

Summary
Idiopathic pulmonary fibrosis (IPF) is a form of progressive interstitial lung disease with unknown etiology. Due to a lack of effective treatment, IPF is associated with a high mortality rate. The hallmark feature of this disease is the accumulation of activated myofibroblasts that excessively deposit extracellular matrix proteins, thus compromising lung architecture and function and hindering gas exchange. Here we investigated the origin of activated myofibroblasts and the molecular mechanisms governing fibrosis formation and resolution. Genetic engineering in mice enables the time-controlled labeling and monitoring of lipogenic or myogenic populations of lung fibroblasts during fibrosis formation and resolution. Our data demonstrate a lipogenic-to-myogenic switch in fibroblastic phenotype during fibrosis formation. Conversely, we observed a myogenic-to-lipogenic switch during fibrosis resolution. Analysis of human lung tissues and primary human lung fibroblasts indicates that this fate switching is involved in IPF pathogenesis, opening potential therapeutic avenues to treat patients.
In Brief
El Agha et al. use genetic engineering in mice to identify precursor cells for activated myofibroblasts and investigate their fate in a reversible model of lung fibrosis. Their findings emphasize the phenotypic plasticity of lipogenic and myogenic lung fibroblasts and indicate that PPARγ agonists might be beneficial in treating IPF.
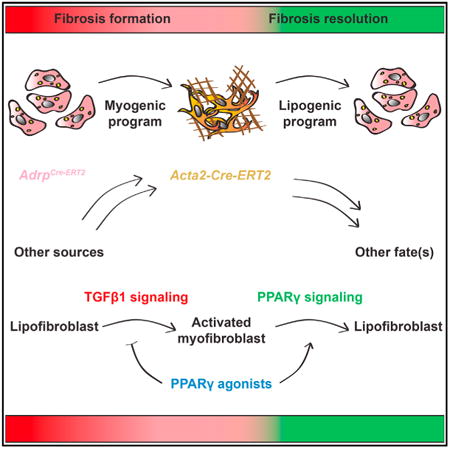
Introduction
Idiopathic pulmonary fibrosis (IPF) is a rare, yet life-threatening disease with very poor prognosis. IPF patients die within 3 to 5 years after diagnosis mostly due to respiratory failure. Lung tissues from these patients display excessive accumulation of alpha smooth muscle actin-positive (ACTA2+) activated myofibroblasts, the effector cells responsible for extracellular matrix (ECM) protein deposition, especially collagen. This pathological phenomenon leads to detrimental consequences such as alveolar collapse and scarring, and consequently the destruction of the lung architecture, rendering the lung similar to a “block of cement” and hindering gas exchange (Gross and Hunninghake, 2001; Homolka, 1987; Thannickal et al., 2004).
Although the exact pathophysiology of IPF remains unclear, extensive research has implicated mechanisms involving both epithelial and mesenchymal cells. Repetitive injury to type 2 alveolar epithelial cells (AEC2), for example, leads to uncontrolled recruitment and activation of mesenchymal cells that cause tissue scarring and ECM protein deposition. Furthermore, misfolding of surfactant proteins A and C (SFTPA and C) leads to endoplasmic reticulum stress and reactive oxygen species production by AEC2, which contributes to aberrant lung remodeling. In addition, transforming growth factor beta 1 (TGFβ1) signaling is believed to drive the fibrotic response in the lungs of IPF patients (Günther et al., 2012; Rafii et al., 2013).
It is widely accepted that the pathogenesis of IPF starts in the alveolar region of the lung, leading to the emergence of spindle-shaped fibroblasts located in fibrotic foci. These foci are regarded as the active fibrotic sites in IPF lungs (Gross and Hunninghake, 2001). Thus, activated myofibroblast progenitors are likely located within the AEC2 stem-cell niche that consists of resident fibroblasts, lipofibroblasts, ECM proteins, and blood capillaries. Many hypotheses have been proposed regarding the cellular origin of activated myofibroblasts in IPF. One hypothesis is that they originate from resident fibroblasts expressing high-affinity type 2 TGFβ receptor (Hoyles et al., 2011). Other studies investigated the possibility that they originate from bone-marrow-derived CD45+ COL1+ CXCR4+ circulating fibrocytes (Phillips et al., 2004) that are recruited to the lungs. However, this scenario remains controversial (Kleaveland et al., 2014). Many studies hypothesized that myofibroblasts derive from epithelial cells undergoing epithelial-to-mesenchymal transition (EMT). Although the involvement of EMT was demonstrated using the active TGFb1 overexpression model of lung fibrosis (Kim et al., 2006), EMT was not a causative mechanism when AEC2 were lineage-traced during bleomycin-induced pulmonary fibrosis (Rock et al., 2011). In this study, we tested the hypothesis that activated myofibroblasts originate from lipofibroblasts.
Lipofibroblasts are lipid-droplet-containing interstitial fibroblasts that are located adjacent to AEC2 and have been well characterized in rodent neonates. Lipofibroblasts are implicated in alveolar maturation and surfactant production (Rehan and Torday, 2014) and have been proposed to contribute to the epithelial stem-cell niche in adult mouse lungs (Barkauskas et al., 2013; McQualter et al., 2013). Interestingly, lipofibroblasts isolated from neonatal rat lungs transdifferentiate to myofibroblasts in response to hyperoxia (Rehan and Torday, 2003) or nicotine exposure (Rehan et al., 2005) in vitro. In a previous study, our group has shown that lipofibroblasts trace back to at least one embryonic population of mesenchymal cells expressing fibroblast growth factor 10 (Fgf10) (El Agha et al., 2014). FGF10 is a potent morphogen that plays a key role in lung organogenesis as demonstrated by Fgf10 knockout mice that suffer from lung agenesis (Bellusci et al., 1997; Sekine et al., 1999). To date, the involvement of lipofibroblasts in lung pathology, particularly lung fibrosis, has not been investigated.
Activated myofibroblasts have been thought to undergo apoptotic clearance after fibrosis resolution (Hinz et al., 2007; Issa et al., 2001). More recently, it was suggested that during fibrosis resolution, myofibroblasts undergo a dedifferentiation event that is controlled by mitogen(s)/ERK/MAPK/CDKs, as opposed to TGFβ1/ALK5/MyoD-dependent myofibroblast differentiation during fibrosis formation (Hecker et al., 2011). In this study, we set out to test a hypothesis that activated myofibroblasts transition to a lipofibroblast-like phenotype during fibrosis resolution.
In the current study, multiple transgenic and knock-in mouse lines were used to lineage-trace lipogenic and myogenic populations of lung fibroblasts during the injury and resolution phases of bleomycin-induced pulmonary fibrosis. We observed remarkable plasticity in resident fibroblastic populations, including lipofibroblasts that served as a source of activated myofibroblasts during fibrosis formation. In addition, a subpopulation of activated myofibroblasts transitioned to a lipofibroblast-like phenotype following fibrosis resolution. Cell sorting followed by gene expression analysis supported our histological observations. Interestingly, our results suggest that activated myofibroblasts do not derive from pre-existing smooth muscle cells (SMCs) in lung fibrosis. The results obtained with the mouse model of lung fibrosis were validated in lung tissues from IPF patients. Finally, functional intervention with the PPARγ agonist rosiglitazone reinforced the lipogenic phenotype and antagonized TGFβ1-mediated fibrogenic response in primary human lung fibroblasts.
Results
Activated Myofibroblasts Originate from ACTA2− Progenitors
Lineage tracing in the context of hypoxia-induced pulmonary hypertension (PH) in mice has shown that SMCs in the remodeled vessels originate from pre-existing SMCs (Sheikh et al., 2014). To determine whether pre-existing (airway and vascular) SMCs serve as a source of activated myofibroblasts in lung fibrosis, Acta2-Cre-ERT2; tdTomatoflox mice were used to label pre-existing SMCs before they were treated with either saline or bleomycin (Figures 1A, 1B, and 1G). Prior to the intratracheal instillation of bleomycin, animals were fed tamoxifen-containing pellets for 2 weeks before being fed normal pellets for 2 weeks in order to ensure enough time for tamoxifen clearance (Figure 1G). As the severity of fibrosis peaks at 14 days post instillation (14 d.p.i.) in this model, 14 d.p.i. served as an end point for this experiment. Lung fibrosis was confirmed by H&E staining and forced oscillation plethysmography, revealing decreased lung compliance in bleomycin-treated mice compared to saline-treated mice (Figure S1). At 14 d.p.i., lungs were harvested and immunostained for ACTA2 (a marker for SMCs and activated myofibroblasts). The labeling efficiency of SMCs using the Acta2-Cre-ERT2; tdTomatoflox transgenic line was 83.5% in saline-treated samples (Figure 1S). While pre-existing airway and vascular SMCs expressing ACTA2 also expressed the lineage label (tdTomato) in both saline groups (Figures 1C–1F) and bleomycin-treated groups (Figures 1H–1K), ACTA2+ activated myofibroblasts located in dense fibrotic areas did not express the lineage label in bleomycin-treated lungs (Figures 1L–1O). The histological observations were supported by FACS-based quantification showing the increase in the number of ACTA2+ cells in bleomycin-challenged lungs (1.8% from total lung suspension) compared to saline-challenged lungs (0.6%) (Figure 1P). Comparable numbers of lineage-labeled cells were observed in both groups, indicating that pre-existing SMCs are not significantly amplified following bleomycin treatment (Figure 1Q) and that lineage-labeled cells were predominantly ACTA2+ (81.6%–92.2%) (Figure 1R). Interestingly, whereas 83.5% of ACTA2+ cells (SMCs) were lineage labeled in the saline-treated group, 52.7% of ACTA2+ cells (SMCs and activated myofibroblasts) were lineage-labeled in the bleomycin-treated group (Figure 1S), indicating that activated myofibroblasts derive from ACTA2− progenitors.
Figure 1. Activated Myofibroblasts Do Not Derive from Pre-existing Smooth Muscle Cells in Lung Fibrosis.
