the evaluation of eating behaviors.
The findings of this study, however, need to be interpreted in light of some limitations. First, the current results derive from a cross-sectional research with a small sample of obese patients. Nevertheless, the sample included all obese patients that consecutively asked for weight reduction therapy in our department. Furthermore, ES and fit indexes of structural equation modeling demonstrated that the results were not influenced by the sample size. A second limitation of the current study is that we have evaluated insulin sensitivity by the HOMA-IR index. Although the euglycaemic–hyperinsulinaemic clamp, which is considered the gold standard method to measure insulin sensitivity, may provide a more accurate estimate of insulin sensitivity, it is time consuming and expensive, and is not feasible in large-scale studies.
Despite the limitations, our findings may have important clinical implications. The current study confirms and expands the actual knowledge about the effects of specific dysfunctional eating behaviors commonly observed among obese patients on important metabolic and inflammatory alterations. All obese patients should be assessed for BED and it can be done through an easy tool, the BES. This could represent an important clinical target to identify patients potentially at high risk for obesity, T2DM, and/or cardiometabolic disease. Further research and prospective studies, however, are needed to assess and evaluate whether early detection and intervention for this specific disorder could have a positive prognostic impact on the long-term outcome (eg, T2DM and/or cardiovascular risk) of this specific group of obese patients seeking weight reduction.
Acknowledgments
Authors are grateful to Antonio José Ruiz Moruno and Carmelo Nobile for their technical support in performing the statistical analysis.
Footnotes
Abbreviations: ADA = American Diabetes Association, BED = binge eating disorder, BED-CI = Binge Eating Disorder-Clinical Interview, BES = Binge Eating Scale, BMI = body mass index, BP = blood pressure, CFI = comparative fit index, CMIN/DEF = ratio of χ2 and degree of freedom, CVD = cardiovascular disease, ED = eating disorder, ESR = erythrocyte sedimentation rate, GLM = general linear model, HDL = high-density lipoprotein, HOMA-IR = homeostasis model assessment index, hs-CRP = high-sensitive C-reactive protein, IL-6 = interleukin-6, LDL = low-density lipoprotein, OGTT = oral glucose tolerance test, RMSEA = root mean square error of approximation, SEM = structural equation modeling, T2DM = type 2 diabetes mellitus, TLI = Tucker–Lewis index, TNF-α = tumor necrosis factor-α.
ES and CS-G contributed equally to this work
Author Contributions: ES designed the study, acquired data, and wrote the article. CS-G designed the study, performed the statistical analysis, and wrote the article; she has had full access to the data in the study and final responsibility for the decision to submit for publication. MR acquired data. MC acquired data. MR acquired data. MA performed the statistical analysis. GS revised the article. PDF contributed to discussion and revised the article. FA designed the study, wrote the article, revised, and edited the article and approved the final version.
The authors have no funding and conflicts of interest to disclose.
REFERENCES
- 1.Haslam DW, James WP. Obesity. Lancet 2005; 366:1197–1209. [DOI] [PubMed] [Google Scholar]
- 2.Hubert HB, Feinleib M, McNamara PM, et al. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation 1983; 67:968–977. [DOI] [PubMed] [Google Scholar]
- 3.Adams KF, Schatzkin A, Harris TB, et al. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. N Engl J Med 2006; 355:763–778. [DOI] [PubMed] [Google Scholar]
- 4.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed.Washington, DC: Text Revision; 2000. [Google Scholar]
- 5.De Zwaan M. Binge eating disorder and obesity. Int J Obes Relat Metab Disord 2001; 25:S51–S55. [DOI] [PubMed] [Google Scholar]
