

Abstract
Specialized proresolving mediators (SPMs) promote the resolution of inflammation and exert beneficial effects in animal models of chronic inflammatory diseases, including asthma. Previously, we have shown that certain SPMs reduce IgE production in B cells from healthy individuals, which has a critical role in allergic asthma. Here, we investigated the effects of SPMs on B cell IgE production in asthma patients. Peripheral blood mononuclear cells from asthma patients were treated with 17-HDHA or RvD1, and IgE levels were measured. RvD1 and 17-HDHA dampened IgE production in B cells from most asthma patients, whereas B cells from a subset of patients taking oral steroids were refractory to SPM treatment. Molecular mechanisms underlying the interaction between corticosteroids and SPMs were investigated by treating B cells from nonasthmatic donors with corticosteroids in vitro. Corticosteroids blocked the inhibitory effects of 17-HDHA and RvD1 on B cell IgE production by abolishing the suppressive activity of these mediators on IgE class switching. Corticosteroids decreased the expression of transcriptional repressor Bcl-6 as well as its suppressive activity on epsilon germline transcription. We conclude that 17-HDHA and RvD1 can reduce IgE production in asthma patients not taking high doses of steroids but that corticosteroids interfere with the ability of B cells to respond to proresolving mediators.
Oral corticosteroids block the ability of specialized proresolving mediators to reduce IgE production in B cells from asthma patients, suggesting interference with endogenous resolution mechanisms.
Introduction
Asthma is a chronic inflammatory disease, involving chronic lung inflammation and narrowing of the small airways through a combination of excessive mucus production and smooth muscle contraction (1). It is characterized by recurrent dyspnea, wheezing, coughing, edema, and chest tightness and usually is associated with reversible airflow obstruction and airway hyperresponsiveness (1). Moreover, severe asthma attacks can lead to life-threatening emergencies. Conventional treatments for asthma include antiinflammatory products, such as inhaled corticosteroids, leukotriene inhibitors, and short- and long-acting bronchodilators (2, 3). In some cases, symptoms that are difficult to control are managed with oral corticosteroids (OCS) in severe asthma patients (3). It is important to recognize that these treatments focus on managing the symptoms of the disease mainly by suppressing the proinflammatory immune responses and do not address the underlying causes of airway inflammation.
The pathogenesis of asthma involves various inflammatory immune cells and proinflammatory mediators (4). IgE is an antibody produced by B cells; it is critical for the onset and maintenance of both acute and chronic allergic diseases, including asthma (5). The vast majority of individuals with allergic asthma have elevated serum IgE levels (6). Because IgE is a key player in allergic asthma, it represents a prime target for therapeutic intervention. A monoclonal anti-IgE antibody (7), omalizumab, has shown to be effective in severe allergic asthma as well as in people with nonallergic asthma (7, 8). This blocking antibody mainly binds to free IgE antibody, preventing it from triggering an IgE-induced inflammatory reaction. However, this anti-IgE antibody does not suppress the differentiation and maturation process of IgE-producing cells.
Acute inflammation is a protective immune response triggered by trauma, pathogens, toxins, etc., which is initiated within minutes of recognition of “danger” signals by activation of the innate immune system. In contrast, resolution of inflammation is a dynamic and active process that regulates many cellular interactions in affected tissues to restore homeostasis (9). Studies suggest that failure to reestablish homeostasis either by prolonged inflammation or insufficient resolution could lead to chronic inflammatory conditions, such as asthma. Recently, endogenous, lipid-derived, specialized proresolving mediators (SPMs) were identified to be important in promoting resolution of inflammation (10, 11). Once synthesized and secreted, SPMs signal through GPCRs to mediate their effects. Thus far, 5 primary receptor-ligand pairs have been identified: RvD1 binds GPCR 32 (GPR32), RvE1 binds chemokine receptor 23 (ChemR23) or leukotriene receptor B1 (BLT1), LXA4 binds lipoxin A4 receptor/formyl peptide receptor 2 (ALX/FPR2), and RvD2 binds G protein–coupled 18 (GPR18) (12, 13). These receptor-mediated actions of SPMs are cell type specific, and the exact mechanisms are not known.
Given the important role of IgE in the pathophysiology of asthma, our lab has previously shown that RvD1, and the RvD1 precursor 17-HDHA, inhibit human B cell IgE production from healthy individuals (14). This was mediated through enhanced expression of the transcriptional repressor, Bcl-6, which in turn suppressed B cell class switch to IgE. In our current study reported herein, we investigated whether 17-HDHA or RvD1 could reduce IgE production in B cells from asthma patients. To test our hypothesis, we recruited asthma patients, most of whom were taking corticosteroids either in an inhaled or oral form, depending on the severity of the disease, and isolated B cells to test the effects of SPMs. We further explored the mechanism(s) by which SPMs affect IgE synthesis and how corticosteroids might influence the effects of SPMs.
Results
17-HDHA and RvD1 reduce B cell IgE production from asthma patients.
Our lab has previously shown that RvD1 and the RvD1 precursor 17-HDHA directly regulate human B cell function and suppress stimulus-driven IgE production from healthy donors. Here, we investigated the effects of these SPMs on B cells from asthma patients (Table 1). First, we evaluated the effects of 17-HDHA and RvD1 on spontaneous IgE production from unstimulated blood-circulating B cells in asthma patients. B cells from half of our asthmatic donors spontaneously produced IgE, whereas B cells from healthy donors produced undetectable levels of IgE (Figure 1A). This confirmed the presence of blood-circulating IgE-secreting B cells in some of the asthma patients, which were not found in healthy individuals. We then tested whether 17-HDHA and RvD1 could block spontaneous B cell IgE production. Peripheral blood mononuclear cells (PBMCs) were treated with the SPMs daily for 6 days, and cell culture supernatants were collected to measure IgE levels. Both mediators strongly dampened spontaneous IgE production (Figure 1B). Next, we investigated whether 17-HDHA and RvD1 can affect stimulus-driven differentiation of B cells to IgE-secreting cells. 17-HDHA and RvD1 were added to PBMCs isolated from asthma patients, followed by stimulation with an IgE-inducing cocktail for 6 days (14). IgE production in B cells from most patients was significantly reduced by 17-HDHA and RvD1 (Figure 2A); however, B cells from 3 donors were refractory to treatment (Figure 2B). Because we wanted to test the effects of these SPMs on a broad range of patients with asthma, we did not specify criteria for recruiting donors or exclude any disease subpopulations. Interestingly, those with B cells that did not respond to treatment with 17-HDHA or RvD1 had severe asthma with occasional asthma attacks and were taking OCS at the time of collection (Table 1). The responders were taking only inhaled steroids and there were no other factors that could potentially differentiate these two groups.
Table 1. Characteristics of the asthma patients.

Figure 1. 17-HDHA and RvD1 reduce spontaneous B cell IgE production from asthma patients.

