

Abstract
Autoantibodies can be present years to decades prior to the onset of disease manifestations in autoimmunity. This suggests that the initial autoimmune trigger involves a peripheral lymphoid component, which ultimately drives disease pathology in local tissues later in life. Here we show Sjögren’s Syndrome manifestations that develop in aged NOD.H-2h4 mice were driven by and dependent on peripheral dysregulation that arose in early life. Specifically, elimination of spontaneous germinal centers in spleens of young NOD.H-2h4 mice by transient blockade of CD40 ligand (CD40L) or splenectomy abolished Sjögren’s pathology of aged mice. Strikingly, a single injection of anti-CD40L at 4 weeks-of-age prevented tertiary follicle neogenesis and greatly blunted the formation of key autoantibodies implicated in glandular pathology, including anti-muscarinic receptor antibodies. Microarray profiling of the salivary gland characterized the expression pattern of genes that increased with disease progression and showed early anti-CD40L greatly repressed B cell function, while having a broader effect on multiple biological pathways including IL-12 and interferon signaling. Importantly, a single, prophylactic treatment with anti-CD40L also inhibited the development of autoimmune thyroiditis and diabetes in NOD.H-2h4 and NOD mice, respectively, supporting a key role for CD40L in the pathophysiology of several autoimmune models. These results strongly suggest early peripheral immune dysregulation gives rise to autoimmune manifestations later in life and for diseases pre-dated by autoantibodies, early prophylactic intervention with biologics may prove efficacious.
Introduction
Primary Sjögren’s syndrome (pSS) is a chronic autoimmune disorder affecting exocrine glands. The disease is characterized by peri-ductal infiltration of salivary glands (sialadenitis) and lacrimal glands that destroy secretory function leading to xerostomia/dry mouth and keratoconjunctivitis sicca/dry eyes (1). The cells that inflame the salivary gland often organize into tertiary lymphoid structures (TLS). Tertiary lymphoid structures act as inductive sites for autoreactive T cell-B cell interactions that drive plasma cell differentiation and production of autoantibodies resulting in tissue damage. Early pioneering work showed salivary gland TLS present in approximately one quarter of pSS patients (28%) displayed germinal center-like areas with follicular dendritic cell (FDC) networks that associated with increased inflammatory cell infiltration and elevated levels of autoantibodies (2). Furthermore, these germinal center-like areas contained autoantibody producing cells suggesting that these plasma blasts differentiated locally in the TLS of inflamed salivary gland (3). Initiation and maintenance of germinal centers requires the TNF receptor superfamily member, CD40 (4–7) which is expressed in salivary gland TLS in both human disease as well as in animal models of pSS (8, 9). CD40 is also expressed on salivary gland epithelial and endothelial cells (8, 10). Elucidation of the cellular and molecular events leading to TLS neogenesis and the role of CD40 is critical for understanding autoimmune pathophysiology.
Autoantibodies that arise in autoimmunity such as type 1 diabetes (T1D), thyroiditis, lupus, rheumatoid arthritis or pSS can be present years to decades prior to the onset of disease manifestations (11–17). In pSS, autoantibodies to several self-antigens are common. Anti-Ro/SSA and anti-La/SSB autoantibodies directed against nuclear antigens are a hallmark of disease and associated with increased disease activity, salivary gland infiltration and extra-glandular manifestations (18). However, the direct role of these autoantibodies in glandular pathogenesis is unclear (19). Anti-Ro/SSA and anti-La/SSB autoantibodies appear in the blood years before the onset of symptoms (14, 17), though they can also be found in saliva once the disease manifests (20). Autoantibodies to the acetylcholine muscarinic type 3 receptor (M3R), a receptor that regulates exocrine secretion and smooth muscle contraction, are also common in pSS and thought to be directly involved in secretory dysfunction of pSS (21–23). Detection of autoantibodies years before clinical or histologic disease suggests that autoimmunity does not simply arise in local inflamed tissues, but rather develops in peripheral immune compartments such as the spleen where autoreactive cells then traffic to target tissues and drive disease manifestations later in life. It is therapeutically important to understand the mechanisms initiating these early autoimmune events and the resultant pathogenic features that occur.
Non-obese diabetic (NOD) mice are an autoimmune strain of mice that develop spontaneous T1D as well as autoantibodies such as anti-insulin autoantibodies (24). When made congenic for the B10.A(4R) (H-2h4) strain-derived MHC haplotype (NOD.H-2h4), these mice are no longer T1D susceptible but develop a disease closely resembling pSS including linkage to females, characteristic anti-Ro/La autoantibodies and inflammation of the salivary gland resulting in xerostomia/dry mouth (25, 26). We have previously characterized the TLS that develop in the salivary glands of these NOD.H-2h4 mice and found that the TLS organize into defined B cell and T cell regions containing FDC networks and dendritic cells (DC), respectively, with PNAd+ high endothelial venules (HEVs) and occasional germinal centers (27). Furthermore, similar to human disease, autoantibodies arise prior to the emergence of salivary gland TLS in these mice coinciding with the appearance of spontaneous germinal centers of the spleen (27). Aberrant regulation of germinal centers has also been implicated in several other autoimmune strains including NOD mice as well as in human autoimmune syndromes (6, 28), suggesting a pivotal role in disease development. NOD.H-2h4 mice also develop spontaneous autoimmune thyroiditis (SAT) when exposed to sodium-iodide (NaI), resulting in thyroid inflammation, destruction of thyroid follicles, and anti-mouse thyroglobulin (MTg) autoantibodies (29).
Here, we studied diseases that arise in both NOD and NOD.H-2h4 mice to understand the origin of autoreactivity and subsequent transition into fully manifested disease. We hypothesized that autoimmune specificities first arise in early-life lymphoid compartments and are then amplified in TLS of local tissues resulting in overt disease pathology. Collectively, using three different autoimmune disease models, our findings highlight the contribution of CD40 to early-life immune dysregulation culminating in organ-specific autoimmunity.
Results
Early immune activation drives autoimmunity that develops in aged mice
Previously, we have characterized the kinetics of TLS neogenesis of female NOD.H-2h4 mice and shown that full establishment of TLS in the salivary gland does not occur until 24 weeks-of-age, where organized B and T cell regions that contain FDC, DC and occasional germinal centers are noted (27). As CD40 is a key mediator of both immunity and autoimmunity, the requirement of CD40 signaling for the development of salivary gland TLS (as defined by foci of 50 or more hematopoietic cells clustered together often around individual excretory ducts) was evaluated in aged female NOD.H-2h4 mice. Notably, six month-old mice devoid of CD40 expression (CD40−/−) showed a complete absence of salivary gland TLS compared to aged-matched CD40-sufficient (Control) mice where salivary glands were heavily infiltrated with organized B and T cell regions (Fig. 1 A, B left panel, C). To determine if early life CD40/CD40L interactions contributed to the development of TLS, a single injection of anti-CD40L was administered to female NOD.H-2h4 mice at 4–5 weeks-of-age, months before detectable salivary gland TLS appeared. Surprisingly, a single dose of anti-CD40L early in life abolished salivary gland TLS in aged mice (Fig. 1B right panel, C). Consistent with immuno-histology, flow-cytometry confirmed that early life anti-CD40L resulted in a dramatic and significant reduction of B and T cells infiltrating the salivary gland (Fig. S1). In order to understand if early CD40L blockade was abolishing TLS by inhibiting CD40/CD40L interactions on hematopoietic cells, presumably B and T cells, or rather by interfering with CD40 signaling potentially expressed on salivary gland epithelial or endothelial cells, radiation chimeras were established. Consistent with Figure, 1A, transfer of wild type (WT) NOD.H-2h4 bone marrow into lethally irradiated WT NOD.H-2h4 recipients, or CD40−/− NOD.H-2h4 bone marrow into CD40−/− recipients, recapitulated the requirement of CD40 for the development of salivary gland TLS in aged mice (Fig. 1D). Notably, CD40 expression on hematopoietic, but not stromal, cells was required for the development of TLS, as TLS were clearly visible when WT bone marrow was transferred into CD40−/− recipients, but completely absent when CD40−/− bone marrow was transferred into WT recipients (Fig. 1D). Taken together, these data clearly show CD40 on hematopoietic cells is a major driver of salivary gland TLS in NOD.H-2h4 mice.
Figure 1. Early life CD40 signaling is required for TLS neogenesis.
