

Abstract
Chronic urethral obstruction and the ensuing bladder wall remodeling can lead to diminished bladder smooth muscle (BSM) contractility and debilitating lower urinary tract symptoms. No effective pharmacotherapy exists to restore BSM contractile function. Neuropilin 2 (Nrp2) is a transmembrane protein that is highly expressed in BSM. Nrp2 deletion in mice leads to increased BSM contraction. We determined whether genetic ablation of Nrp2 could restore BSM contractility following obstruction. Partial bladder outlet obstruction (pBOO) was created by urethral occlusion in mice with either constitutive and ubiquitous, or inducible smooth muscle–specific deletion of Nrp2, and Nrp2-intact littermates. Mice without obstruction served as additional controls. Contractility was measured by isometric tension testing. Nrp2 deletion prior to pBOO increased force generation in BSM 4 weeks following surgery. Deletion of Nrp2 in mice already subjected to pBOO for 4 weeks showed increased contractility of tissues tested 6 weeks after surgery compared with nondeleted controls. Assessment of tissues from patients with urodynamically defined bladder outlet obstruction revealed reduced NRP2 levels in obstructed bladders with compensated compared with decompensated function, relative to asymptomatic controls. We conclude that downregulation of Nrp2 promotes BSM force generation. Neuropilin 2 may represent a novel target to restore contractility following obstruction.
Loss of neuropilin 2 in mice and humans with urethral obstruction promotes increased bladder smooth muscle contractility, implicating neuropilin 2 as a novel therapeutic target.
Introduction
Reduced smooth muscle (SM) contractility in the bladder stems from numerous etiologies including obstruction secondary to benign prostatic hyperplasia (BPH), posterior urethral valves, diabetes mellitus, multiple sclerosis, spinal cord injury, or idiopathic causes (reviewed in ref. 1). In conditions such as BPH or posterior urethral valves, the bladder contracts against an obstructed outlet. The initial response is adaptive, involving a compensatory phase of SM hypertrophy that enables increased force generation to overcome the increased outlet resistance. When the demand outstrips the adaptive capability of the bladder, contractile performance becomes less efficient, residual volumes increase, and the bladder remodels, ultimately leading to a loss of detrusor contractility as the bladder decompensates (reviewed in ref. 2). The prevalence of underactive detrusor function is reported to be as high as 48% in adults (3). Pharmacological treatments for restoration of SM contraction such as muscarinic agonists or cholinesterase inhibitors have shown limited efficacy and adverse effects (4). Thus, investigation of novel therapeutic strategies is warranted.
Studies in rodents in which the urethra is occluded by surgical means to achieve partial bladder outlet obstruction (pBOO) have been shown to recapitulate the salient features of bladder wall remodeling, fibrosis, urodynamic alterations, and eventual loss of contractility evident in humans with urinary tract obstruction (5–7). Furthermore, the creation of pBOO in genetically engineered mouse models has enabled the investigation of specific proteins and associated signaling pathways in the response to obstruction (8–13). In those studies, target gene deletion was typically ubiquitous such that the activity of protein function in a specific tissue could not be assessed. In addition, the constitutive nature of gene deletion precluded investigation of the relative impact of protein loss on the compensation and decompensation phases following obstruction.
In a recent study, we reported a novel role for neuropilin 2 (Nrp2) as a regulator of SM contractility (14). Nrp2 is a transmembrane receptor that binds VEGF family members and class 3 semaphorins (SEMA3) to regulate angiogenesis and axon guidance. Signaling downstream of Nrp2 and its coreceptors impinges on a variety of effectors including PLC-γ/MEK/ERK (15), RhoA/Rho kinase (ROCK)/p-MLC (16), and PI3K/AKT/mTOR pathways (17) that lead to changes in cytoskeletal tension, proliferation, cell survival, and migration (18, 19). Using in vitro and in vivo analyses in our previous study, we (a) identified bladder SM as a major site of Nrp2 expression; (b) demonstrated inhibition of RhoA and cytoskeletal stiffness in primary bladder SM cells treated with the NRP2 ligand, SEMA3F; and (c) observed increased contractility of bladder SM strips from mice with ubiquitous or SM-specific deletion of Nrp2 in vivo, when compared with tissues from Nrp2-intact littermate controls. Together, these findings implicated Nrp2 as an inhibitor of SM contractility (14). In the current study, we tested the hypothesis that Nrp2 deletion in vivo improves bladder SM contractility under conditions of decompensation following pBOO.
Results
Structural and functional changes in WT mice subjected to pBOO.
WT mice subjected to pBOO displayed an increase in the bladder-to-body weight ratio compared with sham-operated animals, consistent with prior reports describing bladder hypertrophy and distension following obstruction (6, 12) (Table 1). Temporal changes in contractility were also observed. Bladders from obstructed mice displayed increased contractility with all agonists compared with those from shams at 1 to 2 weeks following obstruction, but decreased contractility at 4 weeks, consistent with compensated followed by decompensated SM function (Figure 1, A–D).
Table 1. Bladder/body weight ratio in sham versus obstructed mice.

Figure 1. Partial bladder outlet obstruction (pBOO) in wild-type mice reveals time-dependent compensation and decompensation phases.
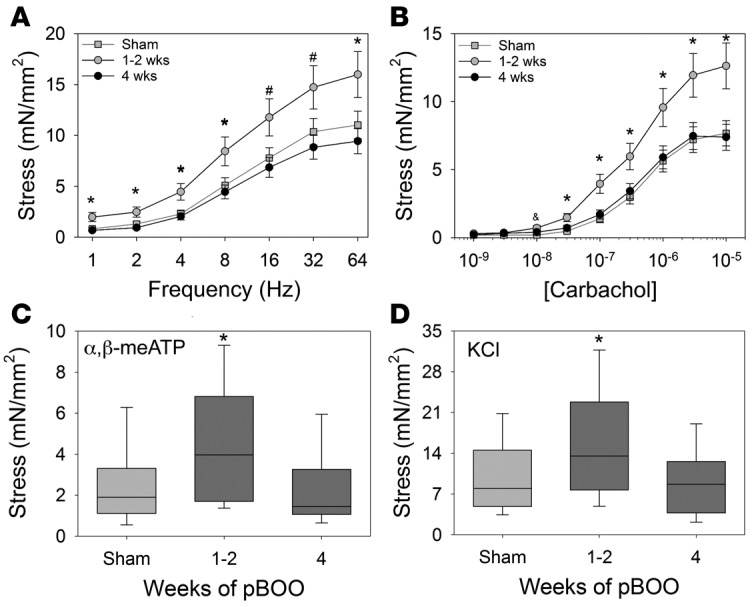
(A–D) Contractile responses of bladder muscle strips from wild-type male mice subjected to pBOO (dark gray boxes) for 1 to 2 weeks (n = 11), 4 weeks (n = 10), or sham operation (light gray box, n = 15) were determined by isometric tension testing in response to electrical field stimulation (A), carbachol (B), α,β-methyl-adenosine triphosphate (α,β-meATP) (C), and potassium chloride (KCl) (D). Line graphs in A and B show mean ± SEM; box-and-whisker plots in C and D show median (line within the box), upper and lower quartiles (bounds of the box), and 90th and 10th percentiles (whiskers). Contractile responses from obstructed mice were higher than sham at 1 to 2 weeks after obstruction, but trended to lower contractility at 4 weeks after obstruction. *P < 0.05, significantly greater than sham and 4-week pBOO; #P < 0.05, significantly greater than 4-week pBOO; &P < 0.05, significantly different than sham; ANOVA followed by Holm-Sidak multiple comparison test. mN, millinewtons.