- 6.Hudson JI, Hiripi E, Pope HG, Jr, et al. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry 2007; 61:348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Preti A, Vilagut GD, Alonso J, et al. The epidemiology of eating disorders in six European countries: results of the ESEMeD-WMH project. J Psychiatr Res 2009; 43:1125–1132. [DOI] [PubMed] [Google Scholar]
- 8.Abraham TM, Massaro JM, Hoffmann U, et al. Metabolic characterization of adults with binge eating in the general population: the Framingham Heart study. Obesity 2014; 22:2441–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudson JI, Lalonde JK, Coit CE, et al. Longitudinal study of the diagnosis of components of the metabolic syndrome in individuals with binge-eating disorder. Am J Clin Nutr 2010; 91:1568–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blomquist KK, Milsom VA, Barnes RD, et al. Metabolic syndrome in obese men and women with binge eating disorder: developmental trajectories of eating and weight-related behaviors. Compr Psychiatry 2012; 53:1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meneghini LF, Spadola J, Fiorez H. Prevalence and associations of binge eating disorder in a multiethnic population with type 2 diabetes. Diabetes Care 2006; 29:2760. [DOI] [PubMed] [Google Scholar]
- 12.Wilfley D, Berkowitz R, Goebel-Fabbri A, et al. TODAY Study Group. Binge eating, mood, and quality of life in youth with type 2 diabetes. Diabetes Care 2011; 34:858–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wing RR, Nowalk MP, Marcus MD, et al. Subclinical eating disorders and glycemic control in adolescents with type I diabetes. Diabetes Care 1986; 9:162–167. [DOI] [PubMed] [Google Scholar]
- 14.Scheuing N, Bartus B, Berger G, et al. DPV Initiative; German BMBF Competence Network Diabetes Mellitus. Clinical characteristics and outcome of 467 patients with a clinically recognized eating disorder identified among 52,215 patients with type 1 diabetes: a multicenter German/Austrian study. Diabetes Care 2014; 37:1581–1589. [DOI] [PubMed] [Google Scholar]
- 15.Bastard JP, Maachi M, Lagathu C, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw 2006; 17:4–12. [PubMed] [Google Scholar]
- 16.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2011; 34:S62–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gormally J, Black S, Daston S. The assessment of binge eating severity among obese persons. Addict Behav 1982; 7:47–55. [DOI] [PubMed] [Google Scholar]
- 18.Lobbestael J, Leurgans M, Arntz A. Inter-rater reliability of the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID I) and Axis II Disorders (SCID II). Clin Psychol Psychoter 2011; 18:75–79. [DOI] [PubMed] [Google Scholar]
- 19.Spitzer RL, Yanovsky SZ, Marcus MD. Binge Eating Clinical Interview. Pittsburgh, PA: HaPI Record; 1994. [Google Scholar]
- 20.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28:412–419. [DOI] [PubMed] [Google Scholar]
- 21.Amato MC, Giordano C, Galia M, et al. AlkaMeSy Study Group. Visceral Adiposity Index: a reliable indicator of visceral fat function associated with cardiometabolic risk. Diabetes Care 2010; 33:920–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen J. Statistical Power Analysis for the Behavioral Sciences. Hillsdale, MI: Lawrence Erlbaum Associates Inc; 1988. [Google Scholar]
- 23.Hu L, Bentler PM. Cutoff criteria for fit indexes in covariance structure analysis: conventional criteria versus new alternatives. Struct Equ Modeling 1999; 6:1–55. [Google Scholar]
- 24.Taylor AE, Hubbard J, Anderson EJ. Impact of binge eating on metabolic and leptin dynamics in normal young women. J Clin Endocrinol Metab 1999; 84:428–434. [DOI] [PubMed] [Google Scholar]
- 25.Kral JG, Buckley MC, Kissileff HR, et al. Metabolic correlates of eating behavior in severe obesity. Int J Obes Relat Metab Disord 2001; 25:258–264. [DOI] [PubMed] [Google Scholar]
- 26.Stricker EM. Biological bases of hunger and satiety: therapeutic implications. Nutr Rev 1984; 42:333–340. [DOI] [PubMed] [Google Scholar]