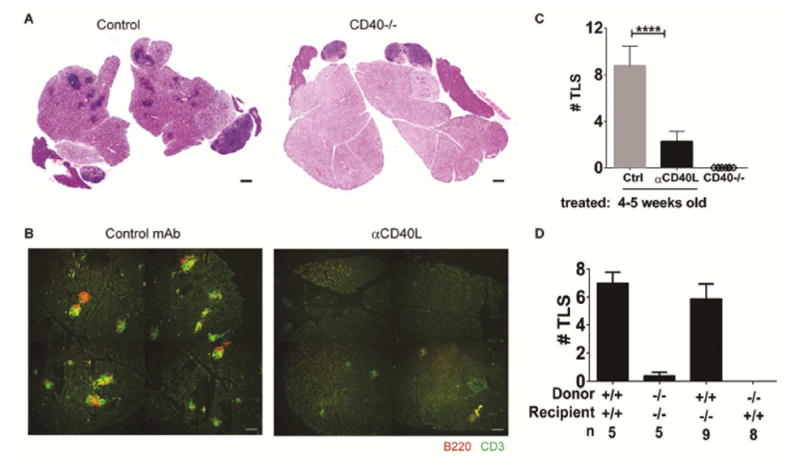
(A) Salivary gland TLS of 24 week-old control or CD40−/− female NOD.H-2h4 mice were visualized by haematoxylin and eosin. (B) Young 4–5 week-old female NOD.H-2h4 mice were treated with a single dose of control (Ctrl) or anti-CD40L mAb and salivary glands were evaluated for B and T cell infiltrates at 24 weeks-of-age. (C) Enumeration (mean +/− SEM) of whole salivary gland TLS from mice that received a single dose of Ctrl or anti-CD40L mAb at 4–5 weeks-of-age and were evaluated at 24–28 weeks of age (n=22 for control or n=24 for anti-CD40L from 6 independent experiments). Salivary gland TLS of 22–24 week-old CD40−/− mice were also enumerated (n=8 from 2 independent experiments). ****p<0.0001. Scale bars indicate 200 um. (D) T cell-depleted bone marrow from either wild type (+/+) or CD40−/− (−/−) NOD.H-2h4 mice was transferred into lethally irradiated 8 week-old recipients as described in methods and the number of TLS in the salivary gland was determined at 24–26 weeks-of age. Data (mean +/− SEM) represents one of two independent experiments. ****p<0.0001
In order to understand the molecular pathways associated with diminished TLS neogenesis induced by CD40 blockade, gene expression was determined from whole salivary gland RNA of 8–24 week-old mice that received a single injection of anti-CD40L at 4 weeks-of-age. At week 8 (4 weeks post-treatment), no genes showed statistically significant expression difference (Fold change ≥ 2, False discovery rate (FDR) ≤ 0.05) between control (Ctrl) and anti-CD40L-treated mice. At 16 and 24 weeks, 48 and 55 genes showed statistically significant down-regulation in anti-CD40L-treated mice compared to control (Fold change ≥ 2, False discovery rate (FDR) ≤ 0.05) (Fig. 2, Table S1). Notably, approximately half of all down-regulated genes were related to B cell phenotype or function. B cell related genes included Cd19, Cd20, Cd79a/b, Bcma, Taci, as well as Pou2af1, required for germinal center formation (30), and Lpxn, a negative regulator of B-cell signaling and function (31) (Fig. 2A). In control mice, expression of the B cell-related genes increased with time and disease progression, where strikingly, 8 Ig genes and Mzb1, which promotes IgM assembly and secretion (32), were among the earliest responsive genes. Despite only one early anti-CD40L treatment, these B cell-related genes remained low throughout the study, indicating suppression of B cell numbers and function (Fig. 2B). Also down-regulated were genes that exert function on multiple lymphocyte types, such as CD3d, expressed on all T cells; Maf, a gene, among other functions, that is involved in T follicular helper cells (Tfh) differentiation (33); CD52, the Campath-1 antigen expressed on all lymphocytes and CD53, expressed on leukocytes, including B, T and NK cells and involved in cell signaling. Importantly, genes repressed by anti-CD40L, such as CD53 and Ighm, are significantly upregulated in pSS salivary gland in human disease and have been reported to be potential biomarkers of disease (34).
Figure 2. Early-life CD40L blockade significantly reduced gene expression in the salivary glands of aged mice.

(A) Heatmap showing expression of lymphocyte-related genes (of total salivary gland RNA) that were significantly down-regulated (fold change ≥2 and FDR ≤0.05) following one injection of anti-CD40L mAb at 4 weeks-of-age compared to control mAb (Ctrl) (n=5 for each group). (B) Median expression value +/− standard deviation of the B cell related genes in (A) at each time point. (C) Heatmap showing expression of additional significantly down-regulated genes in the salivary gland of mice as described in A. (A, C) *Indicates differentially expressed genes detected by >1 probe. Brackets indicate the age when these genes were down-regulated at week 16, week (wk) 16 & 24, or week 24.
Additional genes were found to be significantly repressed by anti-CD40L, but interestingly their expression showed distinct temporal patterns. The first group of genes, such as Prg2, Slpi and Glipr1, showed increased expression in control-treated mice at 16 and 24 weeks, with sustained inhibition at both time points in anti-CD40L treated mice (Fig. 2C, Table S1). The second group of genes also showed increased expression in control-treated mice at 16 and 24 weeks, but were inhibited transiently only at 16, but not 24 weeks in anti-CD40L treated mice, and included genes involved in circadian rhythm, such as Per3 and Dbp, suggesting a previously unappreciated role of CD40L in pSS pathology. A third group of genes did not abundantly appear in control-treated mice until week 24 and remarkably, despite their late expression in the salivary glands, systemic anti-CD40L administration at 4 weeks-of-age inhibited gene expression 20 weeks following treatment (Fig. 2C). Included in these late appearing genes are Plac8, Fam26f 9, Serpina3g, Clec7a, Arhgap15, and MHC H2-M3. Taken together, these results suggest that transient blockade of CD40L in young animals has downstream impacts on several pathways and not simply a reflection of loss of B cell phenotype and function.
To assess the biological processes and signaling pathways that are impacted following early-life CD40L neutralization, Gene Set Enrichment Analysis (GSEA) was performed. Importantly, systemic anti-CD40L treatment at 4 weeks-of-age resulted in a down-regulation of the CD40 signaling pathway in the salivary gland of 16 and 24 week-old mice (Fig. 3, Fig. S2A). CD40L blockade in young mice also resulted in down-regulation of other immunologic pathways in the salivary gland including chemokine, lymphocyte activation, lymphocyte/non-lymphocyte interaction and IL-12 signaling at 16 and 24 weeks-of-age (Fig. 3, Fig. S2A). Anti-CD40L also significantly down-regulated interferon (IFN)-responsive genes and TH1/cytotoxic related genes as early as week 8 and persisted at week 24, where additional genes were found to contribute to the enrichment (Fig. 3, Fig. S2B). Thus, the effect of anti-CD40L extends far beyond B cells themselves and speaks to the interconnection of these cell types and pathways to their involvement in pSS immunopathology.
Figure 3. Early anti-CD40L treatment repressed multiple biological pathways involved in inflammation and autoimmunity.
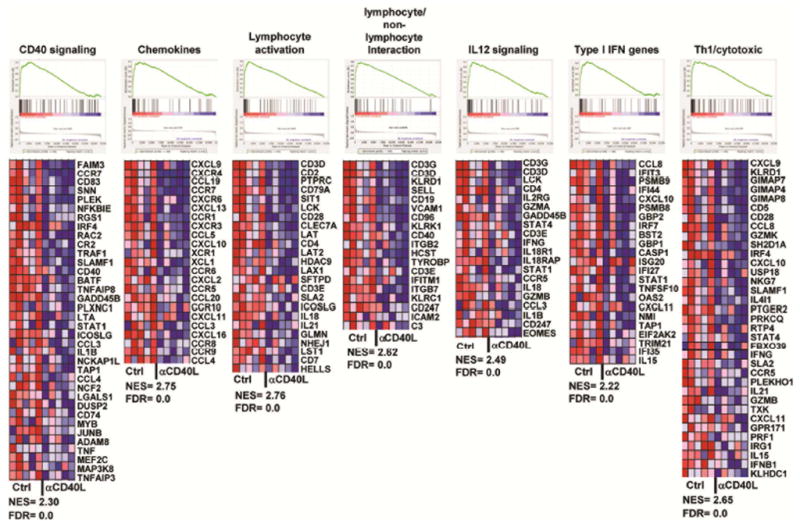
Gene Set Enrichment Analysis (GSEA) showing pathways down-regulated by a single treatment of anti-CD40L at 4 weeks-of–age compared to control. Data derived from whole salivary gland RNA of 24 week-old mice. The heatmaps show expression of the core genes that contribute to pathway enrichment. Red indicates high expression; blue indicates low expression. NES: Normalized enrichment score; FDR: False discovery rate.
Autoimmune germinal centers of the spleen pre-date and are required for salivary gland infiltration of aged mice
Previously we reported that spontaneous splenic germinal centers emerge in female NOD.H-2h4 mice at 8 weeks-of-age, prior to the detection of autoantibodies and TLS in the salivary gland (27). We have now further found that splenic germinal centers were detected in these mice as early as 3.5 weeks of age (Fig. 4A). Notably, these germinal centers did not resolve, as do germinal centers that form after immunization, but rather persisted for the life of the animal (Control Fig. 4B, C), suggesting aberrant regulation of molecules involved in these processes. Germinal center formation to T-dependent antigens requires CD40/CD40L interactions (5). To determine if there was a connection between the emergence of spontaneous germinal centers in the spleens of young autoimmune female NOD.H-2h4 mice and the ability of early anti-CD40L to abolish salivary gland TLS later in life, the effect of anti-CD40L on splenic germinal center formation was examined. Notably, treatment with a single injection of anti-CD40L between 4 to 6 weeks-of-age was found to effectively disrupt spontaneous germinal centers of the spleen for an extended period, in which a 10-fold reduction of germinal center B cell frequency was observed at 1–2 weeks post-treatment (Fig. 4B–D). Germinal centers remained significantly reduced until re-emergence starting at week 16 and did not revert to pre-dose frequencies until week 24 (Fig. 4C). These data show that early anti-CD40 inhibits both splenic germinal center formation as well as TLS neogenesis.
Figure 4. Contribution of the spleen in early life to the development of TLS.

(A) Spleens from young female NOD.H-2h4 mice were stained for germinal center (GC) B cells as indicated. Data is representative of 4–5 animals per group. (B) Young, 4–6 week-old NOD.H-2h4 mice were treated with one injection of control or anti-CD40L mAb and spleens were evaluated for germinal centers at either 8 or 24 weeks-of-age. Data is representative of 7 animals for 8 week-old group from two independent experiments and 12 animals for the 24 week-old group from three independent experiments. (A, B) Scale bars indicate 100 um. (C) Graphical representation of the number of splenic germinal centers per B cell follicle (mean +/− SEM) at the indicated ages is shown. Data shown in blue line indicates mice that were either untreated or treated with control (Ctrl) mAb at 4 weeks-of-age, red line indicates mice that were treated once with anti-CD40L at 4 weeks-of-age. N= 5 per group, except 24 week-old mice where n= 6. Data shown is from one of 2 independent experiments (except for 4.5–6 week-old mice where data is representative of one independent experiment each). **** p< 0.0001 (D) Mice were treated as in (B), and 1 or 2 weeks after treatment, enumeration of splenic germinal centers (mean +/− SEM) as defined by CD19+GL7+CD95+ B cells was determined by flow cytometry. Data represents 2 independent experiments (combined n=6 for control and 6 for anti-CD40L-treated mice, ***p=0.0004). (E) Salivary glands were harvested from aged, 24 week-old sham control mice or mice whose spleens were surgically removed at 3.5–4 weeks-of-age. Lymphocytic infiltrates of T and B cells were evaluated by immunofluorescence as indicated. Scale bars indicate 200 um. (F) Tertiary lymphoid structures (TLS) were enumerated at 6–6.5 months of age (mean +/− SEM) from whole salivary gland of sham (n=9) or splenectomized (n=8, SplX) mice. Data represents one of two independent experiments. * p< 0.05.