(A) PBMCs from asthma or nonasthmatic donors were cultured in 96-well plates for 6 days, and IgE levels in cell culture supernatants were measured by ELISA. (B) PBMCs from 5 asthma patients were pretreated with 17-HDHA or RvD1 and incubated for 6 days. The SPMs were added daily until day 6 when cell culture supernatants were collected to measure IgE levels. Each symbol represents an individual donor and represents means of triplicates. Data were analyzed by repeated-measures 1-way ANOVA with Tukey’s post test, *P ≤ 0.05, ***P ≤ 0.001. PBMC, peripheral blood mononuclear cell; SPM, specialized proresolving mediator.
Figure 2. 17-HDHA and RvD1 reduce induced B cell IgE production from asthma patients not receiving oral corticosteroid treatment.
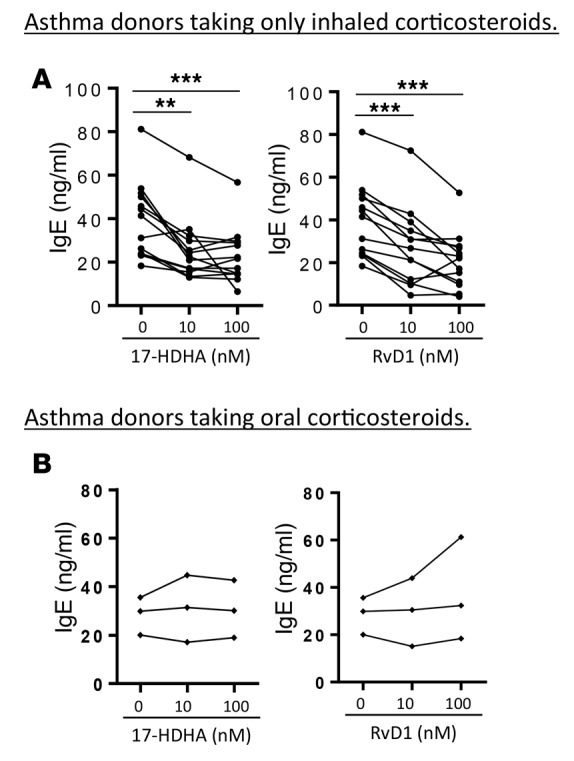
PBMCs from asthma patients taking only inhaled corticosteroids (A), or those taking oral steroids (B), were treated with SPMs, followed by stimulation with an IgE-inducing cocktail. IgE levels were measured at day 6. Each symbol represents an individual donor and represents means of triplicates. Data were analyzed by repeated-measures 1-way ANOVA with Tukey’s post test, **P ≤ 0.01, ***P ≤ 0.001. PBMC, peripheral blood mononuclear cell; SPM, specialized proresolving mediator.
Corticosteroids block the inhibitory effects of SPMs on B cell IgE production in vitro.
It was unexpected that B cells from asthma patients taking OCS would be refractory to SPMs. Therefore, we investigated the direct in vitro effects of corticosteroids on the ability of SPMs to regulate B cell IgE production. PBMCs from asthma patients who were not taking OCS were treated with dexamethasone (10 nM) prior to treatment with 17-HDHA or RvD1. After 24 hours of incubation, the mediators were added to PBMCs, followed by stimulation with an IgE-inducing cocktail for 6 days. All of the asthma patients we tested responded to 17-HDHA and RvD1, and their B cell IgE production was significantly reduced (Figure 3A). However, consistent with the results from donors being treated with OCS, dexamethasone completely abolished the inhibitory effects of 17-HDHA and RvD1 on IgE production (Figure 3B).
Figure 3. Corticosteroids block the inhibitory effects of SPMs on B cell IgE production in vitro.
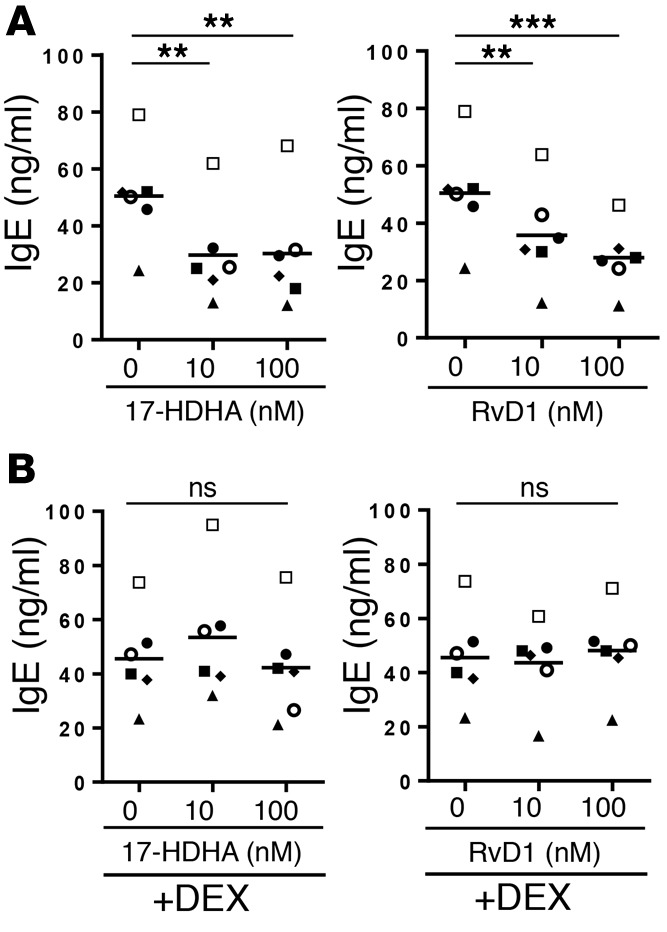
PBMCs from asthma patients who were not taking oral steroids were left untreated (A) or pretreated with dexamethasone (10 nM) (B) for 24 hours. The next day, SPMs were added for 30 minutes, followed by stimulation with an IgE-inducing cocktail for 6 days, after which cell culture supernatant IgE levels were measured. Each symbol represents an individual donor and represents means of triplicates. Data were analyzed by repeated-measures 1-way ANOVA with Tukey’s post test, **P ≤ 0.01, ***P ≤ 0.001. PBMC, peripheral blood mononuclear cell; SPM, specialized proresolving mediator.
Corticosteroids reduce the inhibitory effects of SPMs on B cell class switching to IgE.