Constitutive deletion of Nrp2 prevents decompensation following pBOO.
To extend our published findings showing enhanced force generation in SM from bladders of mice with constitutive Nrp2 knockout (14), we compared the contractile activity of tissue strips from either KO mice (Nrp2–/–, Nrp2–/+) or age-matched WT littermate controls subjected to pBOO. Sham-operated mice were not included in this analysis because of the small number of available mice, a consequence of non-Mendelian breeding ratios in the Nrp2+/gfp strain (20). At 4 weeks, corresponding to the decompensated phase following pBOO in WT mice, tissue strips from KO mice showed higher contractile response to electrical field stimulation (EFS), carbachol, α,β-methyl-ATP, and KCl compared with both WT mice and nonsurgical WT controls (Figure 2). These findings suggest that absence of Nrp2 promotes sustained bladder contractility even in the context of obstruction injury.
Figure 2. Mice with constitutive, ubiquitous deletion of neuropilin 2 (Nrp2) display increased contractility following partial bladder outlet obstruction.
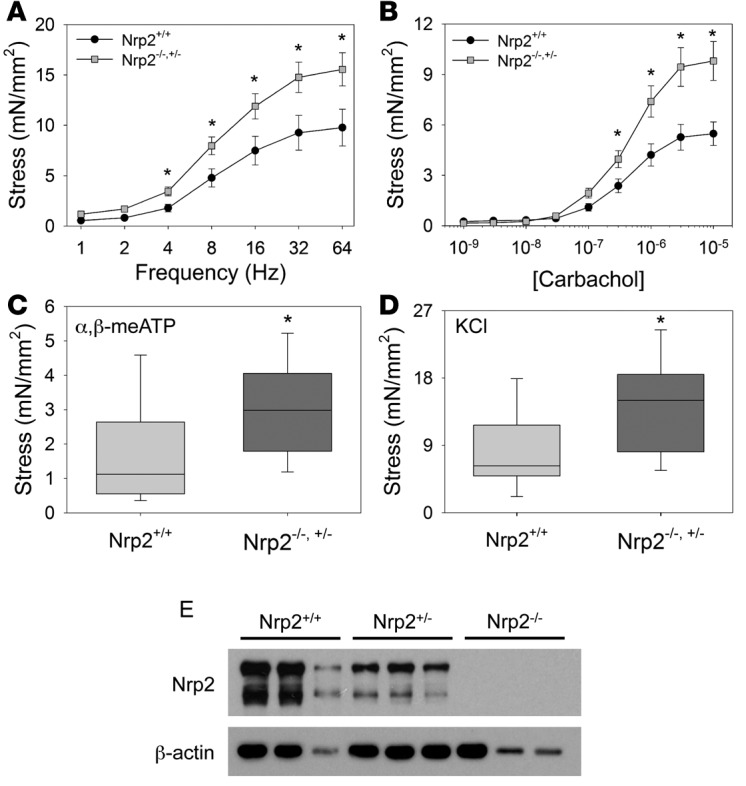
(A–D) Contractile responses of bladder muscle strips from Nrp2-expressing obstructed (Nrp2+/+, n = 10) and Nrp2-deficient obstructed (Nrp2+/–, n = 6, Nrp2–/–, n = 7) mice were determined by isometric tension testing in response to electrical field stimulation (A), carbachol (CCh) (B), α,β-methyl-adenosine triphosphate (α,β-meATP) (C), and potassium chloride (KCl) (D). In each case, force generation was higher in Nrp2-deficient mice compared with wild-type controls. Line graphs in A and B show mean ± SEM; box-and-whisker plots in C and D show median (line within the box), upper and lower quartiles (bounds of the box), and 90th and 10th percentiles (whiskers). *P < 0.05, Mann-Whitney rank-sum test. (E) Immunoblot analysis demonstrates reduction in Nrp2 expression with deletion of one or both Nrp2 alleles. β-Actin was used as a loading control. mN, millinewtons.
SM-specific Nrp2 deletion prevents decompensation following pBOO.
To specifically address the consequence of Nrp2 loss in SM in the response to obstruction, we used Cre-loxP technology to delete Nrp2 in SM, using the 4-hydroxytamoxifen–inducible (4-OHT–inducible) SM22α-CreERT2:Nrp2fl/fl (EXP) strain (14), prior to creation of pBOO. Bladder-to-body weight ratios were increased in obstructed mice compared with sham-operated mice but did not differ between Nrp2fl/fl (CTRL) and EXP genotypes (Table 2). As shown in Figure 3A, at 4 weeks following pBOO, corresponding to the decompensation phase identified in WT mice, SM strips from bladders of EXP mice displayed increased contractility compared with those from CTRL mice. SM-specific loss of Nrp2 protein in the EXP bladders was confirmed by immunoblot (Figure 3B).
Table 2. Bladder/body weight ratio in sham and obstructed CTRL versus EXP mice.

Figure 3. Mice with inducible, smooth muscle–specific deletion of neuropilin 2 (Nrp2) prior to partial bladder outlet obstruction do not undergo decompensation.