To identify if anti-CD40 was acting systemically on both splenic germinal centers and nascent exocrine gland TLS or if effects in the splenic germinal centers were sufficient to abrogate exocrine TLS development, young female mice were splenectomized and the resultant effect on salivary gland TLS of aged mice was determined. Control mice that received an early-life sham splenectomy developed large TLS in their salivary gland by 6 months of age with clear B cell (B220)/T cell (CD3) regions (Fig. 4E). Strikingly, splenectomy of young animals (3.5–4 weeks-of-age) resulted in a dramatic reduction in the size and number of TLS in 6 month-old mice (Fig. 4E, F). Collectively, these findings suggest that TLS in aged mice is a result of early-life immune dysregulation where the splenic microenvironment plays a major role in that early intervention, and anti-CD40L treatment or splenectomy effectively eliminates salivary gland infiltration.
Inhibition of defined autoantibody specificities accompanies loss of TLS
Although the Ro52/SSA autoantigen is upregulated in inflamed salivary glands of pSS patients (35), anti-Ro/SS-A and La/SSB autoantibodies arise years prior to the onset of clinical manifestations (14, 17) suggesting the original response to the autoantigens does not occur in local inflamed tissues, but rather early in peripheral compartments. Our data showing that early germinal centers are present in the spleen prior to disease onset suggest these animals are a good model to study the evolution of exocrine autoimmunity. Therefore, we next characterized the pre-clinical kinetic development of autoantibody specificities by assessing responses against 95 autoantigens longitudinally in NOD.H-2h4 mice. Out of the 95 specificities tested, 40 were found to have a positive signal (Signal-to-Noise Ratio (SNR) > 3 and intensity > 50) (Table S2). Of these, a wide range of autoantibodies were elevated prior to the onset of salivary gland inflammation, including pSS-associated autoantibodies to Ro52/SSA and La/SSB as well as MPO, which is abundantly expressed by neutrophil granulocytes and common in vasculitis, Jo-1, found in polymyositis, and many other autoantibody specificities (Fig. S3A). These antibodies appeared as early as 6–8 weeks-of-age and emerged throughout the life of the animal (Table S2). Reactivity to Fibrinogen IV, ssDNA, PL-12, Histone H-1 and U1-snRNP BB emerged much later in life, coinciding with the full development of TLS at 24 weeks-of age (Fig. S3B, Table S2). These data suggest that while some autoantibody specificities are triggered in inflamed salivary glands during the disease process, reactivity to many self-antigens pre-date disease pathology and arise in peripheral lymphoid compartments.
The effect of early-life anti-CD40L on the kinetics of autoantibody emergence was next evaluated. A single administration of anti-CD40L at 4 weeks-of-age was very effective at reducing autoantibody levels to many but not all self-antigens (31 out of the 40 autoantibodies that were positive in the autoantigen array). All autoantibodies that arose in untreated animals by 6–8 weeks-of age were inhibited by anti-CD40L, such as antibodies to Ro52. Moreover, autoantibodies directed to other nuclear proteins such as Ro60, La and Jo-1 were also significantly inhibited or delayed (Fig. 5A, Table S2). Anti-CD40L also inhibited several autoantibody specificities that did not appear until week 16 and 24, such as antibodies to complement (C1q), Fibrinogen IV and MPO (Fig. 5A). However, there were several autoantibody specificities that were not impacted by early blockade of the CD40L pathway such as antibodies to Sm, U1-snRMP, and ssDNA self-antigens (Fig. 5B, Table S2). Autoantibodies to M3R, which have been shown to play a role in exocrine dysfunction in NOD mice (22, 23), were detected in the serum of female NOD.H-2h4 mice and increased with age coinciding with full development of glandular inflammation (Fig. S3C). Despite the late appearance of this autoantibody specificity at 25 weeks-of age, early administration of anti-CD40L significantly inhibited the development of anti-M3R autoantibodies compared to control mice (Fig. 5C). Taken together, these data demonstrate that the ability of early anti-CD40L to inhibit autoantibodies was not solely a function of when they appeared, but rather dependent on autoantigen specificity.
Figure 5. Anti-CD40L Ab administration to young animals prevents autoantibody development of defined specificities.
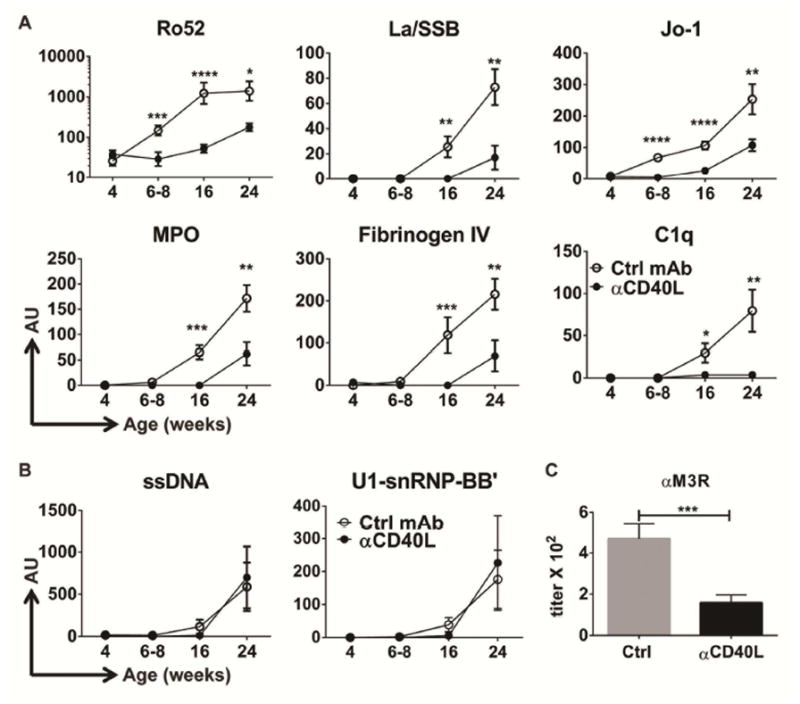
(A–B) Female NOD.H-2h4 mice received a single injection of either isotype control (Ctrl, white circles) or anti-CD40L mAb (black circles) at 4 weeks-of-age, and serum was collected serially at the indicated ages and screened for reactivity to self-antigens using a 95-autoantigen array. (A) Examples of IgG autoantibodies significantly decreased by anti-CD40L treatment compared to control. (B) Examples of IgG autoantibodies unaffected by anti-CD40L treatment compared to control. Data represents n= 11 for control mAb-treated mice and n= 13 for anti-CD40L-treated mice from three independent experiments. AU= Mean normalized fluorescence IgG signal ± SEM. * p< 0.05; ** p<0.01, *** p<0.001, **** p<0.0001. (C) End point titer (mean +/− SEM) of anti-M3R IgG autoantibodies in sera from of 20–26 week-old female NOD.H-2h4 mice that received a single treatment of either isotype control antibody (n=12) or anti-CD40L antibody (n=15) at 4 weeks-of-age. Data represents two independent cohorts.
Early, prophylactic anti-CD40L inhibits development of autoimmune thyroiditis and T1D
NOD.H-2h4 mice also develop autoimmune thyroiditis after receiving NaI in the drinking water (29, 36). In order to understand if early prophylactic blockade of CD40/CD40L interactions also has the ability to inhibit SAT, a single injection of anti-CD40L was given to young female NOD.H-2h4 mice, 1–2 weeks prior to NaI. As shown in Figure 6A, large inflamed thyroids, containing clusters of CD3+ T cells and B220+ B cells that formed TLS, were noted in 14–16 week-old control mice that received NaI 8 weeks prior to examination (Fig. 6A, left panel and Fig. S4). In stark contrast, mice that received early anti-CD40L mAb followed by NaI, were found to have smaller thyroids with significantly reduced SAT severity scores that reflected dramatically reduced lymphocytic infiltrate of T and B cells when evaluated at 14–16 weeks–of–age (Fig. 6A, right panel, Fig. 6B and Fig. S4). Importantly, anti-thyroglobulin autoantibodies that are associated with increased thyroiditis severity scores and disease pathology (37) were also inhibited by early treatment of anti-CD40L mAb (Fig. 6C).
Fig. 6. Early anti-CD40L mAb inhibits Autoimmune Thyroiditis and Diabetes.
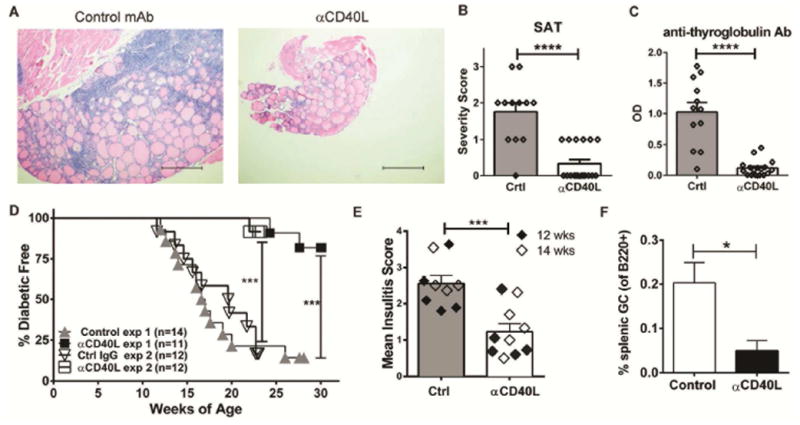
(A–C) Female NOD.H-2h4 mice were treated with a single dose of isotype control (Crtl) or anti-CD40L mAb at 5–6 weeks-of-age and 7–10 days later they were given 0.08% NaI in their drinking water for 8 weeks. (A–B) Thyroids were removed from 14–16 week-old mice and severity scores (mean +/− SEM) were determined from haematoxylin and eosin stained slides. (A) SAT severity scores are 2+ for control and 0 for anti-CD40L mAb. Scale bars = 100μM. (B) SAT severity scores were determined as previously described in detail (29, 37). (C) Autoantibody to thyroglobulin was measured in mice as described in (A) at 14–15 weeks of age. Data (mean +/− SEM) is from three independent experiments of n=12 for control mAb and n=18 for anti-CD40L treated mice. (B–C) **** p<0.0001. (D) Female NOD/LtDvs mice were treated with a single dose of anti-CD40L mAb at 4 weeks-of-age and followed for diabetes incidence starting at week 10. These mice were compared to either non-Ab injected control mice (exp #1, ***p=0.0002) or isotype control injected Ab (exp #2, ***p=0.0001). (E) Female NOD/LtDvs mice were treated with a single dose of anti-CD40L mAb at 4 weeks-of-age and insulitis was compared to age-matched untreated control mice (Ctrl) as described in methods at 12 or 14 weeks-of-age in non-diabetic mice. Data (mean +/− SEM) represents two independent experiments. ***p=0.0005. (F) Mice were treated as in (D), and 10 weeks after treatment, enumeration of splenic germinal centers (mean +/− SEM) as defined by B220+GL7+CD95+ B cells was determined by flow cytometry. Data represents 4 control and 5 anti-CD40L-treated mice, *p=0.036.
