

Abstract
Pathogen-specific polyfunctional T cell responses have been associated with favorable clinical outcomes, but it is not known whether molecular differences exist between polyfunctional and monofunctional cytokine-producing T cells. Here, we report that polyfunctional CD4+ T cells induced during Plasmodium falciparum (P. falciparum) blood-stage infection in humans have a unique transcriptomic profile compared with IFN-γ monofunctional CD4+ T cells and, thus, are molecularly distinct. The 14-gene signature revealed in P. falciparum–reactive polyfunctional T cells is associated with cytokine signaling and lymphocyte chemotaxis, and systems biology analysis identified IL-27 as an upstream regulator of the polyfunctional gene signature. Importantly, the polyfunctional gene signature is largely conserved in Influenza-reactive polyfunctional CD4+ T cells, suggesting that polyfunctional T cells have core characteristics independent of pathogen specificity. This study provides the first evidence to our knowledge that consistent molecular differences exist between polyfunctional and monofunctional CD4+ T cells.
Introduction
T cells contribute to cell-mediated immunity by exerting pleiotropic effects, including helper functions mediated through the production of cytokines. Some T cells produce only one cytokine (monofunctional T cells), while others can simultaneously produce multiple cytokines (polyfunctional T cells) (1). Polyfunctional T cells that simultaneously produce the key Th1 cytokines IFN-γ, IL-2, and TNF-α have been reported following infection or immunization in a number of infectious diseases states including Leishmania major (2), HIV (3, 4), Mycobacterium tuberculosis (5), and Plasmodium (6–10).
Th1 polyfunctional T cells have been associated with favorable disease outcomes (3) and higher protective efficacy after vaccination (2, 5, 11) as compared with IFN-γ monofunctional T cells. More recently, polyfunctional T cells have also been associated with higher effector function compared with monofunctional T cells (12). Thus, T cell polyfunctionality is commonly reported in immunogenicity studies (1). However, there has been no study of polyfunctional T cells at the molecular level, and indeed it remains unknown (apart from a snapshot of their cytokine expression) whether any differences exist that actually distinguish polyfunctional from monofunctional T cells. Additionally, the mechanisms driving their generation are not understood.
The molecular study of polyfunctional T cells has been hampered by technical challenges related to their very low frequency in the peripheral circulation. Furthermore, polyfunctional T cells have been identified using only intracellular cytokine staining, a process that requires cell fixation with pronounced negative effects on RNA quality, thus precluding transcriptomic analysis of the target cell populations. To address this, we developed a 3-color polyfunctional cytokine secretion assay to detect and isolate viable polyfunctional T cells directly from whole blood samples after in vitro stimulation (13). Herein, we successfully applied this assay to isolate and compare the molecular profile of polyfunctional CD4+ T cells and monofunctional CD4+ T cells recovered from human volunteers experimentally infected with Plasmodium falciparum (P. falciparum) and identified for the first time to our knowledge a polyfunctional gene signature that was also conserved in Influenza-specific polyfunctional CD4+ T cells.
Results
Parasite dose-dependent generation of polyfunctional T cells during experimental P. falciparum infection.
The frequency of circulating CD4+ T cells producing different permutations of the 3 key Th1 cytokines — IFN-γ, IL-2, and TNF-α — in response to antigen-specific restimulation over the time course of infection in 19 adult volunteers experimentally infected with P. falciparum–infected erythrocytes was evaluated using multi-parameter flow cytometry (Figure 1A and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.87499DS1). The cytokine combinations produced by CD4+ T cells were dynamically regulated over the time course of infection. While there was minimal change in overall frequencies of antigen-reactive cytokine-producing CD4+ T cells at 7 days after infection (Figure 1A), the frequencies of single positive IFN-γ– (P = 0.050), double positive IFN-γ/TNF-α– (P = 0.004), and triple positive IFN-γ/IL-2/TNF-α–producing cells (P = 0.009) were significantly higher at 4 weeks after infection compared with before infection (Figure 1A). Overall, there was a shift toward a higher polyfunctionality in cytokine-producing CD4+ T cells at 4 weeks after infection compared with 1 week after infection (Figure 1B). IFN-γ was the major cytokine produced at 4 weeks after infection, representing more than 75% of the total cytokine response compared with IL-2 and TNF-α (Figure 1C).
Figure 1. Polyfunctionality of P.

falciparum–reactive CD4+ T cells induced duringP. falciparumblood-stage experimental infection in malaria-naive volunteers. Cytokine production in CD4+ T cells was measured by flow cytometry and intracellular cytokine staining after in vitro stimulation of whole blood samples with Pf prbc parasite antigen extract overnight. (A) Frequency and (B) polyfunctionality index (40) of IFN-γ, IL-2, and TNF-α single (1+), double (2+), or triple positive (3+) CD4+ T cells collected before infection (D0), 1 week after infection (D7), and 4 weeks after infection (D28). (C) Distribution of total cytokine positive cells at 4 weeks after infection according to their polyfunctionality. Frequencies were corrected for antigen nonspecific cytokine production. Data represent means from 19 volunteers from 3 independent cohorts. **P < 0.01, *P < 0.05 (Wilcoxon test).
Next, we explored the relationship between T cell polyfunctionality and clinical outcome by correlating the expansion of polyfunctional or IFN-γ monofunctional CD4+ T cells induced by infection with the blood-stage parasite burden (determined as area under the parasitemia curve until antimalarial treatment at approximately day 7 after infection) in each individual. No significant association was observed between parasite burden and the absolute frequency of IFN-γ single positive or triple positive IFN-γ/IL-2/TNF-α CD4+ T cells circulating at 4 weeks after infection (Figure 2A, Spearman’s correlation coefficient r = –0.05, P = 0.83 for IFN-γ single positive IFN-γ/IL-2/TNF-α and r = –0.44, P = 0.06 for IFN-γ triple positive IFN-γ/IL-2/TNF-α, respectively). When quantifying the cytokine response specifically induced upon infection only (i.e., the fold change frequency between pre-infection and 4 weeks after infection), no association was noted between IFN-γ single positive CD4+ T cells and parasite burden (Figure 2B, Spearman’s correlation coefficient r = -0.15, P = 0.54), whereas there was a highly significant positive correlation between the fold change in frequency of triple positive IFN-γ/IL-2/TNF-α CD4+ T cells and parasite burden (Figure 2B, Spearman’s correlation coefficient r = 0.60, P = 0.0064). Taken together, these results suggest that the amplitude of the induction of polyfunctional T cells upon infection rather than their absolute frequency in the peripheral blood after infection might be a hallmark of protection in P. falciparum blood-stage infection.