We next tested the effect of corticosteroids on several different steps involved in B cell differentiation to IgE-secreting cells. Because corticosteroids might exhibit effects on B cells indirectly through effects on bystander cells, we performed the remaining experiments on purified B cells from healthy donors rather than PBMCs. First, we assessed the effect of dexamethasone and SPMs on IgE secretion in purified B cells. 17-HDHA or RvD1 alone reduced IgE production substantially (Figure 4A). Interestingly, the inhibitory activities of SPMs on IgE production were blocked with dexamethasone pretreatment (Figure 4A), similar to what we observed in the mixed population of PBMCs (Figure 3B). We next investigated the effects of 17-HDHA and RvD1 in combination with dexamethasone on IgE class switching, and as indicators, we measured ɛGLT and mature IgE mRNA levels. Purified B cells were stimulated with the IgE-inducing cocktail for 24 hours for ɛGLT, and for 72 hours for mature IgE (14), following treatment with SPMs and/or dexamethasone. 17-HDHA or RvD1 alone reduced both ɛGLT and mature IgE mRNA levels, whereas dexamethasone pretreatment diminished these effects (Figure 4, B and C). As immune suppressors, corticosteroids are known to dampen cell proliferation and to induce cell apoptosis in high concentrations (μM range) (15, 16). To determine whether the effects of corticosteroids on B cell response to SPMs could be mediated by changes in cell proliferation, a [3H]thymidine incorporation assay was performed. B cells pretreated with dexamethasone showed significantly suppressed proliferation as early as 24 hours after stimulation, and this was maximized at day 3 (Figure 4D). Another well-known effect of dexamethasone is to promote differentiation of many different types of cells, such as adipocytes. Therefore, we next investigated whether SPMs plus dexamethasone could affect B cell differentiation. One of the important signaling pathways involved in B cell differentiation is the activation of PPARγ by PPARγ ligands (17, 18). We measured PPARγ binding activity activated by PPARγ ligands produced by B cells using a PPAR response element–luciferase (PPRE-luciferase) reporter assay. 17-HDHA alone (without dexamethasone) did not affect the levels of PPARγ binding activity (Figure 4E). Interestingly, in B cells pretreated with dexamethasone, 17-HDHA enhanced PPARγ activity compared with dexamethasone alone (Figure 4E).
Figure 4. Corticosteroids reduce the inhibitory effects of SPMs on B cell class switching to IgE.

Purified CD19+ B cells from healthy donors were pretreated with dexamethasone (10 nM) or left untreated for 24 hours. Cells were treated with the 17-HDHA or RvD1, followed by stimulation with an IgE-inducing cocktail for a certain amount of time for each experiment. (A) B cells were incubated for 6 days, and IgE levels were measured by ELISA. B cells were stimulated for 1 day or 3 days, and ɛGLT RNA levels (B), or mature IgE mRNA levels (C), were measured, respectively, using RT-qPCR. (A–C) Experiments were repeated in 2–7 different donors. Each symbol represents an individual donor and represents means of triplicates. (D) One day after B cells were stimulated, [3H]thymidine (1 μCi/well) was added to the culture and incubated for another 48 hours. Cells were harvested at day 3 after stimulation, and incorporation was measured with Topcount Luminometer. Data shown represent 3 different donors. (E) B cells were stimulated for 6 days when cell culture supernatants were collected and applied to PPRE-luciferase assay. Each sample’s luciferase activity was normalized to untreated cells (no 17-HDHA and no dexamethasone). Data shown are from 2 different donors. Each symbol represents an individual donor and represents means of triplicates. Data were analyzed by repeated-measures 2-way ANOVA with Tukey’s post test, *P ≤ 0.05, **P ≤ 0.01, #P ≤ 0.05, ##P ≤ 0.01, ###P ≤ 0.001 (pound signs compare values with or without dexamethasone, asterisks compare values with or without 17-HDHA). ɛGLT, ɛ germline transcript; RT-qPCR, reverse transcriptase quantitative PCR; PPRE, PPAR response element.
Corticosteroids diminish the ability of 17-HDHA to enhance the binding activity of Bcl-6 on the ɛGLT promoter region, Iɛ.
We previously reported that the inhibitory effects of SPMs on B cell IgE production were mediated by enhancing expression of the Bcl-6 transcriptional repressor, which in turn suppressed class switching to IgE by binding to the promoter region of ɛGLT (Iɛ) (14). Here, we investigated the effects of corticosteroids on the expression and activity of Bcl-6 on Iɛ. First, CD19+ B cells were isolated from healthy donors and treated with dexamethasone with or without 17-HDHA or RvD1, followed by stimulation with an IgE-inducing cocktail, and Bcl-6 expression was determined by Western blot. In B cells without dexamethasone, 17-HDHA and RvD1 enhanced Bcl-6 expression in a concentration-dependent manner, but dexamethasone reduced Bcl-6 expression regardless of 17-HDHA treatment in B cells from multiple donors (Figure 5, A and B). To further investigate whether changes in total Bcl-6 expression induced by dexamethasone affected Bcl-6 binding affinity to Iɛ, we performed a ChIP assay. Dexamethasone dramatically reduced the amount of Bcl-6 bound to Iɛ compared with untreated B cells and also diminished the ability of 17-HDHA to enhance Bcl-6 binding to Iɛ (Figure 5, C and D), consistent with dexamethasone’s ability to increase ɛGLT and IgE transcription (Figure 4, B and C) and prevent SPMs from inhibiting IgE production (Figure 4A).
Figure 5. Corticosteroids diminish the ability of 17-HDHA to enhance the binding activity of Bcl-6 on εGLT promoter region (Iε).

(A and B) Purified B cells from healthy donors were pretreated with dexamethasone (10 nM) or left untreated for 24 hours. The next day, cells were treated with 17-HDHA or RvD1, followed by stimulation with an IgE-inducing cocktail for 4 hours. Cell lysates were collected, and total Bcl-6 expression levels were measured using Western blot. β-Tubulin was used as a control. (A) Western blot image of Bcl-6 expression in purified human B cells from a representative healthy donor treated with 17-HDHA or RvD1 (Bcl-6 and β-tubulin were probed in a separate membrane for 17-HDHA treatment). (B) Densitometry analysis for 4 different donors treated with 17-HDHA (mean ± SEM). (C and D) PBMCs from healthy donors were treated with 17-HDHA as described for Western blot, and the amount of Bcl-6 bound to Iɛ was measured using ChIP assay. (C) Representative images of PCR products obtained in PBMCs from healthy donors. (D) Quantification of Bcl-6 binding to Iε for 3 different donors (mean ± SEM). Data were analyzed by repeated-measures 2-way ANOVA with Tukey’s post test, *P ≤ 0.05, #P ≤ 0.05, ##P ≤ 0.01 (pound signs compare values with or without dexamethasone, asterisks compare values with or without 17-HDHA). PBMC, peripheral blood mononuclear cell; Iε, ε germline transcript promoter region; Bcl-6, B cell lymphoma-6 protein.
Inhibition of 5-lipoxygenase activity blocks anti-IgE effects of 17-HDHA and RvD1.