We next asked if this critical early-life immune checkpoint dependent on CD40L/CD40 interactions would also apply to autoimmune T1D that develops spontaneously in NOD mice. Several groups have chronicled the pancreatic infiltrate that develops over time as NOD mice (38–40). While work from the Unanue and Mathis/Benoist laboratories have shown that B and T lymphocytes, as well as myeloid cells infiltrate the pancreas (38, 40), others have reported that organized pancreatic TLS develop including germinal centers that contain insulin-binding autoreactive B cells (39). Thus, young female NOD mice were given a single injection of anti-CD40L at 4 weeks-of-age and the incidence of diabetes was monitored starting at week 10. In controls, mice begin to become diabetic at 12 weeks-of-age, consistent with previous reports (41). Strikingly, at 20 weeks-of-age, when the cumulative incidence of diabetes in the control group was approximately 80% in experiment #1 and 58% in experiment #2, not a single anti-CD40L treated mouse showed signs of glycosuria. At 30 weeks-of-age, when experiment #1 was terminated, 82% (9 out of 11) of anti-CD40L treated mice remained non-diabetic, whereas only 14% (2 out of 14) of control mice remained free of glycosuria (Fig. 6D). For experiment #2, 83% (10 out of 12) of control IgG-injected animals presented with glycosuria at 23 weeks-of-age, where only 8% (1 out of 12) of anti-CD40L treated mice were diabetic at this time (Fig. 6D). In independent experiments, control or anti-CD40L mAb was given as a single injection at 4 weeks-of-age and the ability of anti-CD40L to inhibit insulitis in the pancreas was determined 8–10 weeks later in non-diabetic mice, when mice were either 12 or 14 weeks-of-age. Consistent with its ability to inhibit tissue infiltration of salivary gland and thyroid tissue of NOD.H-2h4 mice, early and transient anti-CD40L mAb in NOD mice significantly reduced pancreatic insulitis scores in 12–14 week-old animals compared to controls (Fig. 6E). Importantly, consistent with NOD.H-2h4 mice, the ability of anti-CD40L to inhibit T1D in NOD mice also corresponded with a statistically significant decrease in spontaneous germinal center B cells in the spleen of these mice (Fig. 6F). These data show that the ability of prophylactic administration of anti-CD40L to inhibit disease manifestations is not limited to pSS, but also proves efficacious for other autoimmune diseases such as thyroiditis and T1D.
In summary, we show that early-life immune dysregulation, which manifests as spontaneous germinal centers of the spleen, played a significant role in the initiation of disease manifestations including autoantibodies, TLS in peripheral tissues and ultimately sialadenitis, thyroiditis, and diabetes in which CD40 was a key driver of immunopathology (Fig. S5)
Discussion
Tertiary lymphoid structures arise in non-lymphoid tissues in response to chronic immune activation as a result of microbial, environmental, inflammatory or autoreactive challenges to the host and can cause extensive pathology (42). Patients with autoimmune conditions that develop TLS are the most difficult to treat using standard biological therapies (43). Here we show that a single injection of anti-CD40L mAb early in life abolished autoimmune manifestations associated with pSS, thyroiditis and T1D.
In our studies, early intervention with anti-CD40L inhibited disease development. Work from others has shown that CD40-Fc was not able to inhibit focus score when delivery directly into the salivary glands of aged NOD mice (44). These data are consistent with our studies that systemic CD40L blockade is not merely inhibiting autoreactivity in local tissues, but rather in the splenic micro-environment early in life.
CD40/CD40L interactions are required for the initiation and maintenance of germinal centers (5) and affinity maturation to T cell-dependent antigens (45), where signaling through both CD40 and CD40L play important roles (46, 47). Aberrant regulation of germinal centers has been implicated in several autoimmune diseases (6, 27, 28), where CD40 presumably is involved. Previous studies showed that Tfh play a critical role in spontaneous germinal center generation and the formation of pathogenic autoantibodies (7, 48, 49). Interestingly, our gene expression profile detected a statistically significant decrease of Pou2af1 and Maf expression in salivary gland upon anti-CD40L treatment. Maf, among its other functions, plays a key role in Tfh development (33) whereas IRF4-induced Pou2af1 regulates germinal center B cells (30). These data suggest that early CD40L blockade may be impacting the presence of Tfh and germinal centers that arise in local tissues, which in control mice may be contributing to disease pathology.
In both the NOD.H-2h4 animal model and in human disease, there is a female predisposition to pSS. It is possible the appearance of spontaneous germinal centers in the spleens of female NOD.H-2h4 mice was prompted by estrogen, as their emergence coincided with the first estrus cycle that begins at 25–30 days of life. Importantly, estrogen has been reported to predispose to autoimmunity by influencing B cell tolerance induction and allowing for the activation of autoreactive B cells that produce high affinity antibodies to self-antigens (50). Here, the preponderance of spontaneous germinal centers that developed in the spleens of young NOD.H-2h4 mice was followed by the emergence of numerous autoantibodies. Many of these autoantibodies react with nuclear antigens, cell surface antigens, components of the extra-cellular matrix (ECM) as well as receptors required for physiological secretion of saliva (M3R). Some autoantibodies were detected as early as 6–8 weeks of age (anti-Ro52) while others arose much later at 24 weeks-of-age (anti-ssDNA). It is likely that antibodies that arose prior to the initiation of salivary gland inflammation exacerbated the development of TLS through immune complex deposition with self-antigens. Release and exposure of other self-antigens in the salivary gland during the initial inflammatory response would in turn result in activation of autoreactive cells and emergence of tissue-specific autoantibodies to antigens such as M3R. The late appearance of anti-M3R autoantibodies, and others such as anti-Fibrinogen IV, anti-ssDNA, anti-Sm and anti-U1-snRNP, suggests that autoreactivity to these self-antigens arose locally in the inflamed salivary gland once TLS were fully formed. This is supported by studies showing anti-Ro/SSA and anti-La/SSB autoantibody-producing cells are present in salivary glands from patients with Sjögren’s syndrome (3, 51) as well as plasma cells present in labial salivary gland that produced inhibitory anti-M3R autoantibodies (52), suggesting a direct pathogenic role of these locally produced autoantibodies.
Clearly the spleen is important for both immunity and autoimmunity. Splenectomy of Immune thrombocytopenic purpura (ITP) patients, where long-lived plasma cells to platelet antigens are found (53), results in a complete remission rate of over 75% (54). Moreover, splenectomy of ITP patients with systemic lupus erythematosus results in a partial or complete response rate of 88% (55). Here, we report splenectomy of young mice also prevented disease manifestations, demonstrating the importance of this lymphoid compartment in early-life to the contribution of autoimmunity that develops in aged mice. Although inflamed tissue is believed to be the site where autoreactivity to local self-antigens is triggered, our data suggest early immune-dysregulation to self-antigens in the spleen is the initial trigger, and this response is then amplified in TLS that develop in local tissues later in life, ultimately resulting in disease progression.
Early-life interventions on TLS in NOD.H-2h4 mice underscores the significance of neonatal immune checkpoints. Others have shown that an early-life window exists where clinical disease can be inhibited by prophylactic therapy. For example, neutralization of TNF in young, but not aged NOD mice inhibits insulitis and delays T1D development (56). Furthermore, prophylactic blockade of IFNα also significantly inhibits T1D (57). Consistent with our data, previous studies reported that blocking CD40L starting early-in-life inhibited T1D development in NOD mice (58). However, in these studies, anti-CD40L was administered for several months with an anti-CD40L hamster antibody capable of depleting CD40L-expressing cells. In contrast, our studies utilized an MR1 anti-CD40L antibody that was engineered as a human IgG1 TM (triple mutant) devoid of effector function (7) where a single injection of anti-CD40L at 4 weeks-of-age was sufficient to statistically significant block T1D development in NOD mice until the experiment was terminated at 30 weeks-of-age. These data support our findings and suggest that in diseases where autoantibodies pre-date clinical signs, there is a treatment window where therapeutic intervention may be able to prevent disease development.
Here, anti-CD40L disrupted early autoreactive immune responses. CD40/CD40L has been clearly implicated in a variety of autoimmune diseases in which B and T cell activation plays an essential role in pathogenesis (4). CD40 and CD40L are both expressed on salivary gland epithelial cells as well as on infiltrating immune cells in pSS that has the potential to contribute to disease pathology (8, 10). Disrupting this pathway could impact the development of autoimmunity through several mechanisms. In radiation chimera experiments, we show that CD40 expression on hematopoietic, but not stromal cells is required for development of TLS. Moreover, early and transient blockade of CD40/CD40L interactions between dendritic cells (or any antigen-presenting cell) and T-helper subsets may contribute to dampening pathology by reducing autoreactive T cell activation and expansion which would in turn limit autoantibody development. In support of this, CD40L blockade has been shown to have an impact on dendritic cell priming of autoreactive T cells (59). In our GSEA studies, a statistically significant decrease in IL-12 signaling was detected in the salivary gland of anti-CD40L-treated mice. IL-12 production is induced during the interaction of dendritic cells with T cells, mainly through CD40/CD40L interaction (60).