Figure 2. Parasite burden is positively correlated with P. falciparum–reactive polyfunctional CD4+ T cells induced during P. falciparum blood-stage experimental infection in malaria-naive volunteers.
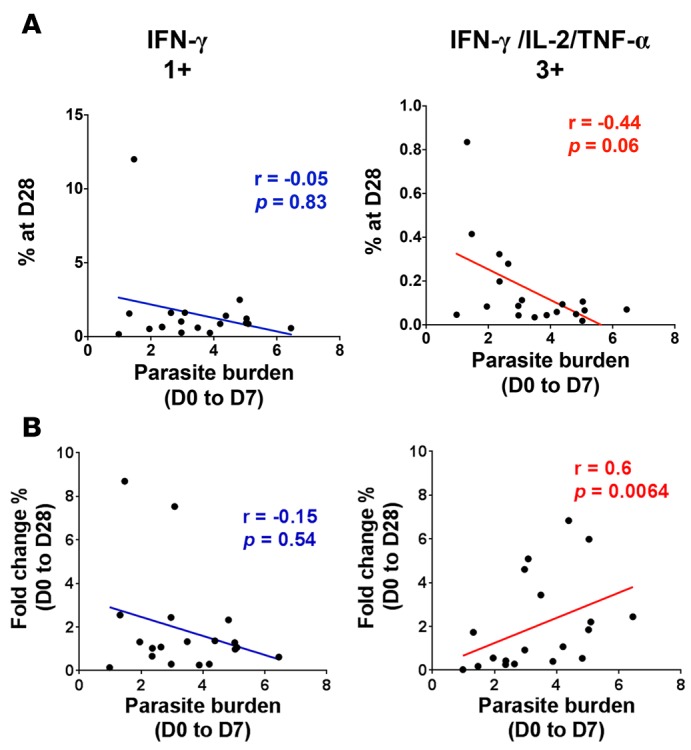
Cytokine production in CD4+ T cells was measured by flow cytometry and intracellular cytokine staining after in vitro stimulation of whole blood samples with Pf prbc parasite antigen extract overnight. Correlation between parasite burden during the first 7 days of infection and (A) absolute frequencies at 4 weeks after infection and (B) fold change between pre-infection (D0) and 4 weeks after infection (D28) in triple positive (3+) and IFN-γ single positive (1+) CD4+ T cell frequencies (nonparametric Spearman’s correlation test). Data represent mean from 19 volunteers from 3 independent cohorts.
Phenotypic differences between P. falciparum–reactive polyfunctional and IFN-γ monofunctional CD4+ T cells.
We then investigated the phenotypic differences between polyfunctional and monofunctional CD4+ T cells by flow cytometry. Triple positive IFN-γ/IL-2/TNF-α CD4+ T cells expressed significantly higher amounts of cytokine per cell as compared with the respective single positive IFN-γ, IL-2, or TNF-α CD4+ T cells (Figure 3A), consistent with observations in other disease models (2, 4, 6). Triple positive CD4+ T cells also had significantly higher expression of the differentiation marker for antigen-experienced T cells, KLRG1 (14), when compared with single positive CD4+ T cells (Figure 3B); these data support the hypothesis that polyfunctional CD4+ T cells are in a distinct differentiation state compared with monofunctional CD4+ T cells, and thus that other significant molecular differences might exist between these two cell populations.
Figure 3. Phenotypic differences in P. falciparum–reactive polyfunctional and monofunctional CD4+ T cells induced during P. falciparum blood-stage experimental infection in malaria-naive volunteers.

Cytokine production in CD4+ T cells was measured by flow cytometry and intracellular cytokine staining after in vitro stimulation of whole blood samples with Pf prbc parasite antigen extract overnight. (A) Comparison of the mean fluorescence intensities for cytokines IFN-γ, IL-2, or TNF-α between triple positive (3+) and IFN-γ single positive (IFN-γ 1+), IL-2 single positive (IL-2 1+), or TNF-α single positive (TNF-α 1+) CD4+ T cells, respectively, matched by volunteers. (B) KLRG1 expression in triple positive (3+) and IFN-γ, IL-2, or TNF single positive (1+) CD4+ T cells circulating 4 weeks after infection. Data represent mean from (A) 19 volunteers from 3 independent cohorts and (B) 8 volunteers from 2 independent cohorts. ****P < 0.0001 (Wilcoxon test). Error bars indicate ±SD.
Transcriptomic differences between P. falciparum–reactive polyfunctional and IFN-γ monofunctional CD4+ T cells.
In order to establish whether differences exist at the mRNA level between Th1 polyfunctional and monofunctional CD4+ T cells, we compared their transcriptomic profile by microarray analysis using the Affymetrix Human Gene ST 2.0 gene array. We applied our newly developed polyfunctional cytokine–secreting assay (13) to sort IFN-γ negative, IFN-γ single positive, and IFN-γ/IL-2/TNF-α triple positive CD4+ T cells from whole blood samples collected 4 weeks after infection with P. falciparum (Supplemental Figure 2). Two independent studies were performed with pooled mRNA samples comprising 4 different volunteers each. Higher expression of IFN-γ, IL-2, and TNF-α was detected in triple positive CD4+ T cells compared with IFN-γ negative CD4+ T cells (Supplemental Figure 3). IFN-γ triple positive CD4+ T cells also had a higher expression of TNF-α and IL-2, but they had similar expression of IFN-γ compared with IFN-γ single positive CD4+ T cells (Supplemental Figure 3). Taken together, these results validated the cytokine secretion assay staining and sorting strategy. Interestingly, IFN-γ single positive cells had higher levels of TNF-α mRNA compared with IFN-γ negative cells, even if this cytokine could not be detected at the protein level. This observation is likely to reflect a recent antigen-specific activation of IFN-γ single positive CD4+ T cells but not of IFN-γ negative CD4+ T cells, as TCR ligation has been shown to trigger the splicing of TNF-α pre-mRNA into mRNA (15).
Comparative analysis of the gene expression profiles of triple positive versus IFN-γ single positive CD4+ T cells identified 61 genes that were differentially regulated in both independent microarray experiments (Figure 4A and Supplemental Table 1). The majority of these 61 genes (n = 44, 72%) were overexpressed in triple positive cells. These genes coded for proteins associated with cytokines/chemokines (n = 12), lymphocyte activation (n = 14), lymphocyte trafficking (n = 4), immunoglobulins (n = 8), T cell receptor (n = 8), and metabolism (n = 4) (Figure 4B) and were significantly associated with immune response (P = 1.26 × 10–5), inflammatory response (P = 7.89 × 10–4), cell motility (P = 1.65 × 10–3), lymphocyte migration (P = 1.10 × 10–3), chemotaxis (P = 2.60 × 10–5), or cytokine/chemokine activity (P = 2.45 × 10–3 and P = 3.55 × 10–2, respectively) (Figure 4C).