17-HDHA can be converted to D-series resolvins by the action of 5-lipoxygenase (5-LOX) (19), which is expressed in B cells (20). In an attempt to investigate whether the activity of 17-HDHA is dependent on its conversion to RvD1 (or other D-series resolvins), we treated B cells or PBMCs with a 5-LOX inhibitor, zileuton. B cells were treated with zileuton and with 17-HDHA or RvD1 at day 0 and day 3 and cultured for 7 days when the cell culture supernatants were collected to measure IgE levels by ELISA. Surprisingly, zileuton suppressed the anti-IgE effects of both 17-HDHA and RvD1 (Figure 6). This was observed in both purified B cells and a mixed population of PBMCs and suggests that the primary method of action of both 17-HDHA and RvD1 on B cells is to stimulate production of other 5-LOX–dependent mediators.
Figure 6. Inhibition of 5-lipoxygenase activity blocks anti-IgE effects of 17-HDHA and RvD1.

CD19+ B cells (A) or PBMCs (B) from a healthy donor were treated with zileuton (10 μM) prior to 17-HDHA or RvD1 treatment, followed by stimulation with the IgE-inducing cocktail. Cell culture supernatants were collected at day 7, and IgE levels were measured. The experiment was done in one representative donor (mean ± SEM). Data were analyzed by 1-way ANOVA with Tukey’s post test, **P ≤ 0.01, ***P ≤ 0.001. PBMC, peripheral blood mononuclear cell.
Discussion
Omega-3 fatty acids have been touted to be effective in inflammatory diseases, including asthma (21). Recently, it was discovered that omega-3–derived SPMs are key mediators that possess proresolving activities (22). Evidence supports the hypothesis that asthma represents, at least in part, a failure to resolve chronic inflammation, with an imbalance between proinflammatory and proresolving mediators (23–27). For example, severe asthma patients have lower levels of LxA4 in BAL fluid and in exhaled breath condensate compared with patients with mild asthma (24, 25). Additionally, the expression of the SPM receptor ALX/FPR2 on neutrophils and eosinophils is reduced in severe asthma patients (25). Exogenous treatment with RvD1 or RvE1 ameliorated asthma symptoms in mouse models of allergic airway diseases by reducing production of inflammatory mediators and recruitment of inflammatory cells in the lung (28–30). In our study, we show that the DHA-derived mediators, 17-HDHA and RvD1, reduced spontaneous as well as stimulated IgE production in B cells from asthma patients with a broad spectrum of disease severity.
Unexpectedly and interestingly, there was a subset of asthma patients whose B cells were refractory to SPM treatment, which was not observed in healthy donors. These refractory asthma patients were taking OCS to treat asthma attacks at the time of blood donation, whereas responding asthma patients were taking only inhaled steroids. Inhaled corticosteroids are known to have localized activity in lung, with low absorption rate and rapid clearance, which minimizes systemic side effects, while OCS are used only for patients who have uncontrolled symptoms or in cases of asthma exacerbations. The estimated serum peak concentration of inhaled fluticasone, a commonly used inhaled steroid, is 40–100 times lower than OCS, such as methylprednisolone or prednisolone (31–33). Therefore, peripheral blood immune cells including B cells in patients taking oral steroids would be exposed to much higher concentrations of corticosteroids. Corticosteroids are by far the most effective treatments available to control the symptoms of allergic diseases such as asthma, atopic dermatitis, and seasonal allergies (34). Their beneficial effects include suppressing the production of proinflammatory mediators and inducing apoptosis of eosinophils (35, 36). However, corticosteroids only manage the symptoms of asthma; they do not eliminate the underlying root cause of the disease. Moreover, there is evidence that corticosteroids can enhance B cell IgE production in vitro and in vivo (37–41). One study reported that allergic asthma patients had higher serum allergen-specific IgE levels after receiving corticosteroid treatment, while half of patients on long-term corticosteroid treatment developed sensitization to new allergens (37). Some in vitro studies have shown that corticosteroids, at micromolar concentrations, promote human B cell IgE production when added with IL-4 (40, 41). Here, we found that nanomolar amounts of dexamethasone, which are in the therapeutic range (42), prevented anti-IgE activities of 17-HDHA and RvD1 on both PBMCs and purified B cells. This was, at least in part, through increased B cell class switching to IgE, based on εGLT and mature IgE transcript levels. B cell class switching to IgE is tightly regulated, and one of the key players is the transcriptional repressor Bcl-6 (43, 44). We previously reported that 17-HDHA and RvD1 inhibited human B cell IgE production by enhancing Bcl-6 expression, which in turn suppresses class switching to IgE by reducing STAT6 activity on Iε (14). Here, dexamethasone dampened basal expression of Bcl-6 even in unstimulated B cells, which also led to reduced Bcl-6 binding affinity to the Iε promoter. Bcl-6 regulates transcription of many different genes related to cell proliferation and proinflammatory cytokines (45). Due to its broad activity, the effects of 17-HDHA and RvD1 or dexamethasone were stronger on Bcl-6 binding activity specific to Iε, compared with total Bcl-6 protein expression. Even with dexamethasone pretreatment, 17-HDHA and RvD1 did enhance Bcl-6 binding affinity to Iε, but this was not enough to suppress class switching, as seen in Figures 4 and 5. It is known that corticosteroids can upregulate CD40 expression on the surface of B cells, leading to enhanced production of IgE in humans (41). In addition, activation of CD40 by mouse CD40 ligands reduces Bcl-6 expression in B cells (46), thus providing a likely mechanism whereby dexamethasone neutralizes the protective effects of SPMs on Bcl-6 expression.
It is interesting to note that in this study dexamethasone exhibited two counteractive activities on IgE class switching: downregulation of Bcl-6, leading to increased εGLT (Figure 4B and Figure 5), and suppression of proliferation (Figure 4D), which is a key step preceding class switch recombination in B cells. The end result of these counterbalancing signals is that dexamethasone itself has no effects on net IgE production (Figure 4A). On the other hand, dexamethasone changes the clearly defined anti-IgE activities of 17-HDHA and RvD1. Dexamethasone reduced the inhibitory effects of SPMs on εGLT transcription by dampening basal Bcl-6 expression and enhanced autologous PPARγ binding activity synergistically with SPMs (Figure 4E). PPARγ signaling is required for B cell differentiation to antibody-secreting cells, and exogenous treatment of B cells with PPARγ ligands enhanced antibody production. These complex interactions could explain why total IgE secretion is enhanced with dexamethasone plus 17-HDHA but not dexamethasone alone. Moreover, this might explain the observation that stress could trigger allergic reactions (47). It is likely that endogenous corticosteroids secreted in response to stress interact with SPMs produced in the B cell microenvironment and, all together, promote IgE production.
In general, PPARγ ligands show antiinflammatory and antiproliferative activities. In this context, some studies reported the beneficial effects of exogenous treatment of PPARγ ligands in preclinical mouse models of asthma, with reduced serum levels of Th2 cytokines that regulate B cell IgE production (48, 49). However, our lab has previously shown direct effects of PPARγ ligands on human B cells by enhancing differentiation and promoting IgG antibody production (17). Here, we show that 17-HDHA in combination with dexamethasone promoted autologous PPARγ ligand production, which is associated with increased IgE production. Therefore, our study suggests that PPARγ ligands might have different effects on different cell types and also provides insights on the physiological role of PPARγ ligands on B cell IgE production in asthma.