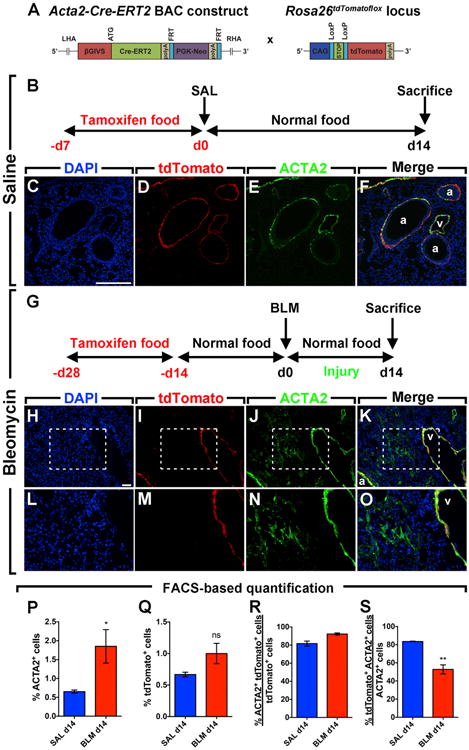
(A) Schematic representation of the Acta2-Cre-ERT2 and tdTomatoflox constructs.
(B) Timeline of tamoxifen and saline treatments. Mice were fed tamoxifen-containing pellets before saline was administered intratracheally. Lungs were harvested at 14 d.p.i.
(C–F) Immunofluorescent staining showing DAPI, tdTomato, and ACTA2 single channels in addition to a merged image.
(G) Timeline of tamoxifen and bleomycin treatments. Mice were fed tamoxifen-containing pellets for 2 weeks followed by 2 weeks of normal pellets before bleomycin was administered intratracheally.
(H–K) Immunofluorescent staining showing DAPI, tdTomato, and ACTA2 single channels in addition to a merged image.
(L–O) High-magnification images of the boxes in (H)–(K).
(P–S) FACS-based quantification of tdTomato+ and ACTA2+ cell populations in saline and bleomycin-treated lungs at 14 d.p.i.
Scale bars: (C)–(F), 250 μm; (H)–(K), 50 μm. a, airway; BLM, bleomycin; ns, not significant; SAL, saline; v, vessel. SAL d14, n = 4; BLM d14, n = 6; n represents biological replicates. Data are presented as mean values ± SEM. *p < 0.05, **p < 0.01.
Activated Myofibroblasts Persist in the Lung and Undergo a Phenotypic Switch after Fibrosis Resolution
In order to validate the idea that activated myofibroblasts can be lineage-traced during bleomycin-induced pulmonary fibrosis, Acta2-Cre-ERT2; tdTomatoflox mice were treated with bleomycin and then fed tamoxifen-containing pellets between 5 and 14 d.p.i. (Figure 2A). This experimental setup allowed labeling of activated myofibroblasts as demonstrated by co-localization of the ACTA2 stain and the tdTomato lineage label in the remodeled parenchyma of fibrotic lungs (Figures 2B–2E). Quantification of the immunofluorescent staining showed that 79.2% of lineage-labeled cells were clearly ACTA2+ at 14 d.p.i. (Figure 2K). It has to be mentioned that the main challenge when doing such quantification is that ACTA2 is not uniformly expressed in fibrotic regions and it is difficult to detect cell boundaries and co-localize the lineage label (nuclear and cytoplasmic) with the ACTA2 stain (cytoskeletal). In order for us to better discern this co-localization, FACS-based quantification was carried out and the results showed that the labeling efficiency of SMCs and activated myofibroblasts at 14 d.p.i. was 93.2% (Figure 2S). The same approach determined that 92.2% of lineage-labeled cells were ACTA2+ at 14 d.p.i. (Figure 2R) (compared to 79.2% based on quantification of immunofluorescence).
Figure 2. Activated Myofibroblasts Lose Their Myogenic Phenotype following the Resolution of Fibrosis.
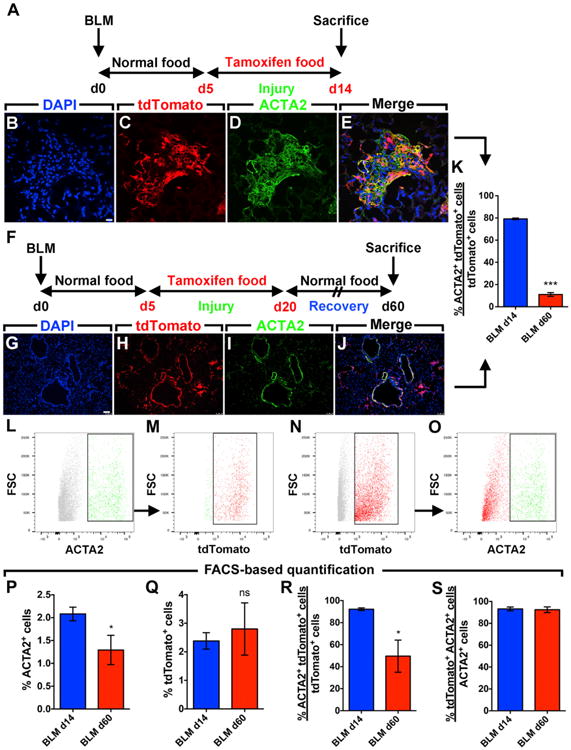
(A) Timeline of bleomycin and tamoxifen treatments. Mice were treated with bleomycin and then fed tamoxifen-containing pellets between 5 and 14 d.p.i. Lungs were harvested at 14 d.p.i.
(B–E) Immunofluorescent staining showing DAPI, tdTomato, and ACTA2 single channels in addition to a merged image.
(F) Timeline of tamoxifen and bleomycin treatments. Mice were treated with bleomycin and then fed tamoxifen-containing pellets between 5 and 20 d.p.i. Lungs were harvested at 60 d.p.i.
(G–J) Immunofluorescent staining showing DAPI, tdTomato, and ACTA2 single channels in addition to a merged image.
(K) Quantification of the immunofluorescence shown in (B)–(E) and (G)–(J).
(L–O) Gating strategy for FACS-based detection of ACTA2+ and tdTomato+ cell populations.
(P–S) FACS-based quantification showing the change in the number of cells expressing ACTA2 and/or tdTomato.
Scale bars: (B)–(E), 10 μm; (G)–(J), 75 μm. FSC, forward scatter. BLM d14, n = 6–8; BLM d60, n = 3; n represents biological replicates. Data are presented as mean values ± SEM. *p < 0.05, ***p < 0.001.
The fate of activated myofibroblasts that appeared at 14 d.p.i. was investigated after fibrosis resolution (Figure 2F). The 60 d.p.i. time point was chosen to ensure that lung fibrosis was adequately resolved. Fibrosis resolution was confirmed by lung function measurement and H&E staining (Figure S2). Interestingly, activated myofibroblast descendants (lineage-labeled cells) were observed in the lung following the resolution of fibrosis. Quantification of the immunofluorescent staining showed that 11% of these cells were ACTA2+ (Figures 2G–2K). Our histological observations were supported by FACS-based quantification (Figures 2L–2O) showing a decrease in the number of ACTA2+ cells at 60 d.p.i. compared to 14 d.p.i. (2% versus 1.2%) (Figure 2P). The total number of tdTomato+ cells was unchanged between the two time points (2.3%–2.8%) (Figure 2Q), indicating that these cells are not cleared following the resolution phase. However, whereas 92.2% of lineage-labeled cells were ACTA2+ at 14 d.p.i., only 49.6% of lineage-labeled cells were ACTA2+ at 60 d.p.i (Figure 2R), indicating that myofibroblast descendants lose Acta2 expression significantly. The majority of ACTA2+ cells at both 14 d.p.i. (SMCs and activated myofibroblasts) and 60 d.p.i. (SMCs and activated myofibroblast descendants) were lineage-labeled (93.2% and 92.5%, respectively) (Figure 2S), indicating that our lineage-tracing approach allowed the capture of the majority of ACTA2+ cells.
In order to test the hypothesis that activated myofibroblasts transition to a lipofibroblast-like phenotype following fibrosis resolution, immunofluorescence for the lipofibroblast marker adipose differentiation-related protein (ADRP) and the AEC2 marker (SFTPC) was carried out. ADRP+ lineage-labeled cells, adjacent to SFTPC+ cells, were identified at 60 d.p.i. (Figures 3A–3D). Quantification of the immunofluorescence showed that out of the total number of tdTomato+ cells, 30.5% were ADRP+ at 60 d.p.i. compared to 15.8% at 14 d.p.i. (Figure 3E). Collagen staining revealed the absence of collagen at 60 d.p.i. compared to 14 d.p.i. (Figures 3F and 3G). Additionally, activated myofibroblasts (at 14 d.p.i.) and activated myofibroblast-derived cells (at 60 d.p.i.) were isolated by FACS and gene expression was analyzed by qPCR. The results showed a clear 4.7-fold downregulation of Acta2 (Figure 3H) and a significant 4.1-fold upregulation of Adrp (Figure 3I), thus confirming the histological findings. A trend for a 1.25-fold increase in Pparg expression (master regulator of lipogenesis and a key player in lipofibroblast formation) and a 1.75-fold increase in Fgf10 expression was also observed (Figures 3J and 3K). In order to support these findings, FACS-based quantification of the neutral lipid stain (LipidTOX) was carried out (Figures 3L–3N). The results clearly showed an increase in the number of LipidTOX+ cells at 60 d.p.i. (7.7%) compared to 14 d.p.i. (3.2%) (Figure 3O). The proportion of lineage-labeled cells that stained for LipidTOX also increased at 60 d.p.i. (13.3%) compared to 14 d.p.i. (6.2%) (Figure 3P).
Figure 3. Activated Myofibroblasts Transition to a Lipofibroblast-like Phenotype after Fibrosis Resolution.