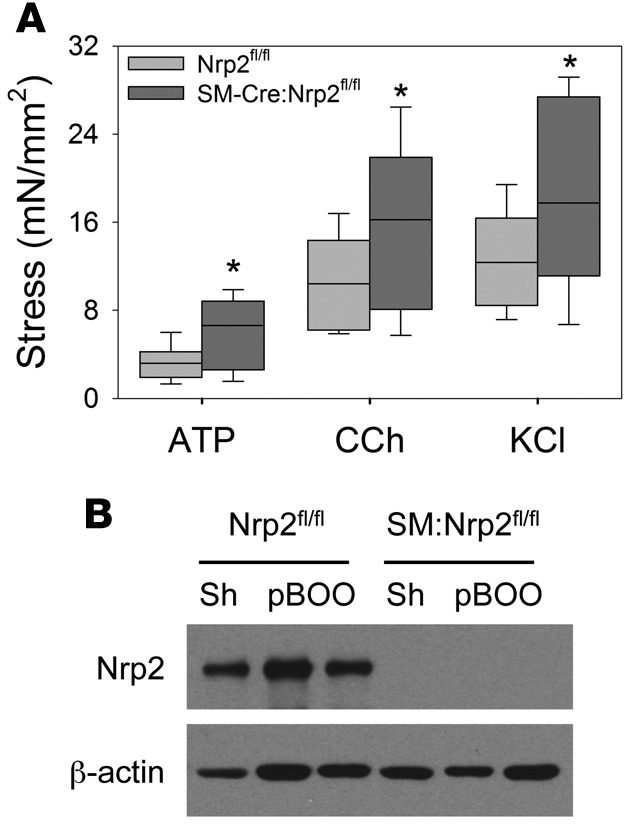
(A) Contractile responses of bladder muscle strips from Nrp2-intact mice (Nrp2fl/fl, light gray boxes, n = 9) and those with smooth muscle–specific Nrp2 deletion (SM-Cre:Nrp2fl/fl, dark gray boxes, n = 8) were determined by isometric tension testing in response to α,β-methyl-adenosine triphosphate (ATP), carbachol (CCh), and potassium chloride (KCl). Box-and-whisker plots show median (line within the box), upper and lower quartiles (bounds of the box), and 90th and 10th percentiles (whiskers). In each case, force generation was higher in Nrp2-deficient mice compared with wild-type littermate controls at the 4-week decompensated time point. *P < 0.05, Mann-Whitney rank-sum test. (B) Immunoblot analysis demonstrates inducible deletion of Nrp2 in muscle strips. β-Actin was used as a loading control. mN, millinewtons.
Nrp2 deletion during decompensation promotes recovery of bladder contractility.
We proceeded to determine the consequence of SM-specific Nrp2 deletion in bladders that had already begun to decompensate during ongoing obstruction. Mice were subjected to sham surgery or pBOO as described. At the 4-week time point corresponding to decompensation in WT and CTRL mice, tissue-specific Nrp2 knockout was induced in a subset of mice (EXP-OHT) by administration of 4-OHT, while the remaining EXP mice received vehicle (EXP-Veh). 4-OHT was also administered to CTRL pBOO mice. All mice were maintained for an additional 2 weeks prior to harvest and functional analysis. As shown in Figure 4, bladder strips from the EXP-OHT group (Nrp2-deleted) showed increased contractility in response to stimuli, compared with those from CTRL and EXP-Veh mice (Nrp2-intact).
Figure 4. Obstructed mice with inducible, smooth muscle–specific deletion of neuropilin 2 (Nrp2) during decompensation display enhanced contractility.
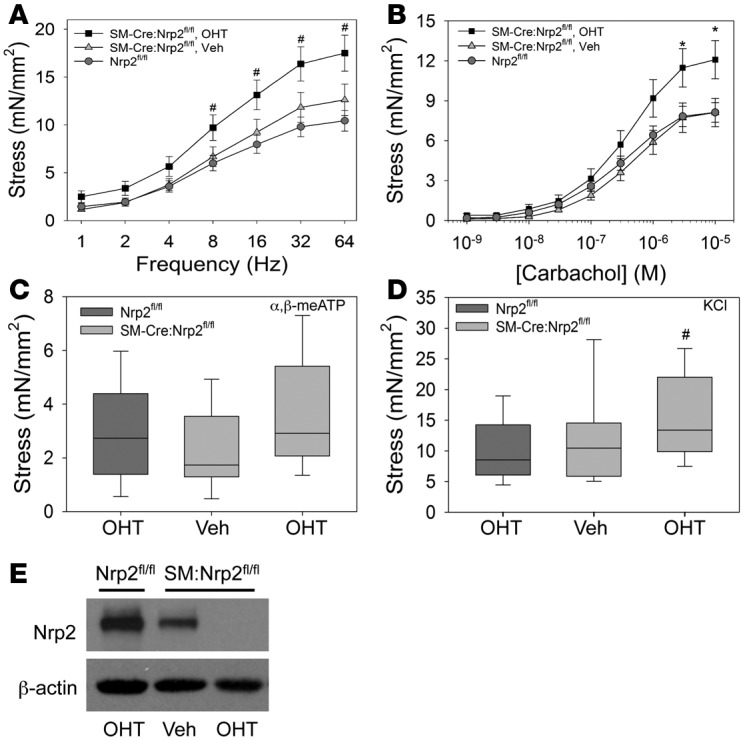
(A–D) Contractile responses of bladder muscle strips from Nrp2-intact mice treated with 4-hydroxytamoxifen (OHT) (Nrp2fl/fl, n = 12, dark gray box), conditional mice treated with vehicle (SM-Cre:Nrp2fl/fl, Veh, n = 6), and conditional mice treated with OHT (SM-Cre:Nrp2fl/fl, OHT, n = 9) were determined by isometric tension testing in response to electrical field stimulation (A), carbachol (B), α,β-methyl-adenosine triphosphate (α,β-meATP) (C), and potassium chloride (KCl) (D). Line graphs in A and B show mean ± SEM; box-and-whisker plots in C and D show median (line within the box), upper and lower quartiles (bounds of the box), and 90th and 10th percentiles (whiskers). In each case, force generation was higher in conditional mice receiving 4-OHT compared with either control group. *P < 0.05, significantly greater than Nrp2fl/fl and SM-Cre:Nrp2fl/fl, Veh; #P < 0.05, significantly greater than Nrp2fl/fl; ANOVA followed by Holm-Sidak multiple comparison test. (E) Immunoblot analysis demonstrates inducible deletion of Nrp2 in muscle strips. β-Actin was used as a loading control. Data are representative of at least 3 trials. mN, millinewtons.
Reduced NRP2 expression in bladders from obstructed patients with compensated function.