Figure 4. Gene expression profile of P. falciparum–reactive IFN-γ single positive versus IFN-γ/IL-2/TNF-α triple positive CD4+ T cells, determined by microarray analysis.

Whole blood was collected 4 weeks after infection with P. falciparum and in vitro stimulated with Pf prbc parasite antigen extract overnight, and cytokine-producing CD4+ T cells were sorted using a 3-color polyfunctional cytokine secretion assay. Gene expression of mRNA pools from sorted IFN-γ single positive (1+) CD4+ T cell and IFN-γ/IL-2/TNF-α triple positive (3+) CD4+ T cell populations was determined using an Affymetrix Human Gene ST 2.0 gene array. (A) Differentially expressed genes (fold change >2) between IFN-γ single positive CD4+ T cell and IFN-γ/IL-2/TNF-α triple positive CD4+ T cell populations. (B) Gene category of differentially expressed genes between IFN-γ single positive and IFN-γ/IL-2/TNF-α triple positive CD4+ T cells determined using GO criteria. (C) GO functions and GO processes significantly enriched within the set of differentially expressed genes determined using the STRING database (corrected P < 0.01). Data derived from 2 independent microarray experiments with 4 independent volunteers per array.
To confirm the existence of a specific transcriptomic signature in polyfunctional CD4+ T cells, we validated the expression of 37 of the differentially expressed genes identified by the Affymetrix microarray analysis using multiplex quantitative PCR (qPCR) on sorted cells isolated from 5 volunteers experimentally infected with P. falciparum who were not included in the primary analysis. Fourteen genes were consistently upregulated in triple positive compared with IFN-γ single positive CD4+ T cells in all 5 volunteers (P < 0.05, Figure 5, left panels, A and B). This polyfunctional gene signature included genes coding for cytokines and chemokines (IL-2, TNF, IL-6, IL-1B, CCL8), signal transduction receptors (MS4A7, IGSF6, CD163, FPR1), catabolic enzymes (PLA2G7, CTSL1, LYZ, PTGS2), and membrane transporter (AQP9).
Figure 5. Differential gene expression profile between IFN-γ single positive and IFN-γ/IL-2/TNF-α triple positive CD4+ T cells with P. falciparum or Influenza reactivity determined by multiplex qPCR.

Fold change in gene expression between IFN-γ single positive and IFN-γ/IL-2/TNF-α triple positive CD4+ T cells that were reactive to P. falciparum (left) or Influenza (right). P. falciparum–reactive CD4+ T cells were isolated after in vitro stimulation, with Pf prbc parasite antigen extract overnight of whole blood samples collected from 5 volunteers at 4 weeks following experimental infection with P. falciparum blood stage. Influenza-reactive CD4+ T cells were isolated after Influenza peptide stimulation of whole blood collected from 5 healthy volunteers at 5 weeks following immunization with seasonal Influenza trivalent vaccine. Cytokine-producing CD4+ T cells were isolated using a 3-color polyfunctional cytokine secretion assay, and the expression of 37 genes selected from the Affymetrix microarray analysis was determined by multiplex qPCR (Fluidigm Biomark HD system). Fold change in gene expression between IFN-γ single positive and IFN-γ/IL-2/TNF-α triple positive CD4+ T cells was determined by the ddCt method. (A) Genes upregulated in both P. falciparum–reactive and Influenza-reactive IFN-γ/IL-2/TNF-α triple positive CD4+ T cells. (B and C) Genes upregulated only in P. falciparum–reactive or Influenza-reactive IFN-γ/IL-2/TNF-α triple positive CD4+ T cells, respectively. (D) Genes with no consistent gene expression across individuals for both antigens.
A core polyfunctional gene signature is conserved between P. falciparum–reactive and Influenza-reactive polyfunctional CD4+ T cells.
Since polyfunctional T cell responses have been observed in several human diseases, we next investigated whether the polyfunctional gene signature identified in the P. falciparum infection model was present in other infections. We elected to explore the Influenza model, using peripheral blood collected from 5 healthy volunteers 5 weeks after immunization with the seasonal Influenza vaccine. The transcriptomic profile of sorted Influenza-reactive IFN-γ single positive and triple positive CD4+ T cells was determined by multiplex qPCR as performed for P. falciparum–reactive CD4+ T cells (Figure 5, right panels). Interestingly, 10 of the 14 genes validated as upregulated in triple positive versus IFN-γ single positive P. falciparum–reactive CD4+ T cells were also upregulated in triple positive CD4+ T cells reactive to Influenza (Figure 5A). Additionally, 4 differentially expressed genes appeared to be specific for P. falciparum–reactive polyfunctional T cells (Figure 5B), and 6 others were overexpressed only in Influenza-reactive polyfunctional CD4+ T cells (Figure 5C). The remaining 17 genes exhibited variable expression in both models (Figure 5D).
Upstream regulators of the polyfunctional CD4+ T cell gene signature.
Finally, we explored whether our newly identified polyfunctional gene signature could inform on the underlying mechanisms controlling the generation of polyfunctional CD4+ T cells in our system. The polyfunctional gene signature did not contain any genes coding for transcription factors (Figure 4A) and was significantly associated with cytokine and chemokine signaling (Figure 4C). Thus, we hypothesized that the process of differentiation of CD4+ T cells into a polyfunctional state may result from the integration of external stimuli, such as cytokine signaling, rather than internal genetic reprogramming. Bioinformatic analysis identified 3 transmembrane receptors (CD47, TNFRSF18, and CLEC7A) and 1 cytokine (IL-27) within the top 20 predicted upstream regulators of our polyfunctional gene signature (Table 1). IL-27 was the only cytokine present in this list (P = 4.86 × 10–13, Table 1), and Ingenuity Pathway Analysis showed that the expression of 8 of the 14 genes contained in the polyfunctional gene signature are regulated by IL-27, either directly (n = 6) or indirectly through STAT1 (n = 2) (Figure 6A). IL-27 is produced by antigen-presenting cells early after activation (16) and plays a key role in initiating Th1 differentiation of CD4+ T cells (17–19). In particular, IL-27 signaling has been found to regulate cytokine production by Th1 CD4+ T cells in Leishmaniasis (20), tuberculosis (21), and malaria (22–24). More recently, IL-27–producing CD4+ T cells have been directly implicated in the regulation of protective immunity to P. berghei infection in mice (25). To investigate the potential role of IL-27 in the generation of polyfunctional T cells, purified CD4+ T cells from healthy volunteers were in vitro Th1 polarized as previously described (26), in the presence or absence of recombinant IL-27. The presence of IL-27 significantly increased the frequency of triple positive CD4+ T cells (average fold change = 2.54, P = 0.0079) but not IFN-γ single positive CD4+ T cells (average fold change = 1.16, P = 0.547), nor total IFN-γ–producing CD4+ T cells (average fold change 1.21, P = 0.569) (Figure 6B).