Another possible mechanism through which corticosteroids diminish the anti-IgE activities of SPMs on B cells is by regulating the expression of SPMs receptors. RvD1 is known to exert its activity through binding to ALX/FPR2 or GPR32 receptors. However, it is not known whether 17-HDHA binds directly to specific receptors or is converted to D-series resolvins, which signal through receptors. Therefore, it will be very important to investigate whether corticosteroids could change B cell expression of different SPM receptors in the future.
Although 17-HDHA has antiinflammatory activity in several experimental systems, it is unclear whether 17-HDHA is directly active as an SPM or whether it must be converted to RvD1 or some other compound. Because 17-HDHA can be converted to RvD1 by the actions of 5-LOX, we attempted to determine whether 17-HDHA has a direct effect as an immunomodulatory SPM or is converted to RvD1 in treated B cells. Unexpectedly, a 5-LOX inhibitor, zileuton, blocked the anti-IgE actions of both 17-HDHA and RvD1 (Figure 6). We suggest that the most likely explanation is that the anti-IgE effects of 17-HDHA and RvD1 are mediated, at least in part, by promoting the synthesis of other 5-LOX–dependent mediators, such as lipoxins or E-series resolvins, that act on B cells in an autocrine fashion. Previous reports have shown that macrophages and neutrophils can produce and respond to SPMs in an autocrine manner to regulate inflammatory responses (50, 51). We have previously reported that lipoxin A4 decreases IgM and IgG production from memory B cells (52), and both lipoxin B4 and resolvin E1 reduce allergic airway inflammation in a mouse model (28, 53), suggesting that these mediators could be involved in autocrine downregulation of IgE production in B cells. While this intriguing finding deserves more study, our results are strong evidence that, regardless of the mechanism of action of 17-HDHA, corticosteroids can disrupt the ability of B cells to respond to proresolving signals in asthma.
These results have important implications for the use of corticosteroids in chronic inflammatory diseases, including asthma. While corticosteroids can be effective at suppressing chronic inflammation, systemic or OCS also have deleterious side effects, including weight gain, osteoporosis, and growth retardation in children (54). As novel asthma therapies are introduced, including CysLT inhibitors and biologics such as omalizumab and mepolizumab, they are positioned as steroid-sparing strategies (55, 56), but when they are ineffective or in the event of acute exacerbations, clinicians return to OCS as the mainstay of treatment (57). While OCS are undoubtedly effective at relieving symptoms and improving airway obstruction, it has been recognized that they may not alter the course of the underlying disease. And there is now a growing body of evidence that immunosuppressive treatments like corticosteroids also suppress resolution mechanisms (58–60). For example, Vachier’s group has shown that polymorphonuclear cells from asthma patients taking long-term OCS treatment showed impaired 5-LOX activity, leading to a decrease production of the metabolites, including lipoxins (58). In addition, steroids had antiresolving effects in rat placental inflammatory responses via inhibiting LXA4 biosynthesis (59). Here, we show that corticosteroids interfere with the IgE inhibitory actions of RvD1 and the RvD1 precursor 17-HDHA on B cells. While this suggests that use of SPMs as therapy may need to be limited to patients not on OCS, it also implies that OCS interfere with endogenous resolution mechanisms and may prolong or worsen underlying disease, given that these mediators are present endogenously in asthma patients, albeit at reduced levels. This has profound implications for the ongoing use of OCS in chronic inflammatory diseases and highlights the need for effective alternatives that do not block endogenous resolution pathways.
In summary, we report the proresolving role of SPMs in regulating human B cell IgE production from asthma patients and their interaction with corticosteroids. Our results suggest that SPMs are important potential therapeutics for most patients with allergic asthma. Further, our results highlight that immunosuppressive therapies like OCS also suppress endogenous resolution pathways and suggest one method by which OCS may actually exacerbate allergic diseases.
Methods
Subjects.
Subject characteristics are listed in Table 1. Seventeen asthma patients were recruited for study at the Mary Parkes Center for Asthma, Allergy and Pulmonary Care, and clinical severity of the disease was assessed according to National Heart, Lung, and Blood Institute guidelines. Because we wanted to test the effects of SPMs on a broad range of patients with asthma, there were no specified criteria for recruiting donors or excluding any disease subpopulations. All subjects with asthma were being treated with inhaled corticosteroids, with β2-agonists as needed. Two subjects were taking oral steroids at the time of blood sample collection, and one had their last OCS 3 weeks before blood collection.
Nonasthmatic donors were recruited for further mechanism(s) study, and none of them presented bronchial or respiratory disease.
Cell culture conditions and reagents.
Human peripheral blood B cells were isolated as previously described (61). Where indicated, some experiments with cells from asthma patients were performed on PBMCs. Purified B cells or PBMCs were cultured in RPMI 1640 (GIBCO/Invitrogen) supplemented with 5% fetal bovine serum, 2 mM L-glutamine, 5 × 10–5 M 2-mercaptoethanol, 10 mM HEPES, and 50 μg/ml gentamicin. mCD40 ligand (CD154) was prepared using an insect membrane culture as previously described (62). The IgE-inducing cocktail was composed of mouse CD40 ligand (1:50 dilution), IL-4 (50 ng/ml, R&D Systems), and CpG ODN2395 (0.5 μg/ml, Invivogen). Resolvin D1 (RvD1, 7S,8R,1-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid) and 17 (R)-hydroxydocosahexaenoic acid (17-HDHA, 17R-hydroxy-4Z, 7Z, 10Z, 13Z, 15E, 19Z-docosahexaenoic acid) (Cayman Chemical Company) were suspended in ethanol and supplemented in cell culture as previously described (14). Cells were pretreated with either vehicle control or SPMs for 30 minutes and then were treated with an IgE-inducing cocktail or left unstimulated. Additional SPM treatments were added every 24 hours for the duration of the experiment in culture. When necessary, dexamethasone (10 nM) was added to cells for 24 hours prior to SPM treatment. The 5-LOX inhibitors, zileuton (Selleckchem) or MK-886 (Selleckchem), were added to cells 30 minutes prior to SPM treatment.
ELISA.
Purified B cells (5 × 105 cells/ml) or PBMCs were cultured in triplicate in 96-well round-bottom plates for 6 days as previously described (14). Antibody levels in the supernatant were measured by ELISA as specified by the manufacturer (Bethyl Laboratories).
Cell proliferation assay.
Purified CD19+ B cells were cultured in round-bottom plates (5 × 105 cells/ml) in triplicate. [3H]Thymidine (1 μCi/well) was added 48 hours prior to harvest. Incorporation was measured with a Topcount Luminometer (PerkinElmer).
PPRE-luciferase reporter assay.