To assess the relationship between NRP2 levels and bladder function in human tissues, the bladder outlet obstruction index and detrusor contractility index were calculated from pressure-flow studies conducted in male patients with lower urinary tract symptoms (Table 3) and correlated with NRP2 mRNA levels in bladder biopsies from the same patients and from asymptomatic controls. As shown in Figure 5A, NRP2 protein was robustly expressed in muscle bundles of the normal human detrusor; α-smooth muscle actin (α-SMA) served as a positive control. In patients with bladder outlet obstruction, NRP2 mRNA levels were significantly reduced in patients with compensated bladder function compared with those with decompensated function (Figure 5B).
Table 3. Summary of urodynamic findings in obstructed patients.

Figure 5. Neuropilin 2 (NRP2) expression in human bladders.
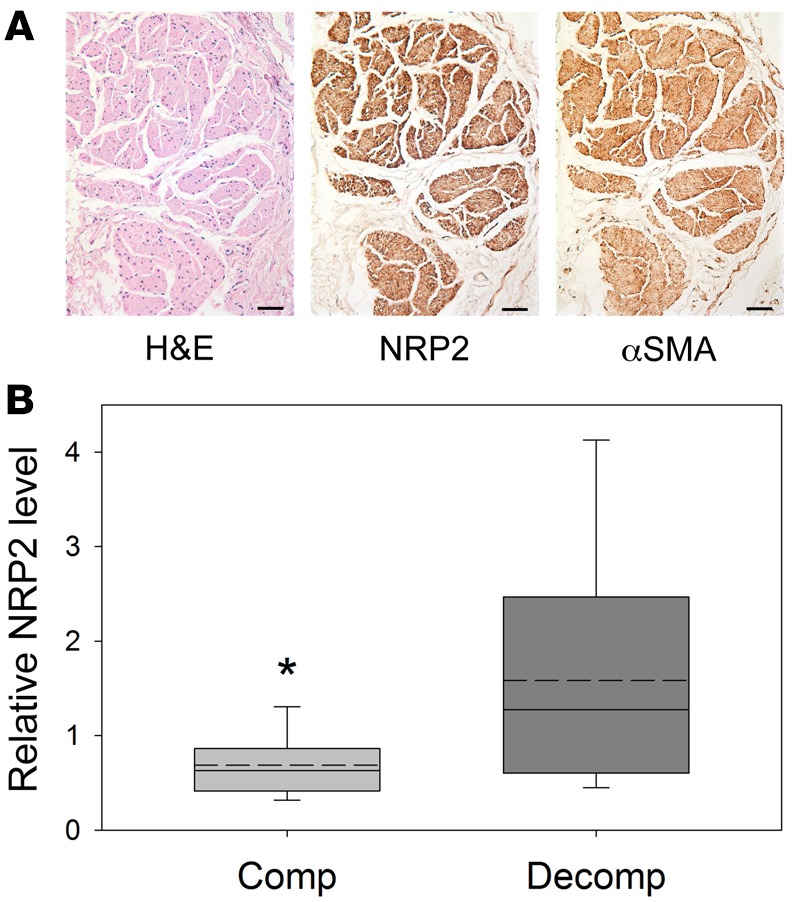
(A) NRP2 is robustly expressed in smooth muscle bundles of the adult human bladder. Scale bars: 50 μm. αSMA, α-smooth muscle actin. (B) Comparison of NRP2 expression in obstructed patients having compensated (n = 18) or decompensated (n = 9) bladder function. NRP2 levels were significantly reduced in obstructed patients with compensated bladder compared with decompensated function, relative to asymptomatic controls (n = 21, relative NRP2 level = 1.2). Box-and-whisker plots show median (solid line within the box), mean (dotted line within box), upper and lower quartiles (bounds of the box), and 90th and 10th percentiles (whiskers). *P < 0.05, Mann-Whitney rank-sum test.
Discussion
In this study, we describe an important association between Nrp2 downregulation in the bladder wall and maintenance of SM contractility following bladder outlet obstruction in mice and humans. The evidence in support of this relationship is as follows: (a) bladder strips from mice with either constitutive, ubiquitous deletion of Nrp2 or inducible, SM-specific Nrp2 deletion displayed increased contractility following obstruction compared with those from Nrp2-intact mice; (b) increased bladder-to-body weight ratios were evident in all mice subjected to pBOO irrespective of Nrp2 status; (c) SM-specific deletion of Nrp2 at a time point corresponding to decompensation resulted in enhanced contractility of bladder strips compared with mice in which Nrp2 remained intact; (d) analysis of urodynamics and pressure-flow data from patients with outlet obstruction revealed that in individuals with obstruction who displayed compensated bladder function NRP2 levels were lower than those in obstructed patients with decompensated function. Collectively, these findings suggest that downregulation of Nrp2 is an important component of the compensatory response to obstruction in experimental animals and in humans with obstruction, and that Nrp2 may represent a novel pharmacological target for maintenance or restoration of detrusor contractility in the decompensated bladder.
Distinct from other reports in the literature, the Cre-loxP–based deletion strategies employed in this study enable us to delineate the impact of Nrp2 deletion specifically in SM and at different times relative to onset of obstruction. Deletion of Nrp2 prior to creation of obstruction was associated with a lack of decompensation in Nrp2-deficient mice, consistent with the higher contractility observed in unobstructed mice reported previously (14). Comparison of results from mice with ubiquitous Nrp2 deletion and those with SM-specific deletion of Nrp2 revealed that the increased contractility at 4 weeks following obstruction was comparable, at least in response to selected agonists, suggesting that much of the protective advantage observed with loss of Nrp2 following obstruction results from its activity in SM and not that in other cell types such as neurons or endothelial cells. Importantly, the inducible-deletion strategy allowed us to investigate the impact of Nrp2 deletion in the setting of existing obstruction, a scenario that more closely models the clinical situation in patients, who present with chronic obstruction and associated lower urinary tract symptoms. Our findings demonstrated that downregulation of Nrp2 during the decompensation phase promoted detrusor contractility by comparison with Nrp2-intact mice, supporting the argument that Nrp2 represents a potentially novel target for inhibition that may have clinical benefit in patients with bladder outlet obstruction. Although beyond the scope of the current project, the inducible-deletion strategy also provides the ability to define the time frame following obstruction after which no recovery of contractility is likely to occur.