Table 1. Top 20 upstream regulators of the polyfunctional gene signature.

Figure 6. Positive association of IL-27 with the generation of polyfunctional T cells.
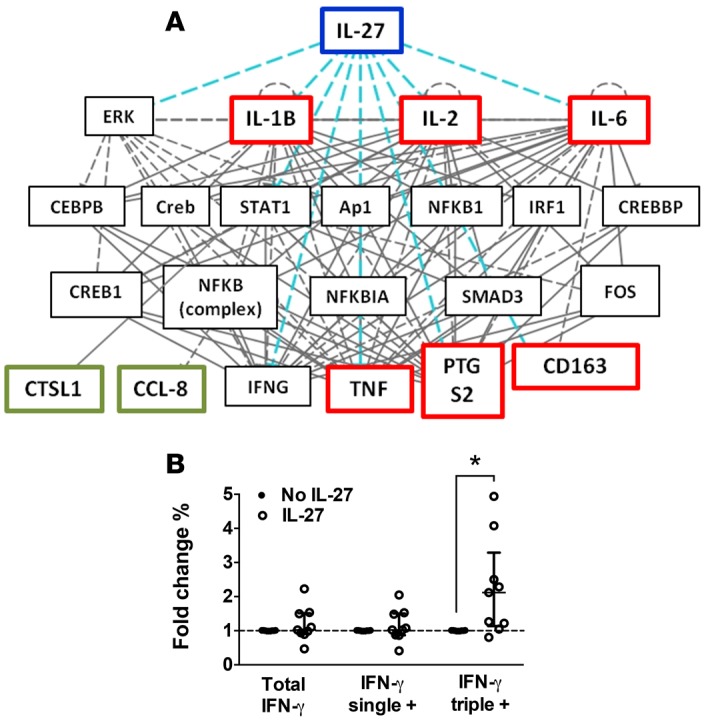
(A) Direct (red) and indirect (green) interactions between IL-27 and the genes differentially regulated between IFN-γ single positive and IFN-γ/IL-2/TNF-α triple positive CD4+ T cells determined by Ingenuity Pathway Analysis. (B) Fold change in total IFN-γ, IFN-γ single positive, and IFN-γ/IL-2/TNF-α triple positive CD4+ T cells in the presence or absence of 200 ng/ml of recombinant human IL-27 during in vitro Th1 polarization of CD4+ T cells isolated from healthy volunteers. Cytokine production was measured by intracellular cytokine staining after PMA/ionomycin restimulation for 4 hours. Graph shows mean from 8 individuals from 3 independent experiments. *P > 0.05 (Wilcoxon test). Error bars indicate ±SD.
Discussion
In this study, we present evidence that polyfunctional CD4+ T cells are induced during experimental blood-stage malaria infection in naive volunteers in a parasite dose-dependent manner and are molecularly distinct from monofunctional CD4+ T cells. This positive association between antigen load and the generation of polyfunctional IFN-γ/IL-2/TNF-α CD4+ T cells is consistent with other reports on malaria (7) and tuberculosis (27). It has been shown, however, that a very high antigen dose can drive the opposite effect, with fewer polyfunctional T cells generated (2). Thus, the amplitude of polyfunctional T cell responses appears to be tightly controlled by the antigen load. The duration of exposure might also affect T cell quality, but unfortunately this could not be assessed here, as all the volunteers in our study were exposed to P. falciparum for a similar period.
We observed that polyfunctional T cells had higher expression per cell of cytokines, as well as higher levels of KLRG1, compared with monofunctional T cells. KLRG1 expression in circulating T cells is restricted to antigen-experienced cells (14) and is, thus, often used as a marker of T cell differentiation. Therefore, the degree of cytokine polyfunctionality might reflect distinct differentiation states within T cell populations, with triple positive polyfunctional T cells more differentiated compared with double positive or monofunctional cytokine-producing T cells.
We also report the first analysis of the transcriptomic profile of polyfunctional human CD4+ T cells compared with IFN-γ monofunctional CD4+ T cells. Using a model of experimental P. falciparum infection in naive volunteers, as well as Influenza vaccination of healthy volunteers, we show that IFN-γ/IL-2/TNF-α triple positive and IFN-γ single positive CD4+ T cells exhibit unique transcriptomic profiles, providing the first evidence that molecular differences exist between cells with distinct Th1 cytokine polyfunctionality and potentially associated with distinct functions. Moreover, we show that the transcriptomic signature of polyfunctional T cells is largely conserved between pathogens.
The P. falciparum polyfunctional gene signature comprised 14 genes that were associated with immune response to infection, cytokine signaling, and lymphocyte chemotaxis; none of these genes have been previously associated with polyfunctional T cells. Many of these genes are also associated with neutrophil chemotaxis (IL-1B, CCL8, TNF-α, PTGS2, AQP9, FPR1; refs. 28–32) and activity (LYZ, CTSL1, IL-6; refs. 33–35), suggesting that polyfunctional CD4+ T cells may be involved in the recruitment and activation of neutrophils to participate in the host response to infection (36). Interestingly, the polyfunctional gene signature did not contain any genes coding for transcription factors, suggesting the process of differentiation of CD4+ T cells into a polyfunctional state may be the result of the integration of external stimuli, such as cytokine signaling, rather than internal genetic reprogramming. A previous study from Han et al. is consistent with this hypothesis, showing that the production of IFN-γ, IL-2, and TNF-α in primary human CD4+ T cells is dynamically regulated after activation through reversible epigenetic reprogramming events (37). Thus, we predict that the comparison of the epigenome of polyfunctional versus monofunctional CD4+ T cells will give useful information into the molecular mechanisms controlling the generation of polyfunctional T cells.