Treatment with anti-CD40L antibody modulated the expression of genes in exocrine tissue not typically associated with immune responses. Our gene expression profile of salivary gland revealed that CD40L blockade had a broader impact on multiple biological processes, suggesting novel mechanisms of pSS pathogenesis. Some of these are likely related to the normalization of the tissue in the absence of fulminant autoimmunity while others may reflect novel CD40/CD40L-regulated pathways. Genes involved in innate immunity and tissue inflammation such as Clec7a (Dectin-1) (61), Arhgap15 (62) and Fam26f9 (63) were inhibited by anti-CD40L. Several genes modulated by anti-CD40L have been described to be involved with bone formation including Ogn (OIF) (64), Igfbp5 (65) and Lpxn (66) where, notably two-thirds of pSS patients have been described with evidence of subclinical impaired bone health (67).
NOD.H-2h4 mice also develop thyroid disease when given NaI in the drinking water (29, 36). Tef, which binds to and transactivates thyroid-stimulating hormone, beta (68), was found to be down modulated by early anti-CD40L, suggesting a novel role by which CD40L blockade inhibits SAT in this animal model. Tef has also been implicated in circadian regulation of insulin production (69). Circadian clock genes are expressed in the salivary gland where salivary flow follows circadian rhythms. Alterations in this pathway have been suggested to contribute to reduced saliva production in pSS patients (70). Additional circadian rhythm-related genes that were repressed by CD40L blockade include Per3 and Dbp. These findings linking CD40 to circadian clock regulation suggest new molecular pathways involved in disease pathogenesis and warrant further investigation.
NOD.H-2h4 mice do not become diabetic due to loss of the pathogenic H2g7 MHC haplotype. However, NOD mice contain over 30 other diabetic susceptibility genes that contribute to the development of autoimmunity(24). Of interest, in salivary gland, several genes that were suppressed by anti-CD40L are genes that have been reported to be associated with T1D and/or insulin regulation including Serpina3g (71), Iapp (72), Maf (73), and Igfbp5 where Igfbp5 is contained within IDDM13 susceptibility locus (74).
While early clinical trials with anti-CD40L showed promising immune-modulatory effects, thromboembolic complications halted trials (75). New strategies are being developed to avoid platelet aggregation, such as monovalent anti-CD40L antibodies (76), CD40L antagonist scaffolds engineered to avoid FcR binding (ClinicalTrials.gov Identifier: NCT02151110), or antibodies to CD40 such as ASKP1240 (77) (ClinicalTrials.gov Identifier: NCT01780844, NCT01585233), or the Fc-silent anti-CD40 antibody CFZ533 (78) that is currently in clinical trial for pSS (ClinicalTrials.gov Identifier: NCT02291029), RA (ClinicalTrials.gov Identifier: NCT02089087), as well as several other indications. The ability of prophylactic anti-CD40L to dissolve germinal centers and halt autoimmune humoral responses in our studies, suggests biologics that target CD40/CD40L interaction may have efficacy in diseases that are predated by the appearance of autoantibodies.
The findings we report in this study have some limitations in regard to the clinic. In order to translate into the clinic, further investigation including examination of longitudinal samples obtained from human subjects before and after the emergence of overt disease manifestations would need to be evaluated to determine specific pharmacodynamic markers. Findings and correlations from such studies could then be mechanistically investigated using mouse models of disease such as the ones we report in this manuscript. Biomarkers that unequivocally indicate early-life immune dysfunction are needed to distinguish those patient populations that will progress to full disease states versus otherwise healthy individuals.
Materials and Methods
Study Design
The objective of this study was to evaluate the contribution of early life CD40-CD40L signaling to downstream autoimmune manifestations. These studies involved either antibody mediated blockade or genetic deletion of CD40-CD40L signaling in several mouse models of autoimmunity including NOD.H-2h4 and NOD mice. To assess the effect of anti-CD40L on TLS development in NOD.H-2h4 mice, a pilot experiment was performed in which mice were treated at 4, 5 or 6 weeks of age. Data obtained from these mice at 6 months of age, a time when TLS fully develop, was then used to estimate the number of animals and experiments needed to achieve statistical significance with 80% power. Female mice within the same age group were randomized into treatment and control groups and quantification of TLS number was performed in a blinded manner. To evaluate the effect of anti-CD40L on autoimmune thyroiditis, mice were randomized to either the control or treatment groups and quantitation of histology was blinded. For T1D studies, the number of mice in the experiments was sufficient to detect the difference between control and anti-CD40L treated groups with 90% power. NOD mice treated with either control IgG or anti-CD40L were from 2 or more litters of different parents that were randomized prior to treatment. The investigator doing the histology scoring was blinded to the groups of mice in any given sample at the time of evaluation. For all studies, the number of independent experiments and the number of mice per experiment are indicated in the figure legends.
Mice
NOD.H-2h4 (26) (stock number 004447) and C57BL/6J mice were obtained from Jackson Laboratories and the colonies were maintained at the MedImmune mouse facility or the animal facility at the University of Missouri. CD40−/− female NOD.H-2h4 mice (MMRRC number 037352) as described previously (29), were maintained at the at the MedImmune mouse facility. NOD/LtDvs (NOD) mice were maintained at The Jackson Laboratory. Mice were housed in pathogen-free conditions. All experiments were performed with female mice. All animal protocols were done in accordance with and approved by Medimmune’s (protocols MI-12-012 and MI-15-0006), University of Missouri (protocol #8195) or The Jackson Laboratory (protocol #99063) Institutional Animal Use and Care Committees.
Anti-CD40L mAb and Treatment
The mouse CD40L triple mutation (TM) mAb used is an in-house modified version of the commercially available MR1 clone that was engineered to contain the Fc region of the human IgG1 with TM in the CH2 region to prevent Fc-mediated effector function (79). For the Sjögren’s studies, NOD.H-2h4 female mice were treated intra-peritoneally at various ages with a single injection of anti-CD40L (TM) or human IgG1 isotype control TM mAb at 20 mg/kg as described previously (7). Mice were serially bled for serum analysis until the end of the study. Four weeks to six months after treatment, mice were sacrificed and spleens and salivary glands were assessed for the effect of anti-CD40L treatment. For the thyroid studies, NOD.H-2h4 female mice were treated intravenously at 5–6 weeks of age with a single injection of 400 μg of either anti-CD40L (TM) or human IgG1 TM isotype control mAb. 7–10 days later, they were given 0.08% NaI in their drinking water, and thyroids were removed 7–8 weeks later. For diabetes studies, NOD/LtDvs mice were injected intra-peritoneally with 400 ug of either anti-CD40L (TM) mAb or human IgG1 TM isotype control mAb at 4 weeks-of-age and monitored for glycosuria and insulitis as described below.
Measurement of salivary gland TLS
Whole salivary glands were removed from female NOD.H-2h4 mice at the indicated ages. Tissues were formalin-fixed and embedded in paraffin for haematoxylin and eosin staining. In all the studies the counts were done in a randomized, blinded fashion. For enumeration, in the majority of experiments, serial sections of both lobes of the salivary glands were cut at least at 3 different planes, each plane being 50 microns apart. Graphed data represents the highest count of TLS, as defined by foci of 50 or more hematopoietic cells clustered together often around individual excretory ducts, in any one of the three section planes examined. In some experiments, TLS were also enumerated from immune-histology sections stained for B and T cells as described below. For these experiments, one TLS was counted when foci of 50 or more T and/or B cells were clustered together.
Bone marrow chimeras
T cell-depleted bone marrow from 8–10 week-old female wild type or CD40−/− NOD.H-2h4 mice was obtained by positively selecting out the murine CD3+ T cells with a magnetic separation kit (Miltenyi Biotec Inc., Auburn). 1 X 10−7 CD3-depleted bone marrow cells were transferred intravenously into lethally irradiated 8 week-old female wild type or CD40−/− NOD.H-2h4 recipients. Salivary glands were removed at 24–26 weeks-of-age and the number of TLS in both lobes of the salivary gland was determined in haematoxylin and eosin stained sections. For enumeration, serial sections through both lobes of the salivary gland was cut at least at 3 different planes, each plane being 50 microns apart. The highest count of TLS in any one of the three section planes examined represents data shown.
Measurement of autoimmune thyroiditis
Following treatment as described above, thyroids were removed and fixed in formalin, sectioned and stained with either haematoxylin and eosin or stained for expression of CD3 and B220 by IDEXX RADIL. SAT severity scores were determined as previously described in detail (29). Serum was obtained at the same time and anti-mouse thyroglobulin (MTg)-specific IgG autoantibodies were measured by ELISA as previously described (29). Results are expressed as OD410 of a 1:50 dilution of duplicate serum samples from individual mice.
Measurement of T1D and insulitis
After mAb treatment at 4 weeks-of-age, NOD/LtDvs mice were monitored for glycosuria with Amnes Diastix (Bayer Diagnostics Division) starting at 10 weeks of age. Mice with two consecutive values of ≥ 3 were scored as diabetic. In a separate experiment, mice received anti-CD40L mAb at 4 weeks-of-age and non-diabetic mice were assessed for insulitis at 12 or 14 weeks-of-age and compared to untreated age match non-diabetic control mice. Pancreas tissues were fixed in Bouin’s solution, sectioned at 3 levels, 70 microns apart and stained with aldehyde fuchsin to detect beta cells. Islets (at least 20 per mouse) were individually scored in a blinded manner as follows: 0, no lesions; 1, peri-insular leukocytic aggregates, usually periductal infiltrates; 2, <25% islet destruction; 3, >25% islet destruction; and 4, >75% or complete islet destruction. An insulitis score for each mouse was obtained by dividing the total score for each pancreas by the number of islets examined.