Evaluation of functional and expression data from a cohort of human patients with obstruction revealed a reduction in NRP2 levels in obstructed patients with compensated bladder function. These findings are consistent with our published report in uninjured Nrp2-deficient mice (14) as well as our current findings from obstructed Nrp2-deficient mice. Although a number of studies have provided correlative evidence for changes in selected genes and/or proteins in bladder tissue from patients with obstruction, to the best of our knowledge our analysis provides the first evidence in humans for an association between expression of the protein of interest — NRP2 — and detrusor function. The mechanisms whereby NRP2 is downregulated in human tissues are still largely unexplored. Evidence from our group suggests that Nrp2 levels are sensitive to mechanical stimuli. Complete urethral occlusion in mice led to reduced Nrp2 expression primarily in detrusor tissue (R.M.A, unpublished observations), whereas activation of the platelet-derived growth factor receptor, shown by us to be mechanosensitive (21), led to significant downregulation of NRP2 in primary human SM cells (14). Bladder distension following outlet obstruction is also known to evoke alterations in blood flow as a result of vascular compression, leading to hypoxia within the bladder wall (22–24). Hypoxia was shown to reduce NRP2 expression in vitro, resulting in attenuation of the inhibitory effects of NRP2 on RhoA activity and actin polymerization (25). In SM cells, RhoA activates the effector kinase ROCK, which in turn phosphorylates, directly or indirectly, several constituents of the contractile apparatus including myosin light chain (MLC), the MLC phosphatase component MYPT1, and CPI-17, resulting in increased contractility (reviewed in ref. 26). Nrp2-mediated inhibition of RhoA and downstream signaling reduces force generation by promoting cofilin-mediated actin depolymerization (16) and through attenuation of MLC phosphorylation (14). Thus, downregulation of Nrp2 is predicted to increase contractility through relief of inhibition of RhoA-ROCK signaling. Notably, bladder outlet obstruction in both experimental animals and human patients was associated with increased expression of RhoA and the effectors ROCK and CPI-17 in the bladder wall (27). In that study, RhoA, ROCK, and CPI-17 levels were also increased by exposing primary bladder SM cells to mechanical stretch in vitro, consistent with a role for bladder distension as an inducer of signals leading to RhoA activation. Taken together, these findings suggest that the combined mechanical and hypoxic stimulation of bladder SM resulting from increased intravesical pressure secondary to outlet obstruction lead to reduced Nrp2 expression, relief of inhibition on RhoA and its effectors, and subsequent increases in SM contractility.
Pharmacological management of diminished detrusor contractility has focused on stimulation of parasympathetic activity to enhance bladder contractility and reduction of outflow resistance to facilitate bladder emptying (28). However, analysis of 10 randomized clinical trials of parasympathomimetic drugs including the cholinergic agonists bethanechol and carbachol, the α-adrenoceptor antagonist phenoxybenzamine, or the cholinesterase inhibitor distigmine in patients with poorly contractile bladders, revealed either a worsening of symptoms or a lack of significant improvement (4). Experimental studies have suggested that prostaglandin E2 can also enhance detrusor contractility as well as urethral relaxation (29), although when combined with bethanechol, PGE2 had limited efficacy in patients (30). Taken together, these findings emphasize the need to identify novel therapeutic targets for pharmacological management of detrusor underactivity. The increase in contractility following Nrp2 deletion in the decompensating bladder suggests that Nrp2 may be a novel target to mitigate reduced detrusor contractility under conditions of chronic obstruction. Small-molecule inhibitors of Nrp2 binding to ligand have been identified through a cell-free, high-throughput screening approach (31), including the antibiotic actinomycin D, the leukotriene receptor antagonist zafirlukast, and the dopamine receptor agonist dihydrexidine. However, based on the target profile of these agents, it is not clear how specificity for Nrp2 in vivo could be achieved. In contrast, Caunt and colleagues demonstrated specific inhibition of Nrp2 in vivo using a function-blocking antibody (32). In that study, administration of anti-Nrp2 antibody resulted in inhibition of Nrp2-mediated lymphatic endothelial cell migration and a corresponding reduction in lymphangiogenesis in 2 experimental lung tumor models (32), providing proof-of-principle evidence for targeted inhibition of Nrp2 in vivo.
In summary, we provide compelling evidence that downregulation of Nrp2 in mice and humans is associated with improved detrusor function under conditions of chronic bladder outlet obstruction. Furthermore, we showed that targeting Nrp2 in bladders undergoing decompensation has the potential to restore contractility in spite of ongoing obstruction. These findings argue that the Nrp2 axis represents a potentially novel pharmacologic target for restoration of SM contractility and provide an important platform for the development of small-molecule or peptide antagonists of Nrp2.
Methods
Genetically modified mouse models.
Nrp2+/gfp mice (also known as Nrp2tm1.2Mom/MomJ; stock number 006700) and Nrp2fl/fl mice (also known as Nrp2tm1.1Mom/MomJ; stock number 006697) were purchased from the Jackson Laboratory and maintained on the C57BL/6 background. Nrp2gfp/gfp mice were viable and displayed constitutive, ubiquitous deletion of Nrp2. SM-specific deletion of Nrp2 after birth was accomplished by breeding mice harboring an inducible Cre recombinase (CreERT2) under the control of the SM22α promoter (33) (hereafter referred to as SM22α-CreERT2, received as a gift from Robert Feil, University of Tübingen, Germany) with those expressing a floxed allele of Nrp2 (Nrp2fl/fl). Prior to use in obstruction experiments, restriction of Cre recombinase activity to SM was verified by crossing SM22α-CreERT2 with a ROSA26 LacZ reporter mouse (B6.129S4-Gt(ROSA)26Sortm1Sor/J; Jackson Laboratory, stock number 003474) and staining of organs and tissues with X-gal, essentially as described (14) (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.90617DS1). Mice positive for both SM22α-CreERT2 and Nrp2fl/fl were treated with 0.5 mg 4-OHT for 3 days by gavage to delete Nrp2 from SM. Both Nrp2gfp/gfp and SM22α-CreERT2;Nrp2fl/fl as well as littermate controls underwent pBOO and sham surgeries.
Creation of pBOO.