Importantly, the polyfunctional gene signature identified in P. falciparum–reactive polyfunctional CD4+ T cells was predominantly conserved in Influenza-reactive polyfunctional CD4+ T cells, suggesting that polyfunctional T cells have intrinsic characteristics independent of antigen specificity. This implies that the function of polyfunctional T cells and the mechanisms driving their generation might be conserved between different disease states. These data further support our proposal that polyfunctional CD4+ T cells and monofunctional CD4+ T cells are molecularly distinct and, thus, are driven by distinct transcriptional programs beyond the cytokines on which their functional profile is determined. The differential gene expression observed between P. falciparum– and Influenza-reactive polyfunctional T cells (Figure 5, B and C) might reflect prior exposure to each pathogen (all malaria volunteers were malaria naive, whereas the Influenza vaccine volunteers had been vaccinated in previous years and their natural exposure is unknown). Alternatively, in addition to a core polyfunctional gene signature common to both P. falciparum and Influenza, polyfunctional T cells may also present gene expression profiles relevant to specific pathogens. Indeed, this would be consistent with the fact that different pathogens can require distinct immune functions for their control. Finally, we cannot exclude the possibility that some of the differences may be due to the heterogeneous (polyclonal) origins of our samples. This could be addressed by using a clonal cell model such as the one used by Almeida et al. to identify determinants of CD8+ T cell polyfunctionality (38).
Previous studies have suggested that the presence of polyfunctional T cells is correlated to better disease control, leading vaccinologists to speculate on the mechanisms driving the generation of these cells. Herein, exploring upstream regulators of the polyfunctional gene signature with in silico pathway analysis, we identified IL-27 as a potential driver of polyfunctional CD4+ T cell generation. Using an in vitro accessory-cell–free model, we found that exogenous IL-27 preferentially drove the generation of polyfunctional CD4+ T cells over IFN-γ monofunctional CD4+ T cells. These findings give valuable preliminary insights into the molecular mechanisms driving the generation of polyfunctional T cells. IL-27 is produced by antigen-presenting cells early after activation (16) and plays a key role in initiating Th1 differentiation of CD4+ T cells (17-19). In particular, IL-27 signaling has been found to regulate cytokine production by Th1 CD4+ T cells (22–24), and more recently, IL-27–producing CD4+ T cells have been implicated in the regulation of protective immunity in mouse malaria (25). However, our study is the first to associate IL-27 with the generation of polyfunctional CD4+ T cells. Future studies using in vivo models will be necessary to confirm this hypothesis.
The present study was limited to the study of polyfunctional CD4+ T cells. No induction of polyfunctional CD8+ T cells was observed in our P. falciparum blood-stage experimental infection model (data not shown), but it would be of interest to investigate whether the polyfunctional gene signature we identified herein also associates with CD8+ T cells. Additionally, herein, we focused on the molecular profiling of polyfunctional T cells expressing the 3 major Th1 cytokines IFN-γ, IL-2, and TNF-α. However, T cells are able to produce a multitude of other cytokines, so it would be important to determine whether our Th1 polyfunctional gene signature is conserved across T cells with distinct cytokine polyfunctionality.
In conclusion, this study provides direct evidence that T cells with distinct Th1 cytokine polyfunctionality present unique molecular profiles. Using a newly developed assay to isolate viable polyfunctional T cells (13), we identified a core transcriptomic signature in Th1 polyfunctional CD4+ T cells associated with immune activation, cytokine signaling, and lymphocyte chemotaxis that appears to be largely pathogen independent, supporting an important role for these cells in immune defense. This study also provides a methodological framework that could be applied to study any T cell subset of a given cytokine polyfunctionality and antigen specificity. Taken together, our results provide a unique foundation for further studies on polyfunctional T cells, which have been implicated in protective immunity against bacterial, viral, and parasitic pathogens.
Methods
Study design.
Healthy malaria-naive individuals were i.v. inoculated with 1,800 viable P. falciparum 3D7 parasitized erythrocytes and treated with antimalarial drugs at 7–8 days after infection. Inoculum preparation, volunteer recruitment, infection, monitoring, and treatment were performed as described previously (39). Blood samples were collected prior to infection, 7 days after infection, and 4 weeks after infection (corresponding to 3 weeks after treatment). Additionally, we collected blood samples from 5 healthy volunteers 5 weeks after immunization with the seasonal Influenza trivalent vaccine (Fluvax 2014 or Fluvax 2015, Seqirus) under informed consent and approval of the QIMRB-Human Research Ethics Committee. Peripheral blood collected in Lithium Heparin vacutainers (BD Biosciences) was used directly for in vitro stimulation assays, or PBMCs were isolated using standard Ficoll density gradient centrifugation.
Determination of parasite-level kinetics and overall parasite burden.
Parasite levels over the time course of infection were determined using a consensus P. falciparum species-specific qPCR assay as previously described (39). Parasite levels were assessed once daily until day 4 after infection and then twice daily until treatment. All samples were batch tested in triplicate together after each study completion. The total parasite burden within the first 7 days of infection was defined as the AUC of the transformed parasite levels over the time course of infection using the trapezoidal rule. The limit of detection by qPCR was 64 parasites/ml (39).
Generation of a P. falciparum–infected rbc extract (Pf rbc).
Red blood cells and plasma from O+ blood group human donors were obtained from the Australian Red Cross. P. falciparum 3D7 from the same GMP culture used for the in vivo experimental infection studies was cultured at 5% haematocrit in complete media (RPMI 1640 containing phenol red supplemented with 10% plasma and 50 mg/l Gentamicin; Sigma-Aldrich) in 60 mm × 15 mm petri dishes with a working volume of 10 ml. In order to mimic the in vivo anaerobic conditions for optimal parasite growth, petri dishes were stacked into a hermetic chamber that was filled daily with a gas mixture containing 2% O2, 5.5% CO2, and 92.5% N2. Used media was removed by decanting, and fresh media was added to the culture every day. Fresh autologous rbcs were added every 2–3 days. Parasitemia was determined daily with a thin smear and Diff Quick staining kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The culture was expanded for 10 days and stopped once the parasitemia reached 4%. The parasitized rbcs were isolated from the whole culture using magnetic sorting (CS column and VarioMACS; both from Miltenyi Biotec) according to the manufacturer’s instructions. The concentration of packed rbc (prbc) in the final eluate was 60%. Cells were adjusted to 2 × 108 prbc/ml, diluted 1:2 with 30% Glycerol in PBS (for a final concentration of 1 × 108 prbc/ml), and aliquoted into cryovials before storage into liquid nitrogen.
Intracellular cytokine staining.