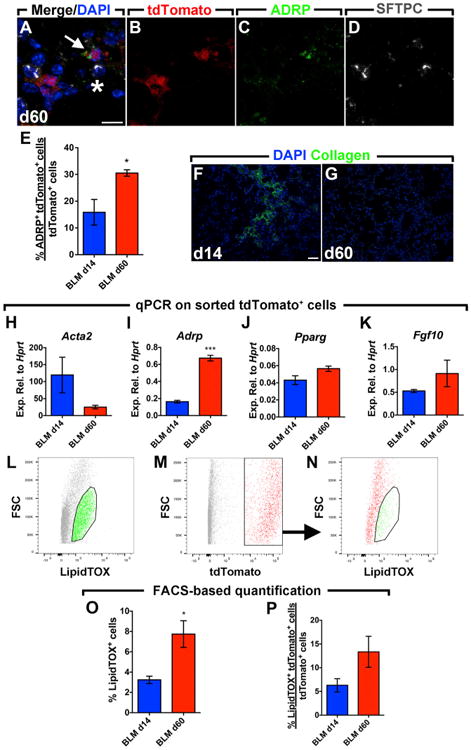
(A–D) Immunofluorescent staining showing a merged image with DAPI in addition to tdTomato, ADRP, and SFTPC single channels. The arrow indicates an ADRP+ tdTomato+ cell adjacent to an SFTPC+ cell (asterisk).
(E) Quantification of the immunofluorescence showing gain of ADRP expression in lineage-labeled cells at 60 d.p.i. compared to 14 d.p.i.
(F and G) Immunofluorescent staining for collagen type 1 at 14 and 60 d.p.i.
(H–K) qPCR for Acta2, Adrp, Pparg, and Fgf10 on lineage-labeled cells sorted from bleomycin-treated lungs at 14 and 60 d.p.i.
(L–N) Gating strategy for the detection of LipidTOX+ and tdTomato+ cell populations by FACS.
(O and P) FACS-based quantification of LipidTOX+ and tdTomato+ cell populations in lung suspensions at 14 and 60 d.p.i.
Scale bars: (A)–(D), 10 μm; (F) and (G), 50 μm. BLM d14, n = 3–4; BLM d60, n = 2–4; n represents biological replicates. Data are presented as mean values ± SEM. *p < 0.05, ***p < 0.001.
Immunofluorescent staining of lung sections revealed that only ∼1.5% of airway and vascular SMCs were ADRP+ in saline-treated lungs (data not shown), indicating that under homeostatic conditions, SMCs and lipofibroblasts are distinct mesenchymal lineages. At 14 d.p.i., a considerable proportion of lineage-labeled activated myofibroblasts were ADRP+ and LipidTOX+, indicating that lipofibroblasts might contribute to the accumulation of activated myofibroblasts during the course of fibrosis.
Lipofibroblasts Are a Source of Activated Myofibroblasts in Lung Fibrosis
To test whether lipofibroblasts contribute to the activated myofibroblast pool during fibrosis formation, a recently generated AdrpCre-ERT2 knock-in mouse line was used to label pre-existing lipofibroblasts before bleomycin injury (Figure 4A). AdrpCre-ERT2; mT/mG mice were fed tamoxifen-containing pellets for 2 weeks followed by 2 weeks of normal pellets before being challenged with either saline or bleomycin (Figure 4B). Lung fibrosis was confirmed by lung function measurement and H&E staining (data not shown). Immunofluorescent staining showed minimal co-localization of the lineage label (mGFP) and ACTA2 in saline-treated lungs at 14 d.p.i. (Figures 4C–4G). In contrast, mGFP+ cells were located in dense fibrotic areas where ACTA2+ myofibroblasts were also aggregated in bleomycin-treated lungs (Figures 4H–4L). In order to quantify the contribution of lipofibroblasts to activated myofibroblast formation, FACS analysis was used (Figures 4M–4O) and any hematopoietic, endothelial, or epithelial cells that were collaterally labeled were excluded from the analysis in order to enhance the specificity of the measurements and restrict the analysis to resident fibroblasts expressing ADRP. The results showed that our lineage-tracing tool labeled 27.2%–34.8% of LipidTOX+ fibroblasts in the lung (Figure 4V) and there was a dramatic decrease in the number of LipidTOX+ cells after bleomycin injury (from 9.4% to 1.5%) (Figure 4T), hinting at a lipofibroblast-to-activated-myofibroblast transdifferentiation. An increase in the number of ACTA2+ cells from 2.3% to 5.7% was observed upon bleomycin injury (Figure 4P), concomitantly with an increase in the number of mGFP+ cells that co-stained for ACTA2 (Figure 4R) and a decrease in the number of mGFP+ that co-stained for LipidTOX (Figure 4U). Interestingly, while the number of lineage-labeled cells remained unchanged (Figure 4Q), these cells contributed to 19.7% of the total ACTA2+ cells in bleomycin-treated lungs (Figure 4S).
Figure 4. Lipofibroblasts Give Rise to Activated Myofibroblasts during Fibrosis Formation.
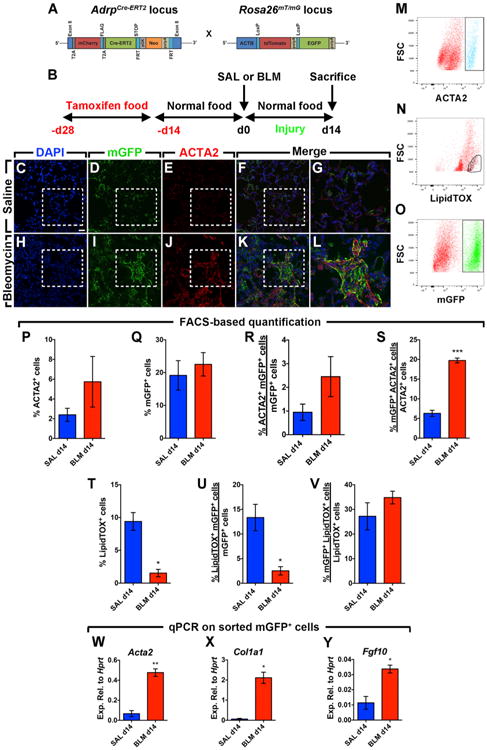
(A) Schematic representation of the AdrpCre-ERT2 and mT/mG constructs.
(B) Timeline of tamoxifen and saline or bleomycin treatments. Mice were fed tamoxifen-containing pellets before saline or bleomycin was administered intratracheally. Lungs were harvested at 14 d.p.i.
(C–F) Immunofluorescent staining of saline-treated lungs showing DAPI, mGFP, and ACTA2 single channels in addition to a merged image.
(G) A high-magnification image of the region marked by the box in the merged image (F).
(H–K) Immunofluorescent staining of bleomycin-treated lungs showing DAPI, mGFP, and ACTA2 single channels in addition to a merged image.
(L) A high-magnification image of the region marked by the box in the merged image (K).
(M–O) Gating strategy for the detection of ACTA2+, LipidTOX+, and mGFP+ cell populations by FACS.
(P–V) FACS-based quantification of ACTA2+, mGFP+, and LipidTOX+ cell populations at 14 d.p.i.
(W–Y) qPCR for Acta2, Col1a1, and Fgf10 on mGFP+ cells sorted from saline- and bleomycin-treated lungs at 14 d.p.i.
Scale bar: 25 μm. SAL d14, n = 3; BLM d14, n = 3–4; n represents biological replicates. Data are presented as mean values ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
mGFP+ cells were also sorted by FACS and gene expression was analyzed by qPCR. Activated myofibroblast markers (Acta2 and Col1a1) were drastically upregulated in lineage-labeled cells derived from fibrotic lungs compared to control lungs (7.8-fold and 35.1-fold, respectively) (Figures 4W and 4X), indicating that these cells acquired an activated myofibroblast phenotype. Fgf10 was also significantly upregulated (3-fold) in lineage-labeled cells upon bleomycin treatment (Figure 4Y).
FGF10+ Cells Contribute to Activated Myofibroblast Formation and Resolution
Since Fgf10 expression was elevated in both lipofibroblast-derived cells after bleomycin treatment (Figure 4Y) and activated myofibroblast-derived cells after fibrosis resolution (Figure 3K), we decided to use Fgf10-lacZ mice (enhancer trap Fgf10 reporter line) to monitor the status of FGF10+ cells during the peak (14 d.p.i.) and early resolution of lung fibrosis (28 d.p.i.) (Figures 5A and 5B). Immunostaining for lacZ and ACTA2 revealed that a proportion of activated myofibroblasts expressed Fgf10 at 14 d.p.i. (Figure 5C). FACS analysis revealed that 30%–53.9% of total ACTA2+ cells were FGF10+ at 14 and 28 d.p.i. (Figure 5H) and confirmed the beginning of the resolution phase as the number of ACTA2+ cells declined from 3.4% at 14 d.p.i. to 1.4% at 28 d.p.i. (Figure 5E). FDG staining showed that the number of FGF10+ cells increased from 2.3% to 7.5% (Figure 5F), concomitantly with an increase in LipidTOX+ cells from 1.5% to 3.8% (Figure 5I). Interestingly, whereas 43.9% of FGF10+ cells expressed ACTA2 at 14 d.p.i., 12.5% of these cells expressed ACTA2+ at 28 d.p.i. (Figure 5G). This was accompanied by an increase in lipid-droplet accumulation in FGF10+ cells (18% versus 1.8%) (Figure 5J). Finally, FDG+ cells represented 37% of all LipidTOX+ cells at 28 d.p.i. (Figure 5K).
Figure 5. Fgf10 Expression Marks Activated Myofibroblast Formation and Resolution.
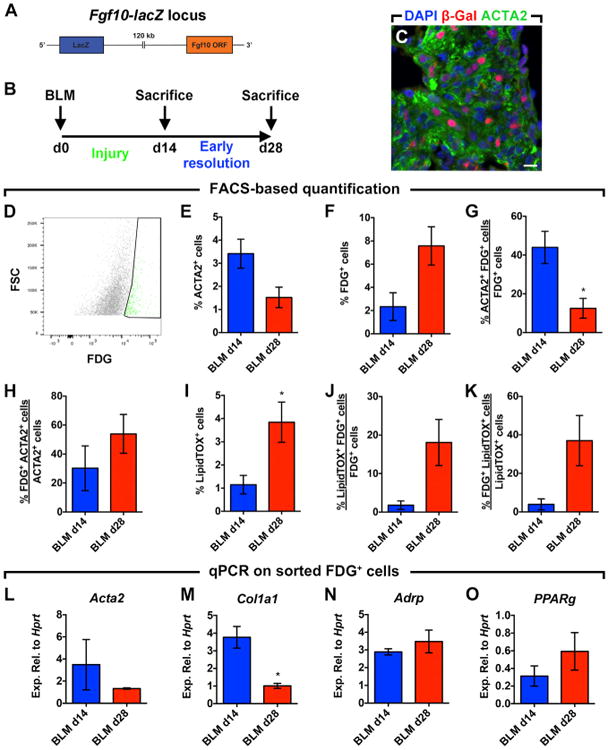
(A) Schematic representation of the Fgf10-lacZ construct.