All experiments were performed using 10- to 12-week-old male mice that were anesthetized with isoflurane and underwent microsurgical creation of pBOO essentially as described (6). Briefly, anesthetized mice underwent a 1-cm laparotomy to expose the bladder and prostate. The prostatic lobes were bisected in the midline to expose the proximal urethra and bladder neck. A right-angle instrument was used to isolate the urethra and pass a 5-0 silk suture behind the urethra approximately 1 mm inferior to the bladder neck. Polyethylene tubing (PE10) with an outer diameter of 0.024 inches was placed peri-urethrally to prevent complete occlusion of the urethra. The 5-0 silk suture was then tied down over the tubing. Once the tubing was removed, the bladder was manually expressed to ensure a complete obstruction was not inadvertently created. The muscle layer was closed with 4-0 monocryl suture, and skin was closed using 9-mm stainless steel clips. Meloxicam was administered for 72 hours. The sham procedure was identical except the 5-0 silk suture was not secured around the urethra. Urethral obstruction was applied in 4 types of experiments: (a) pBOO or sham surgery was performed in WT C57BL/6 mice. Mice were harvested 1, 2, or 4 weeks after pBOO for evaluation of contractility. (b) pBOO surgery was performed in Nrp2gfp/gfp or Nrp2+/gfp (KO) and littermate control Nrp2+/+ mice (WT). Bladders were harvested 4 weeks after pBOO. (c) Using the SM22α-CreERT2:Nrp2fl/fl (EXP) mouse strain and Nrp2fl/fl (CTRL) mice as littermate controls, SM-specific Nrp2 knockout was induced by administration of 4-OHT 1 week prior to surgery. pBOO (CTRL n = 15; EXP n = 9) and sham (CTRL n = 10, EXP n = 8) surgeries were performed, and bladders were harvested at 2 and 4 weeks to assess contractility at both the compensated and decompensated phases. (d) pBOO was created in SM22α-CreERT2:Nrp2fl/fl (EXP) and Nrp2fl/fl (CTRL) littermate controls. Sham surgery was performed only in CTRL mice (n = 3). At 4 weeks after surgery, Nrp2 deletion was induced by administration of 4-OHT in a subset of EXP (EXP-OHT, n = 8) and CTRL mice (n = 11). Vehicle was given to the remaining EXP mice (EXP-Veh, n = 7) to serve as a second control group. Bladders were harvested 2 weeks after 4-OHT/vehicle administration to assess maintenance or recovery of bladder contractility following Nrp2 deletion.
Tissue harvest.
Mice were euthanized at 1, 2, 4, and 6 weeks following surgery. At the time of sacrifice, total body weight and bladder mass were recorded. Bladders for contractility testing were immediately immersed in ice-cold Krebs’ solution (120 mM NaCl, 5.9 mM KCl, 25 mM NaHCO3, 1.2 mM Na2H2PO4, 1.2 mM MgCl2. 6H2O, 2.5 mM CaCl2, and 11.5 mM dextrose) with minimal handling. All tissues used for molecular analysis were immediately snap-frozen in liquid nitrogen.
Contractility testing.
Functional analysis of muscle strips was performed as described previously (14, 34). Briefly, bladder SM strips were prepared by removing the mucosa under stereomicroscope vision. Bladder tissue was attached to a force transducer (Grass Technologies), suspended in Krebs solution in an organ bath, and maintained at 37°C to equilibrate for 1 hour bubbled with a mixture of 95% O2 and 5% CO2 under a force of 0.5 grams. Contractile responses to carbachol (1 nM to 10 μM), 120 mM KCl, 10 μM α,β-methyl-ATP, and to EFS (1 to 64 Hz, 40 V, 0.5-ms pulse width, 10-second duration) were measured. Data were calculated as force (millinewtons) normalized by tissue cross-sectional area and are expressed as means ± SEM.
Immunoblot analysis.
Total protein was extracted from frozen bladder tissue using Fast-Prep matrix beads (MP Biomedicals) in 300 to 500 μl cell lysis extraction buffer, which contained 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM NaPPi, 1 mM β-glycerophosphate, 1 mM Na3VO4, and protease inhibitors. Protein concentration was determined using the MicroBCA protein assay (ThermoFisher Scientific). Equal amounts of total protein (5–20 μg) were resolved in 10% SDS-PAGE gels, electrotransferred to nitrocellulose membrane, and protein transfer verified by Ponceau S stain. After rinsing in PBS/Tween (phosphate-buffered saline, 0.05% Tween-20), and then blocking with 10% fat-free milk in PBS/Tween for 1 hour, membranes were incubated with primary antibodies overnight at 4°C, including rabbit monoclonal anti-Nrp2 antibody (D39A5, 1:1,000 dilution; catalog 3366, Cell Signaling Technology) or mouse monoclonal anti–β-actin antibody (AC-15, 1:10,000 dilution; catalog A5441, Sigma-Aldrich). Membranes were washed 3 times for 15 minutes each in PBS/Tween prior to incubation with HRP-conjugated species-specific secondary antibodies (HRP-anti-rabbit IgG, catalog 31460 and HRP-anti-mouse IgG, catalog 31430; ThermoFisher Scientific) for 1 hour at room temperature. After membranes were washed in PBS/Tween, target proteins were detected using enhanced chemiluminescence (Pierce SuperSignal West Pico chemiluminescent substrate; ThermoFisher Scientific) and signals were visualized after exposure of membranes to x-ray film.
Immunostaining.
Paraffin-embedded adult human normal bladder tissue sections (catalog T2234010) were purchased from Biochain. Sections were dewaxed in xylene and rehydrated through a graded series of alcohols to water. Tissues were incubated sequentially in proteinase K (10 μg/ml) for antigen retrieval, 3% H2O2 in methanol to block endogenous peroxidase, and in protein blocking buffer (Tris-NaCl-Blocking, Perkin Elmer) to block endogenous proteins. Antibody staining included a rabbit Prestige anti–human NRP2 polyclonal antibody (catalog HPA039980, Sigma-Aldrich), validated by the Human Protein Atlas project, and mouse monoclonal anti–human SMA antibody (clone 1A4, Biocare). Primary antibodies were followed by biotinylated anti-rabbit or anti-mouse secondary antibodies (Vector Laboratories) and horseradish peroxidase-conjugated avidin (Vectastain Elite ABC kit, PK-6100; Vector Laboratories). Antibody staining was visualized with a 3,3′-diaminobenzidine peroxidase chromogenic substrate kit (SK-4100; Vector Laboratories) and counterstained with Mayer’s hematoxylin (Sigma-Aldrich). Control specimens exposed to each secondary antibody alone showed no specific staining. For determination of tissue specificity of Cre recombinase activity following breeding of SM22α-CreERT2 and ROSA26 LacZ reporter mice, whole organs and frozen sections of tissues were stained with X-gal according to standard protocols. To better illustrate X-gal staining in images of frozen sections, input signal levels were adjusted to 0, 0.70, 220. Output signal levels were maintained at 0, 255.
Determination of detrusor contractility in human specimens.