Whole blood (300 μl) was diluted 2:1 (v/v) in RPMI 1640 containing 25 mM Hepes (Thermo Fisher Scientific) and 2 mM L-glutamine (Invitrogen), and it was supplemented with 10 units/ml of Penicillin (Invitrogen) and 10 μg/ml of Streptomycin (Invitrogen). It was cultured in polypropylene sterile capped tubes, in the presence of Pf rbc at 1 × 106 prbc/ml or equivalent cell number of uninfected rbc extract, along with costimulatory antibodies anti–human CD28 and anti–human CD49d at 1 μg/ml (BD Biosciences). Cultures were incubated for 20 hours at 37°C, in an atmosphere of 5% CO2. Brefeldin A at 1 μg/ml (GolgiPlug, BD Biosciences) was then added and culture reincubated for a further 4 hours at 37°C, 5% CO2. At the end of stimulation, rbcs were lysed and lymphocytes were fixed simultaneously with BD Lyse/Fix solution (BD Biosciences), followed by lymphocyte permeabilization using BD Perm 2 solution (BD Biosciences) according to the manufacturer’s instructions. Cells were then stained with 50 μl of an antibody cocktail containing anti–human CD4-APC (BD Biosciences), anti–human CD8-APC-H7 (BD Biosciences), anti–human KLRG1-PE (BioLegend), anti–human IFN-γ-FITC (BD Biosciences), anti–human IL-2-AF700 (BioLegend), and anti–human TNF-PE-Cy7 (BD Biosciences) at previously determined optimum dilution, along with 1 μl of human Fc receptor blocking solution (Human TruStain FcX, BioLegend) for 30 minutes at room temperature. It was washed and resuspended in buffer (PBS with 0.5% BSA and 2 mM EDTA) until acquisition. Further details on antibody clones are available in Supplemental Table 2. Acquisition was performed with BD LSR Fortessa 4 (BD Biosciences) and DIVA software. Boolean gates and cytokine positive cell frequencies were determined with FlowJo. Background was determined as the frequency of cytokine positive T cells after uninfected rbc extract stimulation. Polyfunctionality index was calculated as previously described (40).
Three-color polyfunctional cytokine secretion assay.
Whole blood (3 ml) was diluted 2:1 (v/v) in RPMI 1640 containing 25 mM Hepes and 2 mM L-glutamine, and it was supplemented with 10 units/ml of penicillin (Invitrogen) and 10 μg/ml of Streptomycin (Invitrogen) and cultured in 6-well plates, in the presence of 1 × 106 prbc/ml P. falciparum–infected rbcs extract (Pf rbc) or a pool of synthetic Influenza peptides (5 μg/ml each of 9-mer peptides derived from Influenza matrix protein epitope [residues 58–66]and Influenza nucleoprotein epitope [residues 265–274] obtained from Mimotopes Pty Ltd; both peptides are known to induce CD4+ T cell responses in healthy individuals [data not shown], consistent with other reports; ref. 41) together with 1 μg/ml of costimulatory antibodies anti–human CD28 and anti–human CD49d (both BD Biosciences; antibody catalog numbers listed in Supplemental Table 2). Cultures were incubated overnight (Pf rbc stimulation) or for 6 hours (peptide stimulation) at 37°C in an atmosphere of 5% CO2. Following whole blood stimulation, the 3-color polyfunctional cytokine secretion assay was performed as previously described (13). IFN-γ-FITC, IL-2-PE, and TNF-APC cytokine secretion assay detection kits were purchased from Miltenyi Biotec. Briefly, rbcs were lysed with a hypotonic solution (1.55 M NH4Cl, 0.1 M NaHCO3 and 0.5 M EDTA adjusted to pH 7.3) for 10 minutes at room temperature under continuous rotation (0.05 g) using a rotary tube mixer. Cells were washed twice in cold wash buffer (PBS with 0.5% BSA and 1 mM EDTA), resuspended in 100 μl of cocktail containing 20 μl of each catch antibody reagent (Miltenyi Biotec) and incubated for 5 minutes on ice. Cells were resuspended in 5 ml of prewarmed media (media used for whole blood antigen stimulation supplemented with 5% AB human serum; Sigma-Aldrich) and incubated for 1 hour at 37°C under continuous rotation (0.05 g) using a rotary tube mixer. Immediately after this, cells were put on ice for 1 minute and rinsed twice with 5 volumes of cold wash buffer followed by a centrifugation at 300 g and 4°C for 10 minutes. Cells were then resuspended into 100 μl of cocktail containing 20 μl of each cytokine detection antibody reagent, together with 20 μl of anti–human CD4-V500 and 2 μl of anti–human CD8-APC-H7 antibodies (both from BD Biosciences) and 2 μl of anti–human CD14-PE Texas Red (Beckman Coulter). Further details on antibody clones are available in Supplemental Table 2. Cells were incubated for 10 minutes on ice in the dark; rinsed with 10 volumes of cold wash buffer, followed by a centrifugation at 300 g and 4°C for 10 minutes; resuspended into 500 μl of cold wash buffer; and stored at 4°C in the dark. Immediately prior to sorting, 1 μg/ml of propidium iodide (PI, Sigma-Aldrich) was added to allow for assessment of viability. Cells were sorted using a BD Aria III cell sorter (BD Biosciences) directly into 75 μl of RLT lysis buffer (Qiagen) supplemented with 1% of 2-mercaptoethanol (Sigma-Aldrich), and cell lysates were stored at –70°C. Cytokine-producing CD4+ T cells were gated as PI–/CD14–/CD8–/CD4+ cells.
Microarray analysis.
mRNA was extracted from freshly thawed cell lysates using the RNEasy micro kit (Qiagen) according to the manufacturer’s instructions and stored at –70°C. mRNA samples were sent to the Ramaciotti Centre (University of NSW, Sydney, Australia) for quality control and microarray services. Sample quality was assessed using the total RNA pico assay and the 2100 Bioanalyzer (Agilent Technologies). Pooled mRNA samples were obtained by mixing mRNA samples obtained from 4 volunteers (2 independent experiments of 4 volunteers each) and probed onto an Affymetrix Human Gene ST 2.0 gene array with preamplification. Hybridization results were analyzed using the software Gene Spring (Agilent Technologies), and data were normalized with the Robust Multi-array Average (RMA) approach (42). Fold changes in gene expression were calculated with GeneSpring version 12 (Agilent Technologies), and differentially expressed genes were defined using an absolute fold change higher than 2 as a significant cut-off. Gene Ontology (GO) enrichment analysis was determined with STRING (43). The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO; GSE93664).
High-throughput multiplex qPCR.
mRNA was extracted from freshly thawed cell lysates using the RNEasy micro kit (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized using oligo-dT and Superscript III RT (Invitrogen) according to the manufacturer’s instructions. Forward and reverse primers for each of the 37 selected genes for validation and 4 reference genes were designed using the online software Primer 3 Plus and synthesized by Integrated DNA technologies (primers sequences available in Supplemental Table 3). Specificity of the primers was verified using the melting curve analysis and by running each PCR product on a 2% agarose gel. Specific target amplification was performed with Taqman PreAmp master mix (Invitrogen) and a pool of the 41 designed primers according to the “Fast gene expression analysis using EvaGreen on the Biomark HD system” protocol (Fluidigm Corporation). qPCR was performed using SsoFast EvaGreen Supermix with Low ROX (Bio-Rad), the 48 × 48 gene expression IFC dynamic array chip, the IFC mX controller, and the Biomark HD system (all from Fluidigm Corporation) according to the “Fast gene expression analysis using EvaGreen on the Biomark HD system” protocol (Fluidigm Corporation). Cycling conditions used were: hot start 2 minutes at 95°C, 40 cycles of 95°C for 15 seconds and 60°C for 1 minute, followed by a melting curve. Ct values were determined using the Fluidigm Real-Time PCR Analysis software (Fluidigm Corporation). Fold changes were calculated using the ddCt value method (44). Hierarchical gene clustering with Pearson correlation and average-link clustering was done with Multi Experiment Viewer (http://mev.tm4.org/) (45).