- 27.Sysko R, Devlin MJ, Walsh BT, et al. Satiety and test meal intake among women with binge eating disorder. Int J Eat Disord 2007; 40:554–561. [DOI] [PubMed] [Google Scholar]
- 28.Ramacciotti CE, Coli E, Marazziti D, et al. Therapeutic options for binge eating disorder. Eat Weight Disord 2013; 18:3–9. [DOI] [PubMed] [Google Scholar]
- 29.Meule A, Allison KC, Platte P. Emotional eating moderates the relationship of night eating with binge eating and body mass. Eur Eat Disord Rev 2014; 22:147–151. [DOI] [PubMed] [Google Scholar]
- 30.Wadden TA, Foster GD, Letizia KA, et al. Metabolic, anthropometric psychological characteristics of obese binge eaters. Int J Eat Disord 1993; 1:17–25. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Shu X, Li H, et al. Visceral adiposity and risk of coronary heart disease in relatively lean Chinese adults. Int J Cardiol 2013; 168:2141–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fox CS, Massaro JM, Hoffmann U, et al. Abdominal visceral and subcutaneous adipose tissue compartments. Circulation 2007; 116:39–48. [DOI] [PubMed] [Google Scholar]
- 33.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation and insulin resistance. Gastroenterology 2007; 132:2169–2180. [DOI] [PubMed] [Google Scholar]
- 34.Monteleone P, Fabrazzo M, Martiadis V, et al. Opposite changes in circulating adiponectin in women with bulimia nervosa or binge eating disorder. J Clin Endocrinol Metab 2003; 88:5387–5391. [DOI] [PubMed] [Google Scholar]
- 35.Duncan BB, Schmidt MI, Pankow JS, et al. Atherosclerosis Risk in Communities Study. Low-grade systemic inflammation and the development of type 2 diabetes. The arteriosclerosis risk in communities study. Diabetes 2003; 52:1799–1805. [DOI] [PubMed] [Google Scholar]
- 36.Perticone F, Maio R, Sciacqua A, et al. Endothelial dysfunction and C-reactive protein are risk factors for diabetes in essential hypertension. Diabetes 2008; 57:167–171. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt MI, Duncan BB, Sharrett AR, et al. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet 1999; 353:1649–1652. [DOI] [PubMed] [Google Scholar]
- 38.Succurro E, Marini MA, Arturi F, et al. Elevated one-hour post-load plasma glucose levels identifies subjects with normal glucose tolerance but early carotid atherosclerosis. Atherosclerosis 2009; 207:245–249. [DOI] [PubMed] [Google Scholar]
- 39.Sesti G, Fiorentino TV, Succurro E, et al. Elevated 1-h post-load plasma glucose levels in subjects with normal glucose tolerance are associated with an unfavorable inflammatory profile. Acta Diabetol 2014; 51:927–932. [DOI] [PubMed] [Google Scholar]
- 40.Facchini F, Chen YD, Hollenbeck CB, et al. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. J Am Med Assoc 1991; 266:3008–3011. [PubMed] [Google Scholar]
- 41.Perticone F, Sciacqua A, Perticone M, et al. Serum uric acid and 1-h postload glucose in essential hypertension. Diabetes Care 2012; 35:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolfe BE, Baker CW, Smith AT, et al. Validity and utility of the current definition of binge eating. Int J Eat Disord 2009; 42:674–686. [DOI] [PubMed] [Google Scholar]
- 43.Bowen PE, Borthakur G. Postprandial lipid oxidation and cardiovascular disease risk. Curr Atheroscler Rep 2004; 6:477–484. [DOI] [PubMed] [Google Scholar]
- 44.Patel C, Ghanim H, Ravishankar S, et al. Prolonged reactive oxygen species generation and nuclear factor-kappaB activation after a high-fat, high-carbohydrate meal in the obese. J Clin Endocrinol Metab 2007; 92:4476–4479. [DOI] [PubMed] [Google Scholar]
- 45.Hudson JI, Lalonde JK, Berry JM, et al. Binge-eating disorder as a distinct familial phenotype in obese individuals. Arch Gen Psychiatry 2006; 63:313–319. [DOI] [PubMed] [Google Scholar]
- 46.Bulik CM, Sullivan PF, Kendler KS. Genetic and environmental contributions to obesity and binge eating. Int J Eat Disord 2003; 33:293–298. [DOI] [PubMed] [Google Scholar]
- 47.Davis C. The epidemiology and genetics of binge eating disorder (BED). CNS Spectr 2015; 10:1–8. [DOI] [PubMed] [Google Scholar]