Contractility analysis was performed on a subset of patients undergoing treatment at the University Hospital Bern for management of lower urinary tract dysfunction, from whom urodynamics data and bladder biopsies were obtained. Patients in urinary retention were excluded from analysis. In addition, bladder biopsies were obtained from asymptomatic patients undergoing cystoscopy for kidney stones. Tissue was used for isolation of RNA, expression profiling, and quantitative RT-PCR as described (35). Bladder outlet obstruction (BOO) in this subset of patients was defined from pressure-flow studies as the BOO index (BOOI), calculated as Pdet Qmax – 2Qmax (where Pdet Qmax is detrusor pressure at the peak flow rate and Qmax is peak flow rate), greater than 40 cmH2O (36). Bladder contractility was quantified as the detrusor contractility index (DCI) determined from the formula Pdet Qmax + 5Qmax (37). For this analysis, bladder compensation was defined as DCI greater than 110 cmH2O, with voiding efficiency greater than 40% for patients with moderate-to-severe outlet obstruction, and DCI greater than 80 cmH2O, with voiding efficiency greater than 40% for patients with mild obstruction. NRP2 mRNA levels in obstructed patients were expressed relative to those in asymptomatic controls.
Statistics.
Differences in contractile responses among groups were determined by ANOVA followed by a Holm-Sidak multiple comparison test, or by the 2-tailed Mann-Whitney U test. P less than 0.05 was considered significant.
Study approval.
All animal studies were performed with approval from the Boston Children’s Hospital Animal Care and Use Committee and with strict adherence to US Public Health Service and Office of Laboratory Animal Welfare guidelines. Approval to perform human urodynamics analyses and bladder biopsies was granted from the local Ethics Committee, University Hospital Bern, Bern, Switzerland (KEK 146/05), and all subjects provided written informed consent.
Author contributions
EV performed animal surgeries, tissue harvest, and immunoblot analysis. VC performed tension testing. SL assisted with animal husbandry and surgeries, and tissue harvest. FCB performed urodynamics analysis in patients. AHG analyzed RNA expression data. KM performed RNA expression profiling and data analysis. DRB performed tissue staining, contributed to experimental design, and contributed to writing the manuscript. MPS analyzed tension data, contributed to experimental design, and contributed to writing the manuscript. RMA conceived of and coordinated the study, contributed to experimental design, performed immunoblot analysis, and contributed to writing the manuscript.
Supplementary Material
Acknowledgments
The authors would like to thank members of the Urological Diseases Research Center at Boston Children’s Hospital for helpful discussions. The authors acknowledge grant support from the NIH (R01 DK077195 to RMA, R01 DK104641 to RMA and DRB, P50 DK65298 to RMA and DRB) and the Department of Veterans Affairs Medical Research Service (BX001790 to MPS).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2017;2(3):e90617. doi:10.1172/jci.insight.90617.
Contributor Information
Evalynn Vasquez, Email: evalynn.vasquez@gmail.com.
Vivian Cristofaro, Email: Vivian_Cristofaro@hms.harvard.edu.
Stefan Lukianov, Email: Stefan.Lukianov@childrens.harvard.edu.
Ali Hashemi Gheinani, Email: ali.hashemi@dkf.unibe.ch.
Katia Monastyrskaya, Email: monastyk@dkf.unibe.ch.
Diane R. Bielenberg, Email: diane.bielenberg@childrens.harvard.edu.
References
- 1.Drake MJ, Williams J, Bijos DA. Voiding dysfunction due to detrusor underactivity: an overview. Nat Rev Urol. 2014;11(8):454–464. doi: 10.1038/nrurol.2014.156. [DOI] [PubMed] [Google Scholar]
- 2.Zderic SA, Chacko S. Alterations in the contractile phenotype of the bladder: lessons for understanding physiological and pathological remodelling of smooth muscle. J Cell Mol Med. 2012;16(2):203–217. doi: 10.1111/j.1582-4934.2011.01368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osman NI, et al. Detrusor underactivity and the underactive bladder: a new clinical entity? A review of current terminology, definitions, epidemiology, aetiology, and diagnosis. Eur Urol. 2014;65(2):389–398. doi: 10.1016/j.eururo.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Barendrecht MM, Oelke M, Laguna MP, Michel MC. Is the use of parasympathomimetics for treating an underactive urinary bladder evidence-based? BJU Int. 2007;99(4):749–752. doi: 10.1111/j.1464-410X.2006.06742.x. [DOI] [PubMed] [Google Scholar]
- 5.Lemack GE, Burkhard F, Zimmern PE, McConnell JD, Lin VK. Physiologic sequelae of partial infravesical obstruction in the mouse: role of inducible nitric oxide synthase. J Urol. 1999;161(3):1015–1022. doi: 10.1016/S0022-5347(01)61838-X. [DOI] [PubMed] [Google Scholar]
- 6.Austin JC, Chacko SK, DiSanto M, Canning DA, Zderic SA. A male murine model of partial bladder outlet obstruction reveals changes in detrusor morphology, contractility and myosin isoform expression. J Urol. 2004;172(4 Pt 1):1524–1528. doi: 10.1097/01.ju.0000138045.61378.96. [DOI] [PubMed] [Google Scholar]
- 7.Metcalfe PD, et al. Bladder outlet obstruction: progression from inflammation to fibrosis. BJU Int. 2010;106(11):1686–1694. doi: 10.1111/j.1464-410X.2010.09445.x. [DOI] [PubMed] [Google Scholar]
- 8.Lemack GE, Zimmern PE, Vazquez D, Connell JD, Lin VK. Altered response to partial bladder outlet obstruction in mice lacking inducible nitric oxide synthase. J Urol. 2000;163(6):1981–1987. doi: 10.1016/S0022-5347(05)67614-8. [DOI] [PubMed] [Google Scholar]
- 9.Felsen D, et al. Inducible nitric oxide synthase promotes pathophysiological consequences of experimental bladder outlet obstruction. J Urol. 2003;169(4):1569–1572. doi: 10.1097/01.ju.0000054885.51858.99. [DOI] [PubMed] [Google Scholar]
- 10.Schröder A, Newgreen D, Andersson KE. Detrusor responses to prostaglandin E2 and bladder outlet obstruction in wild-type and Ep1 receptor knockout mice. J Urol. 2004;172(3):1166–1170. doi: 10.1097/01.ju.0000134186.58854.2c. [DOI] [PubMed] [Google Scholar]
- 11.Taylor JA, et al. Null mutation in macrophage migration inhibitory factor prevents muscle cell loss and fibrosis in partial bladder outlet obstruction. Am J Physiol Renal Physiol. 2006;291(6):F1343–F1353. doi: 10.1152/ajprenal.00144.2006. [DOI] [PubMed] [Google Scholar]