(B) Timeline for bleomycin treatment. Lungs were harvested at 14 or 28 d.p.i.
(C) Immunofluorescent staining showing β-Gal+ cells in fibrotic (ACTA2+) areas of bleomycin-treated lungs at 14 d.p.i.
(D) Gating strategy for the detection of the fluorescent lacZ substrate (FDG) by FACS.
(E–K) FACS-based quantification of ACTA2+, FDG+, and LipidTOX+ cell populations at 14 and 28 d.p.i.
(L–O) qPCR for Acta2, Col1a1, Adrp, and Pparg on FDG+ cells sorted from bleomycin-treated lungs at 14 and 28 d.p.i.
Scale bar: 10 μm. BLM d14, n = 3; BLM d28, n = 3–4; n represents biological replicates. Data are presented as mean values ± SEM.*p < 0.05.
qPCR on sorted FDG+ cells showed a 2.6-fold and 3.5-fold downregulation of activated myofibroblast markers Acta2 and Col1a1 at 28 d.p.i. compared to 14 d.p.i., respectively (Figures 5L and 5M). This was accompanied by a 1.2-fold and 1.9-fold upregulation of lipofibroblast markers Adrp and Pparg, respectively (Figures 5N and 5O).
Elevated FGF10 and Reduced Lipofibroblast Marker Expression in End-Stage IPF Lungs
In order to validate our findings in the human context, LipidTOX staining was performed on frozen lung tissue samples from donors and IPF patients. The staining clearly revealed the presence of lipid-droplet-containing cells adjacent to AEC2 in both groups (Figures 6A and 6B). These cells did not stain for the hematopoietic cell marker CD45, suggesting that they are indeed resident cells in the human lung (Figure 6C). In order to determine whether the involvement of the lipofibroblast-activated myofibroblast axis observed in the mouse model of lung fibrosis is relevant to the human condition, gene expression analysis was carried out using lung tissues from non-IPF donors and end-stage IPF patients. The fibrotic nature of IPF samples was evident based on elevated ACTA2 and COL1A1 expression levels compared to donors (8.8-fold and 4-fold, respectively) (Figures 6D and 6E). Lipofibroblast differentiation markers ADRP, C/EBPa, and PPARg were significantly downregulated (1.8-fold, 1.5-fold, and 1.9-fold downregulation, respectively) (Figures 6F–6H). These results indicate the loss of lipofibroblasts and accumulation of activated myofibroblasts in IPF lungs in comparison to donor controls.
Figure 6. Human IPF Lungs Show Decreased Lipofibroblast Marker Expression and Increased FGF10 Expression.
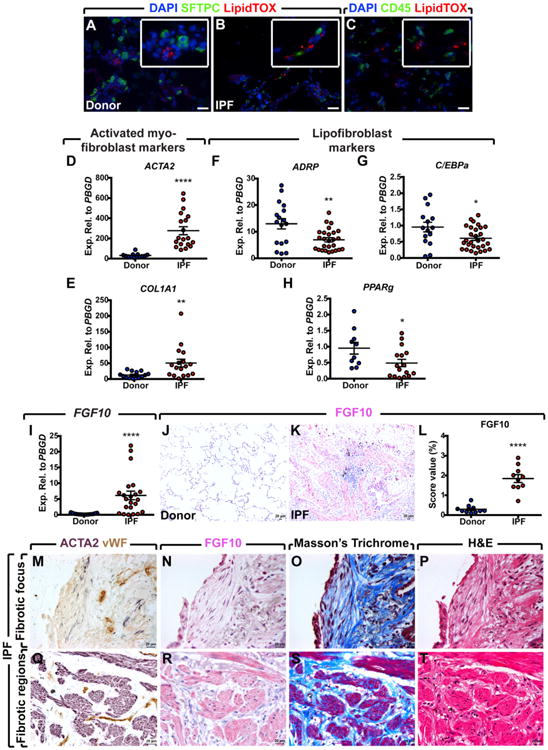
(A and B) LipidTOX staining of frozen lung tissue samples showing the presence of lipid-droplet-containing cells in close proximity to AEC2 in both donors and IPF patients.
(C) Double staining for CD45 and LipidTOX showing that LipidTOX+ cells are CD45−.
(D–H) qPCR analysis on human lung homogenates showing significant upregulation of myofibroblast markers ACTA2 and COL1A, and significant downregulation of lipofibroblast differentiation markers ADRP, C/EBPa, and PPARg in IPF lungs compared to donor lungs.
(I) qPCR analysis showing a significant increase in FGF10 expression in IPF lungs compared to donor lungs.
(J and K) Immunohistochemical staining for FGF10 showing increased expression levels in IPF lungs compared to donor lungs.
(L) Quantification of FGF10 immunoreactivity shown in (J) and (K).
(M–P) Serial sections of a fibrotic focus stained with anti-ACTA2, anti-vWF, and anti-FGF10 antibodies in addition to Masson's trichrome and H&E stains. A similar staining is shown for dense fibrotic regions.
(Q–T) Weaker FGF10 immunoreactivity is observed in the fibrotic focus compared to fibrotic regions.
Scale bars: 20 μm. Donors: n = 12–17; IPF: n = 21–29 (D–H). n = 10 per group (J–L), n represents biological replicates. Data are presented as mean values ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001.
Since Fgf10 was upregulated in lipofibroblast-derived activated myofibroblasts (Figure 4Y) and FGF10+ cells contributed to the activated myofibroblast pool in the mouse model of lung fibrosis (Figure 5), we decided to investigate the expression levels of FGF10 in IPF versus non-IPF donor lung tissue samples by qPCR. The results showed substantial upregulation of FGF10 in IPF lungs compared to donor lungs at both the mRNA (22.7-fold) (Figure 6I) and protein (6.3-fold) levels (Figures 6J–6L). Immunohistochemical staining of paraffin-embedded IPF lung tissues showed FGF10 immunoreactivity in dense fibrotic islands where ACTA2+ cells were accumulated (Figures 6Q–6T). Interestingly, FGF10 was detected at lower levels in fibrotic foci (Figures 6M–6P).
Activation of PPARγ Signaling Antagonizes TGFβ1-Mediated Fibrogenic Response
In order to investigate whether PPARγ activation antagonizes TGFβ1 activity in IPF by reinforcing the lipogenic phenotype, human lung fibroblasts were cultured in the presence of the PPARγ agonist rosiglitazone (20 μM) and/or recombinant TGFβ1 (1 ng/mL), and cells were harvested after 72 hr for qPCR analysis (Figure 7). TGFβ1 treatment strongly inhibited PPARg (Figure 7A) and ADRP expression (Figure 7B) (18.6-fold and 6.8-fold downregulation, respectively) and upregulated ACTA2 (Figure 7C) and COL1A1 expression (Figure 7D) (7-fold and 9.5-fold, respectively) compared to the control group. Interestingly, rosiglitazone treatment significantly upregulated ADRP expression (3.3-fold compared to the control group) and attenuated its downregulation by TGFβ1 (Figure 7B) (with 2.9-fold upregulation in the TGFβ1+Rosi group compared to the TGFβ1-treated group). In parallel to the inhibition of TGFβ1-mediated downregulation of lipogenic markers, rosiglitazone treatment significantly attenuated TGFβ1-mediated upregulation of myogenic markers ACTA2 and COL1A1 (Figures 7C and 7D) (with 2.2-fold and 1.25-fold downregulation, respectively, in the TGFβ1+Rosi group compared to the TGFβ1-treated group).
Figure 7. Rosiglitazone Reinforces the Lipogenic Phenotype in Human Lung Fibroblasts and Attenuates TGFβ1-Mediated Fibrogenesis.
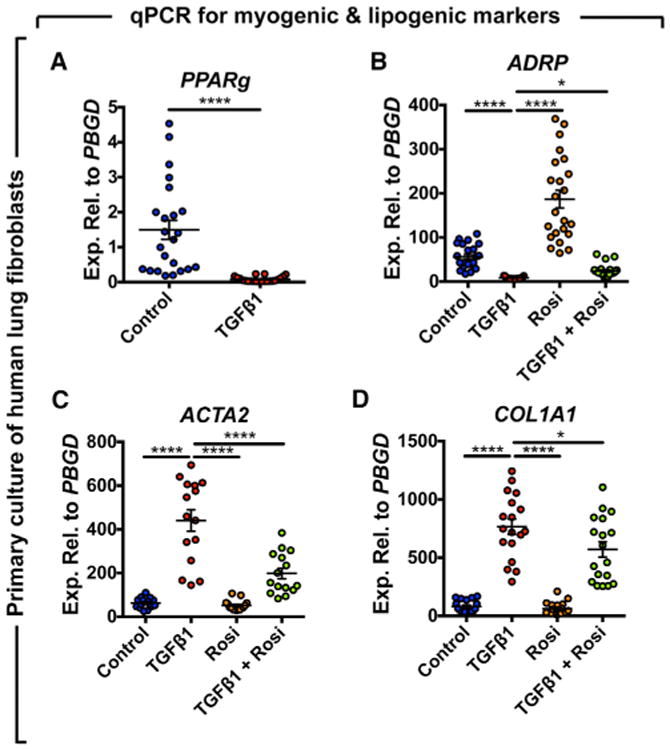
Cells were starved for 24 hr before being treated with 1 ng/mL recombinant TGFβ1 and/or 20 μM rosiglitazone. After 72 hr, cells were harvested and gene expression was analyzed by qPCR.
(A) TGFβ1 strongly inhibits PPARg expression.