Splenectomy
Three and a half to four week-old young mice were anesthetized with 2–3% isofluorane and injected with 0.05 mg/kg Buprenorphine. A one cm skin incision on the left lateral surface of the abdomen was made before separating skin from peritoneal wall. A similar incision was made in the peritoneal wall and the spleen was pulled out with forceps. The splenic artery at the base of the spleen was tied off before the spleen was removed by cutting distally to the suture. For the sham splenectomy control group, mice were anesthetized and the body cavity was opened as described above. Skin was closed with staples and mice were allowed to recover on a heating pad. Mice were monitored for 48 hours for signs of distress and received two doses of Buprenorphine on the day of surgery and a single dose the following morning. Salivary glands were then harvested at 6–6.5 months of age for evaluation of TLS enumerated as described above..
Immunofluorescence Microscopy
Salivary gland and splenic tissue were removed from female NOD.H-2h4 mice at the indicated ages. Tissues were embedded in O.C.T. compound (Tissue-Tek) and frozen on dry ice. Frozen tissue sections (5–7 microns) were fixed in acetone for 5 minutes and stained with the following primary antibodies: anti-IgM-Texas Red (Southern Biotech), Anti-B220 (rat), anti-CD3 (hamster), and biotinylated PNA (Vector Laboratories). All primary antibodies were from BD Biosciences except where indicated. The following secondary antibodies were used: anti-hamster Alexa Fluor 488, anti-rat-Oregon Green, and streptavidin-Oregon Green. All secondary antibodies were purchased from Life Technologies unless otherwise indicated. Anti-B220 (clone RA3-6B2, Biolegend) was labeled with AlexaFluor555 using an antibody labeling kit from Molecular Probes according to the manufacturer’s instructions. Fluorescence images were visualized using a Nikon-Eclipse microscope and NIS Elements AR software.
Flow Cytometry
Splenocytes were processed and stained according to standard protocols. Salivary glands were cut into small pieces and incubated in digestion buffer (PBS, Liberase TL at 0.04 WU/mL, 0.1% DNase) at 37 degrees for one hour in a shaker. The reaction was stopped by addition of RPMI 1640 supplemented with 10% FCS. Cells were then filtered and washed with PBS before further downstream applications. The following antibodies were used: anti-B220-PECy7, anti-GL7-FITC, anti-CD19-V450, CD45-FITC, CD8 PercpCy5.5 (eBiosciences) and CD4 APC. All antibodies were obtained from BD Biosciences except where indicated. Cells were analyzed on an LSR II flow cytometer (BD Biosciences) using FACSDiva software. Data were analyzed using Flowjo software.
Autoantigen arrays
Serial serum samples were collected from anti-CD40L or isotype control-treated mice and screened for reactivity to a panel of 96 autoantigens (UT-Southwestern Microarray Core Facility, Dallas, TX) as described (80). Data values were only considered positive when the signal to noise ratio was greater than 3. In addition, signal intensities with values less than 50 were excluded. Of all 96 autoantibodies screened, 40 tested positive, 2 of which were also detected in C57BL/6 mice at levels equivalent to NOD.H-2h4 mice.
Anti-M3R ELISA
Samples of serum from NOD.H-2h4 mice were analysed for reactivity to M3R peptide to determine the levels of anti-M3R IgG antibodies. Appropriate positive (NOD/Lt serum) and negative (C57BL/6 normal serum) assay controls were included on each plate. Antibody quantities were determined as serum end-point titres. The assay was performed by KWS Biotest, Cambridge, UK.
Microarrays
Salivary glands were isolated and immersed in All-Tissue Protect reagent (Qiagen) at 4 degrees for 24 hours before storage at −20°C. At the time of processing, the samples were thawed and excess All-Tissue Protect reagent was removed prior to tissue homogenization and RNA extraction per the manufacturer’s protocol (Qiagen). Microarray gene expression studies were performed on Affymetrix® GeneChip Mouse Genome 430 2.0 Array and data were analyzed using the Limma package from Bioconductor. False discovery rate was calculated using the Benjamini–Hochberg procedure. Genes with fold change ≥2 and false discovery rate (FDR) ≤0.05 were defined as significant. Gene Set Enrichment Analysis (http://www.broadinstitute.org/gsea/index.jsp) was performed on gene expression of anti-CD40L antibody treated versus control treated samples at each time point. Pathways from Molecular signatures database (MSigDB) were used. Titles of the pathways were modified for simplicity: Type I IFN genes: BROWNE_INTERFERON_RESPONSIVE_GENES; Lymphocyte activation: LYMPHOCYTE_ACTIVATION; TH1/cytotoxic: BOSCO_TH1_CYTOTOXIC MODULE; CD40 signaling: BASSO_CD40_SIGNALING_UP; IL12 signaling: PID_IL12_2PATHWAY; lymphocyte/non-lymphocyte interaction: REACTOME_IMMUNOREGULATORY_INTERACTIONS_BETWEEN_A_LYMPHOID_AND_A_NON_LYMPHOID_CELL; Chemokines: REACTOME_CHEMOKINE_RECEPTORS_BIND_CHEMOKINES. Heatmaps of gene expression and other statistical analysis were performed using R programming software. Microarray data have been deposited at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE83862) with accession code GSE83862.
Statistics
Two-tailed unpaired Mann–Whitney U test was used for group comparisons between control and anti-CD40L treated or splenectomized NOD.H-2h4 mice (Fig. 1C, 4F and 6C). The “Age of Detection” for autoantibodies in NOD.H-2h4 mice (Table S2) and significance for Fig. S3 was determined when autoantibodies were significantly elevated compared to baseline (4 weeks-of-age). Statistical significance for both “Age of Detection” (Table S2) and (Fig. S3) was determined using a one-tailed paired Students t-test when the data followed normal distribution or paired Mann–Whitney U test when the data did not follow normal distribution as determined by D’Agostino & Pearson omnibus normality test. The effect of anti-CD40L on GC B cell frequency was determined using a two-tailed unpaired Student’s t-test (Fig. 4D and 6F). The ability of anti-CD40L to inhibit autoantibody levels compared to control in NOD.H-2h4 mice (Fig. 5A, Table S2) was determined using one-tailed unpaired Students t-test if the data passed F test for equal variance or Mann–Whitney U test if the data did not pass F test. For thyroid experiments in NOD.H-2h4 mice (Fig. 6B, C), two-tailed unpaired Students t-test was used for thyroglobulin autoantibody and unpaired Mann-Whitney U test was used for SAT severity scores. Incidence of diabetes in NOD mice (Fig. 6D) was compared using the Log-rank (Mantel-Cox) test and insulitis score scores (Fig. 6E) were compared using a two-tailed unpaired Students t-test. Statistical tests and plots were performed using Graphpad Prism software.
Supplementary Material
Early life anti-CD40L inhibits lymphocyte infiltration of salivary gland of aged mice
Gene Set Enrichment Analysis (GSEA) of gene pathways down-regulated in salivary gland by early anti-CD40L treatment of NOD.H-2h4 mice
Autoantibodies (IgG) are detected after emergence of spontaneous splenic germinal centers
Greatly reduced B and T cell clusters in thyroids following early anti-CD40L in NOD.H-2h4 mice
Autoimmune manifestations in aged mice arise from early-life germinal centers
Significantly down-regulated salivary gland genes in response to early CD40L blockade
Early-life treatment of anti-CD40L inhibits specific IgG autoantibody profiles
Acknowledgments
From MedImmune, the authors would like to thank Drs. Geoffrey Stephens and Devon Taylor as well as Carlos Gonzalez, Diana Pao, Shonda Hawkins, Julie Bakken and Stephanie Oldham.
Funding: The thyroid work was supported by NIH grant AI RO1 76395; Helen Mullen, P.I., The diabetes work was supported by NIH grants DK46266 and DK95735, David Serreze, P.I.
Abbreviations used in this paper
- BAFF
B-cell Activating Factor
- CD40L
CD40 Ligand
- Ctrl
Control
- DC
Dendritic Cells
- FDR
False Discovery Rate
- FDC
Follicular Dendritic Cells
- GSEA
Gene Set Enrichment Analysis
- ITP
Immune Thrombocytopenic Purpura
- KO
Knock Out
- M3R
Muscarinic Type 3 Receptor
- NOD
Non-Obese Diabetic
- NS
Non-Significant
- pSS
Primary Sjögren’s Syndrome
- SNR
Signal-to-Noise Ratio
- NaI
Sodium-Iodide
- SplX
Splenectomized
- SAT
Spontaneous Autoimmune Thyroiditis
- Tfh
T Follicular Helper Cells
- TLS
Tertiary Lymphoid Structures
- T1D
Type 1 Diabetes
- WT
Wild Type
Footnotes
Competing Interests: JW, BN, JLK, SW, PB, CM, TM, RH, RK and RE are full-time employees and shareholders of MedImmune/AstraZeneca.
Data and materials availability: All data pertaining to this study are published in the paper or included in the detailed Supplementary Materials. Microarray data have been deposited at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE83862) with accession code GSE83862.
Author Contributions: T.I.M. designed, conducted, analyzed and interpreted the majority of experiments. J.W. analyzed and interpreted the microarray data. J. L. K. and B.N. performed, supported and interpreted experiments. S. W. and P. G. supported several experiments. P.B., C.M. and J.A. supported, executed, and analyzed the gene expression experiments. H. B-M. performed and interpreted thyroiditis experiments. Q. W. performed and interpreted the diabetes experiments under the guidance of D.V.S. C. W. performed diabetes experiments under the guidance of M.A.A., R.E, under the guidance of R. K., L.C., R.H. and T.M, conceived, designed, and supervised the project. T.I.M, J.W, J.L.K and R.E wrote and revised the manuscript with input from all authors.