In vitro generation of polyfunctional T cells.
CD4+ T cells were isolated from fresh PBMC using human anti-CD4 microbeads (Miltenyi Biotec) and the “possel” program from AutoMACS Pro separator (Miltenyi Biotec), cultured at 1 × 106 to 2 × 106 cells/ml in medium RPMI 1640 containing 25 mM Hepes, 2 mM L-glutamine supplemented with 10 units/ml of penicillin, 5% human AB serum (Sigma-Aldrich), recombinant human IL-2 and recombinant human IL-12 at 10 ng/ml (Prospec), and — when applicable — recombinant human IL-27 (Biolegend) at 200 ng/ml for 2 weeks at 37°C, 5% CO2. Cells were first stimulated for 1 week with plate-bound anti–human CD3 (BioLegend) and anti–human CD28 (BD Biosciences) antibodies at 1 μg/ml and 2 μg/ml, respectively, rested for 5 days, and restimulated for 2 days with plate-bound antibodies. Medium was changed every 2–3 days and culture split when cell concentration was higher than 5 × 106 cells/ml. After 2 weeks, cells were restimulated with PMA 5 ng/ml (Sigma-Aldrich) and Ionomycin 500 ng/ml (Sigma-Aldrich) along with Brefeldin A at 1 μg/ml (Golgi Plug, BD Biosciences) for 4 hours at 37°C, 5% CO2. Cells were then stained with surface antibodies anti–human CD4-BV510 (BioLegend), anti–human CD8-APC-H7 (BD Biosciences), and live/dead eF450 fixable dye (eBiosicence), fixed with 1% PFA and stained with intracellular antibodies anti–human IFN-γ-FITC (BD Biosciences), anti–human IL-2-AF700 (BioLegend), and anti–human TNF-PE-Cy7 (BD Biosciences) in BD Perm/Wash (BD Biosciences). Acquisition was performed using BD LSR Fortessa 4 (BD Biosciences).
Statistics.
Statistical analyses were performed using GraphPad Prism Software (version 6). Normality was assessed using D’Agostino and Pearson Omnibus normality test and showed the datasets were not normally distributed. Therefore, paired datasets were compared using the nonparametric Wilcoxon test, while unpaired datasets were compared using the nonparametric Mann Whitney test. P values less than 0.05 were considered significant. Correlation between datasets was determined using the nonparametric Spearman’s correlation test.
Study approval.
Experimental infection of malaria-naive healthy volunteers was undertaken at QPharm Pty Ltd (Brisbane, Australia; clinical trial numbers ACTRN12612000814875, ACTRN12613000565741, and ACTRN12613001040752), with written informed consent and approval of the QIMR Berghofer Medical Research Institute Human Research Ethics Committee (QIMRB-HREC).
Author contributions
JGB, SHA, and DLD designed the study experiments. JSM conducted the clinical study. JGB and PLG performed the experiments and data analysis. JGB, SHA, and DLD wrote the manuscript with input from JSM.
Supplementary Material
Acknowledgments
We thank the Q-Pharm staff who conducted the human infection studies, in particular Suzanne Elliot, Nannette Douglas, and Gem Mackenroth. We thank Katharine Trenholme, Silvana Sekuloski, and Paul Griffin for their critical role in the study management and Fiona Amante for coordinating the substudy. We also thank Paula Hall and the Flow Cytometry Facility of QIMR Berghofer Medical Research Institute for technical assistance with cell sorting. The clinical study was supported by The Medicines for Malaria Venture, and the laboratory research was supported by National Health and Medical Research Council of Australia (NHMRC) Program grant 1037304. JGB was supported by an International Research Tuition Award from the University of Queensland. DLD is supported by a Principal Research Fellowship from the NHMRC. JSM is supported by a Government of Queensland Clinical Research Fellowship.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2017;2(3):e87499. doi:10.1172/jci.insight.87499.
References
- 1.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8(4):247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 2.Darrah PA, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13(7):843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 3.Betts MR, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107(12):4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duvall MG, et al. Polyfunctional T cell responses are a hallmark of HIV-2 infection. Eur J Immunol. 2008;38(2):350–363. doi: 10.1002/eji.200737768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindenstrøm T, et al. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol. 2009;182(12):8047–8055. doi: 10.4049/jimmunol.0801592. [DOI] [PubMed] [Google Scholar]
- 6.Roestenberg M, et al. Protection against a malaria challenge by sporozoite inoculation. N Engl J Med. 2009;361(5):468–477. doi: 10.1056/NEJMoa0805832. [DOI] [PubMed] [Google Scholar]
- 7.Seder RA, et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science. 2013;341(6152):1359–1365. doi: 10.1126/science.1241800. [DOI] [PubMed] [Google Scholar]
- 8.Reyes-Sandoval A, et al. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect Immun. 2010;78(1):145–153. doi: 10.1128/IAI.00740-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ewer KJ, et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat Commun. 2013;4:2836. doi: 10.1038/ncomms3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sedegah M, et al. Sterile immunity to malaria after DNA prime/adenovirus boost immunization is associated with effector memory CD8+T cells targeting AMA1 class I epitopes. PLoS ONE. 2014;9(9):e106241. doi: 10.1371/journal.pone.0106241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin L, et al. COMPASS identifies T-cell subsets correlated with clinical outcomes. Nat Biotechnol. 2015;33(6):610–616. doi: 10.1038/nbt.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyd A, et al. Pathogen-Specific T Cell Polyfunctionality Is a Correlate of T Cell Efficacy and Immune Protection. PLoS One. 2015;10(6):e0128714. doi: 10.1371/journal.pone.0128714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burel JG, Apte SH, Doolan DL. Development of a cytokine-secreting-based assay for the identification, sorting and transcriptomic analysis of polyfunctional human T cells. Eur Cytokine Netw. 2015;26(4):67–72. doi: 10.1684/ecn.2015.0369. [DOI] [PubMed] [Google Scholar]