(B) Rosiglitazone induces the expression of ADRP and attenuates TGFβ1-mediated downregulation.
(C and D) TGFβ1 significantly upregulates ACTA2 and COL1A1 and rosiglitazone attenuates this effect. Rosi, rosiglitazone.
Control, n = 18–23; TGFβ1, n = 15–22; Rosi, n = 15–23; TGFβ1+Rosi, n = 15–24; n represents biological replicates. Data are presented as mean values ± SEM. *p < 0.05, ****p < 0.0001.
Discussion
The accumulation of collagen-secreting activated myofibroblasts is a main feature in IPF. Since IPF patients are end stage at the time of diagnosis, the mechanisms involved in the initial trigger and the subsequent remodeling process in the lung parenchyma of these patients remain unclear. Mouse models of lung fibrosis have given hints about possible sources of activated myofibroblasts in IPF. In this study, we identify the resident lipofibroblast as a novel contributor to the activated myofibroblast pool in the pathogenesis of IPF.
Excessive vascular SMCs that accumulate in hypoxia-induced PH in mice originate from pre-existing SMCs (Sheikh et al., 2014). The contribution of pre-existing SMCs to lung fibrosis was investigated in this study and lineage-labeled cells were not detected in fibrotic areas. This indicates that activated myofibroblasts do not originate from pre-existing SMCs in lung fibrosis, but rather from ACTA2− progenitors.
The reversibility of the fibrotic response that is triggered after bleomycin administration was used to investigate the fate of activated myofibroblasts following fibrosis resolution. Our results show that activated myofibroblasts and their descendants are not cleared from the lung during fibrosis resolution, and a subset of these cells transitions to a lipofibroblast-like phenotype, thus supporting the recently proposed myofibroblast dedifferentiation model (Hecker et al., 2011). Our gene arrays on sorted tdTomato+ cells show 1.2-fold, 1.6-fold, and 1.3-fold downregulation of caspase 3 (Casp3), Casp6, and Casp7, respectively, at 60 d.p.i. compared to 14 d.p.i. TUNEL staining of lung sections did not reveal any significant apoptotic event in activated myofibroblast descendants at 60 d.p.i (Figures S3I–S3L).
Lipofibroblasts are adipocyte-like cells that assist AEC2 in surfactant production and support their clonogenic growth in vitro. Similarly to conventional adipocytes, lipofibroblasts express the lipid-droplet-trafficking protein ADRP and secrete leptin in addition to other adipokines (Schultz et al., 2002). In this study, we demonstrate that lipofibroblasts serve as a source of activated myofibroblasts in lung fibrosis. TGFβ has been shown to induce subcutaneous adipocyte-to-myofibroblast differentiation in skin fibrosis. WNT signaling, particularly through the WNT3a ligand, seems to mediate TGFβ-induced mesenchymal fibrogenic responses and loss of adipose tissue in skin fibrosis (Akhmetshina et al., 2012; Wei et al., 2012). Furthermore, the differentiation of adipose-derived stem cells to contractile SMCs has shown to be dependent on TGFβ1 and BMP4 (Wang et al., 2010). Under culture conditions permissive for adipogenesis, skeletal muscle is capable of transdifferentiating to mature adipocytes via PPAR activation and upregulation of lipogenic markers Pparg and C/ebpa (Hu et al., 1995). Our gene arrays indicate that TGFβ signaling is involved in pre-existing lipofibroblast-to-activated-myofibroblast transdifferentiation during fibrosis formation through the upregulation of Smad2 (1.8-fold), Smad3 (3.6-fold), Sp1 (2.6-fold), and c-Myc (1.9-fold) whereas PPARγ signaling is involved in the reversal of this event via the activation of genes involved in lipogenesis, cholesterol metabolism, and adipocyte differentiation (Figure S4). Pathway analysis also revealed the activation of survival pathways in lipofibroblast-derived activated myofibroblasts through the upregulation of Ciapin1 (1.9-fold) and Bcl2l1 (1.8-fold) and a 1.25-fold downregulation of genes involved in intrinsic stress signals such as Aifm1 and Endo-g. Gene arrays also showed a significant 2.5-fold upregulation of Ccnd1, indicating that lipofibroblast-derived cells are actively transitioning from G1 to S phase of the cell cycle at 14 d.p.i. Ccne1 was slightly upregulated (1.2-fold) while Ccnb1 was slightly downregulated (1.1-fold downregulation). We did not observe an upregulation of proliferation markers such as Ki67 (1.4-fold downregulation), E2f1 (2-fold downregulation), Top2a (2-fold downregulation), or Pcna (1.25-fold downregulation) in lipofibroblast-derived cells at 14 d.p.i., which confirmed the FACS data showing that the number of mGFP+ did not change after bleomycin treatment (Figure 4Q). Ki67 and TUNEL staining of lung sections from these mice did not show significant proliferation or apoptosis in these cells (Figures S3M–S3P and data not shown). Interestingly, we also show that activated myofibroblasts do not proliferate at 14 d.p.i. (Figures S3A–S3H). These findings, in addition to the observation that lipofibroblast-derived cells lose their lipid content (demonstrated by LipidTOX staining), acquire Acta2 expression, and produce collagen at the peak of fibrosis, indicate that these cells undergo a transdifferentiation process during fibrosis formation.
One of the intriguing questions is whether pre-existing lipofibroblasts that transdifferentiate to activated myofibroblasts during fibrosis formation are the same cells that revert to a lipofibroblast-like phenotype following fibrosis resolution. The other possibility would be that the latter event is a general phenomenon of activated myofibroblast dedifferentiation after recovery. Unfortunately, due to the patchy and heterogeneous pattern of lung fibrosis, it cannot be concluded with certainty that either of these two scenarios is true. Yet, two extended time points corresponding to injury/early recovery (21 d.p.i.) and mid-recovery phases (42 d.p.i.) were investigated with AdrpCre-ERT2; mT/mG mice (Figure S5). In this experiment, lipofibroblasts were labeled between P7 and P9 and mice were challenged with saline or bleomycin at 9 weeks of age. FACS analysis showed that in bleomycin-treated lungs, lipofibroblast-derived cells transiently acquired ACTA2 expression (3.6-fold increase) and lost their lipid content (6.3-fold decrease) at 21 d.p.i. compared to the saline group, before losing ACTA2 expression (3.4-fold decrease) and regaining lipid content (20-fold increase) at 42 d.p.i. compared to 21 d.p.i. (Figures S5E and S5F). Interestingly, the number of mGFP+ cells tended to increase by 1.9-fold at 42 d.p.i. compared to 21 d.p.i. This indicates that lipofibroblast-derived activated myofibroblasts are not cleared during the resolution phase of lung fibrosis and suggests that pre-existing lipofibroblasts that transdifferentiate to activated myofibroblasts during fibrosis formation might be the same cells that revert to the lipofibroblast-like phenotype during the resolution phase. The data attained with the Fgf10-lacZ reporter line agree with our suggestion, as FGF10+ cells started to lose ACTA2 expression and acquire lipofibroblast characteristics during the early resolution phase.
PPARγ, the master switch of lipogenic differentiation in preadipocytes as well as mesenchymal stem cells, is also involved in lipofibroblast formation (Rehan and Torday, 2012) and PPARγ agonists have been shown to protect mice from developing fibrosis (Fang et al., 2012; Genovese et al., 2005). Furthermore, adiponectin, which is a direct transcriptional target for PPARγ, has shown a similar effect in primary culture of skin fibroblasts isolated from scleroderma patients (Fang et al., 2012). Here, we show that PPARγ signaling is inhibited in IPF, likely due to hyperactive TGFβ1 signaling. We also provide further mechanistic insights into the mode of action of rosiglitazone. We show that TGFβ1 represses the lipogenic program by inhibiting PPARg expression in favor of the activation of the myogenic program in primary human lung fibroblasts. Conversely, rosiglitazone reinforces the lipogenic phenotype and inhibits TGFβ1-mediated fibrogenesis. Thus, it is likely that in IPF, endogenous PPARγ signaling is unable to counteract TGFβ1 signaling without an exogenous stimulus. Our data emphasize the phenotypic plasticity of lung fibroblasts and the potential use of PPARγ agonists to induce lipogenic differentiation at the expense of TGFβ1-mediated myogenic differentiation.
A recent study has reported the absence of lipid-droplet-containing cells in the human lung (Tahedl et al., 2014) although a previous study clearly demonstrated the presence of Oil Red O+ cells in both infant and adult human lungs (Rehan et al., 2006). In this study, we used the neutral lipid fluorescent stain LipidTOX and we clearly demonstrate the presence of resident lipogenic cells that are located adjacent to AEC2 in human lungs. The discrepancy between the aforementioned findings might be related to the unsaturated fatty acid composition of lipid droplets in the human lung and the differences in detection methods (Ahlbrecht and McGowan, 2014).
The process of spontaneous myofibroblast dedifferentiation observed in the mouse model of lung fibrosis is yet to be investigated in human IPF lungs. However, the data attained with human lung fibroblasts indicate that this process can be induced through intervention with PPARγ agonists. Whether myofibroblasts are able to dedifferentiate to cell types other than lipofibroblasts is yet to be established. To date, there is a gap in the knowledge regarding cellular heterogeneity of human lung mesenchyme, apart from the classification of lung fibroblasts into lipogenic and myogenic populations. Thus, there is an urgent need to deploy emerging new technologies such as single-cell RNA-seq to assess mesenchymal heterogeneity in the human lung and identify new cell types based on unique surface markers and molecular signatures. Such an approach might uncover novel fibroblastic populations and open new avenues to cure, or at least attenuate, IPF.