References
- 1.Skopouli FN, Dafni U, Ioannidis JP, Moutsopoulos HM. Clinical evolution, and morbidity and mortality of primary Sjogren’s syndrome. Semin Arthritis Rheum. 2000;29:296–304. doi: 10.1016/s0049-0172(00)80016-5. [DOI] [PubMed] [Google Scholar]
- 2.Jonsson MV, Skarstein K, Jonsson R, Brun JG. Serological implications of germinal center-like structures in primary Sjogren’s syndrome. J Rheumatol. 2007;34:2044–2049. [PubMed] [Google Scholar]
- 3.Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmstrom P, Wahren-Herlenius M, Jonsson R. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren’s syndrome. Arthritis Rheum. 2003;48:3187–3201. doi: 10.1002/art.11311. [DOI] [PubMed] [Google Scholar]
- 4.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G. Cellular interaction in germinal centers. Roles of CD40 ligand and B7-2 in established germinal centers. Journal of immunology. 1995;155:556–567. [PubMed] [Google Scholar]
- 6.Luzina IG, Atamas SP, Storrer CE, daSilva LC, Kelsoe G, Papadimitriou JC, Handwerger BS. Spontaneous formation of germinal centers in autoimmune mice. J Leukoc Biol. 2001;70:578–584. [PubMed] [Google Scholar]
- 7.Yusuf I, Stern J, McCaughtry TM, Gallagher S, Sun H, Gao C, Tedder T, Carlesso G, Carter L, Herbst R, Wang Y. Germinal center B cell depletion diminishes CD4+ follicular T helper cells in autoimmune mice. PloS one. 2014;9:e102791. doi: 10.1371/journal.pone.0102791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohlsson M, Szodoray P, Loro LL, Johannessen AC, Jonsson R. CD40, CD154, Bax and Bcl-2 expression in Sjogren’s syndrome salivary glands: a putative anti-apoptotic role during its effector phases. Scand J Immunol. 2002;56:561–571. doi: 10.1046/j.1365-3083.2002.01168.x. [DOI] [PubMed] [Google Scholar]
- 9.Roescher N, Lodde BM, Vosters JL, Tak PP, Catalan MA, Illei GG, Chiorini JA. Temporal changes in salivary glands of non-obese diabetic mice as a model for Sjogren’s syndrome. Oral Dis. 2012;18:96–106. doi: 10.1111/j.1601-0825.2011.01852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitriou ID, Kapsogeorgou EK, Moutsopoulos HM, Manoussakis MN. CD40 on salivary gland epithelial cells: high constitutive expression by cultured cells from Sjogren’s syndrome patients indicating their intrinsic activation. Clin Exp Immunol. 2002;127:386–392. doi: 10.1046/j.1365-2249.2002.01752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 12.Bingley PJ, Bonifacio E, Williams AJ, Genovese S, Bottazzo GF, Gale EA. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes. 1997;46:1701–1710. doi: 10.2337/diab.46.11.1701. [DOI] [PubMed] [Google Scholar]
- 13.Dayan CM, Daniels GH. Chronic autoimmune thyroiditis. N Engl J Med. 1996;335:99–107. doi: 10.1056/NEJM199607113350206. [DOI] [PubMed] [Google Scholar]
- 14.Jonsson R, Theander E, Sjostrom B, Brokstad K, Henriksson G. Autoantibodies present before symptom onset in primary Sjogren syndrome. JAMA. 2013;310:1854–1855. doi: 10.1001/jama.2013.278448. [DOI] [PubMed] [Google Scholar]
- 15.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 16.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 17.Theander E, Jonsson R, Sjostrom B, Brokstad K, Olsson P, Henriksson G. Prediction of Sjogren’s Syndrome Years Before Diagnosis and Identification of Patients With Early Onset and Severe Disease Course by Autoantibody Profiling. Arthritis Rheumatol. 2015;67:2427–2436. doi: 10.1002/art.39214. [DOI] [PubMed] [Google Scholar]
- 18.Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, Del Pino-Montes J, Calvo-Alen J, Jimenez-Alonso J, Mico ML, Beltran J, Belenguer R, Pallares L, Group GS. Primary Sjogren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore) 2008;87:210–219. doi: 10.1097/MD.0b013e318181e6af. [DOI] [PubMed] [Google Scholar]
- 19.Hernandez-Molina G, Leal-Alegre G, Michel-Peregrina M. The meaning of anti-Ro and anti-La antibodies in primary Sjogren’s syndrome. Autoimmun Rev. 2011;10:123–125. doi: 10.1016/j.autrev.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Halse AK, Marthinussen MC, Wahren-Herlenius M, Jonsson R. Isotype distribution of anti-Ro/SS-A and anti-La/SS-B antibodies in plasma and saliva of patients with Sjogren’s syndrome. Scand J Rheumatol. 2000;29:13–19. doi: 10.1080/030097400750001752. [DOI] [PubMed] [Google Scholar]
- 21.Jin M, Hwang SM, Davies AJ, Shin Y, Bae JS, Lee JH, Lee EB, Song YW, Park K. Autoantibodies in primary Sjogren’s syndrome patients induce internalization of muscarinic type 3 receptors. Biochim Biophys Acta. 2012;1822:161–167. doi: 10.1016/j.bbadis.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen KH, Brayer J, Cha S, Diggs S, Yasunari U, Hilal G, Peck AB, Humphreys-Beher MG. Evidence for antimuscarinic acetylcholine receptor antibody-mediated secretory dysfunction in nod mice. Arthritis Rheum. 2000;43:2297–2306. doi: 10.1002/1529-0131(200010)43:10<2297::AID-ANR18>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 23.Robinson CP, Brayer J, Yamachika S, Esch TR, Peck AB, Stewart CA, Peen E, Jonsson R, Humphreys-Beher MG. Transfer of human serum IgG to nonobese diabetic Igmu null mice reveals a role for autoantibodies in the loss of secretory function of exocrine tissues in Sjogren’s syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7538–7543. doi: 10.1073/pnas.95.13.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol. 2011;33:67–87. doi: 10.1007/s00281-010-0204-1. [DOI] [PubMed] [Google Scholar]
- 25.Cihakova D, Talor MV, Barin JG, Baldeviano GC, Fairweather D, Rose NR, Burek CL. Sex differences in a murine model of Sjogren’s syndrome. Ann N Y Acad Sci. 2009;1173:378–383. doi: 10.1111/j.1749-6632.2009.04760.x. [DOI] [PubMed] [Google Scholar]
- 26.Podolin PL, Pressey A, DeLarato NH, Fischer PA, Peterson LB, Wicker LS. I-E+ nonobese diabetic mice develop insulitis and diabetes. J Exp Med. 1993;178:793–803. doi: 10.1084/jem.178.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karnell JL, Mahmoud TI, Herbst R, Ettinger R. Discerning the kinetics of autoimmune manifestations in a model of Sjogren’s syndrome. Mol Immunol. 2014;62:277–282. doi: 10.1016/j.molimm.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. 2009;9:845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 29.Braley-Mullen H, Yu S. NOD.H-2h4 mice: an important and underutilized animal model of autoimmune thyroiditis and Sjogren’s syndrome. Adv Immunol. 2015;126:1–43. doi: 10.1016/bs.ai.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, Chong AS, Klein U, Dinner AR, Singh H, Sciammas R. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity. 2013;38:918–929. doi: 10.1016/j.immuni.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chew V, Lam KP. Leupaxin negatively regulates B cell receptor signaling. J Biol Chem. 2007;282:27181–27191. doi: 10.1074/jbc.M704625200. [DOI] [PubMed] [Google Scholar]
- 32.Flach H, Rosenbaum M, Duchniewicz M, Kim S, Zhang SL, Cahalan MD, Mittler G, Grosschedl R. Mzb1 protein regulates calcium homeostasis, antibody secretion, and integrin activation in innate-like B cells. Immunity. 2010;33:723–735. doi: 10.1016/j.immuni.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kroenke MA, Eto D, Locci M, Cho M, Davidson T, Haddad EK, Crotty S. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. Journal of immunology. 2012;188:3734–3744. doi: 10.4049/jimmunol.1103246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khuder SA, Al-Hashimi I, Mutgi AB, Altorok N. Identification of potential genomic biomarkers for Sjogren’s syndrome using data pooling of gene expression microarrays. Rheumatol Int. 2014 doi: 10.1007/s00296-014-3152-6. [DOI] [PubMed] [Google Scholar]
- 35.Aqrawi LA, Kvarnstrom M, Brokstad KA, Jonsson R, Skarstein K, Wahren-Herlenius M. Ductal epithelial expression of Ro52 correlates with inflammation in salivary glands of patients with primary Sjogren’s syndrome. Clin Exp Immunol. 2014;177:244–252. doi: 10.1111/cei.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasooly L, Burek CL, Rose NR. Iodine-induced autoimmune thyroiditis in NOD-H-2h4 mice. Clin Immunol Immunopathol. 1996;81:287–292. doi: 10.1006/clin.1996.0191. [DOI] [PubMed] [Google Scholar]
- 37.Braley-Mullen H, Yu S. Early requirement for B cells for development of spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. Journal of immunology. 2000;165:7262–7269. doi: 10.4049/jimmunol.165.12.7262. [DOI] [PubMed] [Google Scholar]
- 38.Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER. Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PloS one. 2013;8:e59701. doi: 10.1371/journal.pone.0059701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kendall PL, Yu G, Woodward EJ, Thomas JW. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. Journal of immunology. 2007;178:5643–5651. doi: 10.4049/jimmunol.178.9.5643. [DOI] [PubMed] [Google Scholar]
- 40.Magnuson AM, Thurber GM, Kohler RH, Weissleder R, Mathis D, Benoist C. Population dynamics of islet-infiltrating cells in autoimmune diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1511–1516. doi: 10.1073/pnas.1423769112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serreze DV, Chapman HD, Niens M, Dunn R, Kehry MR, Driver JP, Haller M, Wasserfall C, Atkinson MA. Loss of intra-islet CD20 expression may complicate efficacy of B-cell-directed type 1 diabetes therapies. Diabetes. 2011;60:2914–2921. doi: 10.2337/db11-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6:205–217. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 43.Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14:447–462. doi: 10.1038/nri3700. [DOI] [PubMed] [Google Scholar]