A PPRE-firefly luciferase reporter construct containing 3 copies of a PPRE (PPRE-firefly luciferase), an SV40 Renilla luciferase construct (Promega), and a PPARγ2 construct (63) were introduced into HEK293FT cells cultured in T25 flasks using Lipofectamine 2000 (Invitrogen). As a negative control, a construct containing constitutively expressed firefly and Renilla luciferase (psiCHECK-2, Promega) was used in place of the PPRE reporter and SV40 reporters. After 4 to 6 hours, reporter cells were washed, trypsinized, plated in 96-well plates, and treated with experimental supernatants or positive or negative controls (20–1,000 nM rosiglitazone or 0.1% DMSO vehicle). Experimental supernatants were collected from human B cell cultures treated with or without dexamethasone for 24 hours, followed by addition of SPMs, and stimulated with an IgE-inducing cocktail for 6 days. After a further 12- to 15-hour overnight incubation, firefly and Renilla luciferase activities were measured using Dual-Glo Luciferase Assay buffer (Promega) and read on a Varioskan Flash luminescent plate reader (Thermo Fisher Scientific). Light output is proportional to PPRE binding activity, which is an indirect readout of PPARγ ligands in the culture medium.
Real-time PCR.
Purified B cells treated with SPMs, with or without dexamethasone pretreatment, were stimulated with an IgE-inducing cocktail for a certain amount of time (see legend for Figure 4). Total RNA was extracted with a Qiagen RNAeasy mini kit using 1 × 106 cells/sample. RNA from each sample was reverse transcribed with SuperScript III and random primers (Invitrogen). The cDNA was amplified in a real-time PCR reaction using iQ SYBR Green Supermix (Bio-Rad) and quantified with Bio-Rad icycler software. Amplification of HPRT was used to normalize and quantify the relative gene expression. The primers used were as follows: εGLT forward, 5′-CACATCCACAGGCACCAAAT-3′; εGLT reverse, 5′-ATCACCGGCTCCGGGAAGTA-3′; mature IgE forward, 5′-ACCCTGGTCACCGTCTCCTCAG-3′; mature IgE reverse, 5′-CAGAGTCACGGAGGTGGCATT-3′; HPRT forward, 5′-ATGACCAGTCAACAGGGGAC-3′; and HPRT reverse, 5′-TGCCTGACCAAGGAAAGCAA-3′.
Western blotting.
Purified human B cells were lysed in RIPA buffer (150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 50 mM Tris, 0.1% SDS, pH 8.0) with protease inhibitor cocktail (Sigma-Aldrich). Protein concentration was determined using the Bio-Rad DC protein assay kit (Bio-Rad). 8% SDS-PAGE gels (Pierce/Thermo Fisher Scientific) were loaded with 10 to 30 μg of protein and transferred to PVDF membranes (Millipore). Western blots were probed with rabbit anti-human Bcl-6 (D65C10, Cell Signaling Technology, 1:1,000) and rabbit anti-human β-tubulin (2146, Cell Signaling Technology). HRP-conjugated goat anti-rabbit secondary antibodies (Jackson ImmunoResearch) were used to detect specific probed antibodies. Western blots were visualized by autoradiography after incubation with ECL (Perkin Elmer Life Sciences Inc.).
ChIP assay.
Human PBMCs isolated from healthy donors were stimulated for 4 hours at a density of 1 × 106 cells/ml with the IgE-inducing cocktail and then cross-linked with 1% formaldehyde for 10 minutes at room temperature. A total of 2 × 107 cells per condition were used. Cross-linking was quenched by incubation with 0.125 M glycine for 5 minutes, and cells were washed with PBS twice, resuspended in 1 ml of ChIP lysis buffer (50 mM Tris-HCl, pH 8.0, 1% SDS, 5 mM EDTA, 1× proteinase inhibitors Complete EDTA-free), and incubated on ice for 5 minutes. The lysates were sonicated on ice to shear DNA using the Misonix Sonicator 3000 at its highest amplitude, with 2 periods of 30-second sonication and then another period of 20-second sonication. DNA fragments of 100–500 bp were obtained. 10 μl of each sample was kept as input control, and the remainder of samples was diluted 5 times with dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0, 1× protease inhibitor Complete EDTA-free) and precleared with 50 μl slurry of protein A-agarose beads (blocked by 0.2 mg/ml salmon sperm DNA in 1 mg/ml of BSA) for 3 hours on a rotating wheel at 4°C. Beads were pelleted in a benchtop centrifuge at 750 g for 1 minute at 4°C, precleared extract was transferred into a new tube, 2 μg of anti-Bcl6 (N-3, Santa Cruz biotechnology) or 1 μg rabbit IgG was added, and samples were incubated rotating overnight at 4°C. 100 μl of blocked protein A beads were added to the ChIP reactions and then incubated rotating for 2 hours at 4°C. Supernatants were removed and beads were washed sequentially in Paro buffer I, II, and III (as shown below) and then 2 times with 1× TE buffer at room temperature. Paro buffer I consists of 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 150 mM NaCl. Paro buffer II consists of 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 200 mM NaCl. Paro buffer III consists of 0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, and 10 mM Tris-HCl, pH 8.1. Washes were 1 ml each, and there were 7–10 minutes of incubation in between Paro washes. Elution was performed twice with 100 μl of Elution buffer (1% SDS, 0.1 M NaHCO3) and tap mixed every couple minutes. 6 μl of 5 mg/ml of proteinase K was added to the combined elutes and the input control samples and then incubated overnight at 65°C. The next day, DNA was purified using Qiagen PCR purification kit and qPCR was performed using primers for Iε germline promoter region: forward, 5′-CTAGAAAGAGGCCTACACCTG-3′ and reverse, 5′-GCCAGACTGTTCTTATTCG-3′.
Statistics.
Each experiment was repeated with cells from at least 3 different human donors. Results are expressed as mean ± SEM. Statistical analyses on normally distributed data were performed using repeated-measures ANOVA. Statistical analyses were performed using Prism version 6 (GraphPad). A P value of less than 0.05 was considered significant.
Study approval.
All donors gave informed written consent in accordance with the Declaration of Helsinki, and the protocol was approved by the University of Rochester Research Subjects Review Board.
Author contributions
NK designed and performed experiments. NK, THT, PJS, and RPP analyzed data. PJS provided clinical expertise. NK, THT, PJS, and RPP prepared the manuscript.
Acknowledgments
This research was supported in part by NIH grants T90DE021985 (University of Rochester Medical school), R21AI103690, and T32HL066988 and the Mary Parkes Center for Asthma, Allergy and Pulmonary Care. We would like to thank the University of Rochester Medical Center Blood Bank for collecting blood samples from healthy donors and Elizabeth Lyda at Mary Parkes Center for Asthma, Allergy and Pulmonary Care for consenting asthma patients to volunteer for the study and collecting blood samples.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2017;2(3):e88588. https://doi.org/10.1172/jci.insight.88588.
Contributor Information
Nina Kim, Email: nina_kim@urmc.rochester.edu.