FGF10 plays a critical role during branching morphogenesis in the developing lung but its role in postnatal homeostasis has not been fully explored. FGF10 is also involved in white adipose tissue formation by activating the PPARγ pathway (Asaki et al., 2004; Sakaue et al., 2002) and our group has shown that Fgf10 expression identifies a subset of lipofibroblast progenitors during embryonic development (El Agha et al., 2014). In this study, we show that FGF10+ cells contribute to the activated myofibroblast population in the mouse model of lung fibrosis and that they acquire lipofibroblast characteristics in the resolution phase. On the other hand, we also show that FGF10 expression levels are significantly increased in lung tissue samples from IPF patients compared to donors and that at the protein level, FGF10 was observed in the fibrotic lesions (areas of mature fibrosis) of IPF lungs, and to a lesser extent in fibrotic foci (areas undergoing active remodeling). Since fibrotic foci are the sites of active fibrosis, it is unlikely that FGF10 is involved in the initial fibrotic response in IPF lungs. Rather, the induction of FGF10 expression might represent an attempt to counteract pro-fibrotic cytokines, particularly TGFβ1. In support of this possibility, mice that overexpress Fgf10 in the lung are protected against bleomycin-induced pulmonary fibrosis (Gupte et al., 2009). Thus, additional studies are needed in order to understand the role of FGF10 in the pathogenesis of IPF.
In summary, our results indicate a phenotypic switch between lipogenic and myogenic fibroblast populations during fibrosis formation and resolution. Lipofibroblast-to-activated myofibroblast transdifferentiation is likely TGFβ1 dependent while activated myofibroblast-to-lipofibroblast dedifferentiation is likely PPARγ dependent. The use of PPARγ agonists, in order to support the lipogenic phenotype and prevent myogenic differentiation, might offer an alternative to pirfenidone and nintedanib in treating IPF patients.
Star★Methods
Contact for reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by, the corresponding author, Dr. Saverio Bellusci (saverio.bellusci@innere.med.uni-giessen.de).
Experimental Model and Subject Details
Animals and bleomycin treatment
All animals were housed in a specific-pathogen-free environment with free access to food and water. Acta2-Cre-ERT2 transgenic mice (Wendling et al., 2009) were kindly provided by Dr. Pierre Chambon. AdrpCre-ERT2 knock-in mice (knock in of Cre-ERT2 in Exon 8 of the endogenous Adrp locus) were generated at the Max Planck Institute in Bad Nauheim, Germany (A.N., unpublished data). Fgf10-lacZ mice (enhancer-trap Fgf10 reporter line) were described previously (Kelly et al., 2001). Tandem dimer Tomato (tdTomatoflox) and membrane-targeted tdTomato/membrane-targeted green fluorescent protein (mT/mG) Cre-reporter mice were purchased from the Jackson laboratory. Both male and female mice at 9 weeks of age were used for lineage-tracing experiments (Acta2-Cre-ERT2; tdTomatoflox and AdrpCre-ERT2; mT/mG mice). Female Fgf10-lacZ mice at 28 to 30 weeks of age were used to analyze FGF10+ cells. Tamoxifen-containing pellets (0.4 g tamoxifen per Kg of pellets) were purchased from Altromin, and intraperitoneal injections of tamoxifen (Sigma-Aldrich) were performed at a dose of 0.4 mg per animal.
Fgf10-lacZ mice received an intratracheal instillation of bleomycin (2.5 U/Kg of body weight) at JLU Giessen, Germany (protocol number 58/2012) and National Jewish Health (Denver, USA) according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institute of Health (IACUC number AS2774-05-16). Acta2-Cre-ERT2; tdTomatoflox mice received an intratracheal instillation of saline or bleomycin (3.5 U/Kg of body weight) at the Ludwig Boltzmann Institute in Graz, Austria (protocol number BMWFW-66.010/0043-WF/V/3b/2016) and National Jewish Health. AdrpCre-ERT2; mT/mG mice received an intratracheal instillation of saline or bleomycin (3.5 U/Kg of body weight) at the Max Planck Institute in Bad Nauheim, Germany (protocol number B2/354).
Human tissues
Human lung tissues and primary fibroblasts from non-IPF donors and IPF patients undergoing lung transplantation were obtained from the Giessen biobank. The study protocol (AZ31/93) was approved by the ethics committee of the University of Giessen that conforms to the principles outlined in the declaration of Helsinki. Tissues were used for RNA extraction, paraffin embedding followed by immunohistochemistry, or cryoembedding followed by LipidTOX staining (Thermo Fischer Scientific). Human lung fibroblasts were starved for 24 hr and then treated with 1 ng/mL recombinant TGFβ1 (R&D Systems) and/or 20 μM rosiglitazone (Sigma-Aldrich). Cells were harvested after 72 hr for RNA extraction and gene expression analysis.
Method Details
Lung function measurement
Mice were anaesthetized, tracheotomized and intubated with a catheter connected to a FlexiVent plethysmograph (Scireq). Lungs were mechanically ventilated at a rate of 150 breaths/min with a tidal volume of 10 μL/g of body weight and lung compliance was measured. After plethysmography, animals were euthanized and lungs were harvested for further processing.
Immunofluorescence and immunohistochemistry
Murine lungs were perfused with PBS and fixed in 4% paraformaldehyde according to standard procedures. Next, they were embedded in paraffin and sectioned at 5 μm thickness. Immunofluorescent staining was performed using monoclonal anti-ACTA2 (Sigma-Aldrich, 1:100), polyclonal anti-ADRP (Abcam, 1:50), polyclonal anti-β-Gal (Rockland Immunochemicals, 1:500), polyclonal anti-collagen type 1 (Rockland Immunochemicals, 1:200), monoclonal anti-Ki67 antibodies (Cell Signaling, 1:200) and polyclonal anti-SFTPC (Santa Cruz, 1:1000). The endogenous tdTomato signal was detected without the use of antibodies. When antigen retrieval was required for double staining, polyclonal anti-RFP antibodies (Rockland Immunochemicals Inc., 1:250) were used. For AdrpCre-ERT2; mT/mG lungs, antigen retrieval was performed using a pressure cooker in the presence of citrate buffer, resulting in the disappearance of endogenous fluorescent signals. The mGFP signal was then detected using polyclonal anti-GFP antibodies (Abcam, 1:1500). Apoptosis was detected using DeadEnd Fluorometric TUNEL assay according to manufacturer's instructions (Promega). Image acquisition was performed using an upgraded version of TCS SP5 × confocal microscope or DM5500 B fluorescence microscope (Leica).
Paraffin-embedded human lung tissues were subjected to immunohistochemistry as previously described (Kosanovic et al., 2015) using polyclonal anti-FGF10 (Antibodies-online, ABIN360398, 1:30), monoclonal anti-ACTA2 (Sigma-Aldrich, 1:900) and polyclonal anti-vWF antibodies (Dako, 1:900). Masson's trichrome and hematoxylin/eosin stainings were performed according to standard procedures. The quantification of the FGF10+ signal was performed using an algorithm that takes into account the intensity as well as the area of immunoreactivity. A special software (QWin) was obtained from Leica for the morphometry and results were expressed as score value (%).
Fluorescence-Activated Cell Sorting
Single-cell suspensions and fluorescence-activated cell sorting (FACS) were performed according to standard procedures. The endogenous tdTomato or mGFP signals were used to detect and sort lineage-labeled cell populations using FACSAria III cell sorter (BD Biosciences). FITC-conjugated monoclonal anti-ACTA2 (Sigma-Aldrich, 1:100) and Alexa Fluor 405-conjugated monoclonal anti-ACTA2 antibodies (Novus Biologicals, 1:100) were used to detect ACTA2+ cells. LipidTOX stain (1:200) was used to detect neutral lipid-containing cells (Thermo Fischer Scientific) and gating was set according to FMO controls. FACS-based quantification of the lacZ signal was carried out using the FluoReporter lacZ flow cytometry kit that utilizes Fluorescein Di-β-D-Galactopyranoside (FDG) as a substrate for lacZ (Thermo Fischer Scientific). Anti-CD45 (1:100), anti-CD31 (1:100) and anti-CD326 antibodies (1:50) were purchased from Biolegend. FACS data were analyzed using FlowJo software (FlowJo LLC).
RNA extraction and gene expression analysis
RNA extraction from sorted murine lung cells, primary human lung fibroblasts and human lung homogenates was done using RNeasy kit (QIAGEN) and quantitative real-time PCR (qPCR) analysis was performed using LightCycler 480 II machine (Roche Applied Science). Primers were designed using the Universal ProbeLibrary Assay Design Center online tool from Roche (available at: https://lifescience.roche.com/webapp/wcs/stores/servlet/CategoryDisplay?tab=Assay+Design+Center&identifier=Universal+Probe+Library&langld=-1). Data were presented as expression relative to hypoxanthine-guanine phosphoribosyltransferase (Hprt) for mouse genes or porphobilinogen deaminase (PBGD) for human genes ± standard error of mean (SEM).
Quantification and Statistical Analysis
For quantification of immunofluorescence, cells were counted in 10 independent 63× fields per sample. GraphPad Prism 6 software (GraphPad Software) was used to analyze and assemble quantitative data. Outliers were identified using the ROUT method. Student's t test (unpaired, two-tailed) was used to compare the means of two groups and one-way ANOVA was used to compare the means of multiple groups. The ‘n’ for each experiment can be found in the figure legends. Data were presented as mean values ± SEM, and results were considered statistically significant if p < 0.05.
Supplementary Material
Supplemental Figure S1, related to Figure 1, Validation of bleomycin-induced lung fibrosis in Acta2-Cre-ERT2; tdTomatoflox mice at 14 d.p.i. (A) H&E stain of a saline-treated lung at 14 d.p.i. A higher magnification is shown in (B). (C) H&E stain of a bleomycin-treated lung at 14 d.p.i. A higher magnification is shown in (D). (E) Lung function measurement showing decreased compliance in bleomycin-treated lungs compared to controls. Scale bars: (A,C) 2 mm, (B,D) 200 μm. SAL d14 n=4, BLM d14 n=7, ‘n’ represents biological replicates. ** P<0.01.
Supplemental Figure S2, related to Figures 2 and 3, Validation of fibrosis resolution in Acta2-Cre-ERT2; tdTomatoflox mice at 60 d.p.i. (A) H&E stain of a saline-treated lung at 60 d.p.i. A higher magnification is shown in (B). (C) H&E stain of a bleomycin-treated lung at 60 d.p.i. A higher magnification is shown in (D). (E) Lung function measurement showing no difference in compliance in bleomycin-treated lungs compared to controls. Scale bars: (A,C) 2 mm, (B,D) 200 μm. n=3 per group, ‘n’ represents biological replicates.