- 12.Lassmann J, Sliwoski J, Chang A, Canning DA, Zderic SA. Deletion of one SERCA2 allele confers protection against bladder wall hypertrophy in a murine model of partial bladder outlet obstruction. Am J Physiol Regul Integr Comp Physiol. 2008;294(1):R58–R65. doi: 10.1152/ajpregu.00477.2007. [DOI] [PubMed] [Google Scholar]
- 13.Anumanthan G, et al. Bladder stromal loss of transforming growth factor receptor II decreases fibrosis after bladder obstruction. J Urol. 2009;182(4 Suppl):1775–1780. doi: 10.1016/j.juro.2009.05.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bielenberg DR, et al. Increased smooth muscle contractility in mice deficient for neuropilin 2. Am J Pathol. 2012;181(2):548–559. doi: 10.1016/j.ajpath.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012;2(7):a006502. doi: 10.1101/cshperspect.a006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu A, et al. ABL2/ARG tyrosine kinase mediates SEMA3F-induced RhoA inactivation and cytoskeleton collapse in human glioma cells. J Biol Chem. 2008;283(40):27230–27238. doi: 10.1074/jbc.M804520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakayama H, Bruneau S, Kochupurakkal N, Coma S, Briscoe DM, Klagsbrun M. Regulation of mTOR signaling by semaphorin 3F-neuropilin 2 interactions in vitro and in vivo. Sci Rep. 2015;5:11789. doi: 10.1038/srep11789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasarre P, Gemmill RM, Drabkin HA. The emerging role of class-3 semaphorins and their neuropilin receptors in oncology. Onco Targets Ther. 2014;7:1663–1687. doi: 10.2147/OTT.S37744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bielenberg DR, Shimizu A, Klagsbrun M. Semaphorin-induced cytoskeletal collapse and repulsion of endothelial cells. Meth Enzymol. 2008;443:299–314. doi: 10.1016/S0076-6879(08)02015-6. [DOI] [PubMed] [Google Scholar]
- 20.Walz A, Rodriguez I, Mombaerts P. Aberrant sensory innervation of the olfactory bulb in neuropilin-2 mutant mice. J Neurosci. 2002;22(10):4025–4035. doi: 10.1523/JNEUROSCI.22-10-04025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramachandran A, Ranpura SA, Gong EM, Mulone M, Cannon GM, Adam RM. An Akt- and Fra-1-dependent pathway mediates platelet-derived growth factor-induced expression of thrombomodulin, a novel regulator of smooth muscle cell migration. Am J Pathol. 2010;177(1):119–131. doi: 10.2353/ajpath.2010.090772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghafar MA, et al. Effects of chronic partial outlet obstruction on blood flow and oxygenation of the rat bladder. J Urol. 2002;167(3):1508–1512. doi: 10.1016/S0022-5347(05)65354-2. [DOI] [PubMed] [Google Scholar]
- 23.Ghafar MA, et al. Hypoxia and an angiogenic response in the partially obstructed rat bladder. Lab Invest. 2002;82(7):903–909. doi: 10.1097/01.LAB.0000021135.87203.92. [DOI] [PubMed] [Google Scholar]
- 24.Iguchi N, Malykhina AP, Wilcox DT. Inhibition of HIF reduces bladder hypertrophy and improves bladder function in murine model of partial bladder outlet obstruction. J Urol. 2016;195(4 Pt 2):1250–1256. doi: 10.1016/j.juro.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coma S, Shimizu A, Klagsbrun M. Hypoxia induces tumor and endothelial cell migration in a semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor neuropilin 2. Cell Adh Migr. 2011;5(3):266–275. doi: 10.4161/cam.5.3.16294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 27.Boopathi E, et al. Mechanical stretch upregulates proteins involved in Ca2+ sensitization in urinary bladder smooth muscle hypertrophy. Am J Physiol, Cell Physiol. 2014;307(6):C542–C553. doi: 10.1152/ajpcell.00033.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chancellor MB, Kaufman J. Case for pharmacotherapy development for underactive bladder. Urology. 2008;72(5):966–967. doi: 10.1016/j.urology.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 29.Morita T, et al. Effects of prostaglandins E1, E2 and F2 alpha on contractility and cAMP and cGMP contents in lower urinary tract smooth muscle. Urol Int. 1994;52(4):200–203. doi: 10.1159/000282608. [DOI] [PubMed] [Google Scholar]
- 30.Hindley RG, Brierly RD, Thomas PJ. Prostaglandin E2 and bethanechol in combination for treating detrusor underactivity. BJU Int. 2004;93(1):89–92. doi: 10.1111/j.1464-410X.2004.04563.x. [DOI] [PubMed] [Google Scholar]
- 31.Parker MW, Vander Kooi CW. Microplate-based screening for small molecule inhibitors of neuropilin-2/vascular endothelial growth factor-C interactions. Anal Biochem. 2014;453:4–6. doi: 10.1016/j.ab.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caunt M, et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell. 2008;13(4):331–342. doi: 10.1016/j.ccr.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 33.Kühbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis. 2000;28(1):15–22. doi: 10.1002/1526-968X(200009)28:1<15::AID-GENE20>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 34.Cristofaro V, Peters CA, Yalla SV, Sullivan MP. Smooth muscle caveolae differentially regulate specific agonist induced bladder contractions. Neurourol Urodyn. 2007;26(1):71–80. doi: 10.1002/nau.20361. [DOI] [PubMed] [Google Scholar]
- 35.Gheinani AH, et al. Molecular biomarkers and hubs of signaling activity identified by integrated mRNA-miRNA profiling of outlet obstruction-induced bladder dysfunction. JCI Insight. 2017;2(2):e89560. doi: 10.1172/jci.insight.89560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffiths D, Höfner K, van Mastrigt R, Rollema HJ, Spångberg A, Gleason D. Standardization of terminology of lower urinary tract function: pressure-flow studies of voiding, urethral resistance, and urethral obstruction. International Continence Society Subcommittee on Standardization of Terminology of Pressure-Flow Studies. Neurourol Urodyn. 1997;16(1):1–18. doi: 10.1002/(SICI)1520-6777(1997)16:1<1::AID-NAU1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 37.van Koeveringe GA, Vahabi B, Andersson KE, Kirschner-Herrmans R, Oelke M. Detrusor underactivity: a plea for new approaches to a common bladder dysfunction. Neurourol Urodyn. 2011;30(5):723–728. doi: 10.1002/nau.21097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.