Patricia J. Sime, Email: patricia_sime@urmc.rochester.edu.
References
- 1.Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002–1014. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- 2.Suissa S, Ernst P. Inhaled corticosteroids: impact on asthma morbidity and mortality. J Allergy Clin Immunol. 2001;107(6):937–944. doi: 10.1067/mai.2001.115653. [DOI] [PubMed] [Google Scholar]
- 3.Wenzel S, Szefler SJ. Managing severe asthma. J Allergy Clin Immunol. 2006;117(3):508–511. doi: 10.1016/j.jaci.2005.12.1316. [DOI] [PubMed] [Google Scholar]
- 4.Busse WW, Lemanske RF. Asthma. N Engl J Med. 2001;344(5):350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 5.Dullaers M, De Bruyne R, Ramadani F, Gould HJ, Gevaert P, Lambrecht BN. The who, where, and when of IgE in allergic airway disease. J Allergy Clin Immunol. 2012;129(3):635–645. doi: 10.1016/j.jaci.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Gergen PJ, Arbes SJ, Calatroni A, Mitchell HE, Zeldin DC. Total IgE levels and asthma prevalence in the US population: results from the National Health and Nutrition Examination Survey 2005-2006. J Allergy Clin Immunol. 2009;124(3):447–453. doi: 10.1016/j.jaci.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gevaert P, et al. Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. J Allergy Clin Immunol. 2013;131(1):110–6.e1. doi: 10.1016/j.jaci.2012.07.047. [DOI] [PubMed] [Google Scholar]
- 8.Di Domenico M, Bisogno A, Polverino M, De Rosa C, Ricci V, Capasso A. Xolair in asthma therapy: an overview. Inflamm Allergy Drug Targets. 2011;10(1):2–12. doi: 10.2174/187152811794352042. [DOI] [PubMed] [Google Scholar]
- 9.Serhan CN. The resolution of inflammation: the devil in the flask and in the details. FASEB J. 2011;25(5):1441–1448. doi: 10.1096/fj.11-0502ufm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 12.Serhan CN, Krishnamoorthy S, Recchiuti A, Chiang N. Novel anti-inflammatory--pro-resolving mediators and their receptors. Curr Top Med Chem. 2011;11(6):629–647. doi: 10.2174/1568026611109060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiang N, Dalli J, Colas RA, Serhan CN. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J Exp Med. 2015;212(8):1203–1217. doi: 10.1084/jem.20150225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim N, Ramon S, Thatcher TH, Woeller CF, Sime PJ, Phipps RP. Specialized proresolving mediators (SPMs) inhibit human B-cell IgE production. Eur J Immunol. 2016;46(1):81–91. doi: 10.1002/eji.201545673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cupps TR, Gerrard TL, Falkoff RJ, Whalen G, Fauci AS. Effects of in vitro corticosteroids on B cell activation, proliferation, and differentiation. J Clin Invest. 1985;75(2):754–761. doi: 10.1172/JCI111757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashwell JD, Lu FW, Vacchio MS. Glucocorticoids in T cell development and function*. Annu Rev Immunol. 2000;18:309–345. doi: 10.1146/annurev.immunol.18.1.309. [DOI] [PubMed] [Google Scholar]
- 17.Ramon S, et al. Peroxisome proliferator-activated receptor γ B cell-specific-deficient mice have an impaired antibody response. J Immunol. 2012;189(10):4740–4747. doi: 10.4049/jimmunol.1200956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Bates TM, Baglole CJ, Bernard MP, Murant TI, Simpson-Haidaris PJ, Phipps RP. Peroxisome proliferator-activated receptor gamma ligands enhance human B cell antibody production and differentiation. J Immunol. 2009;183(11):6903–6912. doi: 10.4049/jimmunol.0900324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohli P, Levy BD. Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol. 2009;158(4):960–971. doi: 10.1111/j.1476-5381.2009.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahshid Y, et al. High expression of 5-lipoxygenase in normal and malignant mantle zone B lymphocytes. BMC Immunol. 2009;10:2. doi: 10.1186/1471-2172-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H, Xun P, He K. Fish and fish oil intake in relation to risk of asthma: a systematic review and meta-analysis. PLoS ONE. 2013;8(11):e80048. doi: 10.1371/journal.pone.0080048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bannenberg GL, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174(7):4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 23.Miyata J, et al. Dysregulated synthesis of protectin D1 in eosinophils from patients with severe asthma. J Allergy Clin Immunol. 2013;131(2):353–60.e1. doi: 10.1016/j.jaci.2012.07.048. [DOI] [PubMed] [Google Scholar]
- 24.Kazani S, et al. Exhaled breath condensate eicosanoid levels associate with asthma and its severity. J Allergy Clin Immunol. 2013;132(3):547–553. doi: 10.1016/j.jaci.2013.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Planagumà A, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med. 2008;178(6):574–582. doi: 10.1164/rccm.200801-061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levy BD, et al. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med. 2005;172(7):824–830. doi: 10.1164/rccm.200410-1413OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vachier I, et al. Severe asthma is associated with a loss of LX4, an endogenous anti-inflammatory compound. J Allergy Clin Immunol. 2005;115(1):55–60. doi: 10.1016/j.jaci.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 28.Flesher RP, Herbert C, Kumar RK. Resolvin E1 promotes resolution of inflammation in a mouse model of an acute exacerbation of allergic asthma. Clin Sci. 2014;126(11):805–814. doi: 10.1042/CS20130623. [DOI] [PubMed] [Google Scholar]
- 29.Rogerio AP, et al. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol. 2012;189(4):1983–1991. doi: 10.4049/jimmunol.1101665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9(8):873–879. doi: 10.1038/ni.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derendorf H, Hochhaus G, Meibohm B, Möllmann H, Barth J. Pharmacokinetics and pharmacodynamics of inhaled corticosteroids. J Allergy Clin Immunol. 1998;101(4 Pt 2):S440–S446. doi: 10.1016/s0091-6749(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 32.Minto C, Li B, Tattam B, Brown K, Seale JP, Donnelly R. Pharmacokinetics of epimeric budesonide and fluticasone propionate after repeat dose inhalation--intersubject variability in systemic absorption from the lung. Br J Clin Pharmacol. 2000;50(2):116–124. doi: 10.1046/j.1365-2125.2000.00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Czock D, Keller F, Rasche FM, Häussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44(1):61–98. doi: 10.2165/00003088-200544010-00003. [DOI] [PubMed] [Google Scholar]
- 34.Boushey HA. Effects of inhaled corticosteroids on the consequences of asthma. J Allergy Clin Immunol. 1998;102(4 Pt 2):S5–16. doi: 10.1016/s0091-6749(98)70001-6. [DOI] [PubMed] [Google Scholar]