- 44.Roescher N, Vosters JL, Lai Z, Uede T, Tak PP, Chiorini JA. Local administration of soluble CD40:Fc to the salivary glands of non-obese diabetic mice does not ameliorate autoimmune inflammation. PloS one. 2012;7:e51375. doi: 10.1371/journal.pone.0051375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J Exp Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen RC, Armitage RJ, Conley ME, Rosenblatt H, Jenkins NA, Copeland NG, Bedell MA, Edelhoff S, Disteche CM, Simoneaux DK, et al. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science. 1993;259:990–993. doi: 10.1126/science.7679801. [DOI] [PubMed] [Google Scholar]
- 47.van Essen D, Kikutani H, Gray D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature. 1995;378:620–623. doi: 10.1038/378620a0. [DOI] [PubMed] [Google Scholar]
- 48.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 49.Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, Craft JE, Nussenzweig MC. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science. 2014;345:1058–1062. doi: 10.1126/science.1257861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bynoe MS, Grimaldi CM, Diamond B. Estrogen up-regulates Bcl-2 and blocks tolerance induction of naive B cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:2703–2708. doi: 10.1073/pnas.040577497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tengner P, Halse AK, Haga HJ, Jonsson R, Wahren-Herlenius M. Detection of anti-Ro/SSA and anti-La/SSB autoantibody-producing cells in salivary glands from patients with Sjogren’s syndrome. Arthritis Rheum. 1998;41:2238–2248. doi: 10.1002/1529-0131(199812)41:12<2238::AID-ART20>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 52.Quadri KKSMS, Harris V, Kurien BT, Scofield RH. Functional Anti-Muscarinic Receptor-3 Monoclonal Antibodies Derived from Salivary Gland in Patients of Sjögren’s Syndrome. Arthritis Rheumatol. 2015;67(suppl 10) [Google Scholar]
- 53.Mahevas M, Patin P, Huetz F, Descatoire M, Cagnard N, Bole-Feysot C, Le Gallou S, Khellaf M, Fain O, Boutboul D, Galicier L, Ebbo M, Lambotte O, Hamidou M, Bierling P, Godeau B, Michel M, Weill JC, Reynaud CA. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J Clin Invest. 2013;123:432–442. doi: 10.1172/JCI65689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP) Am J Hematol. 2003;72:94–98. doi: 10.1002/ajh.10253. [DOI] [PubMed] [Google Scholar]
- 55.You YN, Tefferi A, Nagorney DM. Outcome of splenectomy for thrombocytopenia associated with systemic lupus erythematosus. Ann Surg. 2004;240:286–292. doi: 10.1097/01.sla.0000133182.92780.9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, McDevitt HO. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med. 1994;180:995–1004. doi: 10.1084/jem.180.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12439–12444. doi: 10.1073/pnas.0806439105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balasa B, Krahl T, Patstone G, Lee J, Tisch R, McDevitt HO, Sarvetnick N. CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. Journal of immunology. 1997;159:4620–4627. [PubMed] [Google Scholar]
- 59.Quezada SA, Fuller B, Jarvinen LZ, Gonzalez M, Blazar BR, Rudensky AY, Strom TB, Noelle RJ. Mechanisms of donor-specific transfusion tolerance: preemptive induction of clonal T-cell exhaustion via indirect presentation. Blood. 2003;102:1920–1926. doi: 10.1182/blood-2003-02-0586. [DOI] [PubMed] [Google Scholar]
- 60.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 61.Graham LM, Tsoni SV, Willment JA, Williams DL, Taylor PR, Gordon S, Dennehy K, Brown GD. Soluble Dectin-1 as a tool to detect beta-glucans. J Immunol Methods. 2006;314:164–169. doi: 10.1016/j.jim.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 62.Costa C, Germena G, Martin-Conte EL, Molineris I, Bosco E, Marengo S, Azzolino O, Altruda F, Ranieri VM, Hirsch E. The RacGAP ArhGAP15 is a master negative regulator of neutrophil functions. Blood. 2011;118:1099–1108. doi: 10.1182/blood-2010-12-324756. [DOI] [PubMed] [Google Scholar]
- 63.Julia A, Domenech E, Chaparro M, Garcia-Sanchez V, Gomollon F, Panes J, Manosa M, Barreiro-De Acosta M, Gutierrez A, Garcia-Planella E, Aguas M, Munoz F, Esteve M, Mendoza JL, Vera M, Marquez L, Tortosa R, Lopez-Lasanta M, Alonso A, Gelpi JL, Garcia-Montero AC, Bertranpetit J, Absher D, Myers RM, Gisbert JP, Marsal S. A genome-wide association study identifies a novel locus at 6q22.1 associated with ulcerative colitis. Hum Mol Genet. 2014;23:6927–6934. doi: 10.1093/hmg/ddu398. [DOI] [PubMed] [Google Scholar]
- 64.Kukita A, Bonewald L, Rosen D, Seyedin S, Mundy GR, Roodman GD. Osteoinductive factor inhibits formation of human osteoclast-like cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:3023–3026. doi: 10.1073/pnas.87.8.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andress DL. IGF-binding protein-5 stimulates osteoblast activity and bone accretion in ovariectomized mice. Am J Physiol Endocrinol Metab. 2001;281:E283–288. doi: 10.1152/ajpendo.2001.281.2.E283. [DOI] [PubMed] [Google Scholar]
- 66.Gupta A, Lee BS, Khadeer MA, Tang Z, Chellaiah M, Abu-Amer Y, Goldknopf J, Hruska KA. Leupaxin is a critical adaptor protein in the adhesion zone of the osteoclast. J Bone Miner Res. 2003;18:669–685. doi: 10.1359/jbmr.2003.18.4.669. [DOI] [PubMed] [Google Scholar]
- 67.Gravani F, Papadaki I, Antypa E, Nezos A, Masselou K, Ioakeimidis D, Koutsilieris M, Moutsopoulos HM, Mavragani CP. Subclinical atherosclerosis and impaired bone health in patients with primary Sjogren’s syndrome: prevalence, clinical and laboratory associations. Arthritis Res Ther. 2015;17:99. doi: 10.1186/s13075-015-0613-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drolet DW, Scully KM, Simmons DM, Wegner M, Chu KT, Swanson LW, Rosenfeld MG. TEF, a transcription factor expressed specifically in the anterior pituitary during embryogenesis, defines a new class of leucine zipper proteins. Genes Dev. 1991;5:1739–1753. doi: 10.1101/gad.5.10.1739. [DOI] [PubMed] [Google Scholar]
- 69.Allaman-Pillet N, Roduit R, Oberson A, Abdelli S, Ruiz J, Beckmann JS, Schorderet DF, Bonny C. Circadian regulation of islet genes involved in insulin production and secretion. Mol Cell Endocrinol. 2004;226:59–66. doi: 10.1016/j.mce.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Papagerakis S, Zheng L, Schnell S, Sartor MA, Somers E, Marder W, McAlpin B, Kim D, McHugh J, Papagerakis P. The circadian clock in oral health and diseases. J Dent Res. 2014;93:27–35. doi: 10.1177/0022034513505768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salazar-Olivo LA, Mejia-Elizondo R, Alonso-Castro AJ, Ponce-Noyola P, Maldonado-Lagunas V, Melendez-Zajgla J, Saavedra-Alanis VM. SerpinA3g participates in the antiadipogenesis and insulin-resistance induced by tumor necrosis factor-alpha in 3T3-F442A cells. Cytokine. 2014;69:180–188. doi: 10.1016/j.cyto.2014.05.025. [DOI] [PubMed] [Google Scholar]
- 72.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 73.Pauza ME, Nguyen A, Wolfe T, Ho IC, Glimcher LH, von Herrath M, Lo D. Variable effects of transgenic c-Maf on autoimmune diabetes. Diabetes. 2001;50:39–46. doi: 10.2337/diabetes.50.1.39. [DOI] [PubMed] [Google Scholar]
- 74.Morahan G, Huang D, Tait BD, Colman PG, Harrison LC. Markers on distal chromosome 2q linked to insulin-dependent diabetes mellitus. Science. 1996;272:1811–1813. doi: 10.1126/science.272.5269.1811. [DOI] [PubMed] [Google Scholar]
- 75.Sidiropoulos PI, Boumpas DT. Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus. 2004;13:391–397. doi: 10.1191/0961203304lu1032oa. [DOI] [PubMed] [Google Scholar]
- 76.Tocoian A, Buchan P, Kirby H, Soranson J, Zamacona M, Walley R, Mitchell N, Esfandiari E, Wagner F, Oliver R. First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus. 2015 doi: 10.1177/0961203315574558. [DOI] [PubMed] [Google Scholar]
- 77.Ma A, Dun H, Song L, Hu Y, Zeng L, Bai J, Zhang G, Kinugasa F, Miyao Y, Sakuma S, Okimura K, Kasai N, Daloze P, Chen H. Pharmacokinetics and pharmacodynamics of ASKP1240, a fully human anti-CD40 antibody, in normal and renal transplanted Cynomolgus monkeys. Transplantation. 2014;97:397–404. doi: 10.1097/01.TP.0000440951.29757.bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cordoba F, Wieczorek G, Audet M, Roth L, Schneider MA, Kunkler A, Stuber N, Erard M, Ceci M, Baumgartner R, Apolloni R, Cattini A, Robert G, Ristig D, Munz J, Haeberli L, Grau R, Sickert D, Heusser C, Espie P, Bruns C, Patel D, Rush JS. A novel, blocking, Fc-silent anti-CD40 monoclonal antibody prolongs nonhuman primate renal allograft survival in the absence of B cell depletion. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015;15:2825–2836. doi: 10.1111/ajt.13377. [DOI] [PubMed] [Google Scholar]
- 79.Oganesyan V, Gao C, Shirinian L, Wu H, Dall’Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. 2008;64:700–704. doi: 10.1107/S0907444908007877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, Mohan C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–3439. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Early life anti-CD40L inhibits lymphocyte infiltration of salivary gland of aged mice
Gene Set Enrichment Analysis (GSEA) of gene pathways down-regulated in salivary gland by early anti-CD40L treatment of NOD.H-2h4 mice
Autoantibodies (IgG) are detected after emergence of spontaneous splenic germinal centers
Greatly reduced B and T cell clusters in thyroids following early anti-CD40L in NOD.H-2h4 mice
Autoimmune manifestations in aged mice arise from early-life germinal centers
Significantly down-regulated salivary gland genes in response to early CD40L blockade
Early-life treatment of anti-CD40L inhibits specific IgG autoantibody profiles