Supplemental Figure S3, related to Figures 2, 3 and 4, Analysis of proliferation and apoptosis in lineage-labeled cells at the peak of fibrosis and during the resolution phase. (A-D) Immunofluorescent staining of bleomycin-treated Acta2-Cre-ERT2; tdTomatoflox lungs at 14 d.p.i. showing DAPI, tdTomato and Ki67 single channels in addition to a merged image. High magnification images of the regions marked by the boxes are shown in (E-H). White arrows mark proliferating cells. Note the absence of co-localization between the lineage label and Ki67 stain. (I-L) TUNEL staining of Acta2-Cre-ERT2; tdTomatoflox lungs at 60 d.p.i. showing the absence of apoptosis in lineage-labeled cells. White arrows mark apoptotic cells. (M-P) Immunofluorescent staining of bleomycin-treated AdrpCre-ERT2; mT/mG lungs at 14 d.p.i. showing DAPI, mGFP and Ki67 single channels in addition to a merged image. White arrows mark proliferating cells. Note the absence of co-localization between the lineage label and Ki67 stain. Scale bars: (A-D) 50 μm, (E-P) 25 μm. (A-H) n=3, (I-L) n=2, (M-P) n=3, ‘n’ represents biological replicates.
Supplemental Figure S4, related to Figures 2, 3 and 4, Signaling pathway analysis on lineage-labeled cells isolated from AdrpCre-ERT2; mT/mG and Acta2-Cre-ERT2; tdTomatoflox mice during fibrosis formation and resolution. (A) Analysis of gene arrays performed on sorted mGFP+ cells showing activation of the TGFβ signaling pathway in lipofibroblast-derived cells during fibrosis formation. (B) Analysis of gene arrays performed on sorted tdTomato+ cells showing activation of the PPARγ signaling pathway in activated myofibroblast descendants following fibrosis resolution. (A) n=3 per group, (B) BLM d14 n=3, BLM d60 n=2, ‘n’ represents biological replicates.
Supplemental Figure S5, related to Figure 4, Lipofibroblasts are maintained, regain their lipid content and lose ACTA2 expression during the resolution phase of lung fibrosis. (A) Pre-existing lipofibroblasts were labeled by three intraperitoneal injections of tamoxifen at P7, 8 and 9, before animals were challenged with saline or bleomycin at P63. (B-F) FACS-based quantification shows a transient increase in the number of ACTA2+ cells (B) and a transient loss of LipidTOX+ cells at 21 d.p.i. (D). mGFP+ cells are maintained at 21 d.p.i., and tend to be more abundant at 42 d.p.i. (C). (E,F) Lipofibroblast-derived cells acquire ACTA2 expression and lose their lipid content at 21 d.p.i. and then lose ACTA2 expression and reacquire lipid content at 42 d.p.i. IP: Intraperitoneal injection. SAL d14 n=3, BLM d21 n=3, BLM d42 n=4, ‘n’ represents biological replicates.
Supplemental Table S1, related to STAR Methods, Primers used in this study.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 405-conjugated anti-ACTA2 | Novus Biologicals | IC1420V |
| APC/Cy7-conjugated anti-CD326 | Biolegend | 118218; RRID: AB_2098648 |
| APC/Cy7-conjugated anti-CD45 | Biolegend | 103116; RRID: AB_312981 |
| FITC-conjugated anti-ACTA2 | Sigma-Aldrich | F3777; RRID: AB_476977 |
| PE/Cy7-conjugated anti-CD31 | Biolegend | 102418 |
| Purified anti-ACTA2 | Sigma-Aldrich | SAB1403519; RRID: AB_10737749 |
| Purified anti-ADRP | Abcam | ab52356 |
| Purified anti-β-Gal | Rockland Immunochemicals | 100-4136; RRID: AB_219904 |
| Purified anti-collagen type 1 | Rockland Immunochemicals | 600-401-103-0.5; RRID: AB_217595 |
| Purified anti-FGF10 | Antibodies-online | ABIN360398 |
| Purified anti-GFP | Abcam | ab13970; RRID: AB_300798 |
| Purified anti-Ki-67 | Cell Signaling Technology | 9449s |
| Purified anti-RFP | Abcam | ab62341; RRID: AB_945213 |
| Purified anti-SFTPC | Santa Cruz | sc-7706; RRID: AB_2185507 |
| Purified anti-vWF | Dako | A008202-2 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bleomycin | Hexal | 35913.00.00 |
| Recombinant human TGFβ1 protein | R&D Systems | 240-B |
| Rosiglitazone | Sigma-Aldrich | R2408 |
| Tamoxifen | Sigma-Aldrich | T5648 |
| Critical Commercial Assays | ||
| DeadEnd Fluorometric TUNEL assay | Promega | G3250 |
| FluoReporter lacZ flow cytometry kit | Thermo Fischer Scientific | F1930 |
| HCS LipidTOX Green neutral lipid stain | Thermo Fischer Scientific | H34475 |
| HCS LipidTOX Deep Red neutral lipid stain | Thermo Fischer Scientific | H34477 |
| Experimental Models: Cell Lines | ||
| Primary human lung fibroblasts | Giessen biobank | N/A |
| Experimental Models: Organisms/Strains | ||
| Acta2-Cre-ERT2 transgenic mouse strain | Dr. Pierre Chambon | MGI: 3831907 |
| AdrpCre-ERT2 knock-in mouse strain | A.N., unpublished data | MGI: 5755034 |
| Fgf10-lacZ enhancer-trap reporter mouse strain | Bellusci Lab | MGI: 3629660 |
| mT/mG Cre-reporter mouse strain | Jackson Laboratory | 007576 |
| tdTomatoflox Cre-reporter mouse strain | Jackson Laboratory | 007909 |
| Sequence-Based Reagents | ||
| Primers | See Table S1 | N/A |
| Software and Algorithms | ||
| FlowJo software | FlowJo LLC | N/A |
| GraphPad Prism 6 software | GraphPad Software | N/A |
| QWin software | Leica | N/A |
| Universal ProbeLibrary Assay Design Center online tool | Roche | Available online at: https://lifescience.roche.com/webapp/wcs/stores/servlet/CategoryDisplay?tab=Assay+Design+Center&identifier=Universal+Probe+Library&langId=-1 |
Highlights.
Fate mapping was used to investigate the origin and fate of activated myofibroblasts
Lipofibroblasts are precursors for activated myofibroblasts in lung fibrosis
Activated myofibroblasts dedifferentiate to lipofibroblasts after recovery
PPARγ activation inhibits lipofibroblast-to-myofibroblast transdifferentiation
Acknowledgments
We would like to thank Prof. Thomas Braun, Dr. Martin Szibor, Dr. Robert Voswinckel, and Dr. Friederike Klein for their contribution in generating and validating the AdrpCre-ERT2 mice. We also thank Dr. Athanasios Fysikopoulos for his help with confocal microscopy. We also acknowledge Dr. Jochen Wilhelm for his help with the gene arrays and Stefanie Hezel, Ewa Bieniek, Stephanie Viehmann, Kerstin Goth, and Jana Rostkovius for their technical support. E.E.A. acknowledges the support of the Excellence Cluster Cardio-Pulmonary System (ECCPS) and the Universitätsklinikum Giessen und Marburg (UKGM). S.B. was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, BE4443/4-1, BE4443/6-1, and CRC1213), Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz (LOEWE), UKGM, the Universities of Giessen and Marburg Lung Center (UGMLC), the German Center for Lung Research (DZL), COST (BM1201) and the NIH/NHLBI (1R01HL086322-01A2 and HL107307). J.Q. and S.H. acknowledge the support of the DZL, ECCPS, UGMLC, DFG (SFB1021 C05 and SFB TR84 B2). S.B. and S.H. acknowledge the support of the UKGM (FOKOOPV). S.D.L. acknowledges the support of NIH/NHLBI (R01HL126732, R01HL132156), March of Dimes (1-FY15-352), and the pulmonary fibrosis foundation (The Albert Rose established investigator award).
Footnotes
Supplemental Information: Supplemental Information for this article includes five figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2016.10.004
Author Contributions: E.E.A., A.M., V.K., S.D.L., S.C., F.S., I.H., B.M., J.Q., and A.N. conducted the experiments. E.E.A., G.K., D.K., K.A., and S.B. analyzed the data. E.E.A., R.T.S., S.H., R.E.M., A.G., W.S., and S.B. designed the experiments. E.E.A. and S.B. wrote the manuscript.