- 35.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci. 1998;94(6):557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 36.Ohta K, Yamashita N. Apoptosis of eosinophils and lymphocytes in allergic inflammation. J Allergy Clin Immunol. 1999;104(1):14–21. doi: 10.1016/S0091-6749(99)70107-7. [DOI] [PubMed] [Google Scholar]
- 37.Zieg G, Lack G, Harbeck RJ, Gelfand EW, Leung DY. In vivo effects of glucocorticoids on IgE production. J Allergy Clin Immunol. 1994;94(2 Pt 1):222–230. doi: 10.1016/0091-6749(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 38.Des Roches A, Paradis L, Bougeard YH, Godard P, Bousquet J, Chanez P. Long-term oral corticosteroid therapy does not alter the results of immediate-type allergy skin prick tests. J Allergy Clin Immunol. 1996;98(3):522–527. doi: 10.1016/S0091-6749(96)70085-4. [DOI] [PubMed] [Google Scholar]
- 39.Paterson RL, Or R, Domenico JM, Delespesse G, Gelfand EW. Regulation of CD23 expression by IL-4 and corticosteroid in human B lymphocytes. Altered response after EBV infection. J Immunol. 1994;152(5):2139–2147. [PubMed] [Google Scholar]
- 40.Wu CY, et al. Glucocorticoids increase the synthesis of immunoglobulin E by interleukin 4-stimulated human lymphocytes. J Clin Invest. 1991;87(3):870–877. doi: 10.1172/JCI115092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jabara HH, Brodeur SR, Geha RS. Glucocorticoids upregulate CD40 ligand expression and induce CD40L-dependent immunoglobulin isotype switching. J Clin Invest. 2001;107(3):371–378. doi: 10.1172/JCI10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Czock D, Keller F, Rasche FM, Häussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44(1):61–98. doi: 10.2165/00003088-200544010-00003. [DOI] [PubMed] [Google Scholar]
- 43.Harris MB, Mostecki J, Rothman PB. Repression of an interleukin-4-responsive promoter requires cooperative BCL-6 function. J Biol Chem. 2005;280(13):13114–13121. doi: 10.1074/jbc.M412649200. [DOI] [PubMed] [Google Scholar]
- 44.Harris MB, et al. Transcriptional repression of Stat6-dependent interleukin-4-induced genes by BCL-6: specific regulation of iepsilon transcription and immunoglobulin E switching. Mol Cell Biol. 1999;19(10):7264–7275. doi: 10.1128/MCB.19.10.7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basso K, et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010;115(5):975–984. doi: 10.1182/blood-2009-06-227017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batlle A, et al. CD40 and B-cell receptor signalling induce MAPK family members that can either induce or repress Bcl-6 expression. Mol Immunol. 2009;46(8-9):1727–1735. doi: 10.1016/j.molimm.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 47.Dave ND, Xiang L, Rehm KE, Marshall GD. Stress and allergic diseases. Immunol Allergy Clin North Am. 2011;31(1):55–68. doi: 10.1016/j.iac.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woerly G, et al. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J Exp Med. 2003;198(3):411–421. doi: 10.1084/jem.20021384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Naa MM, El-Refaei MF, Nasif WA, Abduljawad SH, El-Brairy AI, El-Readi MZ. In-vivo antioxidant and anti-inflammatory activity of rosiglitazone, a peroxisome proliferator-activated receptor-gamma (PPAR-γ) agonists in animal model of bronchial asthma. J Pharm Pharmacol. 2015;67(10):1421–1430. doi: 10.1111/jphp.12445. [DOI] [PubMed] [Google Scholar]
- 50.Serhan CN, Krishnamoorthy S, Recchiuti A, Chiang N. Novel anti-inflammatory--pro-resolving mediators and their receptors. Curr Top Med Chem. 2011;11(6):629–647. doi: 10.2174/1568026611109060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shapiro H, Lutaty A, Ariel A. Macrophages, meta-inflammation, and immuno-metabolism. ScientificWorldJournal. 2011;11:2509–2529. doi: 10.1100/2011/397971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramon S, Bancos S, Serhan CN, Phipps RP. Lipoxin A4 modulates adaptive immunity by decreasing memory B-cell responses via an ALX/FPR2-dependent mechanism. Eur J Immunol. 2014;44(2):357–369. doi: 10.1002/eji.201343316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karra L, Haworth O, Priluck R, Levy BD, Levi-Schaffer F. Lipoxin B4 promotes the resolution of allergic inflammation in the upper and lower airways of mice. Mucosal Immunol. 2015;8(4):852–862. doi: 10.1038/mi.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lefebvre P, et al. Acute and chronic systemic corticosteroid-related complications in patients with severe asthma. J Allergy Clin Immunol. 2015;136(6):1488–1495. doi: 10.1016/j.jaci.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 55.Holgate ST. Pathophysiology of asthma: what has our current understanding taught us about new therapeutic approaches? J Allergy Clin Immunol. 2011;128(3):495–505. doi: 10.1016/j.jaci.2011.06.052. [DOI] [PubMed] [Google Scholar]
- 56.Szefler SJ. Advancing asthma care: the glass is only half full! J Allergy Clin Immunol. 2011;128(3):485–494. doi: 10.1016/j.jaci.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scarfone RJ, Fuchs SM, Nager AL, Shane SA. Controlled trial of oral prednisone in the emergency department treatment of children with acute asthma. Pediatrics. 1993;92(4):513–518. [PubMed] [Google Scholar]
- 58.Vachier I, et al. Effects of glucocorticoids on endogenous and transcellular metabolism of eicosanoids in asthma. J Allergy Clin Immunol. 2001;107(5):824–831. doi: 10.1067/mai.2001.113868. [DOI] [PubMed] [Google Scholar]
- 59.Zhang D, et al. Glucocorticoids sensitize rat placental inflammatory responses via inhibiting lipoxin A4 biosynthesis. Biol Reprod. 2014;90(4):74. doi: 10.1095/biolreprod.113.116384. [DOI] [PubMed] [Google Scholar]
- 60.Bhavsar PK, et al. Corticosteroid suppression of lipoxin A4 and leukotriene B4 from alveolar macrophages in severe asthma. Respir Res. 2010;11:71. doi: 10.1186/1465-9921-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernard MP, Phipps RP. CpG oligodeoxynucleotides induce cyclooxygenase-2 in human B lymphocytes: implications for adjuvant activity and antibody production. Clin Immunol. 2007;125(2):138–148. doi: 10.1016/j.clim.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kehry MR, Castle BE. Regulation of CD40 ligand expression and use of recombinant CD40 ligand for studying B cell growth and differentiation. Semin Immunol. 1994;6(5):287–294. doi: 10.1006/smim.1994.1037. [DOI] [PubMed] [Google Scholar]
- 63.Woeller CF, O’Loughlin CW, Pollock SJ, Thatcher TH, Feldon SE, Phipps RP. Thy1 (CD90) controls adipogenesis by regulating activity of the Src family kinase, Fyn. FASEB J. 2015;29(3):920–931. doi: 10.1096/fj.14-257121. [DOI] [PMC free article] [PubMed] [Google Scholar]