- 14.Voehringer D, Koschella M, Pircher H. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1) Blood. 2002;100(10):3698–3702. doi: 10.1182/blood-2002-02-0657. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Chang JF, Parnes JR, Fathman CG. T cell receptor (TCR) engagement leads to activation-induced splicing of tumor necrosis factor (TNF) nuclear pre-mRNA. J Exp Med. 1998;188(2):247–254. doi: 10.1084/jem.188.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pflanz S, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16(6):779–790. doi: 10.1016/S1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 17.Takeda A, et al. Cutting edge: role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J Immunol. 2003;170(10):4886–4890. doi: 10.4049/jimmunol.170.10.4886. [DOI] [PubMed] [Google Scholar]
- 18.Lucas S, Ghilardi N, Li J, de Sauvage FJ. IL-27 regulates IL-12 responsiveness of naive CD4+ T cells through Stat1-dependent and -independent mechanisms. Proc Natl Acad Sci USA. 2003;100(25):15047–15052. doi: 10.1073/pnas.2536517100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brombacher F, Kastelein RA, Alber G. Novel IL-12 family members shed light on the orchestration of Th1 responses. Trends Immunol. 2003;24(4):207–212. doi: 10.1016/S1471-4906(03)00067-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida H, et al. WSX-1 is required for the initiation of Th1 responses and resistance to L. major infection. Immunity. 2001;15(4):569–578. doi: 10.1016/S1074-7613(01)00206-0. [DOI] [PubMed] [Google Scholar]
- 21.Hölscher C, et al. The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J Immunol. 2005;174(6):3534–3544. doi: 10.4049/jimmunol.174.6.3534. [DOI] [PubMed] [Google Scholar]
- 22.Villegas-Mendez A, et al. IL-27 receptor signalling restricts the formation of pathogenic, terminally differentiated Th1 cells during malaria infection by repressing IL-12 dependent signals. PLoS Pathog. 2013;9(4):e1003293. doi: 10.1371/journal.ppat.1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gwyer Findlay E, et al. IL-27 receptor signaling regulates memory CD4+ T cell populations and suppresses rapid inflammatory responses during secondary malaria infection. Infect Immun. 2014;82(1):10–20. doi: 10.1128/IAI.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freitas do Rosário AP, et al. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: a critical mechanism for protection from severe immunopathology during malaria infection. J Immunol. 2012;188(3):1178–1190. doi: 10.4049/jimmunol.1102755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimura D, et al. Interleukin-27-Producing CD4(+) T Cells Regulate Protective Immunity during Malaria Parasite Infection. Immunity. 2016;44(3):672–682. doi: 10.1016/j.immuni.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Dubovsky JA, Powers JJ, Gao Y, Mariusso LF, Sotomayor EM, Pinilla-Ibarz JA. Epigenetic repolarization of T lymphocytes from chronic lymphocytic leukemia patients using 5-aza-2’-deoxycytidine. Leuk Res. 2011;35(9):1193–1199. doi: 10.1016/j.leukres.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aagaard C, et al. Protection and polyfunctional T cells induced by Ag85B-TB10.4/IC31 against Mycobacterium tuberculosis is highly dependent on the antigen dose. PLoS ONE. 2009;4(6):e5930. doi: 10.1371/journal.pone.0005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faccioli LH, Souza GE, Cunha FQ, Poole S, Ferreira SH. Recombinant interleukin-1 and tumor necrosis factor induce neutrophil migration “in vivo” by indirect mechanisms. Agents Actions. 1990;30(3-4):344–349. doi: 10.1007/BF01966298. [DOI] [PubMed] [Google Scholar]
- 29.Smith WB, Gamble JR, Clark-Lewis I, Vadas MA. Interleukin-8 induces neutrophil transendothelial migration. Immunology. 1991;72(1):65–72. [PMC free article] [PubMed] [Google Scholar]
- 30.Menezes GB, Rezende RM, Pereira-Silva PE, Klein A, Cara DC, Francischi JN. Differential involvement of cyclooxygenase isoforms in neutrophil migration in vivo and in vitro. Eur J Pharmacol. 2008;598(1-3):118–122. doi: 10.1016/j.ejphar.2008.08.037. [DOI] [PubMed] [Google Scholar]
- 31.Karlsson T, Glogauer M, Ellen RP, Loitto VM, Magnusson KE, Magalhães MA. Aquaporin 9 phosphorylation mediates membrane localization and neutrophil polarization. J Leukoc Biol. 2011;90(5):963–973. doi: 10.1189/jlb.0910540. [DOI] [PubMed] [Google Scholar]
- 32.Liu M, et al. Formylpeptide receptors mediate rapid neutrophil mobilization to accelerate wound healing. PLoS ONE. 2014;9(6):e90613. doi: 10.1371/journal.pone.0090613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon LI, Douglas SD, Kay NE, Yamada O, Osserman EF, Jacob HS. Modulation of neutrophil function by lysozyme. Potential negative feedback system of inflammation. J Clin Invest. 1979;64(1):226–232. doi: 10.1172/JCI109443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas V, Samanta S, Fikrig E. Anaplasma phagocytophilum increases cathepsin L activity, thereby globally influencing neutrophil function. Infect Immun. 2008;76(11):4905–4912. doi: 10.1128/IAI.00851-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borish L, Rosenbaum R, Albury L, Clark S. Activation of neutrophils by recombinant interleukin 6. Cell Immunol. 1989;121(2):280–289. doi: 10.1016/0008-8749(89)90026-9. [DOI] [PubMed] [Google Scholar]
- 36.Mócsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. 2013;210(7):1283–1299. doi: 10.1084/jem.20122220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han Q, Bagheri N, Bradshaw EM, Hafler DA, Lauffenburger DA, Love JC. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci USA. 2012;109(5):1607–1612. doi: 10.1073/pnas.1117194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Almeida JR, et al. Antigen sensitivity is a major determinant of CD8+ T-cell polyfunctionality and HIV-suppressive activity. Blood. 2009;113(25):6351–6360. doi: 10.1182/blood-2009-02-206557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarthy JS, et al. A pilot randomised trial of induced blood-stage Plasmodium falciparum infections in healthy volunteers for testing efficacy of new antimalarial drugs. PLoS ONE. 2011;6(8):e21914. doi: 10.1371/journal.pone.0021914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsen M, Sauce D, Arnaud L, Fastenackels S, Appay V, Gorochov G. Evaluating cellular polyfunctionality with a novel polyfunctionality index. PLoS ONE. 2012;7(7):e42403. doi: 10.1371/journal.pone.0042403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, et al. HLA class I binding 9mer peptides from influenza A virus induce CD4 T cell responses. PLoS ONE. 2010;5(5):e10533. doi: 10.1371/journal.pone.0010533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 43.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.