References
- El Agha E, Herold S, Al Alam D, Quantius J, MacKenzie B, Carraro G, Moiseenko A, Chao CM, Minoo P, Seeger W, Bellusci S. Fgf10-positive cells represent a progenitor cell population during lung development and postnatally. Development. 2014;141:296–306. doi: 10.1242/dev.099747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlbrecht K, McGowan SE. In search of the elusive lipofibroblast in human lungs. Am J Physiol Lung Cell Mol Physiol. 2014;307:L605–L608. doi: 10.1152/ajplung.00230.2014. [DOI] [PubMed] [Google Scholar]
- Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaki T, Konishi M, Miyake A, Kato S, Tomizawa M, Itoh N. Roles of fibroblast growth factor 10 (Fgf10) in adipogenesis in vivo. Mol Cell Endocrinol. 2004;218:119–128. doi: 10.1016/j.mce.2003.12.017. [DOI] [PubMed] [Google Scholar]
- Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BLM. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124:4867–4878. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- Fang F, Liu L, Yang Y, Tamaki Z, Wei J, Marangoni RG, Bhattacharyya S, Summer RS, Ye B, Varga J. The adipokine adiponectin has potent anti-fibrotic effects mediated via adenosine monophosphate-activated protein kinase: novel target for fibrosis therapy. Arthritis Res Ther. 2012;14:R229. doi: 10.1186/ar4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese T, Cuzzocrea S, Di Paola R, Mazzon E, Mastruzzo C, Catalano P, Sortino M, Crimi N, Caputi AP, Thiemermann C, Vancheri C. Effect of rosiglitazone and 15-deoxy-Delta12,14-prosta-glandin J2 on bleomycin-induced lung injury. Eur Respir J. 2005;25:225–234. doi: 10.1183/09031936.05.00049704. [DOI] [PubMed] [Google Scholar]
- Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- Gu€nther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev. 2012;21:152–160. doi: 10.1183/09059180.00001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte VV, Ramasamy SK, Reddy R, Lee J, Weinreb PH, Violette SM, Guenther A, Warburton D, Driscoll B, Minoo P, Bellusci S. Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2009;180:424–436. doi: 10.1164/rccm.200811-1794OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker L, Jagirdar R, Jin T, Thannickal VJ. Reversible differentiation of myofibroblasts by MyoD. Exp Cell Res. 2011;317:1914–1921. doi: 10.1016/j.yexcr.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka J. Idiopathic pulmonary fibrosis: a historical review. CMAJ. 1987;137:1003–1005. [PMC free article] [PubMed] [Google Scholar]
- Hoyles RK, Derrett-Smith EC, Khan K, Shiwen X, Howat SL, Wells AU, Abraham DJ, Denton CP. An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor β receptor. Am J Respir Crit Care Med. 2011;183:249–261. doi: 10.1164/rccm.201002-0279OC. [DOI] [PubMed] [Google Scholar]
- Hu E, Tontonoz P, Spiegelman BM. Transdifferentiation of myoblasts by the adipogenic transcription factors PPAR gamma and C/EBP alpha. Proc Natl Acad Sci USA. 1995;92:9856–9860. doi: 10.1073/pnas.92.21.9856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa R, Williams E, Trim N, Kendall T, Arthur MJ, Reichen J, Benyon RC, Iredale JP. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut. 2001;48:548–557. doi: 10.1136/gut.48.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleaveland KR, Velikoff M, Yang J, Agarwal M, Rippe RA, Moore BB, Kim KK. Fibrocytes are not an essential source of type I collagen during lung fibrosis. J Immunol. 2014;193:5229–5239. doi: 10.4049/jimmunol.1400753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosanovic D, Luitel H, Dahal BK, Cornitescu T, Janssen W, Danser AHJ, Garrelds IM, De Mey JGR, Fazzi G, Schiffers P, et al. Chymase: a multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur Respir J. 2015;46:1084–1094. doi: 10.1183/09031936.00018215. [DOI] [PubMed] [Google Scholar]
- McQualter JL, McCarty RC, Van der Velden J, O'Donoghue RJJ, Asselin-Labat ML, Bozinovski S, Bertoncello I. TGF-β signaling in stromal cells acts upstream of FGF-10 to regulate epithelial stem cell growth in the adult lung. Stem Cell Res (Amst) 2013;11:1222–1233. doi: 10.1016/j.scr.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafii R, Juarez MM, Albertson TE, Chan AL. A review of current and novel therapies for idiopathic pulmonary fibrosis. J Thorac Dis. 2013;5:48–73. doi: 10.3978/j.issn.2072-1439.2012.12.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan V, Torday J. Hyperoxia augments pulmonary lipofibroblast-to-myofibroblast transdifferentiation. Cell Biochem Biophys. 2003;38:239–250. doi: 10.1385/cbb:38:3:239. [DOI] [PubMed] [Google Scholar]
- Rehan VK, Torday JS. PPARγ Signaling Mediates the Evolution, Development, Homeostasis, and Repair of the Lung. PPAR Res. 2012;2012:289867. doi: 10.1155/2012/289867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan VK, Torday JS. The lung alveolar lipofibroblast:an evolutionary strategy against neonatal hyperoxic lung injury. Antioxid Redox Signal. 2014;21:1893–1904. doi: 10.1089/ars.2013.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan VK, Wang Y, Sugano S, Romero S, Chen X, Santos J, Khazanchi A, Torday JS. Mechanism of nicotine-induced pulmonary fibroblast transdifferentiation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L667–L676. doi: 10.1152/ajplung.00358.2004. [DOI] [PubMed] [Google Scholar]
- Rehan VK, Sugano S, Wang Y, Santos J, Romero S, Dasgupta C, Keane MP, Stahlman MT, Torday JS. Evidence for the presence of lipofibroblasts in human lung. Exp Lung Res. 2006;32:379–393. doi: 10.1080/01902140600880257. [DOI] [PubMed] [Google Scholar]
- Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue H, Konishi M, Ogawa W, Asaki T, Mori T, Yamasaki M, Takata M, Ueno H, Kato S, Kasuga M, Itoh N. Requirement of fibro-blast growth factor 10 in development of white adipose tissue. Genes Dev. 2002;16:908–912. doi: 10.1101/gad.983202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz CJ, Torres E, Londos C, Torday JS. Role of adipocyte differentiation-related protein in surfactant phospholipid synthesis by type II cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L288–L296. doi: 10.1152/ajplung.00204.2001. [DOI] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. Fgf10 is essential for limb and lung formation. Nat Genet. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- Sheikh AQ, Lighthouse JK, Greif DM. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 2014;6:809–817. doi: 10.1016/j.celrep.2014.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahedl D, Wirkes A, Tschanz SA, Ochs M, Mühlfeld C. How common is the lipid body-containing interstitial cell in the mammalian lung? Am J Physiol Lung Cell Mol Physiol. 2014;307:L386–L394. doi: 10.1152/ajplung.00131.2014. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- Wang C, Yin S, Cen L, Liu Q, Liu W, Cao Y, Cui L. Differentiation of adipose-derived stem cells into contractile smooth muscle cells induced by transforming growth factor-beta1 and bone morphogenetic protein-4. Tissue Eng Part A. 2010;16:1201–1213. doi: 10.1089/ten.TEA.2009.0303. [DOI] [PubMed] [Google Scholar]
- Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, Gottardi CJ, Atit R, Whitfield ML, Varga J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012;64:2734–2745. doi: 10.1002/art.34424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendling O, Bornert JM, Chambon P, Metzger D. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis. 2009;47:14–18. doi: 10.1002/dvg.20448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1, related to Figure 1, Validation of bleomycin-induced lung fibrosis in Acta2-Cre-ERT2; tdTomatoflox mice at 14 d.p.i. (A) H&E stain of a saline-treated lung at 14 d.p.i. A higher magnification is shown in (B). (C) H&E stain of a bleomycin-treated lung at 14 d.p.i. A higher magnification is shown in (D). (E) Lung function measurement showing decreased compliance in bleomycin-treated lungs compared to controls. Scale bars: (A,C) 2 mm, (B,D) 200 μm. SAL d14 n=4, BLM d14 n=7, ‘n’ represents biological replicates. ** P<0.01.
Supplemental Figure S2, related to Figures 2 and 3, Validation of fibrosis resolution in Acta2-Cre-ERT2; tdTomatoflox mice at 60 d.p.i. (A) H&E stain of a saline-treated lung at 60 d.p.i. A higher magnification is shown in (B). (C) H&E stain of a bleomycin-treated lung at 60 d.p.i. A higher magnification is shown in (D). (E) Lung function measurement showing no difference in compliance in bleomycin-treated lungs compared to controls. Scale bars: (A,C) 2 mm, (B,D) 200 μm. n=3 per group, ‘n’ represents biological replicates.
Supplemental Figure S3, related to Figures 2, 3 and 4, Analysis of proliferation and apoptosis in lineage-labeled cells at the peak of fibrosis and during the resolution phase. (A-D) Immunofluorescent staining of bleomycin-treated Acta2-Cre-ERT2; tdTomatoflox lungs at 14 d.p.i. showing DAPI, tdTomato and Ki67 single channels in addition to a merged image. High magnification images of the regions marked by the boxes are shown in (E-H). White arrows mark proliferating cells. Note the absence of co-localization between the lineage label and Ki67 stain. (I-L) TUNEL staining of Acta2-Cre-ERT2; tdTomatoflox lungs at 60 d.p.i. showing the absence of apoptosis in lineage-labeled cells. White arrows mark apoptotic cells. (M-P) Immunofluorescent staining of bleomycin-treated AdrpCre-ERT2; mT/mG lungs at 14 d.p.i. showing DAPI, mGFP and Ki67 single channels in addition to a merged image. White arrows mark proliferating cells. Note the absence of co-localization between the lineage label and Ki67 stain. Scale bars: (A-D) 50 μm, (E-P) 25 μm. (A-H) n=3, (I-L) n=2, (M-P) n=3, ‘n’ represents biological replicates.
Supplemental Figure S4, related to Figures 2, 3 and 4, Signaling pathway analysis on lineage-labeled cells isolated from AdrpCre-ERT2; mT/mG and Acta2-Cre-ERT2; tdTomatoflox mice during fibrosis formation and resolution. (A) Analysis of gene arrays performed on sorted mGFP+ cells showing activation of the TGFβ signaling pathway in lipofibroblast-derived cells during fibrosis formation. (B) Analysis of gene arrays performed on sorted tdTomato+ cells showing activation of the PPARγ signaling pathway in activated myofibroblast descendants following fibrosis resolution. (A) n=3 per group, (B) BLM d14 n=3, BLM d60 n=2, ‘n’ represents biological replicates.
Supplemental Figure S5, related to Figure 4, Lipofibroblasts are maintained, regain their lipid content and lose ACTA2 expression during the resolution phase of lung fibrosis. (A) Pre-existing lipofibroblasts were labeled by three intraperitoneal injections of tamoxifen at P7, 8 and 9, before animals were challenged with saline or bleomycin at P63. (B-F) FACS-based quantification shows a transient increase in the number of ACTA2+ cells (B) and a transient loss of LipidTOX+ cells at 21 d.p.i. (D). mGFP+ cells are maintained at 21 d.p.i., and tend to be more abundant at 42 d.p.i. (C). (E,F) Lipofibroblast-derived cells acquire ACTA2 expression and lose their lipid content at 21 d.p.i. and then lose ACTA2 expression and reacquire lipid content at 42 d.p.i. IP: Intraperitoneal injection. SAL d14 n=3, BLM d21 n=3, BLM d42 n=4, ‘n’ represents biological replicates.
Supplemental Table S1, related to STAR Methods, Primers used in this study.